

Abstract
The nature of the regulatory cell types that dominate in any given tumor is not understood at present. Here we addressed this question for Tregs and type II NKT cells in syngeneic models of colorectal and renal cancer. In mice with both type I and type II NKT cells, or in mice with neither type of NKT cell, Treg depletion was sufficient to protect against tumor outgrowth. Surprisingly, in mice lacking only type I NKT cells, Treg blockade was insufficient for protection. Thus, we hypothesized that type II NKT cells may be neutralized by type I NKT cells, leaving Treg cells as the primary suppressor, whereas in mice lacking type I NKT cells, unopposed type II NKT cells could suppress tumor immunity even when Tregs were blocked. We confirmed this hypothesis in three ways by reconstituting type I NKT cells as well as selectively blocking or activating type II NKT cells with antibody or the agonist sulfatide, respectively. In this manner, we demonstrated that blockade of both type II NKT cells and Tregs is necessary to abrogate suppression of tumor immunity, but a third cell, the type I NKT cell, determines the balance between these regulatory mechanisms. As cancer patients often have deficient type I NKT cell function, managing this delicate balance among three T cell subsets may be critical for the success of immunotherapy of human cancer.
Keywords: Tregs, NKT cells, cancer, immune regulation
Introduction
There is increasing evidence suggesting that the immune system plays an important role in eliminating or controlling cancer, and that failure of or escape from this mechanism allows tumors to expand (1). Tumors escape from the immune system by using different regulatory molecules and regulatory cells. At the cellular level, tumors can induce T cell anergy and T cell suppression and recruit regulatory cells such as CD4+T regulatory cells (2), myeloid suppressor cells (3), M2 macrophages (4), and NKT cells (5, 6). One of the most extensively studied negative regulators is the CD4+ T regulatory cell (Treg), characterized by the expression of interleukin 2 receptor α, known as CD25, and the intracellular expression of transcription factor forkhead box p3 (Foxp3). A role for Tregs in tumor immunity was first discovered when anti-tumor T cell immune responses were enhanced in mice inhibited for the function of this T cell subpopulation in vivo by anti-CD25 mAb, clone PC61. The blockade of Tregs was found to induce tumor immunity in many tumor models, including leukemia, myelomas and sarcomas (7). Blockade of Tregs by using other reagents such as Denileukin diftitox (immunotoxin conjugated IL-2, Ontak) and cyclophosphamide also inhibited tumor growth (8, 9) and enhanced vaccine-induced immunity (10, 11).
Another kind of regulator is the NKT cell. NKT cells are a unique subset of T cells capable of recognizing lipid antigens presented by the MHC-like molecule CD1d. They can be divided into at least two subsets. Type I NKT cells express an invariant TCR-α chain utilizing the Vα14Jα18 segment. These cells can be activated by the prototypic lipid antigen α-galactosylceramide (α-GalCer). Type II NKT cells express a diverse TCR repertoire, distinct from Vα14Jα18, and can be activated by other lipids such as sulfatide (12). Each subset of NKT cells can be activated by a specific group of lipids that cannot activate the other subset. There are two strains of NKT cell-deficient mice: CD1d−/− that lack both type I and type II NKT cells, and Jα18−/− that lack type I NKT cells but still retain type II NKT cells. By using these strains it has been shown that type I NKT cells promote tumor immunity (13–15), whereas type II NKT cells can mediate suppression of tumor immunosurveillance in multiple mouse tumor models (16). Previously, we found that these two subsets counteracted each other to regulate tumor immunity when they were simultaneously stimulated, suggesting a new immunregulatory axis (5, 17, 18).
In some tumor models Tregs were found to play a critical role in the suppression of tumor immunity, whereas in other models type II NKT cells were found to be the key suppressive cells. It is unclear why different regulatory cells suppress tumor immunity in different models and what determines which cells control the immune response to tumors. The answers to these questions are still elusive.
Here, by using a widely studied subcutaneous CT26 syngeneic colon tumor model, as well as the R331 renal carcinoma cell line in which tumor immunity was found to be regulated by Tregs in WT mice, we investigated the relative role of two kinds of suppressors – Tregs and type II NKT cells – and the mechanism determining the balance between them. We found that in the absence of both type I and type II NKT cells (CD1d−/− mice), Tregs regulate tumor immunity, similar to the situation in WT mice. However, in the absence of just type I NKT cells (Jα18−/− mice), eliminating or blocking Tregs is not sufficient to overcome immune suppression. Also, by blocking Tregs or type II NKT cells in Jα18−/− mice we discovered that having either one of the suppressors is sufficient to suppress the immune response against tumor formation. Which of these suppressors plays a predominant role in the regulation of tumor immunity depends on the presence of type I NKT cells, as type I NKT cells were found to counteract type II NKT cells. In this study, for the first time we revealed the relative role of Tregs and type II NKT cells in controlling immunity to the same tumor, and discovered that the balance between these regulatory cells is determined by a third cell, the type I NKT cell. This finding may be critical in the therapy of human cancer patients, because they often are deficient in type I NKT cell functions (6, 19–21).
Materials and Methods
Mice
Female BALB/c mice were purchased from Animal Production Colonies, Frederick Cancer Research Facility, National Cancer Institute. BALB/c CD1d−/− mice and BALB/c Jα18−/− mice (Provided by M. Taniguchi, RIKEN Institute, Yokohama, Japan and by D. Umetsu, Harvard Medical School, Boston, MA) were bred at the National Cancer Institute (Bethesda, MD) under specific pathogen-free and Helicobacteria-free conditions. Female mice (at least 6–8 weeks of age) were used for all experiments. All experimental protocols were approved by and performed under the guidelines of the National Cancer Institute’s Animal Care and Use Committee.
Cell lines
A CT26 colon carcinoma cell line was maintained in RPMI 1640 supplemented with 10% FCS, L-glutamine (2mM), sodium pyruvate (1mM), nonessential amino acids, and 2-mercaptoethanol (5 × 10−5 M). R331, a subline of the RENCA BALB/c renal cell carcinoma line, was a kind gift of Dr. Thomas Sayers, NCI, Frederick, MD, and was maintained in RPMI 1640 supplemented with 5% FCS and all of the above supplements.
In vivo tumor assay and antibody treatment
A single cell suspension of 5 × 104 CT26 cells or 5×105 R331 cells in 0.1ml of PBS was injected s.c on day 0. Mice were treated on day -5 with 0.5 mg anti-mCD25 (PC61, Harlan laboratories) and in some experiments with 0.2 mg of anti–mCD1d (1B1, Harlan laboratories) on days 1, 4 and 7. Rat IgG was purchased from Sigma-Aldrich (Sigma, St. Louis, MO) and injected as a control for antibody treatments. Tumor size was measured periodically, starting day 7, by caliper gauge.
Sulfatide treatment
3'-sulfo-C24:1 galactosylceramide (sulfatide) (Avanti polar lipids, Alabaster, AL) was dissolved in PBS+0.5% Tween 20, and 30 µg/mouse were injected s.c 1 hour after tumor challenge.
In vitro Treg suppression assay
The Treg suppression assay was conducted as previously described (22). Briefly, magnetic bead-sorted CD4+CD25− T cells (5×104, responders) and varying numbers of CD4+CD25+ T cells (Tregs) from WT and Jα18−/− lymph nodes (brachial, axillary, inguinal, mesenteric, popliteal and lumbar) were incubated in the presence of CD90.2-depleted splenocytes (5×104, accessory cells) and 0.5 µg/ml anti-mouse CD3 (145-2C11, BD Bioscience). The cells were cultured in a 96 well flat-bottom plate (Corning) for 72 hr in triplicates. Suppression was evaluated by cell proliferation, measured by [3H]-Thymidine incorporation (1 µCi/well, added at the last 16 hr of incubation). Percentage of suppression was calculated according to: 100 × [1- (CPM of Treg culture/ CPM of non-Treg culture)].
Isolation of liver lymphocytes
Liver lymphocytes were prepared as previously described (23, 24). Livers were perfused with liver perfusion medium (Invitrogen), minced and digested with liver digest medium (Invitrogen) at 37°C for 15 minutes. Hepatocytes were removed by centrifugation (500 rpm for 1 minute), and liver lymphocytes were then purified by a 40%/80% gradient of Percoll (Sigma-Aldrich).
