

Abstract
Hypoxia-inducible factors (HIFs) are heterodimeric oxygen-sensitive basic helix-loop-helix transcription factors that play central roles in cellular adaptation to low oxygen environments. The von-Hippel Lindau tumor suppressor (pVHL) is the substrate recognition component of an E3 ubiquitin ligase and functions as a master regulator of HIF activity by targeting the hydroxylated HIF-alpha subunit for ubiquitylation and rapid proteasomal degradation under normoxic conditions. Mutations in pVHL can be found in familial and sporadic hemangioblastomas, clear cell carcinomas of the kidney, pheochromocytomas and inherited forms of erythrocytosis, illustrating the importance of disrupted molecular oxygen sensing in the pathogenesis of these diseases. Tissue-specific gene targeting of pVHL in mice has demonstrated that efficient execution of HIF proteolysis is critically important for normal tissue physiology, and has provided novel insights into the functional consequences of HIF activation on the cellular and tissue level. Here we focus on the contribution of individual HIF transcription factors to the development of VHL phenotypes and discuss how the pVHL/HIF axis could be exploited pharmacologically.
Keywords: von Hippel-Lindau (VHL) tumor suppressor, hypoxia-inducible factor (HIF), renal cell cancer, hemangioblastoma, erythropoietin, anemia, metabolism, kidney cysts, mouse model
INTRODUCTION
Patients with germ line mutations in the von Hippel-Lindau (VHL) tumor suppressor are affected by a rare familial tumor syndrome, which is characterized by the predisposition to develop highly vascularized tumors in multiple organs. These include hemangioblastomas of the retina and central nervous system, renal cancer of the clear cell type (CC-RCC) and pheochromocytomas [1]. VHL disease is grouped into 2 subtypes depending on the presence or absence of pheochromocytoma. Different clinical subtypes are associated with specific VHL mutations, which result in distinct functional and biochemical properties of the mutated VHL gene product, pVHL [2–4]. Biallelic VHL inactivation has been documented in the majority of sporadic CC-RCCs [5], but is less prominent in sporadic hemangioblastomas. pVHL has multiple functions. A major function is serving as the substrate recognition component of an E3 ubiquitin ligase, which ubiquitylates and targets the α-subunit of hypoxia-inducible factor (HIF) for oxygen-dependent proteolysis [6–18] (Fig. (1)). Therefore either loss of VHL expression as a result of gene deletion or promoter hypermethylation, or mutations in pVHL that affect its ability to capture and/or ubiquitylate HIF-α result in constitutive HIF stabilization and activation of HIF controlled transcriptional programs irrespective of oxygen levels. The pVHL/HIF-α interaction is highly conserved between species, underscoring its importance in molecular oxygen sensing. In order to examine the functional role of pVHL-mediated HIF proteolysis and to gain insights into the physiological consequences of constitutive HIF activation during embryonic development, and in the adult, we and other laboratories have used Cre-loxP recombination to generate mice with cell type- and tissue-specific VHL inactivation. Here, we discuss the contribution of individual HIF transcription factors to the development of the VHL phenotype and how the VHL/HIF axis could be exploited therapeutically.
Fig. 1.
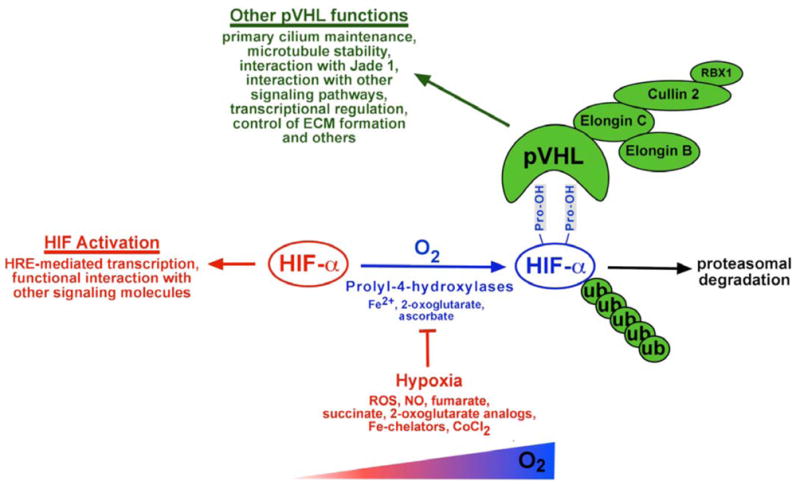
pVHL: master regulator of HIF. Schematic overview of pVHL functions. Aside from targeting HIF-α for proteasomal degradation, pVHL has multiple other functions. These include maintenance of the primary cilium, regulation of microtubule stability and interactions with several other signaling pathways. Binding to hydroxylated HIF-α occurs at the β-domain of pVHL, which spans amino acid residues 64 – 154. The C-terminal α-domain links pVHL via elongin C to the E3 ubiquitin ligase. Indicated are also conditions and molecules, which inhibit HIF prolyl-hydroxylation. Abb.: CoCl2, cobalt chloride; NO, nitric oxide; ROS, reactive oxygen species; ub, ubiquitin.
pVHL: MASTER REGULATOR OF HIF
HIFs belong to the PAS (Per-ARNT-Sim) family of heterodimeric basic helix-loop-helix (bHLH) transcription factors. They regulate gene expression by binding to hypoxia-response elements (HREs), specific DNA recognition sequences located in hypoxia enhancer containing regulatory regions of HIF target genes, and consist of an oxygen-sensitive α-subunit and a constitutively expressed β-subunit, also known as the arylhydrocarbon receptor nuclear translocator (ARNT) or HIF-β [19, 20]. There are three known HIF α-subunits, HIF-1α, HIF-2α and HIF-3α, [18, 21], which are all are targeted by pVHL. Whereas HIF-1α and HIF-2α heterodimers function as transcriptional activators, the role of HIF-3α in transcriptional activation is less clear. Splice variants of HIF-3α have been shown inhibit HIF- dependent transcriptional activation [22, 23]. The spectrum of HIF-1 and HIF-2 regulated biological functions overlaps only partially. Anaerobic glycolysis, for example, appears to be predominantly controlled by HIF-1 [24], whereas HIF-2 appears to be the main regulator of erythropoietin (EPO) production and seems to have a distinct role in VHL-associated tumorigenesis [25–29].
pVHL-mediated polyubiquitylation of HIF-α requires hydroxylation on specific proline residues (Pro402 and Pro564 in human HIF-1α; Pro405 and Pro531 in human HIF-2α) within its oxygen-dependent degradation domain (ODD) [30–36] and is necessary for its rapid proteasomal degradation under normoxic conditions. Hydroxylation of HIF-α is carried out by 2-oxoglutarate-dependent dioxygenases (prolyl-4-hydroxylase domain (PHD) proteins) and requires molecular oxygen, ferrous iron and ascorbate [37]. A defect in the ability to ubiquitylate HIF-α, i.e. as a consequence of mutated pVHL or loss of VHL expression, results in HIF-α stabilization, increased HIF transcriptional activity and up-regulation of HIF target genes such as vascular endothelial growth factor (VEGF), glucose transporter 1 (GLUT-1) and EPO irrespective of oxygen levels. In addition to heterodimerization with HIF-β, HIF-α modulates cellular signaling pathways through functional interaction with non-PAS domain proteins. These include, among others, tumor suppressor protein p53, the c-Myc proto-oncogene and the Notch intracellular domain [38–41]. These and other HIF-α/non-PAS domain protein interactions have to be considered when interpreting phenotypes associated with VHL inactivation. A second hypoxic switch operates in the carboxy-terminal transactivation domain of HIF-α with oxygen dependent asparagine hydroxylation by factor-inhibiting-HIF (FIH) blocking CBP/p300 recruitment. Inhibition of FIH under hypoxic conditions facilitates CBP/p300 recruitment, resulting in increased HIF target gene expression in VHL-deficient cell lines or under pronounced hypoxia [42–45].
