

Abstract
A perspective from the innovative industry is provided in this article about the long awaited legal proposal for a Clinical Trial Regulation (“Proposal”), adopted in July 2012. With this Proposal, the European Commission reacted to a call by all stakeholders for more harmonization and streamlining of the provisions for conducting clinical trials in the EU. Discrepant approaches between Member States, a failure to respect legal timelines, and a lack of formal coordination mechanisms within and between Member States have resulted in an increased workload for the industry and contributed to a decline in Europe’s attractiveness as a place to carry out research and development. The Proposal introduces a concept whereby the sponsor makes a single submission of the clinical trial application dossier to an EU portal, which is followed by a single assessment based on cooperation between Member States. A possibility for the sponsor to choose a ‘reporting Member State’ to take the lead on key aspects of the assessment is expected to support excellence building and work sharing of Competent Authorities in the EU. The Proposal respects the fact that certain aspects need to be reviewed nationally. The new process aims to lead to a single decision per clinical trial per concerned Member State. The rules are built on the principle of strict adherence to timelines for authorization. The timelines are ambitious but at the same time competitive, as the process builds in mechanisms that strengthen compliance. The rules have been designed to encourage sponsors to file complete submission packages, since any substantial modification to a trial would lead to delays in its commencement. Sponsors need to streamline their internal processes accordingly. In the end, streamlining is an effort that needs to be accepted by all parties involved. The Proposal does not detail how Member States organize the involvement of different bodies, such as Competent Authorities and Ethics Committees, because according to the EU Treaty, the EU cannot legislate on aspects falling into pure Member State competence. The Proposal, however, establishes the assessment objectives on the basis of Good Clinical Practices set by the International Conference on Harmonisation (ICH) and the Declaration of Helsinki by the World Medical Association. As such, the new legislation is likely to have implications on Member States’ internal organization. In addition, Ethics Committees in Europe would benefit from an EU platform for best practice exchange—a concept that would need to be requested by the Council and the European Parliament through the legislative process. A single decision system for the entire EU per clinical trial has been discussed as an option, but such an approach was difficult to achieve while respecting national competencies. In this situation, the Proposal represents an acceptable compromise, provided its proposed mechanisms, processes, and timelines are retained upon implementation. As the Proposal is now on the table for discussion by the 27 Member States’ governments and by the European Parliament, co-legislators and stakeholders should be aware that any dilution of these provisions would be detrimental to the objective to ensure patient access and make the conditions for clinical research in Europe attractive and fit for the future.
Introduction
Innovation is a pillar of the EU’s growth strategy [1]. It is essential that legislation affecting innovative sectors, such as the pharmaceutical industry, reflects this. The revision of the Clinical Trials Directive (“Directive”) [2] is an opportunity to improve regulations that are central to pharmaceutical research and thus patients’ access to innovative medicines.
The Directive’s main purposes are to protect participants in clinical trials, ensure the trial’s ethical soundness, guarantee reliable data, and harmonize administrative aspects for clinical trials to facilitate cost-efficient clinical research.
Divergent national implementation of the Directive (applicable from 2004) has led to delays in starting trials and to increased costs and administrative burden for trial sponsors and authorities [3–6]. This has contributed to a steady decline in trials performed in the EU (a 25 % drop in EU clinical trial applications between 2007 and 2011) [7] and is threatening Europe’s competitiveness as investors may seek more favorable locations [8, 9].
Many stakeholders including regulators [10–12], academics [4–6], and patients [5, 13] recognized that the legislation contained some fundamental flaws. It is potentially the most criticized piece of all the EU medicines legislation [14]. Consequently, the European Commission identified the Directive as a key priority in its flagship initiative to reduce the administrative burden stemming from EU legislation [15]. It adopted a legal Proposal for a Clinical Trial Regulation (“Proposal”) on 17 July 2012, which will be discussed by the European Parliament and the Council of the EU (EU Member States) [7].
This article examines the proposed submission, assessment, and decision-making process for clinical trials, from an innovative company’s perspective.