Adoptive transfer of liver type I NKT cells
Lymphocytes from 40 naive BALB/c livers were isolated and stained with PE-PBS57-CD1d tetramer (The NIH tetramer facility, Bethesda, MD) for 40 min at 4°C, followed by staining with anti-PE beads (Miltenyi). PBS57-CD1d-tetramer positive cells (type I NKT cells) were separated by using AutoMACS (Miltenyi). Efficacy of the separation was evaluated after staining the cells with anti-CD3 by FACSCalibur (BD Biosciences), and the data were analyzed using FlowJo (Tree Star). 4×105 type I NKT cells in 0.2 ml PBS were injected i.v a day before tumor challenge into Jα18−/− recipients.
Visualizing liver type II NKT cells with sulfatide-loaded CD1d dimers
Sulfatide was loaded on mCD1d/dimer as described in Parish et al. (in preparation), modified from the sulfatide-CD1d tetramer method of (12). Briefly, mCD1d/Ig fusion protein (CD1d dimers; mouse CD1d dimerX, BD Biosciences) was loaded with sulfatide 24:1 (Avanti Lipids) or PBS at 37°C overnight. Buffer was replaced with PBS using Amicon Ultra Centrifugal Filters 30K (Millipore), followed by the addition of PE–anti-mouse IgG antibodies (BD Biosciences) and incubated for 1 hour at room temperature. Mouse IgG (BD Biosciences) was added for an additional 30 minutes at room temperature to saturate unbound excess anti-IgG antibodies. Liver lymphocytes were stained with CD1d dimers for 1 hour at 4°C, followed by cell surface staining.
Flow cytometry
Total spleen or lymph node cells were incubated with anti-CD16/CD32 (clone 93, Biolegend) and stained with anti-CD3 (145-2C11, Biolegend), anti-CD4 (RM4-5, BD), anti-CD25 (PC61, eBioscience), and anti-Foxp3 (FJK-16S, eBioscience) for the evaluation of Treg frequency. Enriched liver lymphocytes were incubated with anti-CD16/CD32 and stained with anti-CD3, yellow viability dye (LIVE/DEAD Fixable dead cell stain kit, Invitrogen), and PBS57-loaded CD1d-tetramers (NIH Tetramer facility) or sulfatide-loaded CD1d dimers (Prepared as described above). Enriched liver type I NKT cells were also stained with anti-CD25. The cells were visualized on a FACSCalibur flow cytometer using CellQuest software (BD Biosciences) and LSRII using DIVA software (BD Biosciences). Data were analyzed by FlowJo (Tree Star).
Statistics
The data were analyzed using the nonparametric Mann-Whitney test, Wilcoxon rank sum test, t-test with conservative variance estimation or repeated measures analysis of variance test using GraphPad Prism software (version 5; GraphPad software) or JMP software (Version 8, SAS institutes). Significance was determined at p < 0.05.
Results
Tregs are not necessary for the suppression of tumor immunity in Jα18−/− mice
In an attempt to understand the relative roles of Tregs and type II NKT cells in the regulation of tumor immunity, we first addressed the necessity of the two suppressors in NKT cell-deficient mice. We used a CT26 s.c tumor model with three strains of mice, all on the BALB/c background: WT that have both type I and type II NKT cells, CD1d−/− that lack both types of NKT cells, and Jα18−/− that lack type I NKT cells but retain type II NKT cells. In this tumor model, the suppression of tumor immunity in WT mice has been shown to be regulated by Tregs (25). Consistent with this previous report, we found that blocking Tregs by anti-CD25 protected WT mice (Fig 1A). Similarly, protection was also observed by blocking Tregs in CD1d−/− mice, although without treatment, tumors grew significantly more slowly than in wild-type mice (p=0.007). Surprisingly, however, anti-CD25 treatment did not protect Jα18−/− mice (Fig 1A). These results suggest that Tregs are necessary for the regulation of tumor immunity in WT and CD1d−/− mice but not in Jα18−/− mice.
Fig 1. Anti-CD25 treatment is sufficient to induce protection in WT and CD1d−/− mice but not in Jα18−/− mice.
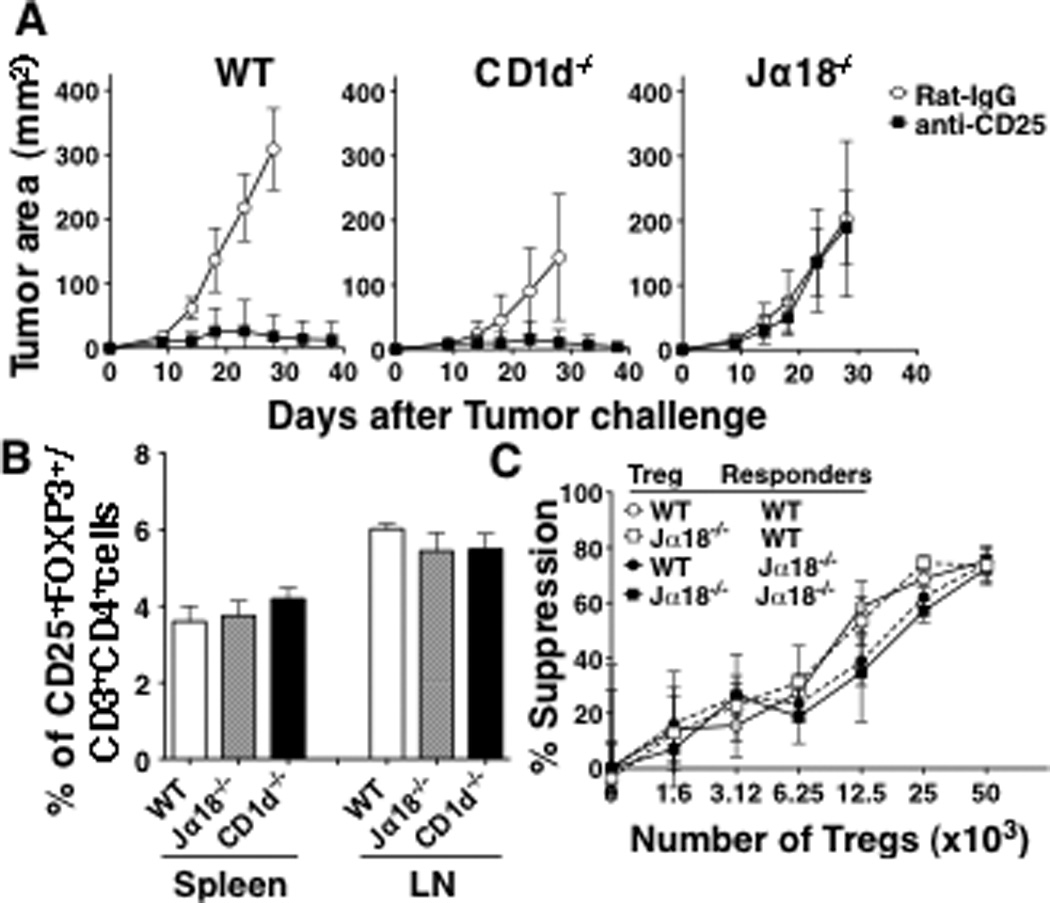
A, CT26 cells (5×104) were injected s.c in the left flank of WT, CD1d−/− and Jα18−/− mice (5 mice/group). Five days before tumor challenge, 0.5 mg of anti-CD25 or Rat IgG was injected i.v. Tumor size was measured twice a week. The experiment was repeated 4 times. Anti-CD25 treatment significantly reduced tumor size in WT mice (p=0.0079 against Rat IgG-treated WT mice) and CD1d−/− mice (p=0.0449 against Rat IgG-treated CD1d−/− mice). Anti-CD25 was not effective in Jα18−/− mice. B, Lymph node and spleen cells were prepared from WT, CD1d−/− and Jα18−/− mice (3 mice/group). Percentage of Tregs (CD3+CD4+CD25+FOXP3+) was evaluated by flow cytometry. The frequency of Tregs was evaluated in 3 mice per group. C, Varying numbers of lymph node CD4+CD25+T cells from WT or Jα18−/− mice were cocultured with 5×104 CD4+CD25− T cells from each strain and 0.5 µg/ml of anti-CD3. Cells were cultured for 72 hr, with the presence of [3H]-thymidine for the last 16 hours. % of suppression was determined as 100 × [1- (CPM of Treg culture/ CPM of non-Treg culture)]. Data are presented as mean ± SD. These experiments were repeated 3 times with comparable results.