Certain mutations in the VHL gene result in the development of familial polycythemia from excessive EPO production [46], but not in tumorigenesis. Chuvash polycythemia, a rare autosomal-recessive disease, which is endemic in central European Russia, is mostly associated with a mutation in VHL codon 200 (C598T ⇒ R200W). Affected individuals, who are usually homozygous for this hypomorphic mutation (compound heterozygotes with other mutations have also been reported), are not predisposed to the development of tumors that are typically associated with VHL disease [47]. When expressed in murine ES cells (R166W) or in VHL-defective renal carcinoma cells the pVHL R200W mutant retained the ability to target HIF-α (HIF-1α > HIF-2α) for proteasomal degradation, but at reduced efficiency [48], whereas VHL alleles (e.g. VHL type 1 mutations) that are highly associated with the development of renal cell cancers have completely lost their ability to capture HIF-α for degradation [3, 4].
HIF-INDEPENDENT FUNCTIONS OF pVHL
The identification of HIF-independent pVHL functions and discovery of novel pVHL interacting proteins has provided important molecular insights into the pathogenesis of VHL-associated clinical phenotypes and represents an important step towards a complete understanding VHL-associated tumorigenesis. pVHL has been shown to regulate microtubule stability and is important for cilia maintenance [49–52]. It furthermore controls the stability of plant homeodomain protein Jade-1 [53, 54]. These observations provide a molecular basis for the pathogenesis of VHL-associated cysts, which are prominent visceral manifestations of VHL disease in the kidney and pancreas. Cystogenesis has been associated with the loss of the primary cilium, a microtubule based organelle, which functions as a luminal flow- and potential chemosensor on epithelial cells, and is also found on many other cell types including neurons, photoreceptors and fibroblasts [55]. Recent studies indicated that pVHL cooperates with glycogen synthase kinase (GSK)-3β in an interlinked signaling pathway that maintains the primary cilium [50]. This notion is supported by findings in knockout mice where inhibition of the PTEN tumor suppressor, which activates PI3K-Akt signaling thereby inhibiting GSK-3 activity, promotes urogenital cyst formation in a pVHL defective background [56, 57]. While these studies suggested HIF independence, Esteban et al. proposed a role for HIF in cilium maintenance [58]. Jade-1 is a recently identified pVHL interacting protein that links pVHL to the cysto- and oncogenic β-catenin signaling pathway (pVHL suppresses β-catenin signaling through Jade-1) [59]. Other pVHL modulated cellular processes or signaling pathways that are likely to be important for VHL-associated tumorigenesis and tumor progression include extra-cellular matrix (ECM) assembly and turnover [60–65], the formation of intracellular junctions [66], signaling through atypical protein kinase C isoforms [67–71] and NF-κB [72], c-Met receptor responsiveness to hepatocyte growth factor (HGF) [63, 73], and signaling through the regulation of p53 transcriptional activity [74]. In addition, pVHL has been shown to interact with KRAB-A domain protein, VHLak, repressing HIF transcriptional activity [75], deubiquitylating enzymes [76], the large subunit of RNA polymerase II [77] and the RNA-binding protein hnRNP A2 [78]. The role of these proteins in the pathogenesis of VHL-associated tumors, however, is unclear and awaits further study.
pVHL IS REQUIRED FOR NORMAL EMBRYONIC DEVELOPMENT
Germ line inactivation of pVHL in mice results in embryonic death during midgestation primarily from abnormal placental vascularization. Placentae from VHL-deficient embryos lack properly developed syncytiotrophoblasts and labyrinths and show evidence of hemorrhage by embryonic day 11.5 to 12.5 [79]. This phenotype appears to be largely HIF-mediated, as germ line inactivation of PHD2 result in similar, but not identical pathology [80], which may indicate HIF independent functions of either pVHL and/or PHD2 during placental development. pVHL is not only essential for normal placental development, but plays a critical role in the development, growth and differentiation of many other tissues. For example, tissue-specific inactivation of pVHL in neuro-epithelial progenitor cells results in abnormal neuronal differentiation and embryonic lethality during late gestation (V.H. Haase, unpublished data), and chondrocyte specific inactivation of pVHL in the growth plate causes stunted bone growth, most likely a result of a HIF dependent increase in cell cycle inhibitor p57kip2 [81].
HIF AND THE VHL PHENOTYPE
VHL haploinsufficincy (VHL +/−) in the murine germ line predisposes mice to the development of cavernous liver hemangiomas [82], which is strongly dependent on genetic background [83], and represents a rare manifestation of VHL disease in human patients [84, 85]. Since hepatocyte-specific deletion of VHL, phenocopied the liver pathology found in heterozygous mice, it is most likely that, following Knudson’s two-hit hypothesis, inactivation of the remaining VHL wild-type allele resulted in the formation of cavernous hemangiomas through uncontrolled and HIF-dependent vascular growth factor production in hepatocytes and not endothelial cells (Fig. (2)). This is mechanistically similar to human VHL-associated hemangioblastomas, in which “stromal cells” and not endothelial cells are VHL-deficient and represent the neoplastic component of these tumors [86]. Inactivation of HIF signaling (pVHL/ARNT double mutants) resulted in a rescue of the liver phenotype and restored normal hepatic gene expression [26], indicating that the effects of VHL inactivation in hepatocytes are entirely HIF dependent. In keeping with this notion, forced expression of non-degradable HIF-1α and HIF-2α phenocopied VHL phenotypes in liver and skin [87].
Fig. 2.
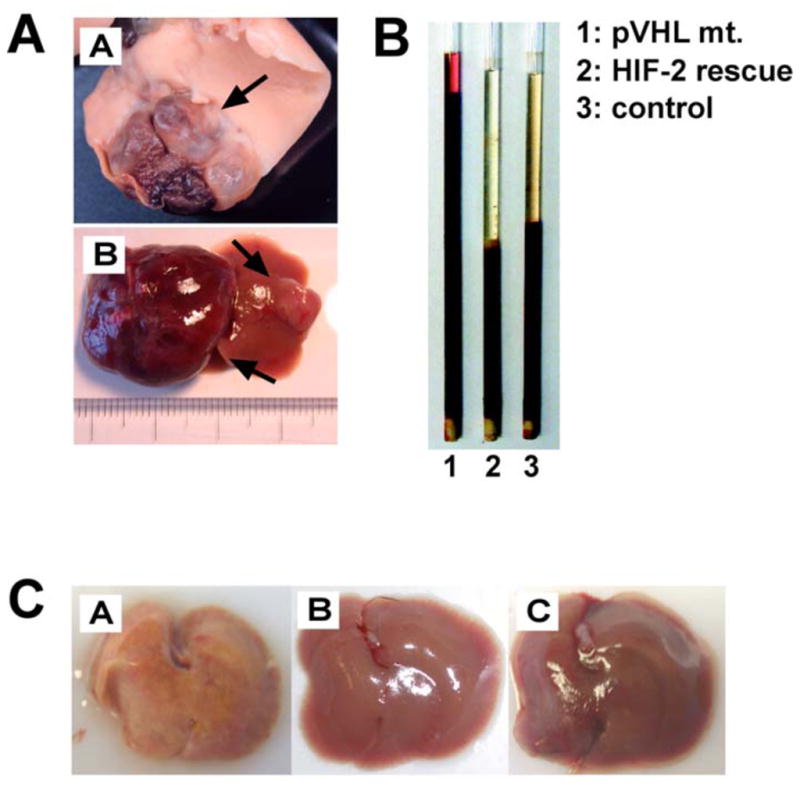
Functional consequences of pVHL inactivation in hepatocytes. Inactivation of pVHL in hepatocytes results in multiple phenotypes. A. VHL-associated hemangiomas. Mice with germ line haploinsufficiency for pVHL (VHL +/−) develop cavernous liver hemangiomas (A). Hemangioma development (arrows) also occurs when pVHL is inactivated conditionally using Cre-loxP recombination. 20–30 % of hepatocytes are targeted in the case shown in (B). B. Erythrocytosis. Hepatocytes are capable of producing EPO when HIF is stabilized. EPO production in the liver is HIF-2-dependent and inactivation of HIF-2 in pVHL deficient livers restores normal erythropoiesis. Shown are spun micro-hematocrits. 1: pVHL mutant mouse (20–30% of hepatocytes are targeted) with hematocrit > 90%; 2: pVHL mutant animal in which HIF-2 was inactivated simultaneously; 3: control mouse. C. Steatohepatitis. Inactivation of pVHL in > 80% of hepatocytes results in severe HIF-2-dependent steatohepatitis, illustrated by the yellow discoloration of the liver. (A), pVHL mutant; (B), pVHL/HIF-2 double mutant liver; (C), control liver.
