

Abstract
The joint sequencing of related genomes has become an important means to discover rare variants. Normal-tumor genome pairs are routinely sequenced together to find somatic mutations and their associations with different cancers. Parental and sibling genomes reveal de novo germline mutations and inheritance patterns related to Mendelian diseases.
Acute lymphoblastic leukemia (ALL) is the most common paediatric cancer and the leading cause of cancer-related death among children. With the aim of uncovering the full spectrum of germline and somatic genetic alterations in childhood ALL genomes, we conducted whole-exome re-sequencing on a unique cohort of over 120 exomes of childhood ALL quartets, each comprising a patient's tumor and matched-normal material, and DNA from both parents. We developed a general probabilistic model for such quartet sequencing reads mapped to the reference human genome. The model is used to infer joint genotypes at homologous loci across a normal-tumor genome pair and two parental genomes.
We describe the algorithms and data structures for genotype inference, model parameter training. We implemented the methods in an open-source software package (QUADGT) that uses the standard file formats of the 1000 Genomes Project. Our method's utility is illustrated on quartets from the ALL cohort.
Background
Acute lymphoblastic leukemia (ALL) is the most common paediatric cancer and the leading cause of cancer-related death among children. Advances in the understanding of the pathobiology of ALL have led to risk-targeted treatment regimes and increased survival rates, but treatment is still far from optimal. Childhood ALL arises after the acquisition of a series of DNA sequence abnormalities. These initiating events, or so-called driver mutations, ultimately confer a selective growth advantage, and are causally implicated in cancer development. A central goal of cancer genome analysis is the identification of cancer genes that, by definition, carry driver mutations.
Next-generation sequencing (NGS) technologies [1] have enabled the genome-wide identification of human disease-related variants. Analysis pipelines have been established for the large-scale sequencing of individual tumor genomes [2]. Briefly, short sequencing reads are collected from the tumor sample, mapped to the reference genome assembly, and the set of aligned reads are used to infer variations across the genome at homologous loci covered with multiple reads. The sequence variants in the tumor genome may be the result of somatic mutations, or constitutional variants preserved in the somatic lineage. In order to distinguish somatic mutations from conserved variants, it is necessary to sequence normal and tumor samples side by side. The Cancer Genome Atlas Network [3] catalogues somatic mutations in different cancers using such normal-tumor pairs.
In general, genetic relationships (like normal-tumor pairs) can be efficiently exploited in genotype inference [4,5]. Inherited and de novo mutations can be traced through jointly sequenced family relatives [6]. Here, we consider variant detection in normal-tumor pairs coupled with parental samples. Such quartet data are used to categorize variants in the tumor and normal genomes by their origin: see Figure 1. One can readily classify inherited variants and de novo germline mutations by comparing the genotypes in the trio of normal and parental genomes. Likewise, somatic mutations correspond to differences between the normal and tumor genomes.
Figure 1.

Alignments for the normal, tumor, and parental reads samples highlight putative somatic and germline mutations. The illustration shows a region along chromosome 12, displayed in the Integrative Genomics Viewer [7]. Mismatched bases are highlighted.
While sequencing an entire human genome is still too expensive for the average research laboratory, various target-enrichment techniques [8] are available for sequencing only regions of interest. In particular, sequencing the so-called exome covering all gene-coding regions, has been a routine step in medical applications [9]. Through our ongoing paediatric oncogenomics study, we conducted whole exome deep re-sequencing of a unique cohort of over 120 exomes of childhood ALL quartets, consisting of the patient's tumor and matched-normal material as well as DNA from both parents.
Existing software tools
Various bioinformatics tools have been developed for genotyping individual genomes from sequencing data, including SNVMix [10], VarScan [11], and The Genome Analysis Toolkit GATK [12,13]. A couple of methods have been developed for the purpose of joint genotyping of paired normal-tumor samples, including SomaticSniper [14], MutationSeq [15], and JointSNVMix [16]. SomaticSniper and MutationSeq employ machine-learning techniques for variant classification; JointSNVMix is based on a full Bayesian model incorporating prior genotype distributions, somatic mutations, and sequencing base call errors. The Strelka software package [17] infers joint tumor-normal genotypes in a Bayesian model that also considers tumor sampling impurity: DNA collected from the tumor sample is usually "contaminated" to some degree with the normal tissue, and therefore the sequencing reads come from a mixture of normal and tumor genomes. To our knowledge, no existing variant caller incorporates somatic and germline mutation models simultaneously to handle quartet data as in our data sets.
