

Abstract
The initial phase of sepsis is characterized by massive inflammatory cytokine production which contributes to multisystem organ failure and death. Costimulatory molecules are a class of receptors capable of regulating cytokine production in adaptive immunity and recent studies describe their presence on neutrophils and monocytes, suggesting a potential role for regulating cytokine production in innate immunity. The purpose of this study is to determine the role for OX40-OX40L interaction in the innate immune response to polymicrobial sepsis. Humans with sepsis demonstrated upregulation of OX40L on both monocytes and neutrophils, with mortality and ICU stay correlating with expression levels. In an animal model of polymicrobial sepsis, a direct role for OX40L in regulating inflammation was evidenced by improved survival, decreased cytokine production and a decrease in remote organ damage in OX40L−/− mice. The finding of similar results with an OX40L antibody suggests a potential future therapeutic role for OX40L blockade in sepsis. The inability of α-OX40L to provide significant protection in macrophage-depleted mice establishes macrophages as an indispensible cell type within the OX40/OX40L axis that helps to mediate the clinical signs of disease in sepsis. Conversely, the protective effect of α-OX40L antibody in RAG1−/− mice, further confirms a T-cell independent role for OX40L stimulation in sepsis. In conclusion, our data provide a novel in vivo role for OX40-OX40L system in the innate immune response during polymicrobial sepsis and suggests a potential beneficial role for therapeutic blockade of OX40L in this devastating disorder.
Introduction
Sepsis, defined as the systemic inflammatory response to infection is a devastating condition with high morbidity and mortality. It remains a significant burden on the health care system, affecting over 700,000 people/year in the United States resulting in 250,000 deaths [#73; #74]. This results in a net cost of $17 billion/year. Over the past decade, mortality from sepsis has remained in excess of 25% in spite of adequate antimicrobial therapy [#58]. This highlights the need for newer adjuvant therapies.
The early phases of sepsis are characterized by activation of the innate immune response with production of multiple pro and anti-inflammatory cytokines. However, numerous attempts have been made to inhibit individual cytokines in sepsis, including TNF-α and IL-1β. These strategies, while effective in limiting LPS mediated inflammation, failed to improve mortality in Phase III clinical trials or in a mouse model of polymicrobial sepsis, such as Cecal Ligation and Puncture (CLP) [#54; #59; #75]. One explanation for their failure is the redundant and overlapping actions of the individual cytokines. Consequently, investigators have begun to focus on cell surface receptors capable of simultaneously regulating multiple inflammatory cascades.
Recent studies attribute a potentially important role for costimulatory molecules in regulating cytokine production and mortality in animal models of polymicrobial sepsis [#30; #314]. The OX40 (CD134)-OX40L (CD252) system has been well described in regulation of T-cell proliferation, effector cell function and survival within the adaptive immune response [#315]. Growing evidence suggests OX40L is present on activated macrophages and mononuclear cells and OX40L is capable of directly activating intracellular signaling cascades and cytokine production [#316; #318]. Furthermore, OX40 has been described on activated neutrophils (PMNs) as well as existing as a shed soluble form, the latter also capable of binding to, and activating, OX40L with subsequent upregulation of IL-6 on airways smooth muscle and c-fos and c-jun in transfected HUVEC cells [#260; #259]. Together these data suggest that OX40-OX40L signaling within innate immune cells could play a role in the innate immune response to sepsis. In this report, we now describe a pivotal role for OX40-OX40L in regulation of mortality and inflammation in the innate immune response to mouse polymicrobial sepsis.
Methods
Mice
WT C57BL/6, MaFIA and RAG1−/− mice were obtained from Jackson Laboratories. OX40L−/− mice on a C57BL/6 background were graciously provided by Dr. Naoto Ishii and bred in our animal facility. All mice were sex (female) and age matched (6–8wks) and allowed to acclimatize for 1 week prior to use. All studies were performed in accordance with the OHSU IACUC.
Cecal Ligation and Puncture
CLP was performed as previously described [#313; #314]. Briefly, mice were anesthetized with 2.5% isoflourane and underwent CLP with a 19 gauge needle. Mice received 1cc 0.9% saline subcutaneously for resuscitation. At specified times, the mice were used to collect plasma, bronchoalveolar lavage (BAL) and peritoneal lavage (PL) (3cc) as previously described [#314]. For survival experiments, mice were monitored for a total of 14 days. For antibody inhibition, 250μg of α-OX40L (hybridoma provided by Dr. Naoto Ishii) were injected ip 4hrs prior to surgery. Rat IgG (Biolegend) was used as an antibody control and PBS as a vehicle control. As there was no difference between the 2, these groups were combined where indicated. For experiments involving MaFIA mice, mice were administered AP20187 ip for 5 days. This results in total macrophage depletion [#377].