Although lack of Treg function in Jα18−/− mice would be expected to have an effect opposite to what we observed, we nevertheless wanted to rule out any differences in frequency and/or function of Tregs among the strains. Therefore, we compared the proportion (Fig 1B) and suppressive activity (Fig 1C) of Tregs among the strains of mice. Gating on CD3+CD4+CD25+Foxp3+ cells in spleens and lymph nodes of mice from each strain, there was no difference in the frequency (Fig. 1B). Also, there was no difference in the suppressive activity of Tregs between Jα18−/− and WT against responders from the same strain (Jα18−/− and WT responders, respectively) (Fig 1C), and both WT and Jα18−/− responders were susceptible to a similar degree to Tregs from different strains. Thus, the fact that Tregs are not necessary for immune regulation in Jα18−/− mice is not due differences in frequency and/or function of Tregs or in susceptibility of conventional CD4+ T cells to suppression by Tregs.
Immune suppression is mediated by both type II NKT cells and Tregs in Jα18−/− mice
It has been shown that type I and type II NKT cells counteract each other’s functions (17). Thus, to explain the surprising difference in the effect of anti-CD25 in WT, CD1d−/− and Jα18−/− mice, we hypothesized that in WT mice, the two types of NKT cells cancel each other’s effects on tumor immunity, leaving Tregs as the dominant suppressor. On the other hand, in CD1d−/− mice, both types of NKT cells are absent, again leaving Tregs as the dominant suppressor. In both circumstances in which Tregs dominate, anti-CD25 treatment is effective to induce tumor rejection, whereas we hypothesize that in Jα18−/− mice anti-CD25 treatment alone is not effective because the lack of type I NKT cells allows unopposed type II NKT cells to suppress tumor immunity as well (Fig 2A). Consistent with this hypothesis, we found a higher frequency of type II NKT cells in livers of Jα18−/− mice than in those of wild-type mice (Fig 2B). The difference is not due to a simple dilution effect in the absence of type I NKT cells because we found that the actual numbers of type II NKT cells were significantly higher in Jα18−/− mice (WT vs Jα18−/−, 1.1±0.6 × 105 vs 3.2±1.3 × 105; p=0.0082). To further test our hypothesis, we examined the necessity of both types of suppressors in Jα18−/− mice. A prediction of our hypothesis is that it is necessary to remove both type II NKT cells and Tregs in order to remove immune suppression in Jα18−/− mice, as removing Tregs alone was not sufficient to induce tumor protection (Fig 1A).
Fig 2. The balance between the 3 kinds of T cells in 3 strains of mice.
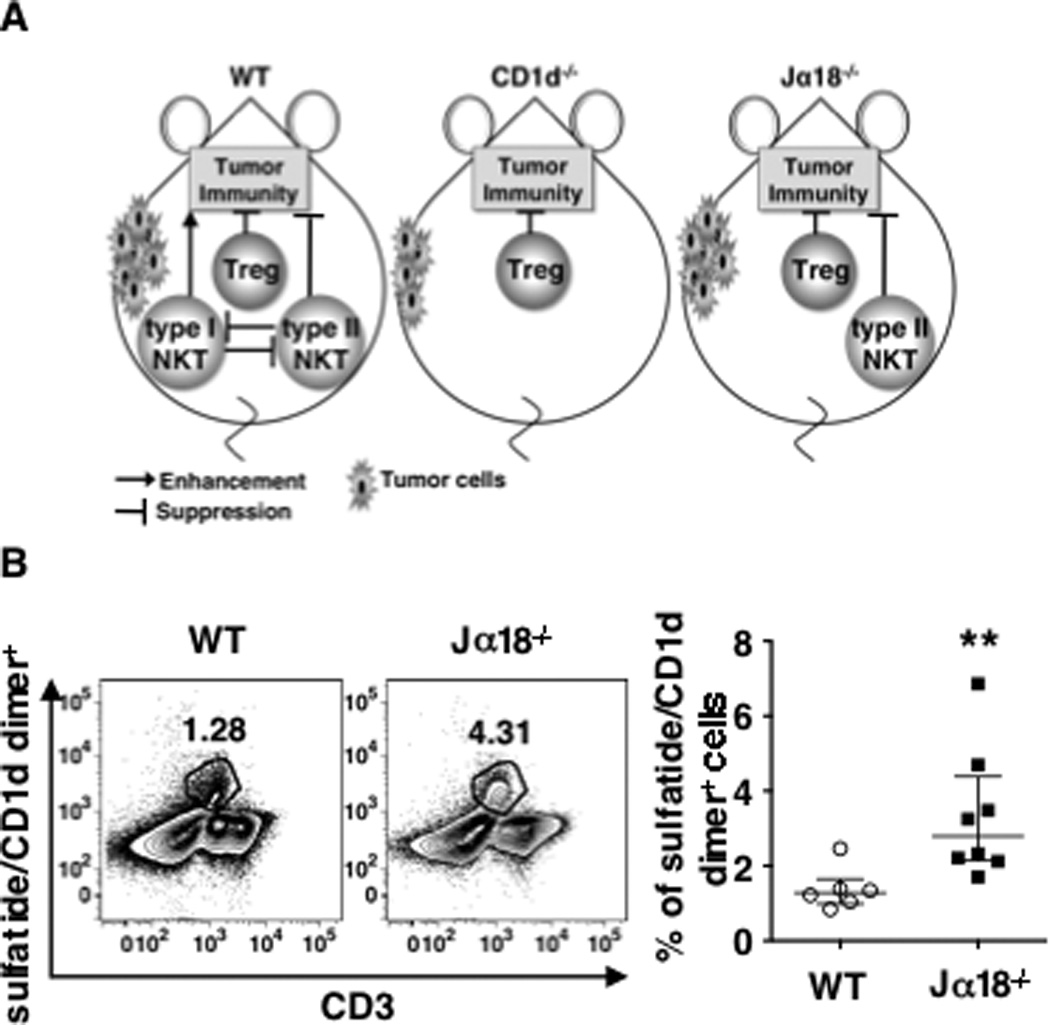
A .Left panel, In WT mice type I and type II NKT cells counteract each other to cancel their effects, leaving Tregs to dominate the suppression. Middle panel, In CD1d−/− mice, neither subset of NKT cells exists, and the dominant suppressors are again the Tregs. Right panel, In Jα18−/− mice the effect of type II NKT cells is not counter-regulated by type I NKT cells, leaving both Tregs and the type II NKT cells able to suppress, so both need to be blocked concurrently to abrogate suppression. B. Livers from naïve BALB/c mice (WT) and Jα18−/− mice were perfused and processed for lymphocyte enrichment. Total liver lymphocytes pooled from 3 mice were stained with anti-CD3 and sulfatide-loaded CD1d-dimer. Unloaded-CD1d-dimer-reactive cells and dead cells were excluded from the analysis. Left panel Representative density plot of sulfatide-loaded CD1d-dimer-reactive cells among total liver lymphocytes. Right panel, The frequency of sulfatide-loaded CD1d-dimer-reactive cells was evaluated in 6 pools of 3 livers each of WT mice and 8 pools of 3 livers each from Jα18−/−. Data, pooled from 6 independent experiments, are presented as values from each pool (symbols) with bars showing mean ± SD. ** p = 0.002
Since the activation of type II NKT cells is CD1d-dependent, and in Jα18−/− mice, the only cells dependent on CD1d are type II NKT cells, we used CD1d blocking antibody to prevent NKT cell activation in vivo. We found that blocking NKT cell activation using anti-CD1d alone did not protect WT or CD1d−/− mice from tumor development, suggesting that blockade of NKT cell activation is not sufficient to induce tumor rejection (Fig 3). In Jα18−/− mice, neither blocking the activation of type II NKT cells alone with anti-CD1d, nor depletion of Tregs alone with anti-CD25, was sufficient to affect the tumor growth. However, when mice were treated with both anti-CD1d and anti-CD25, protection was achieved. This result suggested that either type II NKT cells alone or Tregs alone are sufficient for immune suppression in Jα18−/− mice and that blockade of both types of regulatory T cells is necessary to induce protective tumor immunity.