Our contribution
We infer the four genotypes jointly in a framework that respects the rules of inheritance in the germline and somatic lineages. Aside from assigning belief to de novo and somatic mutations, we hypothesized, constrained patterns in one lineage have an indirect beneficial effect on the inference in other lineages. In particular, the "triangulation" of the normal genome by related genomes means that genotypes and lineage-specific mutations can be resolved more reliably: information from the parental genotypes reinforce the inference of somatic mutations, and tumor sequencing reads help to recognize constitutional mutations. We present a Bayesian framework that incorporates prior parental genotypes, inherited, de novo and somatic mutations, as well as tumor-sampling impurity and sequencing errors. All model parameters are estimated in an expectation-maximization algorithm [18].
Methods
Probabilistic model
Figure 2 illustrates our model of single-nucleotide polymorphisms at homologous loci across four genomes linked by inheritance and somatic mutations. The model quantifies the descent-by-modification relationships between the unknown genotypes via three sets of parameters. First, a genotype frequency model is assumed for the parental genotypes. Second, we assume a standard DNA substitution model for the frequency of germline mutations. The parental diploid genotypes determine the child's normal genotype by Mendelian inheritance. (For simplicity, we discuss only diploid genotypes: our implementation considers sex chromosomes in an analogous manner, but using the appropriate inheritance model.) Finally, another DNA substitution model with its own parameters determines mutations in the tumor genome.
Figure 2.

Probabilistic model for base calls covering a single locus, with dependencies among genotypes and sequencing reads. See Equation (5) for the corresponding likelihood formula.
Base calls are assumed to be independent between different loci. The input for genotype inference at a single locus consists of nucleotide base calls made with their accompanying sequencing error probabilities.
Parental genotype priors
Let π[x] denote the frequency of each allele x ∈ {A; C; G; T} at a given locus. The prior allele frequencies are computed by using a standard DNA substitution model quantifying the divergence from the reference genome assembly. Assuming the simple Jukes-Cantor model with a reference nucleotide genotype for and , where v is the parental genome's divergence from the reference. More general divergence models and known SNP frequencies can be accommodated by using , where is the model's substitution probability from to and is a known allele frequency.
For a diploid locus, let denote the frequency of homozygous and heterozygous genotypes. Allele frequencies immediately determine diploid allele frequencies in standard Hardy-Weinberg equilibrium: for a homozygous locus , and for a heterozygous . In order to accommodate different homozygous-heterozygous ratios, we employ an additional parameter .
| (1) |
(In other words, γ is numerically analogous to the so-called inbreeding coefficient, or F-statistic.) It is easy to verify that haploid allele frequencies are the same at any γ setting.
Mutations and inheritance
The child's normal genotype is determined by Mendelian inheritance and de novo point mutations with probabilities vx→y that occur within the parental germlines. For simplicity, germline mutations in a parental lineage (gF, gM for father and mother, respectively) are conceived of as mutations that result in a diploid genotype, which then determine the child's normal genotype by Mendel's laws.
Germline mutations follow standard molecular evolution model for substitutions in DNA. Let X denote the parent's normal allele at a locus, and X' denote the same allele at the end of the germline before gametogenesis. The mutation model specifies the probabilities that apply to every locus . Let denote the probability of normal genotype gN given the mutated parental genotypes . Then χ may be 0, 1, 1/2 or 1/4, depending on the common alleles between the three genotypes.
Tumor genotype
The tumor genome undergoes mutations following the same type of molecular evolution model as the one used for germline mutations, but has its own parameters.
Sequencing errors
The alignment at the locus is represented as a set of basecall-error probability pairs (zk, ∈k) for k = 1, ..., m. The k-th read calls allele zk with an error probability of 0 <∈k ≤ 1. Typically, the aligned sequencing reads give nucleotide base calls and accompanying error probabilities on a logarithmic integer scale [19,20], called the Phred scale. (In principle, the input SAM- or BAM-format file gives the Sanger-encoded sequencing error in the QUAL column or the OQ tag.) Namely,
where qk is the reported quality score, and . Let Z denote the base call, and Y denote the true nucleotide. We assume that errors are unbiased in the sense that
Allele sampling and sample impurity
Aligned sequencing reads randomly sample the haploid alleles at a given locus. Let yk be the true allele for base call zk. The locus' diploid genotype determines the frequency for each possible allele y. At a homozygous locus xx, ρ[y] = 1 and ρ[x'] = 0 for all x' ≠ x. At a heterozygous locus xx', ρ[y] = 1/2, ρ[x'] = 1/2 and ρ[x"] = 0 for all x" ≠ x, x'.