Challenges of the Existing System
The Directive that governs clinical research in the EU is very complex. Multiple submissions of common documents as well as country-specific information to Member State Competent Authorities and Ethics Committees are needed to start a trial [4, 5, 16]. These processes run mostly in isolation.
The industry has noted that the assessment time to get the authorization to conduct a multinational trial can take as long as 140–301 days compared with the 60-day time limit in the current Directive [17].
A complex system by which ‘single’ Ethics Committee opinions are often based on those of several local committees (of which there are nearly 1,000 in the EU [18]) may have contributed to this situation [3, 8, 9, 16, 19].
Since a Directive sets only basic requirements at EU level, it does not provide a sufficient level of harmonization [16].
Seeking a Remedy
Stakeholder Dialogue
The Commission’s preparations for the legislative Proposal included meetings and public consultations [8, 20, 21] with stakeholders and a report about the impact of European legislation on clinical research [3]. Details around these constructive efforts from 2004 to 2011 are outside the scope of this article; however, the view that the Directive is not functioning properly has been shared by the pharmaceutical industry, small- to medium-sized enterprises (SMEs), academia, and patient organizations.
One policy option discussed during this process was a centralized coordination and assessment by the European Medicines Agency (EMA) that would result in an EU-wide decision, applicable immediately in all Member States [22]. Despite a call from the European Federation of Pharmaceutical Industries and Associations (EFPIA) the Commission did not include this option in the Proposal [22].
Lessons from Voluntary Cooperation
During early talks on the revision of the Directive, stakeholders discussed the idea of building the future framework on an ongoing voluntary cooperation procedure that had been set up between Member States [8, 23–25]. This procedure appeared to offer the advantage of streamlined, well-defined, and common scientific processes [24] and strict adherence to timelines, based on experience from a limited number of cases. However, since it was voluntary and had no legal basis, countries could choose not to participate, which may result in uncertainties for overall clinical development. It also did not address the Ethics Committee reviews, or how to add countries to the trial, which was impractical [23].
The proposed Regulation builds on some of the principles of the voluntary cooperation procedure while attempting to improve upon its weaknesses.
Proposed Regulation
The main concept of the Proposal is that of strictly enforced timelines and complete inputs, from authorities as well as from sponsors.
Another key principle introduced is risk proportionality. The assessment process would take into account the type of intervention and the extent of knowledge/prior experience with the investigational medicinal product (IMP)—particularly whether or not it is authorized in the EU. The provisions on assessment timelines for adding a new country to a trial, safety reporting, conduct of the trial, labeling, and insurance indemnification are likewise risk-adapted.
The new rules are expected to apply at the earliest from 2016. During a 3-year transition phase, many provisions in the old Directive and the new Regulation would apply in parallel [26].
The scope of the Regulation covers interventional clinical trials of medicinal products for human use. Trials requiring no intervention (such as where the research subject is not given a medicine) are excluded.
Submission of a Clinical Trial Application Dossier with Two Distinct Parts
The Commission proposes a clear delineation between the EU elements of the sponsor’s application dossier (Part I) and aspects “of an intrinsic ethical or national/local nature” [7] (Part II) (Fig. 1):
Part I containing ‘general’ scientific/technical data that would be standardized for all Member States concerned
Part II containing national, ethical, and local information (e.g., investigator suitability, insurance) for each concerned Member State
Fig. 1.
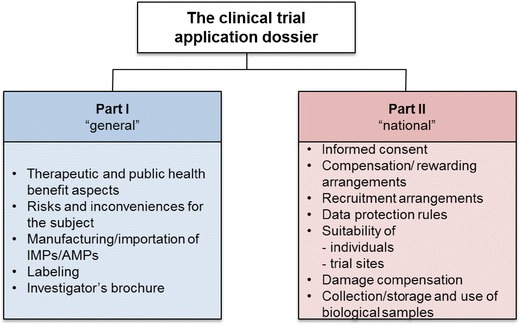
Proposed clinical trial application dossier. AMPs auxiliary medicinal products, IMPs investigational medicinal products
The application dossier and all trial-related information would be submitted to an EU portal linked to a public database and managed by the Commission [7]. Clinical trial data would—as a general rule—be publicly accessible unless confidentiality is justified on specific grounds such as the protection of commercially confidential information and personal data [7].