Fig 3. A combination of Treg blockade and type II NKT cell blockade reduces tumor burden in Jα18−/− mice.
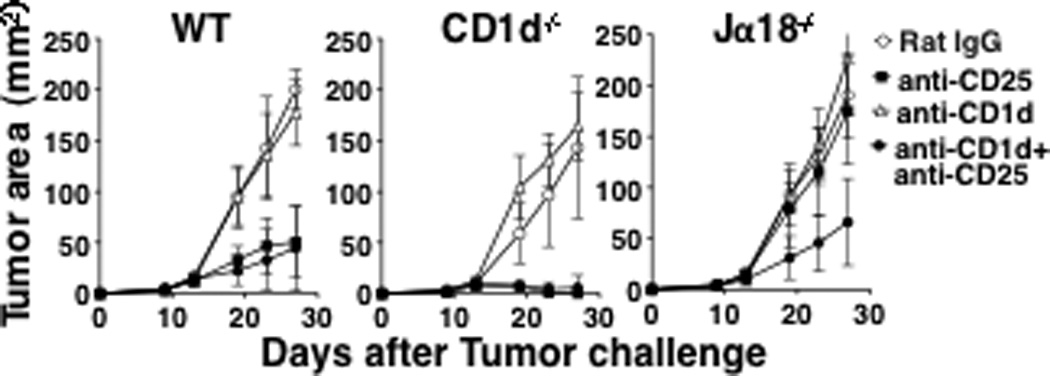
CT26 cells (5×104) and anti-CD25 (0.5 mg) were injected into WT, CD1d−/− and Jα18−/− mice (5 mice/group) as described in Fig 1A. Some mice were also treated with 0.2 mg of anti-CD1d mAb or Rat IgG on days 1, 4, and 7. Tumor size was measured twice a week. Jα18−/− mice that received the combined treatment developed significantly smaller tumors (p=0.0079 against Jα18−/− mice that were treated with anti-CD25 alone or anti-CD1d alone). Mice that received the combined treatment (anti-CD25+anti-CD1d) and mice that received only anti-CD25 treatment developed similar tumor size in WT and CD1d−/− groups. Data are presented as mean ± SD. The experiment was repeated 3 times with comparable results.
Adoptive transfer of Type I NKT cells induces tumor protection in Treg deficient Jα18−/− mice
The results above confirm our hypothesis that in the Jα18−/− mice, both types of regulatory cells are active so both need to be blocked, whereas in the WT mice, only Tregs are active and need to be blocked. The second part of our hypothesis is the explanation for this, namely that in the WT mice that have both type I and type II NKT cells, the type II NKT cells are inhibited by type I, leaving only Tregs active. The only difference between Jα18−/− mice and WT mice is the lack of type I NKT cells, so their absence now reveals the suppressive nature of type II NKT cells. Thus, we suggest that the protective effect of Treg blockade is dependent on the balance between two types of NKT cells. A prediction of this hypothesis is that adoptive transfer of type I NKT cells that counteract type II NKT cells should restore the balance between the NKT cell subsets, neutralizing type II NKT cells and making Treg blockade protective in type I NKT cell-deficient Jα18−/− mice. Therefore, to test this hypothesis, we adoptively transferred enriched type I NKT cells from WT livers into Jα18−/− mice.
In our hands, type I NKT cells comprise approximately 10% of total liver lymphocytes (Fig 4A). These lymphocytes were enriched for type I NKT cells by using PBS57-loaded CD1d-tetramer with magnetic beads. After the enrichment we found that 80% of the cells were PBS57-loaded CD1d-tetramer reactive (Fig 4B).
Fig 4. Adoptive transfer of type I NKT cells and Treg blockade reduce tumor burden in Jα18−/− recipients.
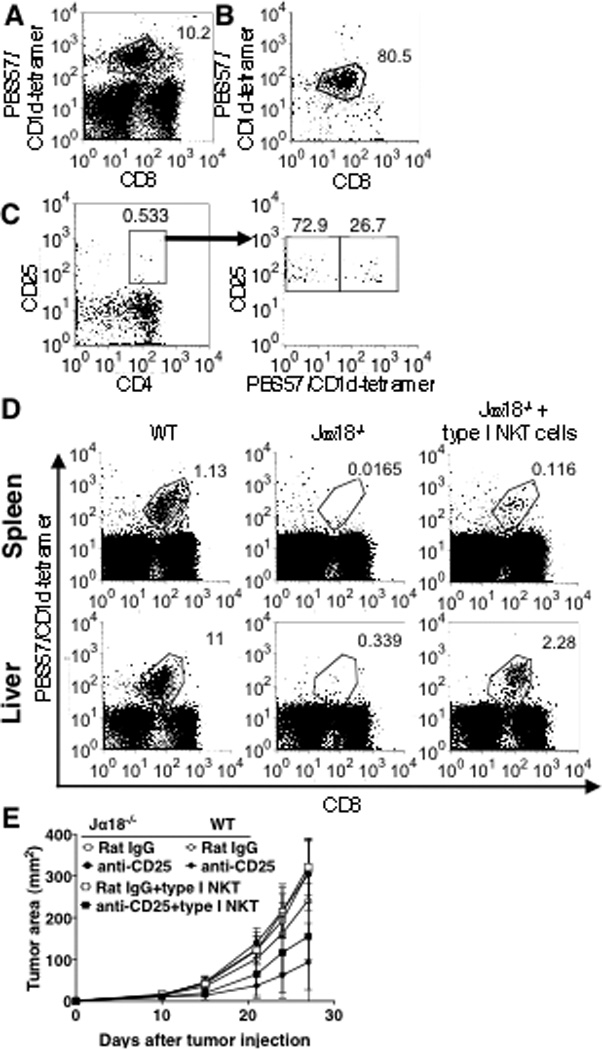
Livers from 40 naïve BALB/c mice were perfused and digested for lymphocyte enrichment. A, Total liver lymphocytes were stained with anti-CD3 and PBS57-CD1d-tetramer. A presented pseudo dot plot represents the entire lymphocyte population. B and C, PBS57-CD1d-tetramer positive cells were isolated by magnetic bead sorting. The sorted cells were stained with anti-CD3, anti-CD4, anti-CD25 and analyzed by flow cytometry. Presented pseudo dot plots represent the entire positive fraction (B and left panel of C) or CD4+CD25+ gated population (right panel of C). D and E, On day -1, 4×105 PBS57-CD1d-tetramer positive cells were adoptively transferred into the tail vein of Jα18−/− mice that were already treated on day -5 with 0.5 mg anti-CD25 i.v (5 mice/group). On day 0 mice were challenged s.c. with CT26 cells (5×104). D, on day 5, liver lymphocytes and spleen cells of WT or Jα18−/− mice which did or did not receive adoptively transferred NKT cells (3 mice/group) were stained with anti-CD3 and PBS57-CD1d-tetramer and analyzed by flow cytometry. Presented pseudo dot plots represent the entire lymphocyte population in each tissue. E, Tumor size was measured twice a week. Adoptive transfer of type I NKT cells into anti-CD25-treated Jα18−/− mice significantly reduced tumor size (p=0.0035 against Jα18−/− mice treated with Rat-IgG; p=0.0007 against Jα18−/− mice treated with anti-CD25; p=0.0002 against Jα18−/− mice adoptively transferred type I NKT cells). Data are presented as mean ± SD. The experiment was repeated 4 times with comparable results.
Next we examined the enriched population of type I NKT cells for possible contamination by Tregs. This is important since the cells are transferred into mice already treated with anti-CD25. If transferred Tregs were to contaminate the enriched type I NKT cells, this could result in the suppression of tumor immunity regardless of the activity of type II NKT cells. We found that approximately 0.5% of the enriched type I NKT cells are CD4+CD25+. Among those cells, some of them were CD1d-tetramer reactive NKT cells that may have been activated by the tetramer staining during the purification process. Thus, less than 3/4 of the gated double positive population was presumably Tregs, which represents less than 0.4% of the enriched type I NKT cells (Fig 4C). Therefore, the vast majority of the cells that were transferred into Jα18−/− mice were type I NKT cells, and these cells contained very few Tregs.