To determine the role of individual HIF transcription factors (HIF-1 versus HIF-2) in the development of VHL-associated liver hemangiomas, we performed genetic studies with VHL knockout mice, which also lacked HIF-1α, HIF-2α or both in hepatocytes. Analysis of these mice demonstrated that inactivation of HIF-2α was sufficient to prevent the development of cavernous hemangiomas, whereas deletion of HIF-1α did not affect vascular tumorigenesis [29]. This HIF-2-dependent vascular tumor phenotype correlated with a HIF-2-dependent increase in angiogenic gene expression [27, 29], and is in contrast to other VHL-deficient organs, where vascular phenotypes (increased vascular density, but not tumors) were found to be HIF-1-dependent [88], indicating that the ability of HIF-1 and HIF-2 to regulate common target genes is tissue- and context-dependent [89]. Whether preferential recruitment of HIF-1 or HIF-2 to hypoxia enhancer elements requires tissue-specific transcriptional co-activators, certain DNA or protein modifications, absence of a recently postulated titratable repressor or other signaling events awaits further investigation [90]. The findings from the genetic analysis of mouse hemangioma development are in line with clinical studies in human patients, which have demonstrated a correlation between HIF-2α expression and the development of VHL-associated angiogenic lesions [91], and identify HIF-2 as a potential therapeutic target for the treatment of these tumors.
Inactivation of pVHL in hepatocytes furthermore results in the development of HIF-2-dependent steatosis (accumulation of neutral fat in hepatocytes) and erythrocytosis (Fig. (2)). Lipid accumulation is also found in stromal cells, the neoplastic components of VHL-associated hemangioblastomas [92], and in CC-RCCs, which are distinguished histologically from other types of renal cancer by the presence of ‘clear’ cytoplasm resulting from the washout of lipids during tissue processing.
Renal cysts, a major visceral manifestation of VHL disease in human patients [1, 93], were only found at very low frequency in VHL +/− mice (<5%) [82]. VHL-associated renal cysts can be malignant or benign, and should be viewed as pre-neoplastic lesions. Mice with tissue-specific inactivation of pVHL in the proximal renal tubule develop renal cysts at a frequency of ~20%, however CC-RCCs were not observed in these studies [82, 83, 94] (Fig. (3)). Renal cystogenesis was found to be HIF-, but not HIF-1-dependent, suggesting that HIF-2 may act as a cystogenic transcription factor [94]. The notion of HIF having cystogenic properties is supported by in vivo experiments with CC-RCC cell lines where re-introduction of wild-type pVHL restored primary cilia in a HIF dependent manner [58], and by a mouse model of hereditary leiomyomatosis and renal cancer syndrome (HLRCC), where a genetic defect in fumarate hydratase, which results in HIF-α stabilization (fumarate inhibits HIF prolyl-hydroxylation, see Fig. (1)), led to kidney cyst development in aged mice [95]. These findings, however, would have to be reconciled with reports that suggest HIF-independent mechanisms by which pVHL suppresses cyst formation [50–52].
Fig. 3.

Proposed HIF-dependent and HIF-independent functions of pVHL in VHL-associated tumorigenesis. A clinical hallmark of VHL disease is the development of CNS hemangioblastomas and clear cell carcinoma of the kidney (CC-RCC). Panel A: Pathogenesis of VHL-associated hemangiomas/hemangioblastomas. Mice with germ line mutations in pVHL (VHL+/−) are predisposed to the development of liver hemangiomas (photograph). Genetic studies in mice demonstrated exclusive HIF-2-dependence, which is in line with HIF expression studies in human hemangioblastomas. Panel B: Pathogenesis of VHL-associated renal cancer. The pathogenesis of CC-RCC is more complex and most likely involves both, HIF-dependent and HIF independent functions of pVHL, as well as other genetic events that lead to malignant transformation. Shown is the macroscopic (A) and microscopic (B) appearance of large cortical renal cysts in mouse kidneys with pVHL inactivation (arrows and stars). (B), magnification x100. (C) and (D), human CC-RCC; (C), gross photography; (D), clear cell histology (arrows), magnification x400. Images (C) and (D) were kindly provided by Dr. John Tomaszewski, Department of Pathology, University of Pennsylvania, Philadelphia, PA.
The absence of CC-RCC development in VHL-deficient mice in conjunction with genetic data from human patients indicates that transformation of cystic epithelium into CC-RCC requires additional genetic events, such as mutations in other tumor suppressor genes or oncogenes. Loss of pVHL function and stabilization of HIF-α, however, represent the earliest detectable molecular events in renal tumorigenesis [96], resulting in cellular alterations, which ultimately facilitate transformation to and progression of CC-RCC. These include HIF-dependent and HIF-independent loss of intercellular junctions and epithelial de-differentiation resulting from E-cadherin suppression [66, 97–99], HIF-dependent and HIF-independent alterations in p53, c-Myc or NF-κB activity [38, 39, 72, 74, 100], HGF signaling [63, 73, 101], increased susceptibility to transforming growth factor (TGF)-α/epidermal growth factor (EGF) signaling [89, 102–106], as well as modifications in ECM turnover and re-modeling [60–65].
The importance of HIF activation in CC-RCC pathogenesis and growth is underscored by reports, which demonstrated that inhibition of HIF-α translation correlated with reduced tumor growth [107] and that the expression of certain HIF target genes, such as CXC chemokine receptor-4 (CXCR4) was associated with disease progression [108]. Since a bias towards HIF-2α expression was found in clinical CC-RCC samples with confirmed VHL defect and in CC-RCC cell lines [18, 109], VHL-associated tumor development may depend on de novo expression of HIF-2α or a shift in the ratio of HIF-1α versus HIF-2α levels towards an increase in HIF-2α (HIF-2α is not detectable in non-transformed renal epithelial cells following hypoxia/ ischemic injury [110]). In support of this notion are studies in CC-RCC cell lines, which indicate that HIF-2 is oncogenic and is able to override pVHL’s tumor suppressor function by regulating molecular pathways that are critical for renal cell growth, such as signaling through the TGF-α/EGF receptor pathway, cyclin D1 and the c-Myc proto-oncogene [89, 100, 103–106, 111–115]. Taken together, there is substantial evidence that HIF-1 and HIF-2 have diverse functions with regard to VHL-associated renal tumorigenesis, which offers potential for therapeutic exploitation.
THE VHL/HIF AXIS AS A THERAPEUTIC TARGET
Pharmacological targeting of the VHL/HIF/PHD axis offers enormous opportunities for the treatment of anemia and ischemic disorders, since HIF activation induces EPO and appears to mediate the tissue protective effects of ischemic preconditioning [116]. HIF hydroxylation is a prerequisite for binding to the pVHL-E3 ubiquitin ligase complex and proteasomal degradation in the presence of oxygen. Stabilization of HIF-α under normoxia results from the inability to hydroxylate HIF-α or to efficiently ubiquitylate hydroxylated HIF-α, which can be inherited or somatically acquired as is the case in HLRCC, VHL disease and in sporadic CC-RCCs, or it can result from pharmacological inhibition. Analogs of 2-oxoglutarate, which is the substrate for PHD enzymes (PHD1, PHD2, PHD3 are the three major HIF prolyl-4-hydroxylases) and FIH, have been successfully used for the stimulation of endogenous EPO production in vivo, and furthermore have the potential to improve the clinical outcome of acute ischemic injuries as demonstrated in animal models [117–119]. Some compounds have entered clinical trials for the treatment of renal anemia (patients with chronic kidney disease loose the ability to produce adequate amounts of EPO) and are awaiting safety and efficacy evaluations.