Assessment
Increased Cooperation Between Countries and Adherence to Timelines
The Proposal is based on two key principles: ‘a single decision’ per country and adherence to timelines, meaning that each Member State is responsible for reaching a decision on whether to authorize the clinical trial, within a set time frame.
Each Member State will decide how it organizes itself to reach this decision, which shall be based on an assessment of scientific, technical, national, and ethical aspects. While the EU sets conditions for the assessment, e.g., involvement of patients and the absence of conflicts of interest, it is not entitled to adopt rules on who—Competent Authorities or Ethics Committees—in a Member State assesses which aspect of a trial application.
The Proposal would facilitate European cooperation through the Clinical Trials Coordination and Advisory Group (CTCAG) comprised of national contact points, which would be set up to address conceptual issues that may arise during the authorization procedure [7].
A Process Where One Member State Takes the Lead
Although the final decision to authorize a trial lies with each individual Member State, the Commission proposes that the sponsor name a ‘reporting Member State’ to lead the scientific, technical, and benefit–risk aspects of the assessment. If the Member State does not wish to take on this role, it can offer the responsibility to other Member States. If there are no candidates, however, the Member State proposed by the sponsor is obliged to assume the responsibility of the reporting Member State.
The reporting Member State assesses Part I and prepares a report with conclusions. This is generally expected to be accepted by other concerned Member States (Fig. 2), which would have the opportunity to ask questions. All such questions are channeled through the reporting Member State, which contacts the sponsor via the portal. The need for a further coordination mechanism to facilitate input from Member States on Part I is likely to be subject to debate as the Proposal moves through the EU legislative process.
Fig. 2.
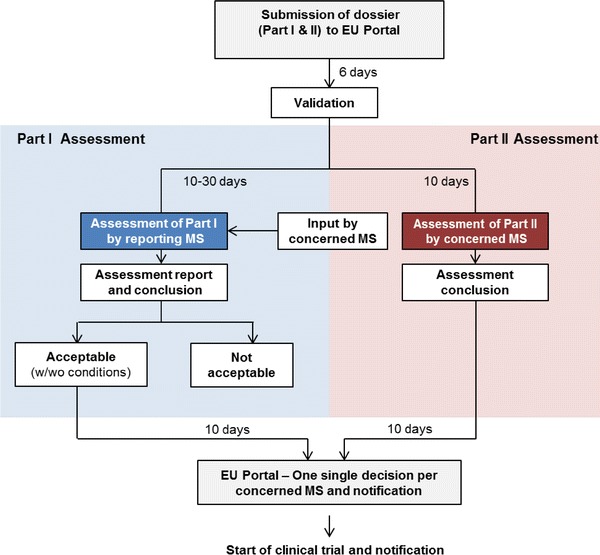
Proposed clinical trial authorization process (simplified). MS Member State, w/wo with/without
As for Part II, each concerned Member State assesses it individually and is allowed, “with justified reasons” [7], to request explanations from the sponsor within a predefined period.
Ethics Committees
Ethical aspects of a clinical trial include, for example, the process and documents to collect informed consent. The Proposal includes, for the first time, clear references to universal ethical principles that shall guide conduct of a trial: the International Conference on Harmonisation (ICH) Good Clinical Practice (GCP) guidelines and the World Medical Association’s Declaration of Helsinki—Ethical Principles for Medical Research Involving Human Subjects.
The actual ethical assessment will continue to be handled at national level. Each Member State decides how to organize itself to perform it. The assessment must be carried out within the time frame that applies to the application. They may introduce national procedures or legislation on the mandate and organization of Ethics Committees and Competent Authorities. Depending on the country, there may be one national Ethics Committee involved or a larger network of several local Ethics Committees.
There is no mechanism proposed to guide the functioning of Ethics Committees or to support systematic cooperation between them. Thus there is no EU forum for information/best practice exchange that may help reduce duplication of work.