In order to evaluate the efficacy of the adoptive transfer of type I NKT cells into Jα18−/− mice, we examined the frequency of type I NKT cells in the spleens and livers 6 days after the i.v injection of 4×105 PBS57-CD1d-tetramer reactive cells. There was 20% and 10% type I NKT cell reconstitution in livers and spleens, respectively, of recipient mice compared to wild-type mice (Fig 4D), suggesting that the adoptive transfer was efficient, but reconstitution was incomplete. 24 hours after the transfer, mice were challenged s.c. with CT26 cells and monitored for tumor growth (Fig 4E). The adoptive transfer of type I NKT cells into Jα18−/− mice by itself did not affect tumor burden. However, in anti-CD25 treated Jα18−/− mice that were adoptively transferred with type I NKT cells, protection was achieved despite only partial reconstitution. This result demonstrated that even with only 10% reconstitution in the spleen and 20% in the liver (that could be achieved with cells from 40 donor livers), the presence of type I NKT cells made Treg blockade effective to reduce tumor burden in Jα18−/− mice. We infer that suppression of type II NKT cells by type I NKT cells left Tregs as the major suppressor, so that Treg blockade was now sufficient to remove the suppression of tumor immunity.
Type II NKT cell activation abrogates the protective effect of Treg blockade in wild-type mice
A further prediction of our hypothesis is that activating regulatory type II NKT cells after Treg blockade should shift the balance of NKT cell subsets in WT mice toward immunosuppression, changing the outcome of tumor growth. We previously reported that activation of a subset of type II NKT cells by sulfatide can suppress the protective effect of type I NKT cells (17). Thus, as a further test of the hypothesis, we treated tumor-bearing mice with or without anti-CD25 with sulfatide (Fig 5).
Fig 5. Sulfatide treatment after Treg blockade suppresses tumor immunity in WT mice.
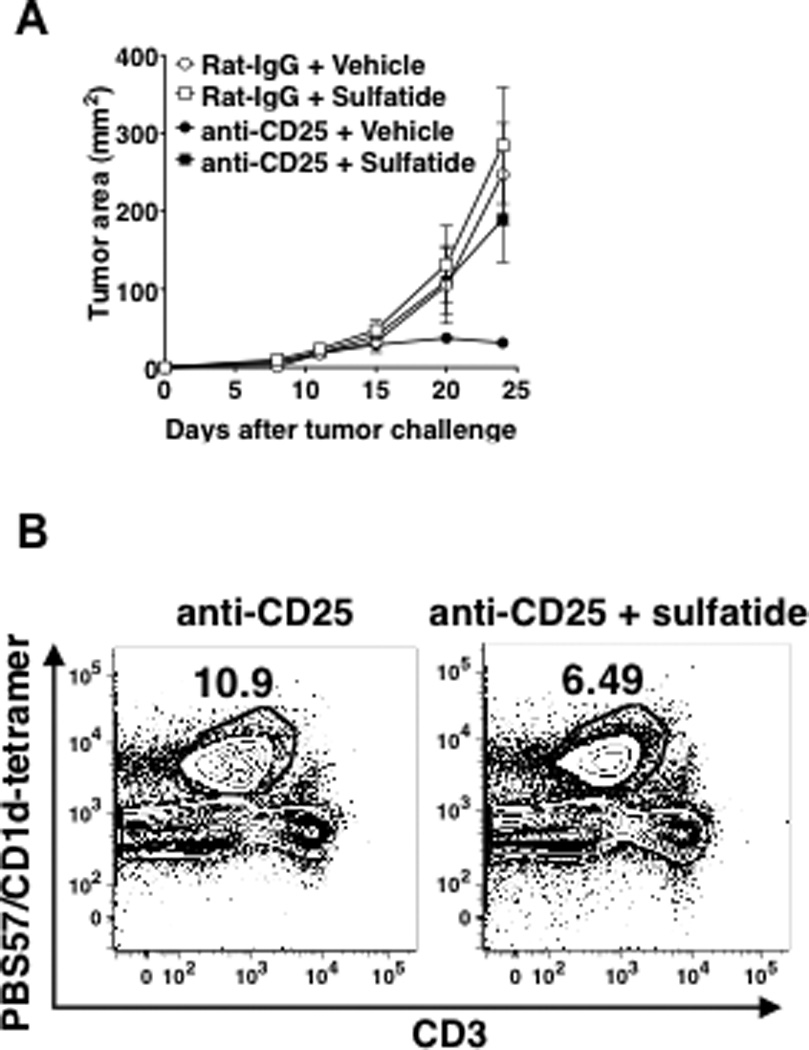
A. CT26 cells (5×104) and anti-CD25 mAb (0.5 mg) were injected into WT mice as described in Fig 1A (5 mice/group). One hour after tumor challenge, mice were injected s.c with 30 µg sulfatide or control vehicle at a site adjacent to that of the tumor injection. Tumor size was measured twice a week. Sulfatide significantly increased tumor size in anti-CD25-treated mice (p=0.0079 vs vehicle+anti-CD25-treated mice). Data are mean ± SD. The experiment was repeated 3 times. B. Anti-CD25-treated mice were challenged with CT26 cells and injected with vehicle or 30 µg/mouse sulfatide. 72 hours after the injections, livers from the mice were perfused and processed to enrich lymphocytes. Total liver lymphocytes were stained with PercCP-Cy5.5- anti-CD3 and PE-PBS57-CD1d tetramer to enumerate type I NKT cells. The proportion of type I NKT cells in the mice with anti-CD25+sulfatide significantly lower than in the mice with anti-CD25 alone (mean 41% reduction, range 32–46%, p<0.01 by t-test with conservative variance estimation). Presented density plots are representative of four independent experiments.
Treating Treg-intact WT mice with sulfatide (30 µg/mouse) did not affect tumor growth compared to mice treated with vehicle or untreated mice (Fig 5A). However, in mice treated with anti-CD25, sulfatide treatment abrogated the protective effect of anti-CD25. Consistent with the effect on tumor growth, the sulfatide treatment in the anti-CD25-treated mice significantly reduced the percentage of type I NKT cells (mean 41% reduction, range 32–46%, p < 0.01) in livers of tumor-challenged animals 72 hours after injections (Fig 5B). These results demonstrate that shifting the balance between type I and type II NKT cells at the level of their activity and numbers by stimulating type II NKT cells can overcome the neutralizing effect of type I NKT cells on type II, and thereby reveal the immunoregulatory potential of type II NKT cells even in the presence of type I NKT cells. Thus, overall, we conclude that the protective effect of Treg blockade in WT mice relies on the absence of suppression by type II NKT cells, and that either selectively stimulating type II NKT cells or removing their natural inhibitor, the type I NKT cells, unmasks the presence of a second immunoregulatory T cell, the type II NKT cell, that acts in parallel with Tregs to control tumor immunity.
Blockade of both Tregs and type II NKT cells is necessary to induce protection in a R331 renal cell carcinoma model
Recently Teng et al., (26) reported that the R331 renal cell carcinoma model behaves similarly to the s.c. CT26 model, in which anti-CD25 treatment induces tumor rejection, whereas the absence of NKT cells in CD1d−/− mice or blockade of their activation does not result in tumor rejection. Therefore, we hypothesized that a similar mechanism might apply, and so asked whether anti-CD25 treatment induces rejection of tumors in Jα18−/− mice, and if not, whether a combination of anti-CD1d and anti-CD25 can protect. Consistent with the previous report, anti-CD25 treatment induced tumor rejection in wild-type mice. In contrast, the same treatment did not affect tumor growth in Jα18−/− mice (Fig 6A). However, as in the s.c. CT26 model, when combined with CD1d blockade, now anti-CD25 treatment protected Jα18−/− mice (Fig 6B). Thus, we conclude that the observations that we made in this study are not unique to one tumor model, but are applicable to at least two different mouse tumor models.
Fig 6. Blockade of Treg and type II NKT cells decreases tumor burden in R331 tumor-challenged Jα18−/− mice.