In theory, it is conceivable that small molecule compounds could also be used to disrupt the VHL/HIF-α interaction, resulting in temporary HIF-α stabilization, while HIF-independent tumor suppressor functions of pVHL would be left intact. Hydroxylated HIF-α interacts with the β-domain of pVHL, which spans amino acid residues 64–154. This strategy was successfully used to stabilize HIF-α in cell lines using an engineered protein that contained the HIF-ODD fused to green fluorescent protein and resulted in disruption of the pVHL interaction with endogenous HIF-α [112].
In mouse models, our laboratory has shown that genetic inactivation of pVHL in hepatocytes results in HIF-2-dependent erythrocytosis from constitutive EPO production (a 2- to 3-fold rise in serum EPO levels is sufficient to produce erythrocytosis) [27]. This finding indicated that the VHL/HIF axis could be exploited therapeutically in clinical situations, where the ability of the kidney to generate adequate amounts of EPO is impaired, i.e. anemia of chronic kidney disease (the kidney is the main physiologic source of EPO in adults). Since constitutive activation of hepatic HIF can result in severe organ pathology, such as steatohepatitis and hemangioma development, pharmacological targeting of the VHL/HIF-α interaction would have to be intermittent in order to suppress these non-desirable on-target effects. Central questions in the design of strategies that aim at harnessing the VHL/HIF/PHD pathway for therapeutic purposes relate to whether intermittent normoxic HIF stabilization and activation of HIF controlled transcriptional programs is oncogenic or has other adverse clinical effects on the human body, such as alterations in glucose and fat metabolism. Both, inhibiting HIF prolyl-4-hydroxylation with 2-oxoglutarate analogs and disruption of the pVHL/ HIF-α interaction face the same clinical challenges and questions as far as HIF activation and patient safety is concerned. Aside from safety issues relating to HIF, disruption of the pVHL/HIF-α interaction has the potential to affect HIF-independent tumor suppressor functions of pVHL, which would have to be evaluated carefully before this strategy to activate HIF can be considered as an alternative to PHD inhibition.
CONCLUDING REMARKS AND FUTURE DIRECTIONS
Over the last 10 years major advances have been made in understanding molecular functions of the VHL tumor suppressor as they relate to tumorigenesis and normal physiology. Although targeting of HIF-α for proteolysis appears to be the dominant biological function of pVHL, there is growing evidence for pVHL regulated cellular processes that are critical for tumorigenesis and do not involve HIF signaling. It is likely that a direct comparison of PHD and pVHL deficiency phenotypes in the absence or presence of HIF will help to identify additional HIF-independent biological functions of either pVHL or PHDs.
Mouse models, clinical and in vitro studies have been used to define the role of individual HIF transcription factors in the development of VHL phenotypes, and studies with pVHL knockout mice have helped to identify novel HIF regulated cellular processes and target genes. These investigations have established that HIF-1 and HIF-2, although structurally closely related, have distinct cell type- and context-dependent biological functions with potential for therapeutic exploitation. In the non-oncologic setting, pharmacological targeting of the VHL/HIF/PHD axis with the goal to activate HIF under normoxic conditions, may be useful in the treatment of anemia and ischemic disorders in the short term and is currently undergoing clinical evaluation. Whether long-term treatment with HIF stabilizing compounds is safe will have to be carefully established.
Acknowledgments
VHH is supported by the Krick-Brooks chair in Nephrology and by grants from the National Cancer Institute (NCI) and the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK).
Footnotes
AUTHOR STATEMENT
The author states that no conflict of interest exists.
References
- 1.Lonser RR, Glenn GM, Walther M, Chew EY, Libutti SK, Linehan WM, et al. von Hippel-Lindau disease. Lancet. 2003;361:2059–67. doi: 10.1016/S0140-6736(03)13643-4. [DOI] [PubMed] [Google Scholar]
- 2.Neumann HP, Bender BU. Genotype-phenotype correlations in von Hippel-Lindau disease. J Intern Med. 1998;243:541–5. doi: 10.1046/j.1365-2796.1998.00336.x. [DOI] [PubMed] [Google Scholar]
- 3.Clifford SC, Cockman ME, Smallwood AC, Mole DR, Woodward ER, Maxwell PH, et al. Contrasting effects on HIF-1alpha regulation by disease-causing pVHL mutations correlate with patterns of tumourigenesis in von Hippel-Lindau disease. Hum Mol Genet. 2001;10:1029–38. doi: 10.1093/hmg/10.10.1029. [DOI] [PubMed] [Google Scholar]
- 4.Hoffman MA, Ohh M, Yang H, Klco JM, Ivan M, Kaelin WG., Jr von Hippel-Lindau protein mutants linked to type 2C VHL disease preserve the ability to downregulate HIF. Hum Mol Genet. 2001;10:1019–27. doi: 10.1093/hmg/10.10.1019. [DOI] [PubMed] [Google Scholar]
- 5.Gnarra JR, Tory K, Weng Y, Schmidt L, Wei MH, Li H, et al. Mutations of the VHL tumour suppressor gene in renal carcinoma. Nat Genet. 1994;7:85–90. doi: 10.1038/ng0594-85. [DOI] [PubMed] [Google Scholar]
- 6.Kibel A, Iliopoulos O, DeCaprio JA, Kaelin WG., Jr Binding of the von Hippel-Lindau tumor suppressor protein to Elongin B and C [see comments] Science. 1995;269:1444–6. doi: 10.1126/science.7660130. [DOI] [PubMed] [Google Scholar]
- 7.Pause A, Lee S, Worrell RA, Chen DY, Burgess WH, Linehan WM, et al. The von Hippel-Lindau tumor-suppressor gene product forms a stable complex with human CUL-2, a member of the Cdc53 family of proteins. Proc Natl Acad Sci USA. 1997;94:2156–61. doi: 10.1073/pnas.94.6.2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kamura T, Koepp DM, Conrad MN, Skowyra D, Moreland RJ, Iliopoulos O, et al. Rbx1, a component of the VHL tumor suppressor complex and SCF ubiquitin ligase [see comments] Science. 1999;284:657–61. doi: 10.1126/science.284.5414.657. [DOI] [PubMed] [Google Scholar]
- 9.Stebbins CE, Kaelin WG, Jr, Pavletich NP. Structure of the VHL-ElonginC-ElonginB complex: implications for VHL tumor suppressor function. Science. 1999;284:455–61. doi: 10.1126/science.284.5413.455. [DOI] [PubMed] [Google Scholar]
- 10.Feldman DE, Thulasiraman V, Ferreyra RG, Frydman J. Formation of the VHL-elongin BC tumor suppressor complex is mediated by the chaperonin TRiC. Mol Cell. 1999;4:1051–61. doi: 10.1016/s1097-2765(00)80233-6. [DOI] [PubMed] [Google Scholar]