Decision
Conclusions of the reporting Member State on Part I would generally be accepted by other Member States, which would only be able to disagree with the conclusions on two grounds:
Significant differences in normal clinical practice compared to that in the reporting Member State
Infringement of national legislation restricting the use of human or animal cells and national legislation on drugs containing, consisting of, or derived from such cells
This is called the ‘qualified opt out’ and obliges the Member State to communicate its disagreement and a detailed scientific and socioeconomic justification to the Commission, all Member States, and the sponsor (Fig. 2).
Each concerned Member State would give one single decision on the trial’s authorization or rejection through the EU portal. If the Member State fails to transmit its decision within the specified timelines, the reporting Member State’s conclusion on Part I of the assessment report would be considered as accepted. According to the Commission, this mechanism should ensure compliance with assessment timelines [7].
Changes to a Clinical Trial: Pre- and Post-approval
The procedures assessing changes to a trial are not consistent under the current Directive. According to the Proposal, a change to a trial application may only be submitted by the sponsor before the assessment on Part I of the initial submission has been communicated. This approach aims to discipline sponsors to ensure accuracy and completeness of their initial application when it is first submitted.
In line with the proposed risk-adapted approach, ‘substantial’ modifications to an already authorized clinical trial would undergo an authorization similar to that of the initial procedure. ‘Substantial’ modifications are changes that have a substantial impact on the safety or rights of the subjects, or on the reliability and robustness of the data generated [7].
Modifications to a clinical trial that are not ‘substantial’ would not require additional authorization.
Is the Legal Proposal Fit for Purpose? Viewpoints of an Innovative Company
The Proposal aims to make the clinical trial assessment system in the EU more benefit–risk based and the authorization processes more effective and less bureaucratic, while still maintaining a high level of patient protection. It reflects the Commission’s efforts to consider the different needs of many stakeholders, namely patients, academia, healthcare professionals, and the pharmaceutical industry. In the following sections, the perspective of an innovative healthcare company is given on how the Proposal meets its goals and what improvements are still needed to make it a success.
How to Make the New Assessment System a Success in Reality
The Proposal is a major step forward. In particular, the introduction of a system where decisions are made according to the principle of respect and adherence to clearly set review timelines has great potential to make the clinical trial authorization process much more effective. These concepts must be kept intact in the final law. It is essential that Europe regains its attractiveness as a place to conduct clinical research in the future, especially in the face of stronger competition than ever from world regions that have managed to create effective regulatory systems for clinical trials.
The proposed system has the built-in flexibility to still allow Member States to carry out expedited assessments of single-country trials if they choose to do so [7].
The pharmaceutical industry supports [27] the concept of a ‘reporting Member State’ (see Sect. 4.2.2) taking a coordinating role. It has the potential to enhance competition and reinforce the establishment of centers of excellence amongst Competent Authorities in Member States [28].
Subsequent trials on the same new molecular entity should be able to refer to the existing information in the EU portal as long as the information has not changed. This reduces repetitive submissions.
It is recognized that to comply with the new requirements, the industry needs to review its own processes to ensure that the initial submission packages are complete and up-to-date (see Sect. 4.5), as failure to provide this could result in delays in starting trials. There is a high likelihood this will require significant internal coordination efforts within globally acting pharmaceutical companies.
In conclusion, to make the proposed streamlined procedures happen in practice, efforts by national authorities and Ethics Committees on one side and clinical trial sponsors on the other side will be required.
What is Needed to Ensure Legislation is Flexible for the Future?
Innovative healthcare companies are testing new possibilities such as recruitment via social media and remote patient participation. Clinical research leading to personalized healthcare and advanced therapies, as well as new endpoints such as patient-reported outcomes, will influence clinical trials designs and approaches in future.
The future legal framework must actively support the assessment of joint clinical trials on personalized healthcare solutions such as a drug and companion diagnostic. It should not be administratively cumbersome or more complex to conduct such trials. Today, this is a real challenge as different regulatory frameworks exist for drugs and companion diagnostics.