Cytokines and tissue MPO were determined by commercially available immunoassays (R&D systems, Hycult Biotechnology (MPO)) and performed according to the manufacturers’ specifications. NF-κB DNA binding was determined by DNA binding ELISA (Active Motifs). Pulmonary capillary leak was determined by Evan’s Blue dye as previously described [#314].
Flow Cytometry
Flow cytometry was performed as previously described [#314]. Splenocytes, whole blood or peritoneal lavage were collected and 1x106 cells were incubated with 100μl Fc block (mouse cells only) for 15min, then labeled with the following antibodies: OX40, OX40L, CD4, CD8, LY6g (mouse), CD11b, F4/80 (mouse), CD14 (human) at optimal concentration for 45min in the dark. Red blood cells were lysed with RBC lysis buffer then the cells were fixed with 0.1%paraformaldehyde and analyzed on a BD LSRII 8-color analyzer with FloJo software. All reagents were purchased from BDPharmigen. BD compensation beads were used to calibrate the instrument before each use. PMNs were identified by FSC/SSC characteristics as Ly6G+ (mice only), mononuclear cells by FSC/SSC, CD11b+ and F4/80 (mice) and CD14+(human), The T-cells were identified by FSC/SSC and further subgrouped by CD4+ and CD8+ labeling. Isotype antibody labeled cells were used to control for nonspecific staining.
Human studies
All studies were approved by the OHSU Institutional Review Board. All patients meeting SCCM/ACCP criteria for sepsis in the first 24hrs of ICU stay were eligible for inclusion [#314]. ICU controls were selected at random from patients admitted to the medical ICU and did not have a suspected site of infection. Patients were excluded for the following reasons; presence of a Do Not Resuscitate order or decision to institute comfort care measures, Hgb<7g/dl or the presence of active bleeding requiring more than 2 units of packed red blood cells. After obtaining informed consent, 25cc of blood was collected into glass (serum) or EDTA coated tubes (platelet poor plasma) on days 1 (defined as first 24hrs of admission to ICU), 3–5, 7 and 14 or until death or hospital discharge. After obtaining preliminary data on soluble mediators, and optimization of labeling protocols, all subsequent subjects had additional blood collected for flow cytometry. Clinical data including APACHE II scores were recorded at the time of each blood draw. Patients were followed for 28 days or until death or hospital discharge. ICU and Ventilator Free days were defined as number of days alive outside the ICU or off the ventilator during the 28 day observation period.
Cell Culture
THP-1 cells were differentiated with PMA for 24hrs. Cells were subsequently stimulated with LPS (100ng/ml) for 24hrs and harvested for immunoblot as previously described [#30]. For stimulation studies, cells were blocked with CD16/32 for 1hr and then either OX40:Ig (10μg/ml), FC control or PBS added for 24hrs. Supernatants were collected for ELISA.
Statistics
Survival was analyzed by Kaplan-Meier analysis. All comparisons between groups were performed by Mann-Whitney or Kruskal-Wallis (ANOVA) depending on the number of groups analyzed. For multigroup analysis, intergroup comparisons were performed with Dunn’s test. Correlations were made with Spearman’s test. All statistics were done with GraphPad Prism 5.0
RESULTS
Expression of OX40-OX40L in Human Sepsis
We first examined OX40 and OX40L expression in human sepsis. We obtained blood from subjects admitted to the ICU with sepsis, subjects admitted to the ICU for non-septic conditions (ICU controls), and healthy controls. Overall, healthy controls were more likely to be younger than septic patients or ICU controls (Table 1). In concert with prior studies documenting OX40L expression on dendritic cells and macrophages and increased expression of OX40L on circulating monocytes from patients with acute coronary syndromes, patients with sepsis exhibited upregulation of OX40L on circulating monocytes compared to healthy controls (Figure 1B and C) [#329]. Of even greater interest, was the expression of OX40L on circulating PMNs (Figure 1D). The presence of OX40L on PMNs was specific as evidenced by lack of staining for other costimulatory molecules, CD40 and CD80 (Figure 1A). Overall, both monocyte and PMN OX40L expression were higher in septic subjects compared to healthy and ICU controls on day 1 in the ICU (Figures 1C and D) and decreased over time with resolution of symptoms (Supplementary figure 1). Finally, among septic subjects there was no correlation between monocyte or PMN OX40L expression on day 1 and levels of IL-6, IL-10 or IL-12. However, for the entire cohort (septic patients and controls), there was a significant correlation between day 1 OX40L expression and IL-6 (R=0.35; p=0.009) and IL-10 (r=0.35; p=0.02). This is consistent with the known ability of OX40L to regulate IL-6 in other disease systems [#260].