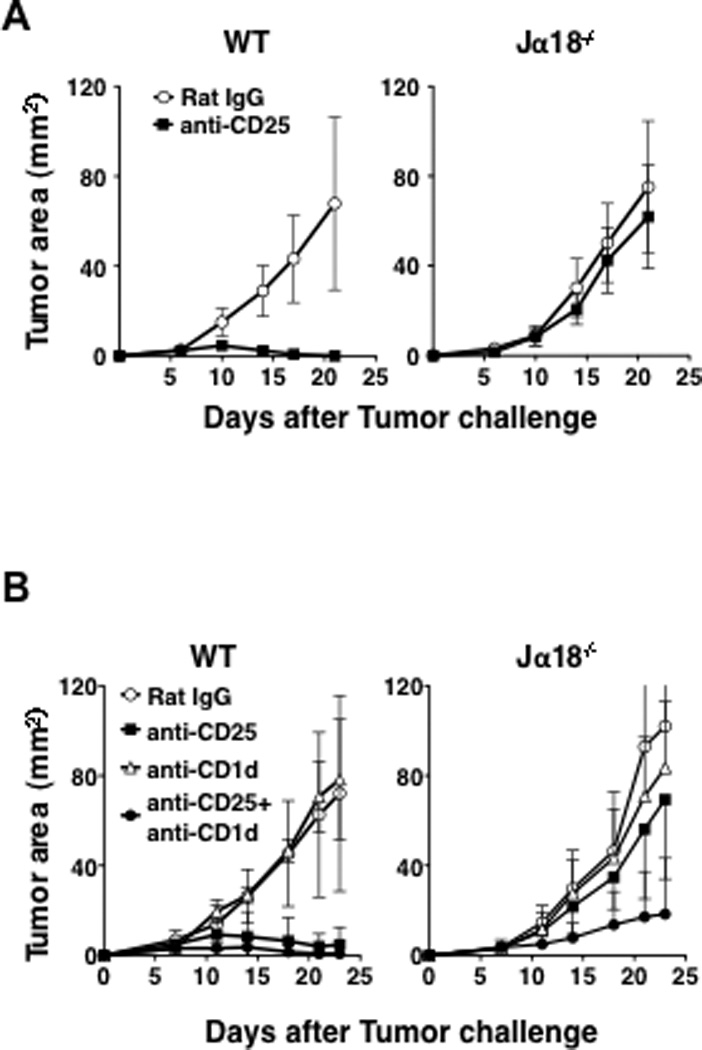
A. R331 cells (5×105) were injected s.c in the left flank of WT and Jα18−/− mice (5 mice/group). Five days before tumor challenge, 0.5 mg of anti-CD25 or Rat IgG was injected i.v. Tumor size was measured every four days starting day 7. Anti-CD25 treatment significantly reduced tumor size in WT mice (p= 0.0075 vs Rat IgG-treated WT mice) but not in Jα18−/− mice. B. R331 cells (5×105) and anti-CD25 (0.5 mg) were injected into WT and Jα18−/− mice (5 mice/group) as described above. Two groups of each strain of mice were also treated with 0.2 mg of anti-CD1d mAb or Rat IgG on days 1, 4, and 7. Tumor size was measured every 2–4 days starting on day 7. Jα18−/− mice that received anti-CD25+anti-CD1d treatment had significantly slower tumor growth than Jα18−/− mice that were treated with anti-CD25 alone or anti-CD1d alone. (p<0.05 by the repeated measures analysis of variance test). All group had five mice each except for the anti-CD25 treated group that had four mice. Data are presented as mean ± SD. The experiment was repeated 2 times with comparable results.
Discussion
In this study, for the first time we revealed the relative role for the two suppressors, Tregs and type II NKT cells, in the same tumor model, and showed that the balance between them is determined by a third cell, the type I NKT cell, that counterbalances type II NKT cells. The role of each suppressor alone has been documented in the past in a variety of tumor models. In some settings type II NKT cells were found to dominate the suppression of tumor immunity (16, 17, 26, 27) whereas in other settings Tregs were found to be the primary regulatory cell (7, 16, 25). In contrast to these studies, we decided to focus on the relative roles of both suppressors in the same tumor model, the s.c. CT26 colon carcinoma. We found that each one of the regulators is sufficient to induce suppression of tumor immunity in the absence of type I NKT cells (Jα18−/− mice) and in order to achieve protection the effects of both regulators need to be abrogated.
Our findings may also be relevant to human cancer. It has been reported that patients with advanced cancers have reduced numbers and function of type I NKT cells, suggesting an immunological status in cancer patients similar to that in type I NKT-cell-deficient Jα18−/− mice (19–21). These observations may provide an explanation why treatments reducing the number of Tregs do not always induce tumor regression in cancer patients (28, 29). In situations like that, it could be that blockade of both Tregs and type II NKT cells may be necessary to overcome the suppression of tumor immunity in humans, as we found in Jα18−/− mice.
By using two mouse tumor models, we found that NKT cell deficient (CD1d−/−) and NKT cell sufficient (WT) mice are protected from tumor formation when their Tregs are blocked/suppressed (Fig 1A, and 6A), suggesting that in these mice, Tregs regulate tumor immunity. However, when we looked carefully at challenged CD1d−/− mice in the CT26 tumor model, we found that the growth rate of solid tumors in the skin (s.c.) is significantly slower in these mice compared to WT mice (Fig 1A, p=0.007; Mann Whitney test). This observation suggests that there may be some role for NKT cells in tumor formation in the subcutaneous tumor. It might be that in WT mice not all the type II NKT cells are counteracted by type I NKT cells and the ones that are not, together with Tregs, can suppress tumor immunity, whereas in CD1d−/− mice type II NKT cells do not exist and Tregs are the only suppressor T cells.
In contrast, when WT or CD1d−/− mice were challenged i.v with the same CT26 tumor cell line, Treg blockade was not sufficient to induce protection in either strain (16) but the number of lung metastases was substantially smaller in CD1d−/− mice than in WT mice, indicating that NKT cells predominantly regulate tumor immunity in that model (17, 30). These contrasting results suggest that the immune responses against the same tumor cell line are regulated differently in different tissues. It seems that NKT cells control tumor immunity in the lungs as well as partly in the skin, whereas Tregs control tumor immunity in the skin but not in the lungs. Although Tregs have been implicated in immune regulation in both lungs and skin, a recent report demonstrating high expression levels of skin homing receptors on a majority of Tregs in the peripheral blood of humans may suggest that Tregs have easier access to the skin (31). The reason why Tregs do not show an apparent role in the regulation of tumor immunity in the lung metastasis model remains elusive. This may provide a potential explanation for our observation in this study.
Our hypothesis is that in WT mice the two subsets of NKT cells balance each other, so their effect on tumor growth is canceled, leaving Tregs to dominate immune regulation. When we shifted the balance between type I and type II NKT cells by activating type II NKT cells with sulfatide (Fig 5), we found that the frequency of type I NKT cells was reduced and that the protective affect of Treg blockade was abrogated in these mice. This suggests that the balance between these two types of NKT cells is the key for protection when Tregs are blocked or depleted. These results are consistent with our previous observations that activation of type II NKT cells with sulfatide diminishes the protective effect or proliferative response of type I NKT cells (17). We also found that adoptive transfer of type I NKT cells into Jα18−/− mice partially protected the mice from tumor growth only after Treg blockade, suggesting that blockade of both suppressors is needed (Fig 4E). This result strengthens our hypothesis that the protective effect of Treg blockade relies on the balance between the two types of NKT cells as we saw in Fig 5. However, the protection was only partial. Analyzing the efficacy of the adoptive transfer revealed that only 20% reconstitution was observed in the liver and 10% in the spleen. It could be that the reason for the partial protection is the low percentage reconstitution of type I NKT cells and the higher frequency of type II NKT cells in recipient Jα18−/− mice. Yet, even with such a relatively low level of reconstitution that was feasible to achieve with type I NKT cells from 40 livers, we could see a substantial protective effect.