This reality demands a regulatory framework that is flexible enough to adapt to technological advances. Therefore, the Regulation should be assessed every 5 years. The innovative industry would like to maintain an ongoing close dialogue with policy makers and legislators about advances in clinical research (purposes and methods) with the goal of contributing to a regulatory framework that is truly fit for purpose.
What Can Be Done to Further Strengthen the Ethics Review Processes?
The Commission’s decision to explicitly transfer key ethical principles, guiding the assessment of clinical trials, from the introductory section of the law into the actual provisions is a positive development. The ICH GCP guidelines and the World Medical Association’s Declaration of Helsinki—Ethical Principles for Medical Research Involving Human Subjects must be followed by all pharmaceutical companies and applied when designing, conducting, recording, and reporting on clinical trials inside and outside the EU. The more prominent placing of these principles in the proposed Regulation is promising.
There is an unfortunate misunderstanding in the medical community that the Proposal weakens the ethical assessment of clinical trials. As explained above, on the contrary, it strengthens the ethical aspect through references to common ethical standards.
While it is understood that further legislation on Ethics Committees falls under Member States’ exclusive competence, the industry would be supportive of further cooperation and harmonization across the EU. In a modern regulatory framework that is moving towards universally defined principles for ethical assessment, it is essential to ensure that the decision processes are likewise adjusted to this international dimension.
A system where national Ethics Committees work in isolation may not be equipped to serve those needs. Therefore, it would be beneficial to ensure further information and best practice exchange and cooperation, at European level, between local and national Ethics Committees. This could be achieved by greater networking opportunities or even by the creation of an EU-funded platform for Ethics Committees. Discussions may help Ethics Committees avoid duplication of work, and ensure that the universal nature of ethical standards is truly respected in national decision making.
Could Contract Negotiations Cause Delays in Trial Commencement?
In addition to the delays in the regulatory assessment (see Sect. 2), some companies have experienced delays to the start of clinical trials in large institutions, like universities. This is partly because the institutions’ contract signature processes have required additional reviews, by that institution, on top of the legal requirements. It is well noted that this is a point for discussion between sponsors and institutions, and cannot be regulated at European level. Therefore, institutions should ensure that contracting processes remain reasonable to avoid delaying the start of clinical trials. This aspect should also be regarded in the legislative and stakeholder debates.
What Level of Transparency is Needed?
The principle of transparency must also apply to the pharmaceutical field to ensure that each stakeholder can take informed decisions in the EU.
The publication of results of clinical studies is currently subject to debate by the public, companies, and the EMA. The Proposal addresses some aspects of this theme, such as submission, decision, and information on inspection planning, through the EU database.
In line with EU access to documents legislation, information held by the EU institutions may be made available, unless publication would undermine commercial interests of companies and the privacy of individuals. Therefore, guidance is needed, based on an open stakeholder discussion, on what constitutes commercially confidential information and personal protected data in the envisaged EU database on clinical trials, while taking into account the market authorization status of the product. In general, a clinical trial application includes a great deal of proprietary information, such as preclinical and manufacturing data but no unpublished clinical trial results. Owing to the primarily confidential nature of the application, establishing the transparency will be a challenge.
Are Provisions for Clinical Trials in Third Countries Sufficient?
Innovative companies are committed to applying the highest ethical and regulatory standards to serve the needs of global patient population. Companies are obliged to perform clinical trials to address needs of local or regional patient populations in third countries (i.e., countries outside the EU/European Economic Area) [29] if they plan to apply for marketing authorizations in these countries and have adopted standards. However, there are limitations on how far EU legislators can support international conditions in third countries since any such regulation is outside their remits. In the EU, any clinical trial report which is submitted with an application for a marketing authorization needs to have been conducted in accordance with ICH GCP. According to the Proposal, the Commission plans to conduct controls and inspections in third countries to verify that the international standards are indeed applied and complied with in those countries. Existing initiatives, such as by the European and Developing Countries Clinical Trials Partnership Programme (EDCTP), must be continued and enhanced, to ensure that international standards are implemented in practice in these countries to respect the specific vulnerability of patients there.