Table 1.
Clinical Characteristics of enrolled Human Subjects.
| Sepsis (N=31) | ICU Controls (N=8)* | Healthy Controls (N=9) | |
|---|---|---|---|
|
| |||
| Age (years) | 58.2 ± 14.5 | 52.2 ± 22.8 | 35.7 ± 9.3 |
|
| |||
| Sex (% Female) | 41.9% | 37.5%S | 67% |
|
| |||
| APACHE II | 17.7 ± 6.5 | 14.5 ± 9.3 | N/A |
|
| |||
| Mechanical Ventilation (%) | 54.8 | 50% | N/A |
|
| |||
| Vasopressors (%) | 67.7 | 25% | N/A |
|
| |||
| Bacteremia (%) | 54.8 | N/A | N/A |
|
| |||
| 28 Day Mortality (%) | 16.1% | 12.5% | N/A |
|
| |||
| Source of Infection | N/A | N/A | |
| Abdomen | 9% | ||
| Blood | 9% | ||
| Lung | 45% | ||
| Genitourinary | 21% | ||
| Brain | 9% | ||
| Skin | 6% | ||
N/A=Not applicable-
Diagnoses of ICU controls were congestive heart failure and cardiogenic shock (N=3), hypovolumic shock (N=2), pancreatitis (N=2) and airway obstruction (N=1)
Figure 1. Upregulation of OX40 and OX40L in human sepsis.
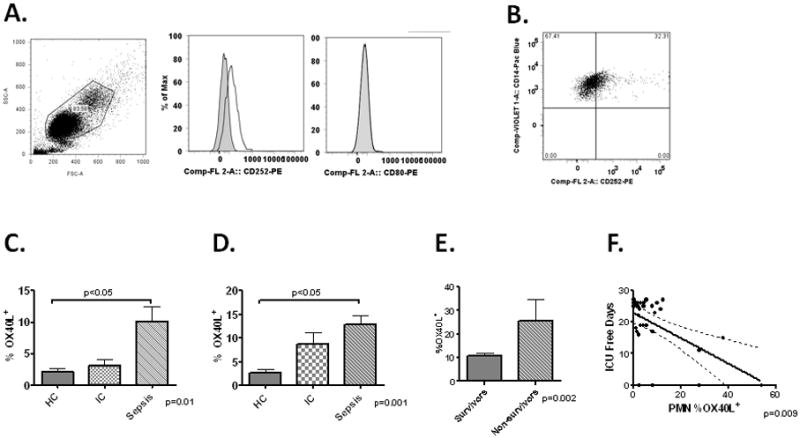
Panel A-Representative scattergram of whole blood gated from a septic patient on Day 1 gated on PMNs by FSC/SSC. Histogram of PMN OX40L (CD252-middle panel) and CD80 expression (right panel). Panel B –Representative dot plot of CD14+ monocytes (identified by FSC/SSC) expression of OX40L in a septic subject. Gates determined by isotype stained controls. Panel C-Quantification of PMN OX40L expression (MFI) from healthy controls (HC-N=8), ICU controls (IC-N=8) and septic patients and day 1 (Sepsis-N=32). Panel D- Quantification of OX40L MFI on circulating CD14+ monocytes in the same groups. Panel E- Monocyte OX40L expression (MFI) on admission to the ICU in survivors (N=26) and non-survivors (N=6). Panel F.-Correlation between day 1 PMN OX40L expression (MFI) and ICU free days among septic patients.