Impure tumor samples have a mixed distribution, which is the linear combination of the normal and tumor genotype distributions. For tumor reads, ρ[y] comes from a mixture (0 ≤ ω ≤ 1; ω = 1 for pure tumor sampling) between the normal and tumor genotypes:
| (3) |
Likelihood for aligned reads given the genotypes
Suppose we are given the set of basecall-error probability pairs (zk, ∈k) for k = 1, ..., m, representing the alignment at a locus. Let Zk be the random variable for base call in read k at a fixed error rate ∈k, and let Yk be the random variable for the true sampled allele. Define the base call probability
Hence, .
The read likelihood for a given allele distribution ρ is defined as
Since base calls are independent across reads when conditioned on the allele mixture in the sample,
| (4) |
Complete likelihood
Let , , , denote the diploid genotypes for father, mother, normal, and tumor samples, respectively. Let and denote the parental genotypes after germline mutations. These six random variables constitute the hidden variables in our probabilistic model. The input is a set of aligned sequenced bases from each of the four samples:, , , . Every set consists of base call and error pairs (zk, ∈k). The likelihood for the aligned base calls is then
| (5) |
The factor is the likelihood for the reads, given the genotypes. By the independence of the sequencing runs,
| (6) |
The four factors are defined by (4), via the allele frequencies ρ that are determined by genotypes and tumor sampling purity, as discussed above (see Allele sampling and sample impurity).
The factor in (5) covers all mutation and inheritance events, as well as the parental genotypes. By the dependencies depicted in Figure 2,
| (7) |
Algorithmic techniques and data structures
Our algorithmic solutions address the efficient calculation of the likelihood formula of (5), and its use in an Expectation-Maximization (EM) framework for model parameter setting. First, we examine a straightforward decomposition of the likelihood formula dictated by the assumed probabilistic graphical model.
For the EM algorithm, we need to recompute likelihoods and posterior probabilities in a number of iterations, which can be directly achieved by storing all sequencing reads in memory, but such an approach may be costly. We scrutinize the computation of read likelihoods, in order to arrive at an economical data structure, also discussed in some detail, that eliminates the need to store all base calls in memory.
Likelihood decomposition
The summation formula for the full likelihood in Equation (5) is rearranged for efficiency, using the independencies apparent in (6) and (7). In addition, the germline mutations can be combined with Mendelian inheritance: define as
Then
| (8) |
If there are G possible diploid genotypes (G = 10 for DNA with four alleles), Equation (8) shows that the likelihood can be computed in O(G3) time, instead of O(G6) suggested by the definition of (5). In particular, the likelihood computation proceeds by calculating the following values.
Read likelihoods
Equation (4) is conveniently rearranged by different base calls:
| (9) |
Each subproduct is calculated separately as
| (10) |
by Equation (2). Note that only the called base's frequency β = ρ[z] appears in the formula. Define
If no reads call z, then Tβ[z] = 1. Equation (10) becomes Lz(ρ) = Tρ[z][z]. For pure diploid samples, ρ[z] may be 0, 1 or 1/2, corresponding to the possible subproducts for diploid samples E[y] = T0[y] (sequencing error), C[y] = T1[y] (homozygote yy), and H[y] = T1/2[y] (heterozygote with y). Likelihood formulas become even more economical with normalized subproducts C'[y] = C[y]/E[y], H'[y] = H[y]/E[y], and scaling factor
Homozygous sample. For a pure homozygous sample with genotype yy (ρ[y] = 1 and ρ[z] = 0 for z ≠ y),
| (11) |
Heterozygous sample. For a pure heterozygous sample yy' (y ≠ y'; ρ[y] = ρ[y'] = 1/2 and ρ[z] = 0 for z ≠ y, y'), the likelihood becomes
| (12) |
Impure tumor sample. The pure tumor and normal genotypes are proper diploid genotypes. Tumor sequencing reads come from an impure sample: they sample the tumor genotype with probability ω, and the normal genotype with probability (1 - ω). By Eq. (3), identical genotypes correspond to identical allele frequencies ρ, no matter what the purity level ω is. Suppose, however, that the locus has a mixture of divergent normal and tumor genotypes. Figure 3 shows that there are up to four correct base calls appearing in the reads, depending on the tumor mutation pattern . There are 6 possible queried allele frequencies β ≠ 0, 1, 1/2 (Figure 3):
Figure 3.