Across the Atlantic
Given the global nature of clinical trials, regions use their systems and regulations to compete for research investment. Companies often report that in the USA, investigational new drug applications for a particular trial are cleared in 30 days by the Food and Drug Administration (FDA). Ethics approvals by the US equivalent of Ethics Committees, Institutional Review Boards (IRBs), are also obtained faster than in the EU. This has resulted in earlier initiation of clinical trials and earlier access to investigational drugs in the USA [17].
The regulation of IRBs has been under increasing scrutiny in recent years. The system has been criticized for “not adequately calibrating the review process to the risk of research” [30]. The FDA is therefore planning to change its regulations and streamline the review of multi-site studies.
Conclusion
Streamlined processes for clinical trial authorizations are essential for the innovative industry to conduct effective clinical research with the goal of developing treatments for patients. The ideal scenario would be one single submission, assessment, and decision for the conduct of a trial in all EU Member States. Owing to the complexity of the EU jurisdiction in relation to national jurisdictions and cultural aspects, such a truly harmonized system would be challenging to achieve in the EU but should be the long-term goal. That said, the Proposal already offers a vast improvement over the current situation. Emphasizing common objectives and adherence to clear timelines, it provides for maximum coordination between Member States and implies a streamlining of national cooperation of authorities and Ethics Committees. In line with these goals, Ethics Committees should be offered an EU-funded platform for best practice exchange.
Sponsors, on their side, need to be prepared to submit comprehensive, complete application dossiers since substantial modifications may prolong the procedure. This may require readjustment of processes within global companies.
The reinforced focus on adherence to timelines and the updated assessment concepts must be supported by stakeholders as the Proposal moves through the EU legislative process. All parties involved in the conduct of clinical trials must be ready to adapt to future changes, in the interest of effective assessment of ethical and safety aspects of trials, patient access to treatment, and the EU’s competitiveness.
Acknowledgments
The authors were employed by F. Hoffmann-La Roche Ltd in Basel and San Francisco at the date of the submission.
The writing of the manuscript was supported by Anna Hallersten and Karen Finn (both SFL Regulatory Affairs & Scientific Communication Ltd, Switzerland) and funded by F. Hoffmann-La Roche Ltd.
The authors interpreted the analysis and the proposed Clinical Trials Regulation, reviewed and edited the manuscript, and made the decision for submission independently.
Contributor Information
Sabine Atzor, Phone: +41-61-6889286, Phone: +41-79-9090280, FAX: +41-61-6881560, Email: sabine.atzor@roche.com.
Surendra Gokhale, Phone: +41-61-6881844, Phone: +41-79-7887211, FAX: +41-61-6881560, Email: surendra.gokhale@roche.com.
Michael Doherty, Phone: +1-650-2258066, Phone: +1-415-2057689, FAX: +41-61-6881560, Email: doherty.michael@gene.com.
References
- 1.European Commission. Europe 2020. http://ec.europa.eu/europe2020/index_en.htm. Accessed 13 Jul 2012.
- 2.European Parliament and the Council. Directive 2001/20/EC of 4 April 2001 on the approximation of the laws, regulations and administrative provisions of the Member States relating to the implementation of good clinical practice in the conduct of clinical trials on medicinal products for human use. http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2001:121:0034:0044:EN:PDF. Accessed 7 Jan 2013. [PubMed]
- 3.European Forum for Good Clinical Practice. Impact on Clinical Research of European Legislation (ICREL). Final report—second version. http://www.efgcp.be/downloads/icrel_docs/Final_report_ICREL.pdf. Accessed 12 July 2012.