We next sought to determine whether day 1 expression of OX40L could potentially serve as a biomarker for outcome among septic patients. Levels of monocyte OX40L were higher in non-survivors compared to survivors (Figure 1E) and expression levels loosely correlated with severity of illness as determined by APACHE II score (R=0.35; p=0.04). Further, PMN OX40L negatively correlated with ICU free days (Figure 1F). However, there was no association between either monocyte or PMN OX40L expression levels on day 1 and the presence of bacteremia, renal failure, circulatory failure or respiratory failure (data not shown).
In contrast to our findings with OX40L, the typical reservoir of OX40, CD4+ T-cells, expressed only low levels of OX40 (less than 10% of cells stained positive), with no difference between septic subjects and either control group at any time point (data not shown). However levels of the soluble isoform of OX40 (sOX40) were increased in septic patients compared to controls on day 1 (Figure 2A). We further demonstrated a pro-inflammatory role for sOX40 on macrophages. We first established that OX40L could be upregulated on human macrophages in vitro with stimuli consistent to what is observed with human sepsis. LPS stimulation of THP-1 cells significantly upregulated OX40L, both by immunoblot and flow cytometry, compared to vehicle controls (Figure 2B). Further stimulation of LPS primed cells with an OX40:Ig fusion protein significantly upregulated IL-6 compared to vehicle or Ig treated controls confirming that OX40L is capable of reverse signaling in macrophages via stimulation with soluble ligands (Figure 2C).
Figure 2. Soluble OX40 is expressed in human sepsis and activates OX40L.
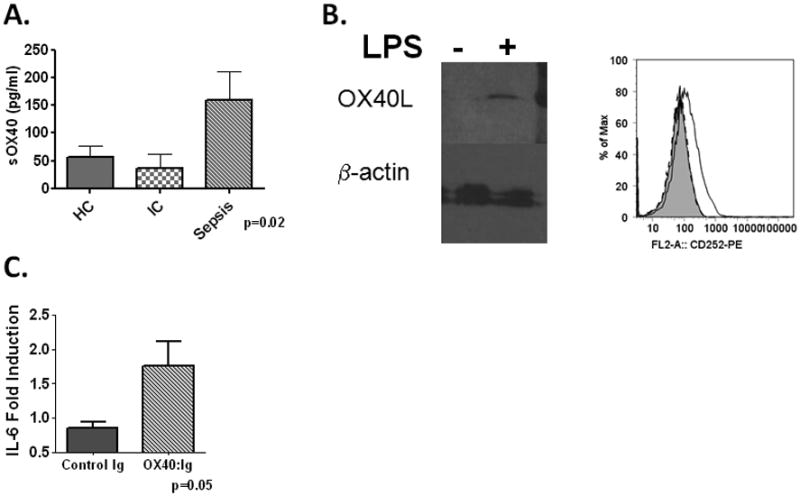
Panel A-Expression of soluble OX40 in healthy controls, ICU controls and Septic patients within 24hrs of admission to the ICU. Panel B- THP-1 monocytes were treated with LPS (100ng/ml) or PBS for 24hrs and cells lysed for immunoblot or stained for OX40L via flow cytometry (right panel). Lanes normalized for protein (75μg/lane). Experiment performed 3 times. Panel C- THP-1 macrophages were primed with LPS for 24hrs after which supernatant was removed and cells were treated with either vehicle (PBS), OX40:Ig (10ug/ml) or control Ig for 24hrs. Supernatants were collected and assayed for IL-6 via ELISA. Experiment was performed 3 times.
OX40L Regulates Lethality of Polymicrobial Sepsis
To better understand the biologic role of the OX40/OX40L system in sepsis, we employed a mouse model of polymicrobial sepsis, cecal ligation and puncture (CLP). We first wished to confirm expression of OX40 and OX40L on mouse innate immune effector cells in polymicrobial sepsis, thereby confirming our human observations and further validating the mouse model as a useful pre-clinical model for OX40L targeted therapeutic interventions. Eighteen hours after CLP, OX40L was upregulated on peritoneal macrophages and PMNs compared to controls (Figure 3A). We confirmed PMN expression of OX40L via immunoblot (Figure 3B). PMNs were also the predominant source of OX40 in CLP, with peritoneal PMNs expressing high levels of OX40 (Figure 3C). This is similar to what has been observed by others with human PMNs [#259]. Finally, similar to our human observations, circulating and splenic CD4+ T-cells expressed low levels of OX40 (less than 10% positive) with no difference between septic and control mice (data not shown).
Figure 3. Expresion and function of OX40L in mouse sepsis.