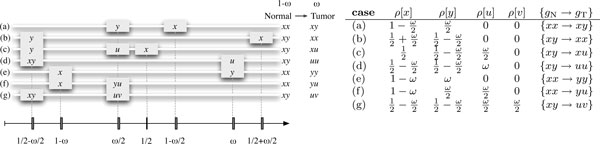
Allele frequencies with impure normal-tumor genotypes that differ from each other. Reads sample the tumor with probability ω, and the normal genotype with 1 - ω.
| (13) |
If the normal and tumor genotypes are identical, then the purity is immaterial. The formulas with somatic mutations are:
| (14a) |
| (14b) |
| (14c) |
| (14d) |
| (14e) |
| (14f) |
| (14g) |
Data structure for storing base calls
Storing individual base calls at each locus is costly, because the sequencing coverage may be large across the four samples. It suffices, however, to store the partial likelihood factors appearing in Equations (11), (12) and (14). In particular, for each locus with mapped base calls, it is enough to store the sample-specific H'[x], C'[x], and values for each called base x and the six possible values of β, in addition to a single scaling value E. For a sample with m base calls at the locus, (1 + 8m) variables thus suffice, independently of read coverage. The stored partial likelihoods are reused throughout the iterations optimizing the model parameters at a fixed tumor purity level ω.
Recurrent base calls
In our experience, loci with identical sets of base calls reoccur at an appreciable frequency, especially at lower coverages (less than about 15×) that characterize exon boundaries in exome sequencing. We exploit recurrent patterns in the piled-up base calls to achieve even better memory usage and speed. Namely, we sort the base calls at a given locus for a given sample (by allele and quality score), and use run-length encoding to achieve a compact characterization . The encoding is not used for higher coverages or widely varying quality scores, where h would take too many bits. Information-theoretic considerations [21] suggest that compactly encoded occur more often ( is our proxy for Kolmogorov complexity). Short codes are placed in a small hash table to find recurrent calls (in our experiments with 20-30× total coverage by AB SOLiD sequencing reads, 20-30% savings can be achieved this way).
Parameter optimization and genotype inference
The genotypes, germline and tumor mutations are inferred by carrying out the summation of (5) for a restricted set of genotypes in order to calculate posterior probabilities. For example, in order to infer the child's normal genotype, calculate first
Then the child's normal genotype has posterior probabilities .
Model parameters are optimized using the EM algorithm [18]. In one iteration, likelihoods and various posterior probabilities are computed across all loci in the so-called E-step, which are then used to set the model parameters for the next iteration in the so-called M-step. The iterations continue until convergence is achieved. Among the optimized model parameters, the parental genotype priors, the germline and tumor mutation parameters are optimized through multiple iterations using the same set of precalculated partial likelihoods.
Setting the single parameter of the Jukes-Cantor model for the germline and tumor mutations is fairly straightforward by using posterior probabilities. For example, the tumor mutation parameter is set by summing the posterior probabilities for allele substitutions across all loci:
| (15) |
where N is the number of loci, is the posterior probability for a normal-tumor genotype pair at locus j, and α(g, g') is the expected number of substitutions for the two alleles given the diploid genotypes.
In order to set the tumor purity ω, the partial likelihoods need to be recomputed (for different Tβ values) by reading the input read-mapping files at each iteration. At the same time, we compute a calibrated map from reported base-calling qualities to sequencing errors in the same framework. Note that both μ and ω have well-estimated initial values (μ starts with the canonical Phred-scaled values, and ω is estimated experimentally).
Decomposing zygosity and divergence
For the purposes of parameter inference, consider the following machine realizing the formulas for the parental genotype priors of (1). Upon receiving a heterozygous genotype xy, it flips either allele to output homozygous xx or yy with γ /2 probability each. Otherwise, with probability (1 - γ), the output is the same heterozygote xy. Homozygous genotypes are output without any change. Clearly, if the input genotype distribution is for Hardy-Weinberg, then the machine's output is distributed by the probabilities of (1). Accordingly, the divergence and heterozygosity parameters for the parental genotype prior ϕ are inferred by treating the machine's input genotype as a hidden variable. Expected frequencies for divergent input genotypes are used to estimate the divergence parameter, and expected frequencies for heterozygous → homozygous "flips" are used to estimate γ.
Sequencing data
Exome sequencing
We conducted validation experiments using exome-sequencing reads for two sets of quartets (A and B) generated on the Child Health Genomics Platform of the Sainte-Justine UHC Research Center. Sequencing reads were produced on Applied Biosystem's SOLiD sequencer and mapped with the accompanying software. Table 1 summarizes statistics on the mapped sequencing reads.
Table 1.