- 11.Iwai K, Yamanaka K, Kamura T, Minato N, Conaway RC, Conaway JW, et al. Identification of the von Hippel-lindau tumor-suppressor protein as part of an active E3 ubiquitin ligase complex. Proc Natl Acad Sci USA. 1999;96:12436–41. doi: 10.1073/pnas.96.22.12436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lisztwan J, Imbert G, Wirbelauer C, Gstaiger M, Krek W. The von Hippel-Lindau tumor suppressor protein is a component of an E3 ubiquitin-protein ligase activity. Genes Dev. 1999;13:1822–33. doi: 10.1101/gad.13.14.1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Salceda S, Caro J. Hypoxia-inducible factor 1alpha (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J Biol Chem. 1997;272:22642–7. doi: 10.1074/jbc.272.36.22642. [DOI] [PubMed] [Google Scholar]
- 14.Cockman ME, Masson N, Mole DR, Jaakkola P, Chang GW, Clifford SC, et al. Hypoxia inducible factor-alpha binding and ubiquitylation by the von Hippel-Lindau tumor suppressor protein. J Biol Chem. 2000;275:25733–41. doi: 10.1074/jbc.M002740200. [DOI] [PubMed] [Google Scholar]
- 15.Tanimoto K, Makino Y, Pereira T, Poellinger L. Mechanism of regulation of the hypoxia-inducible factor-1 alpha by the von Hippel-Lindau tumor suppressor protein. EMBO Jl. 2000;19:4298–309. doi: 10.1093/emboj/19.16.4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ohh M, Park CW, Ivan M, Hoffman MA, Kim TY, Huang LE, et al. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat Cell Biol. 2000;2:423–27. doi: 10.1038/35017054. [DOI] [PubMed] [Google Scholar]
- 17.Kamura T, Sato S, Iwai K, Czyzyk-Krzeska M, Conaway RC, Conaway JW. Activation of HIF1alpha ubiquitination by a reconstituted von Hippel-Lindau (VHL) tumor suppressor complex. Proc Natl Acad Sci USA. 2000;97:10430–5. doi: 10.1073/pnas.190332597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis [see comments] Nature. 1999;399:271–5. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 19.Semenza GL. Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annu Rev Cell Dev Biol. 1999;15:551–78. doi: 10.1146/annurev.cellbio.15.1.551. [DOI] [PubMed] [Google Scholar]
- 20.Wenger RH, Stiehl DP, Camenisch G. Integration of oxygen signaling at the consensus HRE. Sci STKE. 2005;2005:re12. doi: 10.1126/stke.3062005re12. [DOI] [PubMed] [Google Scholar]
- 21.Maynard MA, Qi H, Chung J, Lee EH, Kondo Y, Hara S, et al. Multiple splice variants of the human HIF-3alpha locus are targets of the VHL E3 ubiquitin ligase complex. J Biol Chem. 2003;278:1032–40. doi: 10.1074/jbc.M208681200. [DOI] [PubMed] [Google Scholar]
- 22.Makino Y, Cao R, Svensson K, Bertilsson G, Asman M, Tanaka H, et al. Inhibitory PAS domain protein is a negative regulator of hypoxia-inducible gene expression. Nature. 2001;414:550–4. doi: 10.1038/35107085. [DOI] [PubMed] [Google Scholar]
- 23.Maynard MA, Evans AJ, Shi W, Kim WY, Liu FF, Ohh M. Dominant-negative HIF-3 alpha 4 suppresses VHL-null renal cell carcinoma progression. Cell Cycle. 2007;6:2810–6. doi: 10.4161/cc.6.22.4947. [DOI] [PubMed] [Google Scholar]
- 24.Hu CJ, Wang LY, Chodosh LA, Keith B, Simon MC. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol Cell Biol. 2003;23:9361–74. doi: 10.1128/MCB.23.24.9361-9374.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Warnecke C, Zaborowska Z, Kurreck J, Erdmann VA, Frei U, Wiesener M, et al. Differentiating the functional role of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha (EPAS-1) by the use of RNA interference: erythropoietin is a HIF-2alpha target gene in Hep3B and Kelly cells. Faseb J. 2004;18:1462–4. doi: 10.1096/fj.04-1640fje. [DOI] [PubMed] [Google Scholar]
- 26.Rankin EB, Higgins DF, Walisser JA, Johnson RS, Bradfield CA, Haase VH. Inactivation of the arylhydrocarbon receptor nuclear translocator (Arnt) suppresses von Hippel-Lindau disease-associated vascular tumors in mice. Mol Cell Biol. 2005;25:3163–72. doi: 10.1128/MCB.25.8.3163-3172.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rankin EB, Biju MP, Liu Q, Unger TL, Rha J, Johnson RS, et al. Hypoxia-inducible factor-2 (HIF-2) regulates hepatic erythropoietin in vivo. J Clin Invest. 2007;117:1068–77. doi: 10.1172/JCI30117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gruber M, Hu CJ, Johnson RS, Brown EJ, Keith B, Simon MC. Acute postnatal ablation of Hif-2alpha results in anemia. Proc Natl Acad Sci USA. 2007;104:2301–6. doi: 10.1073/pnas.0608382104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rankin EB, Rha J, Unger TL, Wu CH, Shutt HP, Johnson RS, et al. Hypoxia-inducible factor-2 regulates vascular tumorigenesis in mice. Oncogene. 2008;27:5354–8. doi: 10.1038/onc.2008.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–72. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 31.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–8. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 32.Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O’Rourke J, Mole DR, et al. C elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107:43–54. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- 33.Hon WC, Wilson MI, Harlos K, Claridge TD, Schofield CJ, Pugh CW, et al. Structural basis for the recognition of hydroxyproline in HIF-1 alpha by pVHL. Nature. 2002;417:975–8. doi: 10.1038/nature00767. [DOI] [PubMed] [Google Scholar]
- 34.Masson N, Willam C, Maxwell PH, Pugh CW, Ratcliffe PJ. Independent function of two destruction domains in hypoxia-inducible factor-alpha chains activated by prolyl hydroxylation. Embo J. 2001;20:5197–206. doi: 10.1093/emboj/20.18.5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu F, White SB, Zhao Q, Lee FS. HIF-1alpha binding to VHL is regulated by stimulus-sensitive proline hydroxylation. Proc Natl Acad Sci USA. 2001;98:9630–5. doi: 10.1073/pnas.181341498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294:1337–40. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- 37.Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol. 2004;5:343–54. doi: 10.1038/nrm1366. [DOI] [PubMed] [Google Scholar]
- 38.An WG, Kanekal M, Simon MC, Maltepe E, Blagosklonny MV, Neckers LM. Stabilization of wild-type p53 by hypoxia-inducible factor 1alpha. Nature. 1998;392:405–8. doi: 10.1038/32925. [DOI] [PubMed] [Google Scholar]
- 39.Ravi R, Mookerjee B, Bhujwalla ZM, Sutter CH, Artemov D, Zeng Q, et al. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia- inducible factor 1alpha. Genes Dev. 2000;14:34–44. [PMC free article] [PubMed] [Google Scholar]
- 40.Koshiji M, Kageyama Y, Pete EA, Horikawa I, Barrett JC, Huang LE. HIF-1alpha induces cell cycle arrest by functionally counteracting Myc. Embo J. 2004;23:1949–56. doi: 10.1038/sj.emboj.7600196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gustafsson MV, Zheng X, Pereira T, Gradin K, Jin S, Lundkvist J, et al. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev Cell. 2005;9:617–28. doi: 10.1016/j.devcel.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 42.Mahon PC, Hirota K, Semenza GL. FIH-1: a novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001;15:2675–86. doi: 10.1101/gad.924501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002;16:1466–71. doi: 10.1101/gad.991402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science. 2002;295:858–61. doi: 10.1126/science.1068592. [DOI] [PubMed] [Google Scholar]