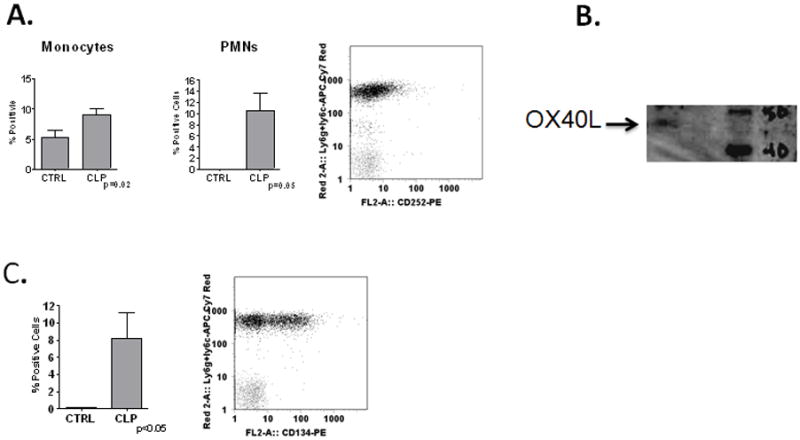
Panel A-WT mice underwent no surgery (N=3) or Cecal Ligation and Puncture (CLP-N=3) and harvested at 18hrs. Peritoneal lavage (PL) analyzed by flow cytometry for OX40L expression on macrophages (F4/80+-left panel) or PMNs (Ly6G+-middle panel). Representative dot plot of PMN expression (right panel). Panel B- Immunoblot for OX40L from TG PMNs. Panel C-Expression of OX40 on peritoneal PMNs (Left panel) and representative dot plot (Right panel).
To test the in vivo significance of OX40L upregulation in sepsis, we employed OX40L−/− mice. OX40L−/− mice had a dramatic improvement in survival compared to WT mice after CLP (Figure 4A). This was associated with decreased levels of IL-6, IL-10 and IL-12 in plasma (Figure 4B), bronchoalveolar lavage (BALF) and peritoneal lavage fluid (IL-6 only) compared to WT mice 6hrs and 18hrs after CLP (Supplementary Figure 2). The attenuation in cytokine production was associated with a decrease in remote organ injury as evidenced by pulmonary capillary leak (Figure 5A), hepatic NF-κB induction (Figure 5B) and hepatic PMN accumulation as assessed by myeloperoxidase (MPO) 18hrs after CLP (Figure 5C).
Figure 4. OX40L contributes to lethality of polymicrobial sepsis.
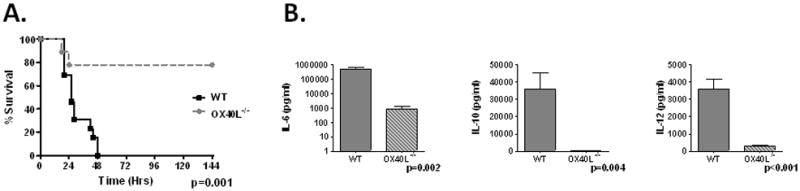
Panel A-WT (N=13) and OX40L−/− mice (N=9) and monitored for survival for 14 days. No additional deaths beyond day 6. Panel B-WT and OX40L−/− underwent CLP and plasma harvest at 18hrs for cytokine analysis (N=4–6/group).
Figure 5. OX40L contributes to remotes organ injury in polymicrobial sepsis.
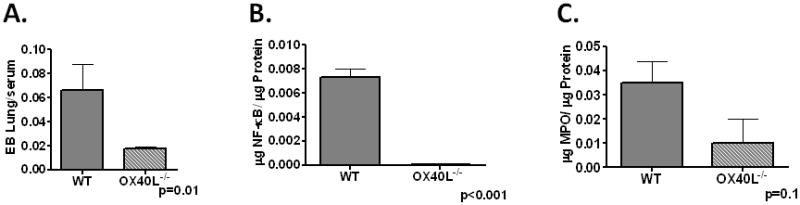
Panel A-WT and OX40L−/− mice underwent CLP and lung leak evaluated by Evan’s Blue dye (N=5/group) at 18hrs post surgery. Panels B–C. WT and OX40L−/− mice underwent CLP and Liver harvested at 18hrs for NF-κB (Panel B) and MPO (Panel C) N=5/group.