Coverage statistics for two quartets used in validation experiments.
| Exome sequencing (AB SOLiD system) | |||||
|---|---|---|---|---|---|
| Total | Normal | Tumor | Father | Mother | |
| Quartet A (chromosome 12 only) | |||||
| Sites: | 11 458 426 | ||||
| Reads: | 5 530 702 | 1 454 529 | 1 396 361 | 1 117 647 | 1 562 165 |
| Depth: | 23.0 | 6.0 | 5.8 | 4.6 | 6.6 |
| Experimental tumor purity: | 0.63 | ||||
| QUADGT's purity estimate: | 0.41 | ||||
| Quartet B (all chromosomes) | |||||
| Sites: | 425 344 130 | ||||
| Reads: | 134 574 732 | 38 156 404 | 34 580 382 | 29 997 426 | 31 840 520 |
| Depth: | 23.0 | 5.4 | 5.1 | 6.5 | 5.9 |
| Experimental tumor purity: | 0.97 | ||||
| QUADGT's purity estimate: | 0.44 | ||||
| Whole-genome sequencing (Illumina HiSeq 2000) | |||||
| Quartet B (whole genome) | |||||
| Total | Normal | Tumor | |||
| Depth: | 123.8 | 48.6 | 75.2 | ||
| Illumina's purity estimate: | 0.46 | ||||
Whole-genome sequencing
Tumor and normal DNA samples from Quartet B were submitted to Illumina, Inc, for deep whole-genome sequencing using the standard operating procedures of the HiSeq 2000 sequencing platform. Table 1 summarizes coverage statistics and tumor impurity. The whole-genome data was further analyzed for somatic mutations with CASAVA and Strelka [17] by Illumina, Inc.
Implementation
We incorporated the presented methods into an open-source Java software package called QUADGT, using the standard file formats of the 1000 Genomes Project (SAM v1.4 [20] for input and VCF v4.1 [22] for output). Any of the input files may be missing, which makes QUADGT suitable to analyze sets with just normal-tumor samples, or just parental-offspring trios.
The probabilistic framework enabled us to couple the inference with confidence measures in the form of quality scores computed from posterior probabilities. The quality scores accompany sample-specific genotype calls (VCF's GQ field), as well as the joint genotyping for the four samples (VCF's QUAL column). The posterior probabilities for germline and somatic mutations are calculated, as well, by summing across all pertinent genotype assignments. The program specifically introduces ambiguity in the genotyping calls to meet prescribed quality scores: a definite x/y base call in a sample is replaced by x/. or ./y, and then by ./. in a greedy manner, in order to achieve high specificity.
Parallelization
The E-step of the optimization, where sums of posterior probabilities are calculated across loci, is well-suited to parallelization. In our implementation, we use a hash key computed at each locus to assign the calculations to different computing threads running in parallel.
Availability
The QUADGT software package is publicly available at http://www.iro.umontreal.ca/~csuros/quadgt/.
Results and discussion
We used two quartet data sets (A and B) to compare independent and joint variant detection. Figure 1 summarizes the coverage statistics for the quartets. Exome-sequencing reads at 5-6× coverage per sample were mapped to the human reference, and we inferred genome variants using our software package QUADGT. The entire analysis pipeline for one quartet set, including model training and genotyping, took about 12 hours (wall-clock time) on standard multi-core computer workstations with 16 Gbytes of memory.
Exome-sequencing data from Quartet A was used to assess the concordance of genotyping calls by QUADGT and a well-established variant caller, The Genome Analysis Toolkit [12]. We used independently produced whole-genome (WG) sequencing reads for the normal-tumor pair in Quartet B (with 124× total coverage, see Table 1) to gauge the two variant callers' sensitivity.
Concordance experiments
Table 2 compares individual genotyping calls made by the Genome Analysis Toolkit [12] and QUADGT on a small example consisting of calls on chromosome 12 (parameters were set to result in a comparable number of genotyping calls). The table illustrates that most calls are made in agreement between the two programs. The known relationships between the samples ensure the consistency of calls made by QUADGT, resulting in only 4 putative de novo germline mutations. GATK, ignorant of the relations, has 327 cases where a normal allele does not appear at either parent, which is by at least two magnitudes higher than what one would expect based on human intergeneration mutation rates [6]. GATK genotypes imply a large number (520) of somatic mutations, as well. As with de novo mutations, the joint calls by QUADGT are more conservative: only 14 somatic mutation calls are made.
Table 2.
Comparison of calls made by the Genome Analysis Toolkit and QUADGT on Quartet A.