- 45.Stolze IP, Tian YM, Appelhoff RJ, Turley H, Wykoff CC, Gleadle JM, et al. Genetic analysis of the role of the asparaginyl hydroxylase factor inhibiting hypoxia-inducible factor (HIF) in regulating HIF transcriptional target genes. J Biol Chem. 2004;279:42719–25. doi: 10.1074/jbc.M406713200. [DOI] [PubMed] [Google Scholar]
- 46.Cario H, Schwarz K, Jorch N, Kyank U, Petrides PE, Schneider DT, et al. Mutations in the von Hippel-Lindau (VHL) tumor suppressor gene and VHL-haplotype analysis in patients with presumable congenital erythrocytosis. Haematologica. 2005;90:19–24. [PubMed] [Google Scholar]
- 47.Ang SO, Chen H, Hirota K, Gordeuk VR, Jelinek J, Guan Y, et al. Disruption of oxygen homeostasis underlies congenital Chuvash polycythemia. Nat Genet. 2002;32:614–21. doi: 10.1038/ng1019. [DOI] [PubMed] [Google Scholar]
- 48.Hickey MM, Lam JC, Bezman NA, Rathmell WK, Simon MC. von Hippel-Lindau mutation in mice recapitulates Chuvash polycythemia via hypoxia-inducible factor-2alpha signaling and splenic erythropoiesis. J Clin Invest. 2007;117:3879–89. doi: 10.1172/JCI32614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hergovich A, Lisztwan J, Barry R, Ballschmieter P, Krek W. Regulation of microtubule stability by the von Hippel-Lindau tumour suppressor protein pVHL. Nat Cell Biol. 2003;5:64–70. doi: 10.1038/ncb899. [DOI] [PubMed] [Google Scholar]
- 50.Thoma CR, Frew IJ, Hoerner CR, Montani M, Moch H, Krek W. pVHL and GSK3beta are components of a primary cilium-maintenance signalling network. Nat Cell Biol. 2007;9:588–95. doi: 10.1038/ncb1579. [DOI] [PubMed] [Google Scholar]
- 51.Schermer B, Ghenoiu C, Bartram M, Muller RU, Kotsis F, Hohne M, et al. The von Hippel-Lindau tumor suppressor protein controls ciliogenesis by orienting microtubule growth. J Cell Biol. 2006;175:547–54. doi: 10.1083/jcb.200605092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lutz MS, Burk RD. Primary cilium formation requires von hippel-lindau gene function in renal-derived cells. Cancer Res. 2006;66:6903–7. doi: 10.1158/0008-5472.CAN-06-0501. [DOI] [PubMed] [Google Scholar]
- 53.Zhou MI, Wang H, Foy RL, Ross JJ, Cohen HT. Tumor suppressor von Hippel-Lindau (VHL) stabilization of Jade-1 protein occurs through plant homeodomains and is VHL mutation dependent. Cancer Res. 2004;64:1278–86. doi: 10.1158/0008-5472.can-03-0884. [DOI] [PubMed] [Google Scholar]
- 54.Zhou MI, Wang H, Ross JJ, Kuzmin I, Xu C, Cohen HT. The von Hippel-Lindau tumor suppressor stabilizes novel plant homeodomain protein Jade-1. J Biol Chem. 2002;277:39887–98. doi: 10.1074/jbc.M205040200. [DOI] [PubMed] [Google Scholar]
- 55.Yoder BK. Role of primary cilia in the pathogenesis of polycystic kidney disease. J Am Soc Nephrol. 2007;18:1381–8. doi: 10.1681/ASN.2006111215. [DOI] [PubMed] [Google Scholar]
- 56.Frew IJ, Thoma CR, Georgiev S, Minola A, Hitz M, Montani M, et al. pVHL and PTEN tumour suppressor proteins cooperatively suppress kidney cyst formation. Embo J. 2008;27:1747–57. doi: 10.1038/emboj.2008.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Frew IJ, Minola A, Georgiev S, Hitz M, Moch H, Richard S, et al. Combined VHLH and PTEN mutation causes genital tract cystadenoma and squamous metaplasia. Mol Cell Biol. 2008;28:4536–48. doi: 10.1128/MCB.02132-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Esteban MA, Harten SK, Tran MG, Maxwell PH. Formation of primary cilia in the renal epithelium is regulated by the von Hippel-Lindau tumor suppressor protein. J Am Soc Nephrol. 2006;17:1801–6. doi: 10.1681/ASN.2006020181. [DOI] [PubMed] [Google Scholar]
- 59.Chitalia VC, Foy RL, Bachschmid MM, Zeng L, Panchenko MV, Zhou MI, et al. Jade-1 inhibits Wnt signalling by ubiquitylating beta-catenin and mediates Wnt pathway inhibition by pVHL. Nat Cell Biol. 2008;10:1208–16. doi: 10.1038/ncb1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kurban G, Duplan E, Ramlal N, Hudon V, Sado Y, Ninomiya Y, et al. Collagen matrix assembly is driven by the interaction of von Hippel-Lindau tumor suppressor protein with hydroxylated collagen IV alpha 2. Oncogene. 2008;27:1004–12. doi: 10.1038/sj.onc.1210709. [DOI] [PubMed] [Google Scholar]
- 61.Kurban G, Hudon V, Duplan E, Ohh M, Pause A. Characterization of a von Hippel Lindau pathway involved in extracellular matrix remodeling, cell invasion, and angiogenesis. Cancer Res. 2006;66:1313–9. doi: 10.1158/0008-5472.CAN-05-2560. [DOI] [PubMed] [Google Scholar]
- 62.Ohh M, Yauch RL, Lonergan KM, Whaley JM, Stemmer-Rachamimov AO, Louis DN, et al. The von Hippel-Lindau tumor suppressor protein is required for proper assembly of an extracellular fibronectin matrix. Mol Cell. 1998;1:959–68. doi: 10.1016/s1097-2765(00)80096-9. [DOI] [PubMed] [Google Scholar]
- 63.Koochekpour S, Jeffers M, Wang PH, Gong C, Taylor GA, Roessler LM, et al. The von Hippel-Lindau tumor suppressor gene inhibits hepatocyte growth factor/scatter factor-induced invasion and branching morphogenesis in renal carcinoma cells. Mol Cell Biol. 1999;19:5902–12. doi: 10.1128/mcb.19.9.5902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Davidowitz EJ, Schoenfeld AR, Burk RD. VHL induces renal cell differentiation and growth arrest through integration of cell-cell and cell-extracellular matrix signaling. Mol Cell Biol. 2001;21:865–74. doi: 10.1128/MCB.21.3.865-874.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bishop T, Lau KW, Epstein AC, Kim SK, Jiang M, O’Rourke D, et al. Genetic analysis of pathways regulated by the von hippel-lindau tumor suppressor in Caenorhabditis elegans. PLoS Biol. 2004;2:e289. doi: 10.1371/journal.pbio.0020289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Calzada MJ, Esteban MA, Feijoo-Cuaresma M, Castellanos MC, Naranjo-Suarez S, Temes E, et al. von Hippel-Lindau tumor suppressor protein regulates the assembly of intercellular junctions in renal cancer cells through hypoxia-inducible factor-independent mechanisms. Cancer Res. 2006;66:1553–60. doi: 10.1158/0008-5472.CAN-05-3236. [DOI] [PubMed] [Google Scholar]
- 67.Pal S, Claffey KP, Dvorak HF, Mukhopadhyay D. The von Hippel-Lindau gene product inhibits vascular permeability factor/vascular endothelial growth factor expression in renal cell carcinoma by blocking protein kinase C pathways. J Biol Chem. 1997;272:27509–12. doi: 10.1074/jbc.272.44.27509. [DOI] [PubMed] [Google Scholar]
- 68.Pal S, Claffey KP, Cohen HT, Mukhopadhyay D. Activation of Sp1-mediated vascular permeability factor/vascular endothelial growth factor transcription requires specific interaction with protein kinase C zeta. J Biol Chem. 1998;273:26277–80. doi: 10.1074/jbc.273.41.26277. [DOI] [PubMed] [Google Scholar]