- 4.Frewer LJ, Coles D, van der Lans IA, et al. Impact of the European clinical trials directive on prospective academic clinical trials associated with BMT. Bone Marrow Transplant. 2011;46:443–447. doi: 10.1038/bmt.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mason E. European Commission considers revisions to clinical trials directive. J Natl Cancer Inst. 2010;102:292–297. doi: 10.1093/jnci/djq052. [DOI] [PubMed] [Google Scholar]
- 6.Hoey R. The EU clinical trials directive: 3 years on. Lancet. 2007;369:1777–1778. doi: 10.1016/S0140-6736(07)60797-1. [DOI] [PubMed] [Google Scholar]
- 7.European Commission. Proposal for a Regulation of the European Parliament and of the Council on clinical trials on medicinal products for human use, and repealing Directive 2001/20/EC. http://ec.europa.eu/health/files/clinicaltrials/2012_07/proposal/2012_07_proposal_en.pdf. Accessed 17 July 2012.
- 8.European Commission. Assessment of the functioning of the “Clinical Trials Directive” 2001/20/EC. Public consultation paper. ENTR/F/2/SF D (2009) 32674. http://ec.europa.eu/health/files/clinicaltrials/docs/2009_10_09_public-consultation-paper.pdf. Accessed 16 Jul 2012.
- 9.Moulton B. Two years later: the impact of the EU Directive. Appl Clin Trials. 2006;1 Aug.
- 10.Medicines and Healthcare products Regulatory Agency (MHRA). UK response to European Commission review of the Clinical Trials Directive. http://ec.europa.eu/health/files/clinicaltrials/docs/responses_2001-20/medicines_and_healthcare_products_regulatory_agency.pdf. Accessed 7 Jan 2013.
- 11.Danish Ministry of Health and Prevention. Comments on assessment of the functioning of the “Clinical Trials Directive” 2001120/EC. http://ec.europa.eu/health/files/clinicaltrials/docs/responses_2001-20/danish_ministry_of_health_and_prevention.pdf. Accessed 7 Jan 2013.
- 12.Bundesinstituts für Arzneimittel und Medizinprodukte (BfArM). Statement of the Federal Institute for Drugs and Medical Devices and the Federal Institute for Vaccines and Biomedicines (Paul Ehrlich Institute) on the public consultation on the “Revision of the Clinical Trial Directive 2001/20/EC” of the European Commission. http://ec.europa.eu/health/files/clinicaltrials/ctresp_2011-06/bfarm_pei_2.pdf. Accessed 7 Jan 2013.
- 13.European Patients’ Forum. EPF’s response to the Commission public consultation on the Clinical Trials Directive 2001/20/EC. http://ec.europa.eu/health/files/clinicaltrials/docs/responses_2001-20/european_patient_forum.pdf. Accessed 7 Jan 2013.
- 14.European Commission. Roadmap. Legislative proposal on a Regulation/Directive amending the Clinical Trials Directive 2001/20/EC. http://ec.europa.eu/governance/impact/planned_ia/docs/47_sanco_clinical_trials_directive_en.pdf. Accessed 16 Jul 2012.
- 15.European Commission. COM (2007) 23 Communication from the Commission to the Council, the European Parliament, the European Economic and Social Committee and the Committee of Regions. Action programme for reducing administrative burdens in the European Union. http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=COM:2007:0023:FIN:EN:PDF. Accessed 10 Oct 2012.
- 16.van Vyve D, Meunier F. Facing the challenges of the European Clinical Trials Directive—The European Organisation for Research and Treatment of Cancer Perspective. Eur Oncol. 2008;4:14–18. [Google Scholar]
- 17.European Federation of Pharmaceutical Industries and Associations (EFPIA). Assessment of the Functioning of the “Clinical Trials Directive” 2001/20/EC. EFPIA response to public consultation paper ENTR/F/2/SF D (2009) 32674. http://ec.europa.eu/health/files/clinicaltrials/docs/responses_2001-20/efpia_1.pdf. Accessed 16 Jul 2012.