| Normal genome | |
|---|---|
| Heterozygous SNPs (ref/alt) | Homozygous SNPs (alt/alt) |
| called by both QUADGT and GATK: 2100 | called by both QUADGT and GATK: 945 |
| called by GATK only: 60 | called by GATK only: 500 |
| QUADGT calls ref/ref: 4 | QUADGT calls ref/ref: 0 |
| QUADGT calls alt/alt: 9 | QUADGT calls ref/alt: 206 |
| called by QUADGT only: 839 | called by QUADGT only: 17 |
| GATK calls alt/alt: 206 | GATK calls ref/alt: 9 |
| De novo mutations | |
| called by both QUADGT and GATK: 4 | |
| called by GATK only: 327 | |
| called by QUADGT only: 0 | |
| Tumor genome | |
| Heterozygous SNPs (ref/alt) | Homozygous SNPs (alt/alt) |
| called by both QUADGT and GATK: 2032 | called by both QUADGT and GATK: 938 |
| called by GATK only: 41 | called by GATK only: 504 |
| QUADGT calls ref/ref: 3 | QUADGT calls ref/ref: 0 |
| QUADGT calls alt/alt: 7 | QUADGT calls ref/alt: 224 |
| called by QUADGT only: 989 | called by QUADGT only: 17 |
| GATK calls alt/alt: 224 | GATK calls ref/alt: 7 |
| Somatic mutations | |
| called by both QUADGT and GATK: 8 | |
| called by GATK only: 512 | |
| called by QUADGT only: 6 |
Sensitivity assessment
The normal-tumor pair in Quartet B was submitted to Illumina, Inc. for deep whole-genome (WG) sequencing and somatic mutation calling. Table 3 tallies the WG somatic calls with coverage by exome data. Based on the WG sequencing data, 1817 loci have somatic mutations, of which 40 are covered by exome reads to sufficient depth (Table 3, b). Some of the 40 WG somatic calls have no or weak support (lines c and e) in exome reads, since with at most one exception, all normal and tumor base calls are identical with the reference. The remaining 24 WG somatic calls (line f) with sufficient exome read coverage and non-reference base calls are not beyond the reach of the variant callers to discover somatic mutations. On closer inspection, 5 WG somatic calls fall into a 150 bp region (line g), which, judged by largely divergent base calls, is likely to be misaligned; 4 WG somatic calls (line h) may even be erroneous. For instance, at chr8:10078796, which has a fairly low WG somatic quality score, the parental exome reads suggest that all four samples contain a heterozygous SNP that is in fact found in dbSNP (rs112078536). The remaining 15 WG somatic calls (line i) have support in the exome reads, and QUADGT discovers them all at some quality threshold cutoffs.
Table 3.
Whole-genome somatic loci and exome genotyping on Quartet B.
| Locus | WG genotyping | Exome base calls (A:C:G:T) | QuadGT call | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| mutation | quality | N | T | F | M | N | T | F | M | |
| chr4:85818319 | GG > AG | 68 | 0:0:8:0 | 2:0:6:0 | 0:0:9:0 | 0:0:9:0 | 0/0 | 0/0 | 0/0 | 0/0 |
| chr6:29965983 | TT > CT | 17 | 0:1:0:27 | 0:1:0:25 | 0:3:0:0 | 0:0:0:43 | 0/0 | 0/0 | 0/1 | 0/0 |
| chr8:10078796 | GG > CG | 15 | 0:2:1:0 | 0:2:3:0 | 0:5:2:0 | 0:2:4:0 | 0/1 | 0/1 | 0/1 | 0/1 |
| chr12:25289551 | CC > TC | 29 | 0:37:0:1 | 0:31:0:2 | 0:57:0:0 | 1:45:0:0 | 0/0 | 0/0 | 0/0 | 0/0 |
Genome coordinates refer to NCBI 36.1/hg18 genome build.
a. WG somatic loci in exome reads: 274
b. exome coverage ≥ 10: 40
c. only ref base calls in normal and tumor: 12
d. at least one non-ref base call in normal and tumor: 28 (= c - d)
e. exactly 1 non-ref base call in normal and tumor: 4
f. at least 2 non-ref base call in normal and tumor: 24 (= e - f)
g. within misaligned region chr19:4463156-4463205: 5
h. strong disagreement between exome and WG reads: 4
i. QUADGT calls SOMATIC: 15 (= f - (g + h)) -- see Table 4
Table 4 compares the sensitivity of joint and independent genotyping using the 15 WG somatic calls with support in exome base calls (Table 3, line i). First, it is notable that QUADGT's tumor purity estimation (by Expectation-Maximization) is close to the Illumina's estimate from whole-genome data (see Table 1).
Table 4.