- 69.Datta K, Nambudripad R, Pal S, Zhou M, Cohen HT, Mukhopadhyay D. Inhibition of insulin-like growth factor-I-mediated cell signaling by the von Hippel-Lindau gene product in renal cancer. J Biol Chem. 2000;275:20700–6. doi: 10.1074/jbc.M909970199. [DOI] [PubMed] [Google Scholar]
- 70.Datta K, Sundberg C, Karumanchi SA, Mukhopadhyay D. The 104–123 amino acid sequence of the beta-domain of von Hippel-Lindau gene product is sufficient to inhibit renal tumor growth and invasion. Cancer Res. 2001;61:1768–75. [PubMed] [Google Scholar]
- 71.Okuda H, Saitoh K, Hirai S, Iwai K, Takaki Y, Baba M, et al. The von Hippel-Lindau tumor suppressor protein mediates ubiquitination of activated atypical protein kinase C. J Biol Chem. 2001;276:43611–7. doi: 10.1074/jbc.M107880200. [DOI] [PubMed] [Google Scholar]
- 72.Yang H, Minamishima YA, Yan Q, Schlisio S, Ebert BL, Zhang X, et al. pVHL acts as an adaptor to promote the inhibitory phosphorylation of the NF-kappaB agonist Card9 by CK2. Mol Cell. 2007;28:15–27. doi: 10.1016/j.molcel.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Peruzzi B, Athauda G, Bottaro DP. The von Hippel-Lindau tumor suppressor gene product represses oncogenic beta-catenin signaling in renal carcinoma cells. Proc Natl Acad Sci USA. 2006;103:14531–6. doi: 10.1073/pnas.0606850103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Roe JS, Kim H, Lee SM, Kim ST, Cho EJ, Youn HD. p53 stabilization and transactivation by a von Hippel-Lindau protein. Mol Cell. 2006;22:395–405. doi: 10.1016/j.molcel.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 75.Li Z, Wang D, Na X, Schoen SR, Messing EM, Wu G. The VHL protein recruits a novel KRAB-A domain protein to repress HIF-1alpha transcriptional activity. EMBO J. 2003;22:1857–67. doi: 10.1093/emboj/cdg173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li Z, Na X, Wang D, Schoen SR, Messing EM, Wu G. Ubiquitination of a novel deubiquitinating enzyme requires direct binding to von Hippel-Lindau tumor suppressor protein. J Biol Chem. 2002;277:4656–62. doi: 10.1074/jbc.M108269200. [DOI] [PubMed] [Google Scholar]
- 77.Kuznetsova AV, Meller J, Schnell PO, Nash JA, Ignacak ML, Sanchez Y, et al. von Hippel-Lindau protein binds hyperphosphorylated large subunit of RNA polymerase II through a proline hydroxylation motif and targets it for ubiquitination. Proc Natl Acad Sci USA. 2003;100:2706–11. doi: 10.1073/pnas.0436037100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pioli PA, Rigby WF. The von Hippel-Lindau protein interacts with heteronuclear ribonucleoprotein a2 and regulates its expression. J Biol Chem. 2001;276:40346–52. doi: 10.1074/jbc.M105391200. [DOI] [PubMed] [Google Scholar]
- 79.Gnarra JR, Ward JM, Porter FD, Wagner JR, Devor DE, Grinberg A, et al. Defective placental vasculogenesis causes embryonic lethality in VHL- deficient mice. Proc Natl Acad Sci USA. 1997;94:9102–7. doi: 10.1073/pnas.94.17.9102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Takeda K, Ho VC, Takeda H, Duan LJ, Nagy A, Fong GH. Placental but not heart defects are associated with elevated hypoxia-inducible factor alpha levels in mice lacking prolyl hydroxylase domain protein 2. Mol Cell Biol. 2006;26:8336–46. doi: 10.1128/MCB.00425-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pfander D, Kobayashi T, Knight MC, Zelzer E, Chan DA, Olsen BR, et al. Deletion of Vhlh in chondrocytes reduces cell proliferation and increases matrix deposition during growth plate development. Development. 2004;131:2497–508. doi: 10.1242/dev.01138. [DOI] [PubMed] [Google Scholar]
- 82.Haase VH, Glickman JN, Socolovsky M, Jaenisch R. Vascular tumors in livers with targeted inactivation of the von Hippel-Lindau tumor suppressor. Proc Natl Acad Sci USA. 2001;98:1583–8. doi: 10.1073/pnas.98.4.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ma W, Tessarollo L, Hong SB, Baba M, Southon E, Back TC, et al. Hepatic vascular tumors, angiectasis in multiple organs, and impaired spermatogenesis in mice with conditional inactivation of the VHL gene. Cancer Res. 2003;63:5320–8. [PubMed] [Google Scholar]
- 84.Rojiani AM, Owen DA, Berry K, Woodhurst B, Anderson FH, Scudamore CH, et al. Hepatic hemangioblastoma. An unusual presentation in a patient with von Hippel-Lindau disease. Am J Surg Pathol. 1991;15:81–6. [PubMed] [Google Scholar]
- 85.McGrath FP, Gibney RG, Morris DC, Owen DA, Erb SR. Case report: multiple hepatic and pulmonary haemangioblastomas--a new manifestation of von Hippel-Lindau disease. Clin Radiol. 1992;45:37–9. doi: 10.1016/s0009-9260(05)81467-9. [DOI] [PubMed] [Google Scholar]
- 86.Vortmeyer AO, Gnarra JR, Emmert-Buck MR, Katz D, Linehan WM, Oldfield EH, et al. von Hippel-Lindau gene deletion detected in the stromal cell component of a cerebellar hemangioblastoma associated with von Hippel-Lindau disease. Hum Pathol. 1997;28:540–3. doi: 10.1016/s0046-8177(97)90075-7. [DOI] [PubMed] [Google Scholar]
- 87.Kim WY, Safran M, Buckley MR, Ebert BL, Glickman J, Bosenberg M, et al. Failure to prolyl hydroxylate hypoxia-inducible factor alpha phenocopies VHL inactivation in vivo. EMBO J. 2006;25:4650–62. doi: 10.1038/sj.emboj.7601300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Biju MP, Neumann AK, Bensinger SJ, Johnson RS, Turka LA, Haase VH. Vhlh gene deletion induces Hif-1-mediated cell death in thymocytes. Mol Cell Biol. 2004;24:9038–47. doi: 10.1128/MCB.24.20.9038-9047.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Raval RR, Lau KW, Tran MG, Sowter HM, Mandriota SJ, Li JL, et al. Contrasting Properties of Hypoxia-Inducible Factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-Associated Renal Cell Carcinoma. Mol Cell Biol. 2005;25:5675–86. doi: 10.1128/MCB.25.13.5675-5686.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hu CJ, Iyer S, Sataur A, Covello KL, Chodosh LA, Simon MC. Differential regulation of the transcriptional activities of hypoxia-inducible factor 1 alpha (HIF-1alpha) and HIF-2alpha in stem cells. Mol Cell Biol. 2006;26:3514–26. doi: 10.1128/MCB.26.9.3514-3526.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Flamme I, Krieg M, Plate KH. Up-regulation of vascular endothelial growth factor in stromal cells of hemangioblastomas is correlated with up-regulation of the transcription factor HRF/HIF-2alpha. Am J Pathol. 1998;153:25–9. doi: 10.1016/s0002-9440(10)65541-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kim WY, Kaelin WG. Role of VHL gene mutation in human cancer. J Clin Oncol. 2004;22:4991–5004. doi: 10.1200/JCO.2004.05.061. [DOI] [PubMed] [Google Scholar]
- 93.Neumann HP, Zbar B. Renal cysts, renal cancer and von Hippel-Lindau disease [editorial] Kidney Int. 1997;51:16–26. doi: 10.1038/ki.1997.3. [DOI] [PubMed] [Google Scholar]