We next wished to establish whether physiologic blockade of OX40L would have a similar effect, both to confirm the genetic model as well as provide important pre-clinical data. Administration of an α-OX40L monoclonal antibody significantly improved survival compared to isotype treated controls (Figure 6A). Similar to results with OX40L−/− mice, mice treated with α-OX40L showed a reduction in plasma IL-6, IL-10 and IL-12 18hrs after CLP compared to controls (Figure 6B). Finally, use of α-OX40L as a rescue therapy, administered 4hrs after the onset of sepsis, resulted in a significant improvement in survival (Figure 6C).
Figure 6. Efficacy of α-OX40L antibody in mouse sepsis.
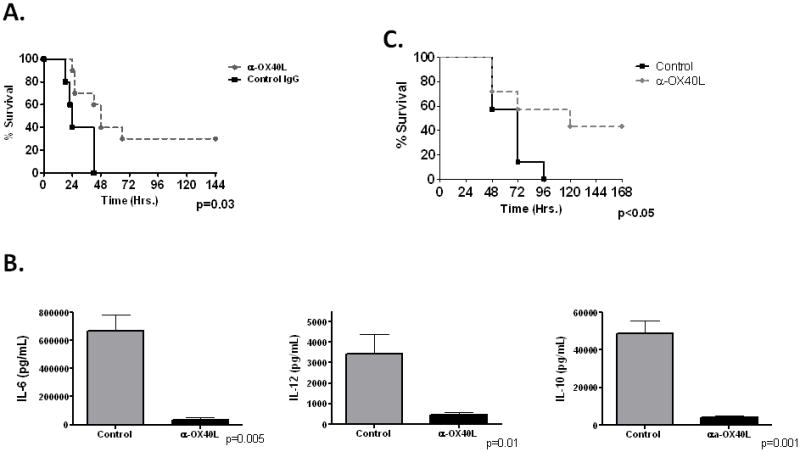
Panel A. WT mice were administered either control IgG or α-OX40L (250μg) intrapertioneally 4hrs pre-CLP and monitored for survival (N=10–13/group). Mice were monitored for 14 days, with no additional deaths after day 6. Panel B- WT mice were administered either vehicle or α-OX40L 4hrs pre-CLP and harvested at 18hrs for plasma cytokine analysis (N=5/group). Panel C- WT mice were administered either vehicle control (N=7) or α-OX40L (250μg) (N=7) intrapertioneally 4hrs post- 22guage CLP and monitored for survival. Mice were monitored for 14 days, with no additional deaths after day 6.
OX40L activation is macrophage dependent and T-cell Independent in vivo
The upregulation of OX40L on PMNs and mononuclear cells suggest a pivotal role for myeloid cells in mediating OX40L inflammation in vivo, However, there are numerous other reservoirs for OX40L described, including vascular endothelium and smooth muscle [#348; #321]. To confirm the requirement of macrophages in our system, we employed Macrophage Fas Induced Apoptosis (MaFIA) mice, which contain a Fas receptor (CD95) expressing transgene driven by the CSF1 promoter [#347]. These mice, when treated with the dimerizing compound AP20187, get targeted Fas receptor dimerization and subsequent systemic macrophage apoptosis and total animal macrophage depletion by day 5 [#347; #377]. Unlike our observations with WT mice or macrophage intact MaFIA mice (not shown), α-OX40L failed to provide any meaningful protection in macrophage depleted MaFIA mice (Figure 7A), suggesting macrophages as a central effector cell type of OX40-OX40L mediated inflammation in sepsis.
Figure 7. Cellular source of OX40/OX40L activity in sepsis.