Exome somatic calls supported by whole-genome data.
| Locus | Whole-genome | Exome | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| mutation | score | Quality score | Rank | Base calls | (A:C:G:T) | ||||||
| qGT | GATK | qGT | GATK | Normal | Tumor | Father | Mother | ||||
| 1 | chr20:577556 | GG > AG | 61 | 282 | 99,99 | 1 | 1 | 0:0:90:0 | 27:0:64:0 | 1:0:104:0 | 1:0:90:0 |
| 2 | chr18:26965596 | AA > GA | 87 | 282 | 99,99 | 1 | 1 | 77:0:1:2 | 63:1:20:0 | 106:0:0:0 | 84:0:0:0 |
| 3 | chr9:139897201 | GG > AG | 59 | 91 | 66,99 | 16 | 73 | 0:0:23:0 | 6:0:18:0 | 0:0:24:0 | 0:0:21:0 |
| 4 | chr2:88942555 | GG > TG | 93 | 53 | - | 84 | - | 0:0:29:0 | 0:0:5:3 | 0:0:36:0 | 0:0:26:0 |
| 5 | chr2:88942554 | AA > CA | 93 | 52 | - | 86 | - | 30:0:0:0 | 0:0:6:3 | 0:0:36:0 | 0:0:26:0 |
| 6 | chr7:150399212 | CC > TC | 93 | 48 | 60,99 | 98 | 104 | 0:20:0:0 | 1:6:0:9 | 0:14:0:0 | 0:13:0:0 |
| 7 | chrX:129017457 | CC > AC | 41 | 46 | 45,99 | 109 | 221 | 0:15:0:0 | 5:6:0:0 | 0:16:0:0 | 0:12:0:0 |
| 8 | chr16:3681082 | CC > TC | 89 | 44 | 30,82 | 118 | 629 | 0:10:0:0 | 0:10:0:4 | 0:19:0:0 | 0:11:0:0 |
| 9 | chr10:107013067 | CC > AC | 30 | 36 | - | 186 | - | 0:52:0:0 | 5:57:0:1 | 0:74:0:0 | 0:72:1:0 |
| 10 | chr8:72396806 | TT > CT | 59 | 24 | - | 398 | - | 0:0:0:6 | 0:3:0:13 | 0:0:0:20 | 0:0:0:11 |
| 11 | chrX:21779651 | GG > TG | 89 | 21 | 36,99 | 483 | 412 | 2:0:13:0 | 0:0:7:8 | 0:0:9:0 | 0:0:14:0 |
| 12 | chr5:125673987 | CC > TC | 97 | 14 | 6,70 | 790 | 7393 | 0:2:0:0 | 0:7:0:3 | 0:6:0:0 | 0:5:0:0 |
| 13 | chr12:10156011 | GG > AG | 89 | 8 | - | - | - | 0:0:16:0 | 3:0:8:0 | 0:0:28:0 | 0:0:9:0 |
| 14 | chr19:43602412 | GG > AG | 32 | 5 | - | - | - | 0:0:10:0 | 2:0:10:0 | 0:1:17:0 | 0:0:10:0 |
| 15 | chr6:112619582 | GG > AG | 68 | 3 | - | - | - | 0:0:5:0 | 2:1:5:0 | 0:0:6:0 | 0:0:5:0 |
Somatic mutation calls of QUADGT (qGT columns) and the Genome Analysis Toolkit (GATK columns) were ranked by quality score (smaller of normal and tumor genotype call qualities in GATK's VCF output, and score of somatic call in QUADGT's VCF output that is q = - 10 log10(1 - p) for a mutation with posterior probability p).
Rank is "-" at loci where no somatic mutations were inferred by QUADGT or GATK. Genome coordinates refer to NCBI 36.1/hg18 genome build.
Table 4 suggests that the joint variant-calling in QUADGT leads to better sensitivity than GATK, which does not consider the relations between the genomes. In particular, 9 out of 276 (3%) SOMATIC calls by QUADGT with quality score at least 30 have support in the WG data, whereas only 7 of GATK's 667 (1%) divergent genotypes of same quality threshold are validated. At a quality score cutoff of 20, 12 out of 555 (2%) and 7 out of 1312 (0.5%) are validated QUADGT and GATK predictions.
Conclusions
Sequencing multiple genomes with known pedigrees or clonal relationships has a great promise for understanding the development of particular diseases. Our experiments with sequenced quartets of parents and normal-tumor pairs illustrate that the joint calls improve the reliability of inferred de novo and somatic mutations. The constraints imposed by the known relationships greatly improve the consistency of calls between different samples, and ultimately help to delineate the single nucleotide polymorphisms that can be associated with the disease.
A future release of the software is now under development that incorporates more nuanced substitution models with variable rates, transition-transversion ratios and nucleotide composition, as well as site-specific priors relying on public variant databases and gene annotations.
Competing interests
The authors declare that they have no competing interests.
Acknowledgements
The authors are indebted to the patients and their parents for participating in this study. This study was supported by research funds provided by the Terry Fox Research Institute, the Canadian Institutes for Health Research, and Canada's National Sciences and Engineering Research Council. JFS is the recipient of a Cole Foundation studentship. RV is the recipient of a post-doctoral research fellowship from the Government of Canada through the "Foreign Affairs and International Trade Canada." DS holds the François-Karl-Viau Research Chair in Pediatric Oncogenomics.