- 18.Druml C, Wolzt M, Pleiner J, et al. Research ethics committees in Europe: trials and tribulations. Intensive Care Med. 2009;35:1636–1640. doi: 10.1007/s00134-009-1544-y. [DOI] [PubMed] [Google Scholar]
- 19.Rice M. New data on clinical trials directive in Europe show few favorable outcomes. J Natl Cancer Inst. 2006;98:159–160. doi: 10.1093/jnci/djj060. [DOI] [PubMed] [Google Scholar]
- 20.European Commission. Revision of the ‘Clinical Trials Directive’ 2001/20/EC. Concept paper submitted for public consultation. http://ec.europa.eu/health/files/clinicaltrials/concept_paper_02-2011.pdf. Accessed 16 Jul 2012.
- 21.European Medicines Agency. European Commission-European Medicines Agency conference on the operation of the Clinical Trials Directive (Directive 2001/20/EC) and perspectives for the future. www.ema.europa.eu/docs/en_GB/document_library/Report/2009/11/WC500011204.pdf. Accessed 12 July 2012.
- 22.European Federation of Pharmaceutical Industries and Associations (EFPIA) and European Vaccine Manufacturers (EVM). Revision of the ‘Clinical Trials Directive’ 2001/20/EC. EFPIA and EVM joint responses and comments in relation to concept paper submitted for public consultation. http://ec.europa.eu/health/files/clinicaltrials/ctresp_2011-06/efpia_evm.pdf. Accessed 16 Jul 2012.
- 23.German Pharmaceutical Industry Assosciation (BPI). Public consultation on the assessment of the functioning of the “Clinical Trials Directive” 2001/20/EC. Public consultation paper (9th October 2009) European Commission. Comments of the German Pharmaceutical Industry Association. http://ec.europa.eu/health/files/clinicaltrials/docs/responses_2001-20/bpi_german_pharmaceutical_industry_assosciation.pdf. Accessed 13 July 2012.
- 24.Gokhale S, Gasser-Stracca M. Voluntary harmonization process for multinational clinical trials in the EU. Right direction, but is it enough? Regulatory focus. http://www.raps.org/focus-online/features/features-article-view/article/1865/voluntary-harmonization-process-for-multinational-clinical-trials-in-the-eu.aspx. Accessed 16 Jul 2012.
- 25.European Organisation for Research and Treatment of Cancer (EORTC). Reply to public consultation revision of the “Clinical Trials Directive” 2001/20/EC concept paper. http://ec.europa.eu/health/files/clinicaltrials/ctresp_2011-06/eortc.pdf. Accessed 7 Jan 2013.
- 26.European Commission. Fostering EU’s attractiveness in clinical research: Commission proposes to revamp rules on trials with medicines IP/12/795. http://europa.eu/rapid/pressReleasesAction.do?reference=IP/12/795. Accessed 4 Sep 2012.
- 27.European Federation of Pharmaceutical Industries and Associations (EFPIA). Position on the legal proposal for a regulation of the European Parliament and of the Council on clinical trials on medicinal products for human use, and repealing Directive 2001/20/EC. http://www.efpia.eu/sites/www.efpia.eu/files/efpia_key_messages_ctr_10.12.2012.pdf. Accessed 30 Jan 2013.
- 28.European Federation of Pharmaceutical Industries and Associations. EFPIA position on the legal proposal for a regulation of the European Parliament and of the Council on clinical trials on medicinal products for human use, and repealing Directive 2001/20/EC (COM (2012) 369 final). 10 December 2012. http://www.efpia.eu/sites/www.efpia.eu/files/efpia_key_messages_ctr_10.12.2012.pdf. Accessed 10 Jan 2013.
- 29.European Medicines Agency. Reflection paper on ethical and GCP aspects of clinical trials of medicinal products for human use conducted in third countries and submitted in marketing authorisation applications to the EMA. EMA/712397/2009. http://www.emea.europa.eu/docs/en_GB/document_library/Regulatory_and_procedural_guideline/2010/06/WC500091530.pdf. Accessed 7 Jan 2013.
- 30.US Department of Health and Human Services Food and Drug Administration. Human subjects research protections: enhancing protections for research subjects and reducing burden, delay, and ambiguity for investigators. Advance notice of proposed rulemaking. http://www.gpo.gov/fdsys/pkg/FR-2011-07-26/pdf/2011-18792.pdf. Accessed 16 Jul 2012.