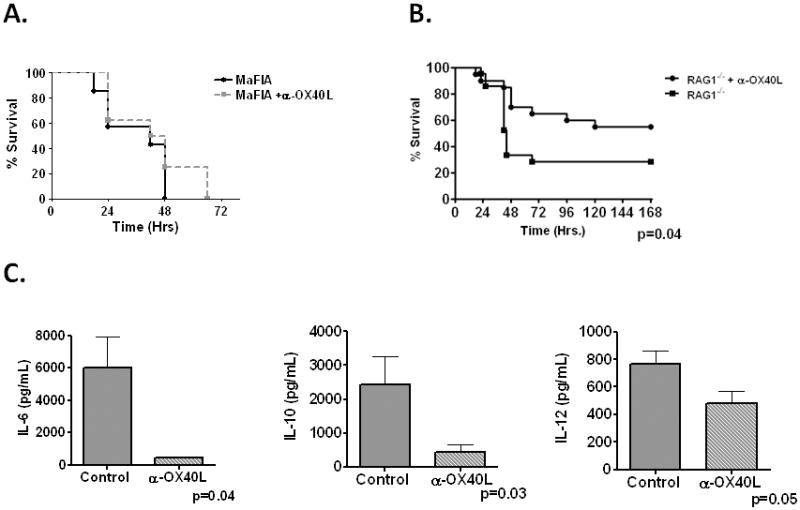
Panel A- MaFIA mice were treated with AP20187 daily for 5 days and allowed 2 days recovery. Mice were administered either vehicle control of α-OX40L (250μg) intrapertioneally 4hrs pre CLP (18ga) and monitored for survival (N=7–8/group). Panel B-RAG1−/− mice were administered either control vehicle or α-OX40L (N=19–20/group) 4hrs pre-CLP and monitored for survival for 14 days. No additional deaths after day 6. Panel C- RAG1−/− mice were administered either control or α-OX40L 4hrs pre-CLP and harvested 18hrs post-CLP for plasma cytokine analysis (N=4–5/group).
We next sought to establish that OX40L stimulation was indeed T-cell independent (either through PMN expressed OX40 or generation of sOX40) and thus further establishing its role in innate immunity. To establish a T-cell independent role for OX40L activation in CLP, we repeated experiments in RAG1−/− mice. Surprisingly, similar to our observations in WT mice, α-OX40L significantly improved sepsis specific survival in RAG1−/− mice compared to controls (Figure 7B). Improvement in survival was associated with attenuation in IL-6, IL-10 and IL-12 in plasma (Figure 7C) and BAL (Data not shown). These data strongly suggest, for the first time, a T-cell independent mechanism of OX40L activation in vivo.
Discussion
Sepsis is a devastating disorder for which there are limited adjuvant therapies. Numerous attempts at anti-cytokine therapy have failed in animal models and humans. The redundant and overlapping affects of many pro-inflammatory cytokines suggests that strategies targeting either transcription factors or upstream receptors which control multiple inflammatory pathways may be more effective, such as observations with Macrophage Migration Inhibitory Factor (MIF) and High Mobility Group Box Protein-1 (HMGB-1) [#51; #351; #354]. Costimulatory molecules represent another potential target based on their presence on macrophages, a potent source of cytokine production in sepsis, and their well documented ability to regulate numerous transcriptional pathways in a bidirectional manner [#30; #314; #358].
One of the major and novel findings of this paper is the improved survival in OX40L−/− mice subjected to polymicrobial sepsis. The ability to recapitulate this with pharmacological inhibition of OX40L implies this is not an epiphenomenon due to congenital absence of OX40L in the knockout mice. However, it remains unclear why such a large discrepancy exists between OX40L−/− and α-OX40L treated mice. Whether this represents incomplete blockade and/or compensatory dysregulation of other pathways with congenital OX40L deficiency remains to be determined. More importantly, the ability of an OX40L antibody to provide protection after the onset of disease further established OX40L inhibition as a potential therapeutic intervention in human sepsis. Enthusiasm for the potential of OX40L blockade in human sepsis is bolstered by the novel finding of increased OX40L expression in our cohort of septic patients. This appeared to be relatively specific for sepsis in a medical ICU population as similar findings were not observed in either healthy or ICU controls and the increase in expression levels returned to that of healthy controls with resolution of illness. The association between OX40L expression levels and mortality, severity of illness and ICU free days further supports the hypothesis that OX40L contributes to the lethality of polymicrobial sepsis. While, numerous studies have investigated the role of the OX40/OX40L system in graft rejection, autoimmune disease and antitumor immunity, to our knowledge this is the first description of OX40L blockade in the innate immune response to and acute bacterial infection (sepsis) [#325; #323; #362].
The mechanism by which OX40L regulates lethality lies in its ability to regulate multiple inflammatory pathways. The OX40/OX40L has been well described to control T-cell proliferation in the adaptive immune response. However, a growing literature describes a putative receptor function to OX40L as well. Stimulation of OX40L on dendritic cells and airway smooth muscle cells results in JNK activation and subsequent IL-6 production [#260; #259]. We now show that macrophages are capable of similar OX40L mediated IL-6 production in vitro. This correlates with the attenuation of IL-6, IL-10 and IL-12 observed in the OX40L−/− and α-OX40L treated mice after CLP. Our in vitro findings and the correlation between OX40L expression levels and circulating IL-6 and IL-10 in our human cohort suggests this is valid in humans as well. Clinically, the attenuation of inflammatory cytokines improves survival through reduction in remote organ injury. In our mice this is represented by a reduction in pulmonary capillary leak and hepatic NF-κB and PMN accumulation in the OX40L−/− mice compared to controls.