We showed that adoptive transfer of type I NKT cells into Jα18−/− mice makes Treg depletion/blockade by anti-CD25 effective to suppress tumor growth, and that activation of type II NKT cells by sulfatide in vivo abrogates the protective effect of anti-CD25 in WT mice. Both results strongly suggest cross regulation between the two types of NKT cells. These results are consistent with our previous reports (32) and with our current findings that type II NKT cells are more frequent in type I-deficient Jα18−/− mice and that conversely, stimulation of type II NKT cells diminishes the frequency of type I NKT cells. The mechanism by which the two types of NKT cells regulate each other remains elusive. The cross regulation can potentially occur in two different ways. One is by direct interaction between the two types of NKT cells, either through soluble factors or by cell-to-cell contact. Although definitive experiments need to be carried out, the finding that the transfer of supernatant from stimulated type II NKT cells did not suppress type I NKT cell activation suggests that cell-to-cell contact may be required for them to directly regulate each other (32). An alternative mechanism could be opposing effects on the same effector cells. We have shown that type II NKT cells can suppress tumor specific CD8+ T cells by producing IL-13 that induces TGF-β production by CD11b+Gr-1+ cells (27, 33). On the other hand, type I NKT cells can facilitate the activity of NK cells and CD8+ T cells (6). Thus, the overall effect of each type of NKT cell on tumor immunity may possibly be the result of opposing effects from the two types of NKT cells on the same effector cells. However, our current finding that each type can diminish the other’s frequency would be more consistent with a direct effect of each on the other.
In this study, we show that blockade of type II NKT cell activation in Jα18 KO mice by anti-CD1d mAb (1B1) made Treg depletion/blockade effective to enhance tumor immunity. Although we used anti-CD1d mAb to block antigen presentation by CD1d to NKT cells, it has been reported that the same treatment can also activate APCs to make IL-12 by transducing the signal from CD1d (34, 35). It is reported that all existing anti-CD1d blocking mAbs have similar activity even if the antibody is in an F(ab’)2 form (34). Teng et al., previously attributed the protective effect of anti-CD1d treatment in some mouse tumor models in part to this activity of anti-CD1d (26). Therefore, the effect of anti-CD1d treatment might be partly due to activation of APCs. However, this treatment by itself did not have any effect on tumor growth in any of the three strains of mice, suggesting that there is a minimal impact of APC activation on the tumor growth in the tumor models used in this study.
Previously it was found in some tumor models, for instance in the i.v CT26 model, that activation of type II NKT cells increases tumor burden (17). Therefore we expected to see an increase in tumor growth after treating CT26 challenged WT mice with sulfatide (Fig 5). However, there was no significant difference in the growth rate of tumors with or without activation of type II NKT cells in the absence of other treatment. It may be that the suppression of tumor immunity by Tregs is already sufficient to prevent any immunosurveillance, so that adding another suppressive activity by another regulatory cell type has no further effect. Consistent with this interpretation, when Tregs were blocked, then an effect of sulfatide induction of type II NKT cells was observable (Fig. 5).
It is important to mention that the anti-CD25 mAb used to block/deplete Tregs in the current study diminishes not only Tregs but also other cells expressing CD25, such as activated conventional T cells. Thus, in this study anti-CD25 mAb was injected into naïve mice before tumor challenge that may activate tumor-specific T cells. Although it would be interesting to know whether our findings in this study could be extended to a spontaneous tumor model and/or to a therapeutic setting where mice with established tumors are treated, it would be difficult to test this hypothesis using anti-CD25 because of this effect on effectors, so a different method to deplete Tregs would be required. In fact, it was shown by others that anti-CD25 treatment after tumor challenge inhibited the efficacy of cancer immunotherapy (36).
Recently, non-Vα14Jα18 NKT cells that recognize αGalCer and utilize a Vα10Jα50 gene segment in their TCRα chain were reported. This NKT cell subset still exists in Jα18−/− mice. Although this novel subset of NKT cell seems to be a very minor population, it may possibly be involved in the immune regulation occurring in Jα18−/− mice (37).
Collectively, our data show, for the first time, a role for both immunosuppressive type II NKT cells and Tregs in the same tumor model, suggesting that both regulate tumor immunity. Which of these suppressors dominates depends on whether type II NKT cells are counter-balanced by a third cell, the type I NKT cell. Thus, not only is there a delicate balance between type I and type II NKT cells, but we reveal here that this balance between type I and type II NKT cells in turn determines the balance between type II NKT cells and T reg cells in cancer. In type I NKT cell-defective cancer settings, found widely in humans as well as studied here in mice, it may be necessary to block both Tregs and type II NKT cells in order to overcome the suppression of tumor immunity. Thus, these findings could be critical for the effective immunotherapy of cancer.
Acknowledgments
The authors gratefully acknowledge Dr. Thomas Sayers for providing the R331 cell line; Drs. Ethan M. Shevach and Howard A. Young for critical reading of the manuscript; The NIH Tetramer Core Facility for supplying the PBS-57/CD1d tetramers.
Grant Support
This work was supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health and the Gui Foundation.
Footnotes
The authors declare no conflict of interest.
Author contributions:.
Conception and design – L. Izhak, E. Ambrosino, J.A. Berzofsky, M. Terabe.
Acquisition of data- L. Izhak, E. Ambrosino, S. Kato, H. Weber, J.J O’Konek, S.T Parish, M. Terabe.
Analysis and interpretation of data- L. Izhak, D. Venzon, J.A. Berzofsky, M. Terabe.
Writing, review and/or revision of the manuscript - L. Izhak, J.A. Berzofsky, M. Terabe.
Administrative, technical or material support – Z. Xia
Study supervision - J.A. Berzofsky, M. Terabe
REFERENCES
- 1.Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. 2004;22:329–360. doi: 10.1146/annurev.immunol.22.012703.104803. [DOI] [PubMed] [Google Scholar]
- 2.Nishikawa H, Sakaguchi S. Regulatory T cells in tumor immunity. Int J Cancer. 2010;127:759–767. doi: 10.1002/ijc.25429. [DOI] [PubMed] [Google Scholar]
- 3.Nagaraj S, Gabrilovich DI. Tumor escape mechanism governed by myeloid-derived suppressor cells. Cancer Res. 2008;68:2561–2563. doi: 10.1158/0008-5472.CAN-07-6229. [DOI] [PubMed] [Google Scholar]
- 4.Vasievich EA, Huang L. The suppressive tumor microenvironment: a challenge in cancer immunotherapy. Mol Pharm. 2011;8:635–641. doi: 10.1021/mp1004228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Terabe M, Berzofsky JA. NKT cells in immunoregulation of tumor immunity: a new immunoregulatory axis. Trends Immunol. 2007;28:491–496. doi: 10.1016/j.it.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 6.Terabe M, Berzofsky JA. The role of NKT cells in tumor immunity. Adv Cancer Res. 2008;101:277–348. doi: 10.1016/S0065-230X(08)00408-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Onizuka S, Tawara I, Shimizu J, Sakaguchi S, Fujita T, Nakayama E. Tumor rejection by in vivo administration of anti-CD25 (interleukin-2 receptor alpha) monoclonal antibody. Cancer Res. 1999;59:3128–3133. [PubMed] [Google Scholar]
- 8.Knutson KL, Dang Y, Lu H, Lukas J, Almand B, Gad E, et al. IL-2 immunotoxin therapy modulates tumor-associated regulatory T cells and leads to lasting immune-mediated rejection of breast cancers in neu-transgenic mice. J Immunol. 2006;177:84–91. doi: 10.4049/jimmunol.177.1.84. [DOI] [PubMed] [Google Scholar]