- 94.Rankin EB, Tomaszewski JE, Haase VH. Renal cyst development in mice with conditional inactivation of the von Hippel-Lindau tumor suppressor. Cancer Res. 2006;66:2576–83. doi: 10.1158/0008-5472.CAN-05-3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pollard PJ, Spencer-Dene B, Shukla D, Howarth K, Nye E, El-Bahrawy M, et al. Targeted inactivation of fh1 causes proliferative renal cyst development and activation of the hypoxia pathway. Cancer Cell. 2007;11:311–9. doi: 10.1016/j.ccr.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 96.Mandriota SJ, Turner KJ, Davies DR, Murray PG, Morgan NV, Sowter HM, et al. HIF activation identifies early lesions in VHL kidneys: evidence for site-specific tumor suppressor function in the nephron. Cancer Cell. 2002;1:459–68. doi: 10.1016/s1535-6108(02)00071-5. [DOI] [PubMed] [Google Scholar]
- 97.Krishnamachary B, Zagzag D, Nagasawa H, Rainey K, Okuyama H, Baek JH, et al. Hypoxia-inducible factor-1-dependent repression of E-cadherin in von Hippel-Lindau tumor suppressor-null renal cell carcinoma mediated by TCF3, ZFHX1A, and ZFHX1B. Cancer Res. 2006;66:2725–31. doi: 10.1158/0008-5472.CAN-05-3719. [DOI] [PubMed] [Google Scholar]
- 98.Esteban MA, Tran MG, Harten SK, Hill P, Castellanos MC, Chandra A, et al. Regulation of E-cadherin expression by VHL and hypoxia-inducible factor. Cancer Res. 2006;66:3567–75. doi: 10.1158/0008-5472.CAN-05-2670. [DOI] [PubMed] [Google Scholar]
- 99.Evans AJ, Russell RC, Roche O, Burry TN, Fish JE, Chow VW, et al. VHL promotes E2 box-dependent E-cadherin transcription by HIF-mediated regulation of SIP1 and snail. Mol Cell Biol. 2007;27:157–69. doi: 10.1128/MCB.00892-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gordan JD, Bertout JA, Hu CJ, Diehl JA, Simon MC. HIF-2alpha promotes hypoxic cell proliferation by enhancing c-myc transcriptional activity. Cancer Cell. 2007;11:335–47. doi: 10.1016/j.ccr.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pennacchietti S, Michieli P, Galluzzo M, Mazzone M, Giordano S, Comoglio PM. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell. 2003;3:347–61. doi: 10.1016/s1535-6108(03)00085-0. [DOI] [PubMed] [Google Scholar]
- 102.Wang Y, Roche O, Yan MS, Finak G, Evans AJ, Metcalf JL, et al. Regulation of endocytosis via the oxygen-sensing pathway. Nat Med. 2009;15:319–24. doi: 10.1038/nm.1922. [DOI] [PubMed] [Google Scholar]
- 103.Smith K, Gunaratnam L, Morley M, Franovic A, Mekhail K, Lee S. Silencing of epidermal growth factor receptor suppresses hypoxia-inducible factor-2-driven VHL−/− renal cancer. Cancer Res. 2005;65:5221–30. doi: 10.1158/0008-5472.CAN-05-0169. [DOI] [PubMed] [Google Scholar]
- 104.Bindra RS, Vasselli JR, Stearman R, Linehan WM, Klausner RD. VHL-mediated hypoxia regulation of cyclin D1 in renal carcinoma cells. Cancer Res. 2002;62:3014–9. [PubMed] [Google Scholar]
- 105.Zatyka M, da Silva NF, Clifford SC, Morris MR, Wiesener MS, Eckardt KU, et al. Identification of cyclin D1 and other novel targets for the von Hippel-Lindau tumor suppressor gene by expression array analysis and investigation of cyclin D1 genotype as a modifier in von Hippel-Lindau disease. Cancer Res. 2002;62:3803–11. [PubMed] [Google Scholar]
- 106.Wykoff CC, Sotiriou C, Cockman ME, Ratcliffe PJ, Maxwell P, Liu E, et al. Gene array of VHL mutation and hypoxia shows novel hypoxia-induced genes and that cyclin D1 is a VHL target gene. Br J Cancer. 2004;90:1235–43. doi: 10.1038/sj.bjc.6601657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Thomas GV, Tran C, Mellinghoff IK, Welsbie DS, Chan E, Fueger B, et al. Hypoxia-inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nat Med. 2006;12:122–7. doi: 10.1038/nm1337. [DOI] [PubMed] [Google Scholar]
- 108.Staller P, Sulitkova J, Lisztwan J, Moch H, Oakeley EJ, Krek W. Chemokine receptor CXCR4 downregulated by von Hippel-Lindau tumour suppressor pVHL. Nature. 2003;425:307–11. doi: 10.1038/nature01874. [DOI] [PubMed] [Google Scholar]
- 109.Turner KJ, Moore JW, Jones A, Taylor CF, Cuthbert-Heavens D, Han C, et al. Expression of hypoxia-inducible factors in human renal cancer: relationship to angiogenesis and to the von Hippel-Lindau gene mutation. Cancer Res. 2002;62:2957–61. [PubMed] [Google Scholar]
- 110.Rosenberger C, Mandriota S, Jurgensen JS, Wiesener MS, Horstrup JH, Frei U, et al. Expression of hypoxia-inducible factor-1alpha and -2alpha in hypoxic and ischemic rat kidneys. J Am Soc Nephrol. 2002;13:1721–32. doi: 10.1097/01.asn.0000017223.49823.2a. [DOI] [PubMed] [Google Scholar]
- 111.Kondo K, Klco JM, Nakamura E, Lechpammer M, Kaelin WG. Inhibition of HIF is necessary for tumor suppression by the von Hippel-Lindau protein. Cancer Cell. 2002;1:237–46. doi: 10.1016/s1535-6108(02)00043-0. [DOI] [PubMed] [Google Scholar]
- 112.Maranchi JK, Vasselli JR, Riss J, Bonifacio JS, Linehan WM, Klausner RD. The contribution of VHL subtrate binding and HIF-1α to the phenotype of vhl loss in renal cell carcinoma. Cancer Cell. 2002;1:247–53. doi: 10.1016/s1535-6108(02)00044-2. [DOI] [PubMed] [Google Scholar]
- 113.Zimmer M, Doucette D, Siddiqui N, Iliopoulos O. Inhibition of hypoxia-inducible factor is sufficient for growth suppression of VHL−/− tumors. Mol Cancer Res. 2004;2:89–95. [PubMed] [Google Scholar]
- 114.Kondo K, Kim WY, Lechpammer M, Kaelin WG., Jr Inhibition of HIF2alpha is sufficient to suppress pVHL-defective tumor growth. PLoS Biol. 2003;1:E83. doi: 10.1371/journal.pbio.0000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gordan JD, Lal P, Dondeti VR, Letrero R, Parekh KN, Oquendo CE, et al. HIF-alpha effects on c-Myc distinguish two subtypes of sporadic VHL-deficient clear cell renal carcinoma. Cancer Cell. 2008;14:435–46. doi: 10.1016/j.ccr.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Loor G, Schumacker PT. Role of hypoxia-inducible factor in cell survival during myocardial ischemia-reperfusion. Cell Death Differ. 2008;15:686–90. doi: 10.1038/cdd.2008.13. [DOI] [PubMed] [Google Scholar]
- 117.Ivan M, Haberberger T, Gervasi DC, Michelson KS, Gunzler V, Kondo K, et al. Biochemical purification and pharmacological inhibition of a mammalian prolyl hydroxylase acting on hypoxia-inducible factor. Proc Natl Acad Sci USA. 2002;99:13459–64. doi: 10.1073/pnas.192342099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hsieh MM, Linde NS, Wynter A, Metzger M, Wong C, Langsetmo I, et al. HIF prolyl hydroxylase inhibition results in endogenous erythropoietin induction, erythrocytosis, and modest fetal hemoglobin expression in rhesus macaques. Blood. 2007;110:2140–7. doi: 10.1182/blood-2007-02-073254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bernhardt WM, Campean V, Kany S, Jurgensen JS, Weidemann A, Warnecke C, et al. Preconditional activation of hypoxia-inducible factors ameliorates ischemic acute renal failure. J Am Soc Nephrol. 2006;17:1970–8. doi: 10.1681/ASN.2005121302. [DOI] [PubMed] [Google Scholar]