Macrophages appear to be critical for the mediating the lethal effects of OX40L in sepsis in vivo. The ability of LPS to upregulate OX40L on macrophages provides a mechanism for the specificity of OX40L upregulation in septic humans. However, it is likely that LPS is not the only mediator of OX40L upregulation in vivo. The known ability of CD154 and IL-12, both of which are known to be upregulated in sepsis, to upregulate OX40L expression on dendritic cells and T-cells respectively suggests multiple potential pathways towards the increased expression seen in our patients and mice [#316; #314; #375]. Finally, the failure of OX40L blockade to rescue macrophage depleted mice suggests macrophages are necessary for mediating the toxic effects of OX40L in sepsis in vivo. However, we acknowledge that the induction of systemic macrophage apoptosis as a method of macrophage depletion may result in dysregulation of other pathways which could have potentially been OX40L dependent in our model. Our finding of OX40L on circulating PMNs in septic mice and humans provide another potential and novel source of OX40L. The significance of this remains unclear and will be the subject of future investigations. Finally, the description of OX40L expression on endothelial cells provides another mechanism by which OX40L can regulate lethality [#348].
The source of OX40 in our model remains less clear. The ability of α-OX40L to rescue RAG1−/− mice suggests there is a T-Cell independent source of OX40 in sepsis and firmly places OX40 in the innate immune response to polymicrobial sepsis. One potential source is cell bound OX40 on circulating PMNs. Baumann et al described OX40 expression on human PMNs and our finding of OX40 expression on mouse PMNs is consistent with this result [#259]. It is unclear why we did not observe high levels of OX40 expression on human PMNs. Soluble OX40 represents another potential means of OX40L activation in sepsis. Recently, studies have documented that OX40 can also be shed as a soluble isoform (sOX40) which, similar to its cell-bound form, is able to activate and induce cytokine production from OX40L expressing airway smooth muscle and now as described in this paper, macrophages as well [#321]. It is believed this is generated through alternative splicing, but the source(s) of it remain less clear [#331]. The presence of sOX40 also establishes its potential as a circulating biomarker of the presence and/or outcome in human sepsis, similar to its ability to discriminate between patients with Systemic Sclerosis and Systemic Lupus Eyrthematosus [#332]. However, greater numbers of subjects will be required to validate this.
However, it is important to point out some limitations to our study. First, and most important, we are looking at the initial phases of sepsis and septic shock. While these early phases are characterized by a robust pro-inflammatory response, recovery is associated with a compensatory immunoparalysis and lymphocyte apoptosis. This is thought to be in part T-cell mediated and other costimulatory molecules have been shown to regulate this phase of sepsis as well [#74; #224]. We cannot exclude a role for the OX40 system in the transition to this phase due to the high early lethality of our animal model. In addition, both our animal model and our human cohort had predominantly bacterial sepsis with multiple bacterial species represented. It should be acknowledged that the contribution of OX40L to the host inflammatory response may be pathogen dependent. This is even more of an issue for invasive fungal diseases which were not represented in our studies and comprise up to 5% of isolateable pathogens in human sepsis and its incidence is continuing to rise [#37]. Finally, we acknowledge that the ability of OX40L and sOX40 expression to serve as biomarker for either presence or outcome in sepsis is preliminary and must be validated in a larger cohort with a multivariate analysis to better control for other confounders. A recent study suggests monocytes OX40L upregulation may be present in patients with acute coronary syndromes and other studies describe sOX40 upregulation in patients with autoimmune disease [#330; #332]. Neither of these populations were represented in our cohort.
In conclusion, our data describes an integral and novel role for the OX40/OX40L system in the regulation of inflammation and mortality in the early stages of the innate immune response to polymicrobial sepsis. The results of this study provide important pre-clinical information in terms of OX40/OX40L specific biomarker development and therapeutic interventions for the treatment of sepsis and septic shock in the future.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
We would like to thank Meghan Lindauer for her assistance with experiments with MaFIA mice.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.