- 9.Ge Y, Domschke C, Stoiber N, Schott S, Heil J, Rom J, et al. Metronomic cyclophosphamide treatment in metastasized breast cancer patients: immunological effects and clinical outcome. Cancer Immunol Immunother. 2011 doi: 10.1007/s00262-011-1106-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gritzapis AD, Voutsas IF, Baxevanis CN. Ontak reduces the immunosuppressive tumor environment and enhances successful therapeutic vaccination in HER-2/neu-tolerant mice. Cancer Immunol Immunother. 2011 doi: 10.1007/s00262-011-1113-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Litzinger MT, Fernando R, Curiel TJ, Grosenbach DW, Schlom J, Palena C. IL-2 immunotoxin denileukin diftitox reduces regulatory T cells and enhances vaccine-mediated T-cell immunity. Blood. 2007;110:3192–3201. doi: 10.1182/blood-2007-06-094615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jahng A, Maricic I, Aguilera C, Cardell S, Halder RC, Kumar V. Prevention of Autoimmunity by Targeting a Distinct, Noninvariant CD1d-reactive T Cell Population Reactive to Sulfatide. J Exp Med. 2004;199:947–957. doi: 10.1084/jem.20031389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Toura I, Kawano T, Akutsu Y, Nakayama T, Ochiai T, Taniguchi M. Cutting edge: inhibition of experimental tumor metastasis by dendritic cells pulsed with alpha-galactosylceramide. J Immunol. 1999;163:2387–2391. [PubMed] [Google Scholar]
- 14.Crowe NY, Smyth MJ, Godfrey DI. A critical role for natural killer T cells in immunosurveillance of methylcholanthrene-induced sarcomas. J Exp Med. 2002;196:119–127. doi: 10.1084/jem.20020092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smyth MJ, Thia KY, Street SE, Cretney E, Trapani JA, Taniguchi M, et al. Differential tumor surveillance by natural killer (NK) and NKT cells. J Exp Med. 2000;191:661–668. doi: 10.1084/jem.191.4.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Terabe M, Swann J, Ambrosino E, Sinha P, Takaku S, Hayakawa Y, et al. A nonclassical non-Va14Ja18 CD1d-restricted (type II) NKT cell is sufficient for down-regulation of tumor immunosurveillance. J Exp Med. 2005;202:1627–1633. doi: 10.1084/jem.20051381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ambrosino E, Terabe M, Halder RC, Peng J, Takaku S, Miyake S, et al. Cross-regulation between type I and type II NKT cells in regulating tumor immunity: A new immunoregulatory axis. J Immunol. 2007;179:5126–5136. doi: 10.4049/jimmunol.179.8.5126. [DOI] [PubMed] [Google Scholar]
- 18.Berzofsky JA, Terabe M. NKT cells in tumor immunity: opposing subsets define a new immunoregulatory axis. J Immunol. 2008;180:3627–3635. doi: 10.4049/jimmunol.180.6.3627. [DOI] [PubMed] [Google Scholar]
- 19.Tahir SM, Cheng O, Shaulov A, Koezuka Y, Bubley GJ, Wilson SB, et al. Loss of IFN-gamma production by invariant NK T cells in advanced cancer. J Immunol. 2001;167:4046–4050. doi: 10.4049/jimmunol.167.7.4046. [DOI] [PubMed] [Google Scholar]
- 20.Fujii S, Shimizu K, Klimek V, Geller MD, Nimer SD, Dhodapkar MV. Severe and selective deficiency of interferon-gamma-producing invariant natural killer T cells in patients with myelodysplastic syndromes. Br J Haematol. 2003;122:617–622. doi: 10.1046/j.1365-2141.2003.04465.x. [DOI] [PubMed] [Google Scholar]
- 21.Molling JW, Kolgen W, van der Vliet HJ, Boomsma MF, Kruizenga H, Smorenburg CH, et al. Peripheral blood IFN-gamma-secreting Valpha24+Vbeta11+NKT cell numbers are decreased in cancer patients independent of tumor type or tumor load. Int J Cancer. 2005;116:87–93. doi: 10.1002/ijc.20998. [DOI] [PubMed] [Google Scholar]
- 22.Terabe M, Ambrosino E, Takaku S, O'Konek JJ, Venzon D, Lonning S, et al. Synergistic enhancement of CD8+ T cell-mediated tumor vaccine efficacy by an anti-transforming growth factor-beta monoclonal antibody. Clin Cancer Res. 2009;15:6560–6569. doi: 10.1158/1078-0432.CCR-09-1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Terabe M, Shimizu M, Mabuchi A, Matui S, Morikawa H, Kaneda K, et al. Unresponsiveness of intrahepatic lymphocytes to bacterial superantigen: rapid development of suppressive Mac-1(high) cells in the mouse liver. Hepatology. 2000;32:507–513. doi: 10.1053/JHEP.2000.9875. [DOI] [PubMed] [Google Scholar]
- 24.O'Konek JJ, Illarionov P, Khursigara DS, Ambrosino E, Izhak L, Castillo BF, 2nd, et al. Mouse and human iNKT cell agonist beta-mannosylceramide reveals a distinct mechanism of tumor immunity. J Clin Invest. 2011:683–694. doi: 10.1172/JCI42314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Golgher D, Jones E, Powrie F, Elliott T, Gallimore A. Depletion of CD25+regulatory cells uncovers immune responses to shared murine tumor rejection antigens. Eur J Immunol. 2002;32:3267–3275. doi: 10.1002/1521-4141(200211)32:11<3267::AID-IMMU3267>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 26.Teng MW, Yue S, Sharkey J, Exley MA, Smyth MJ. CD1d activation and blockade: a new antitumor strategy. J Immunol. 2009;182:3366–3371. doi: 10.4049/jimmunol.0802964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Terabe M, Matsui S, Noben-Trauth N, Chen H, Watson C, Donaldson DD, et al. NKT cell-mediated repression of tumor immunosurveillance by IL-13 and the IL-4R-STAT6 pathway. Nature Immunology. 2000;1:515–520. doi: 10.1038/82771. [DOI] [PubMed] [Google Scholar]
- 28.Powell DJ, Jr, Attia P, Ghetie V, Schindler J, Vitetta ES, Rosenberg SA. Partial reduction of human FOXP3+ CD4 T cells in vivo after CD25-directed recombinant immunotoxin administration. J Immunother. 2008;31:189–198. doi: 10.1097/CJI.0b013e31815dc0e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Powell DJ, Jr, Felipe-Silva A, Merino MJ, Ahmadzadeh M, Allen T, Levy C, et al. Administration of a CD25-directed immunotoxin, LMB-2, to patients with metastatic melanoma induces a selective partial reduction in regulatory T cells in vivo. J Immunol. 2007;179:4919–4928. doi: 10.4049/jimmunol.179.7.4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Park JM, Terabe M, van den Broeke LT, Donaldson DD, Berzofsky JA. Unmasking immunosurveillance against a syngeneic colon cancer by elimination of CD4+ NKT regulatory cells and IL-13. International J of Cancer. 2004;114:80–87. doi: 10.1002/ijc.20669. [DOI] [PubMed] [Google Scholar]
- 31.Hirahara K, Liu L, Clark RA, Yamanaka K, Fuhlbrigge RC, Kupper TS. The majority of human peripheral blood CD4+CD25highFoxp3+ regulatory T cells bear functional skin-homing receptors. J Immunol. 2006;177:4488–4494. doi: 10.4049/jimmunol.177.7.4488. [DOI] [PubMed] [Google Scholar]
- 32.Ambrosino E, Berzofsky JA, Terabe M. Regulation of tumor immunity: the role of NKT cells. Expert Opin Biol Ther. 2008;8:725–734. doi: 10.1517/14712598.8.6.725. [DOI] [PubMed] [Google Scholar]
- 33.Terabe M, Matsui S, Park J-M, Mamura M, Noben-Trauth N, Donaldson DD, et al. Transforming Growth Factor-β production and myeloid cells are an effector mechanism through which CD1d-restricted T cells block Cytotoxic T Lymphocyte-mediated tumor immunosurveillance: abrogation prevents tumor recurrence. J Exp Med. 2003;198:1741–1752. doi: 10.1084/jem.20022227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yue SC, Nowak M, Shaulov-Kask A, Wang R, Yue D, Balk SP, et al. Direct CD1d-mediated stimulation of APC IL-12 production and protective immune response to virus infection in vivo. J Immunol. 2010;184:268–276. doi: 10.4049/jimmunol.0800924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yue SC, Shaulov A, Wang R, Balk SP, Exley MA. CD1d ligation on human monocytes directly signals rapid NF-kappaB activation and production of bioactive IL-12. Proc Natl Acad Sci U S A. 2005;102:11811–11816. doi: 10.1073/pnas.0503366102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Curtin JF, Candolfi M, Fakhouri TM, Liu C, Alden A, Edwards M, et al. Treg depletion inhibits efficacy of cancer immunotherapy: implications for clinical trials. PLoS One. 2008;3:e1983. doi: 10.1371/journal.pone.0001983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Uldrich AP, Patel O, Cameron G, Pellicci DG, Day EB, Sullivan LC, et al. A semi-invariant Valpha10+ T cell antigen receptor defines a population of natural killer T cells with distinct glycolipid antigen-recognition properties. Nat Immunol. 2011;12:616–623. doi: 10.1038/ni.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]