Declarations
Funding for the publication of this article was provided by a grant from the National Sciences and Engineering Research Council.
This article has been published as part of BMC Bioinformatics Volume 14 Supplement 5, 2013: Proceedings of the Third Annual RECOMB Satellite Workshop on Massively Parallel Sequencing (RECOMB-seq 2013). The full contents of the supplement are available online at http://www.biomedcentral.com/bmcbioinformatics/supplements/14/S5.
References
- Shendure J, Li H. Next-generation DNA sequencing. Nature Biotechnology. 2008;26(10):1135–1145. doi: 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- Wood LD, Parsons DW, Jones S, Lin J, Sjöblum T. et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318(5853):1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le SQ, Durbin R. SNP detection and genotyping from low-coverage sequencing data on multiple diploid samples. Genome Research. 2011;21(6):952–960. doi: 10.1101/gr.113084.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Chen W, Zhan X, Busonero F, Sanna S, Sidore C, Cucca F, Kang HM, Abecasis GR. A likelihood-based framework for variant calling and De Novo mutation detection in families. PLoS Genetics. 2012;8(10):e1002944. doi: 10.1371/journal.pgen.1002944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach JC, Glusman G, Smit AFA, Huff CD, Hubley R, Shannon PT, Rowen L, Pant KP, Goodman N, Bamshad M, Shendure J, Drmanac R, Jorde LB, Hood L, Galas DJ. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science. 2010;328(5978):636–639. doi: 10.1126/science.1186802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Intergrative genomics viewer. Nature Biotechnology. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamanova L, Coffey AJ, Scott CE, Kozarewa I, Turner EH, Kumar A, Howard E, Shendure J, Turner DJ. Target-enrichment strategies for next-generation sequencing. Nature Methods. 2010;7:111–118. doi: 10.1038/nmeth.1419. [DOI] [PubMed] [Google Scholar]
- Teer JK, Mullikin JC. Exome sequencing: the sweet spot before whole genomes. Human Molecular Genetics. 2010;19:R145–R151. doi: 10.1093/hmg/ddq333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goya R, Sun MG, Morin RD, Leung G, Ha G, Wiegand KC, Senz J, Crisan A, Marra MA, Hirst M, Huntsman D, Murphy KP, Aparicio S, Shah SP. SNVMix: predicting single nucleotide variants from next-generation sequencing of tumors. Bioinformatics. 2010;26(6):730–736. doi: 10.1093/bioinformatics/btq040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt DC, Chen K, Wylie T, Larson DE, McLellan MD, Mardis ER, Weinstock GM, Wilson RK, Ding L. VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics. 2009;25(17):2283–2285. doi: 10.1093/bioinformatics/btp373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research. 2010;20(9):1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature Genetics. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson DE, Harris CC, Chen K, Koboldt DC, Abbott TE, Dooling DJ, Ley TJ, Mardis ER, Wilson RK, Ding L. SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics. 2012;28(3):311–317. doi: 10.1093/bioinformatics/btr665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J, Bashashati A, Roth A, Oloumi A, Tse K, Zeng T, Haffari G, Hirst M, Marra MA, Condon A, Aparicio S, Shah SP. Feature based classifiers for somatic mutation detection in tumour-normal paired sequencing data. Bioinformatics. 2011. http://bioinformatics.oxfordjournals.org/content/early/2011/11/13/bioinformatics.btr629.abstract [DOI] [PMC free article] [PubMed]
- Roth A, Ding J, Morin R, Crisan A, Ha G, Giuliany R, Bashashati A, Hirst M, Turashvili G, Oloumi A, Marra MA, Aparicio S, Shah SP. JointSNVMix: a probabilistic model for accurate detection of somatic mutations in normal/tumour paired next-generation sequencing data. Bioinformatics. 2012;28(7):907–913. doi: 10.1093/bioinformatics/bts053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders CT, Wong WSW, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28(14):1811–1817. doi: 10.1093/bioinformatics/bts271. [DOI] [PubMed] [Google Scholar]
- Dempster AP, Laird NM, Rubin DP. Maximum likelihood from incomplete data via the EM algorithm. Journal of the Royal Statistical Society Series B. 1977;39:1–38. [Google Scholar]
- Ewing B, Green P. Base-calling of automated sequencer traces using Phred: II. error probabilities. Genome Research. 1998;8:186–194. [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Subgroup GPDP. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Vitányi P. An Introduction to Kolmogorov Complexity and Its Applications. 3. Springer Science+Business Media; 2008. [Google Scholar]
- Danecek P, Auton A. et al. The variant call format and VCFTools. Bioinformatics. 2011;27(15):2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]