

Abstract
Acetaminophen (APAP) overdose causes severe and fulminant liver injuries. The underlying mechanism of APAP-induced liver injury (AILI), studied by a murine model, displays similar characteristics of injury as those seen in patients. Previous studies suggest that aside from APAP-induced direct damage to hepatocytes, the hepatic innate immune system is activated and may contribute to the overall pathogenesis of AILI. The current study employed the use of two murine NKT cell knockout models (CD1d−/− and Jα18−/−) to elucidate the specific role of NKT cells in AILI. Compared with WT mice, NKT cell-deficient mice were more susceptible to AILI, as indicated by higher serum alanine transaminase (ALT) levels and mortality. Increased levels of cytochrome P450 2E1 (CYP2E1) protein expression and activities, which resulted in increased APAP-protein adduct formation, were observed in the livers of APAP-treated NKT cell-deficient mice compared with WT mice. Compared with WT mice, starvation of NKT cell-deficient mice induced a higher increase of ketone bodies, which up-regulate CYP2E1 through protein stabilization.
Conclusion
our data revealed a novel role of NKT cells in regulating responses to starvation-induced metabolic stress. Elevated ketone body production in NKT cell-deficient mice resulted in increased CYP2E1-mediated APAP biotransformation and susceptibility to AILI.
Keywords: drug-induced liver injury, reactive oxygen species, innate immunity, CYP2E1 stability, ketone bodies
Introduction
Acetaminophen (APAP) is a commonly used anti-pyretic and analgesic known to be safe and effective at therapeutic doses (1–4 grams/day) (1). However, severe liver injuries have been observed following an acute or cumulative overdose of APAP (10–15 grams/day) (1).
APAP-induced hepatocyte damage is initiated by the formation of the reactive metabolite, N-acetyl-p-benzoquinone imine (NAPQI) (2). NAPQI rapidly depletes glutathione (GSH) within the liver and covalently binds to cellular macromolecules. The impairment of macromolecules results in mitochondrial dysfunction, loss of ATP, and centrilobular necrosis (3). In addition to APAP-induced direct hepatotoxicity, activation of innate immune cells and their production of pro-inflammatory and anti-inflammatory mediators may further influence the severity of APAP-induced liver injury (AILI) (4–8).
Natural killer T (NKT) cells are a unique subset of T lymphocytes that express NK cell markers and represent one subset of innate immune cells within the liver (30–50% of liver lymphocytes) (9). NKT cells possess an invariant T cell receptor (Vα14-Jα18) and recognize glycosphingolipids (9). NKT cell activation by glycolipid antigens occurs through the MHC class I-like molecule CD1d, which presents glycolipid antigens to the T-cell receptor (10). Although the role of NKT cells in AILI has not been examined directly, an earlier study using anti-NK1.1 antibody, which depletes both NK and NKT cells, demonstrated a protoxicant role of the combination (11). The data suggested IFN-γ secretion from NK/NKT cells was responsible for the induction of inflammatory mediators, enhanced leukocyte recruitment, and fas ligand expression. A more recent study revealed that this pathogenic role of NK and NKT cells in AILI was likely due to dimethyl sulfoxide that was used as a solvent for APAP (12). It was found that dimethyl sulfoxide increased the number and activation of hepatic NK and NKT cells. As such, the role of NK and/or NKT cells in AILI remains to be elucidated. In the present study, we aim to investigate the specific role of NKT cells in AILI by the use of mouse models of genetic deletion of NKT cells (CD1d−/− and Jα18−/−).
Our data showed that both CD1d−/− and Jα18−/− mice developed higher degrees of liver injury than wild-type (WT) mice following APAP-challenge. This increased susceptibility in NKT cell-deficient mice was due to their increased expression and activity of CYP2E1 resulting in enhanced APAP metabolism and protein adduct formation.
Experimental Procedures
Mice treatment
Female and male Balb/cJ WT, CD1d−/−, C57Bl/6J WT (Jackson Laboratories, Bar Harbor, ME), and Jα18−/− mice (13) (gift from Dr. Laurent Gapin, National Jewish Health) were maintained in the Center for Animal Care. Mice (7–10 weeks old) were allowed food and water ad libitum until experimental use. Before treatment, food was withheld overnight (16h). APAP (Sigma, St. Louis, MO), dissolved in warm PBS, was administered by intraperitoneal (i.p.) injection and food restored. After various time points, blood and liver tissues were collected.
GSH measurement
Livers were sonicated in 0.1 N perchloric acid (1:20, w/v). GSH was measured by HPLC equipped with electrochemical detection, using a CoulArray system (ESA, Chelmsford, MA).
Measurement of mitochondrial membrane potential and reactive oxygen species (ROS)
Mitochondria were isolated by homogenization of liver tissue (0.5 g), followed by two centrifugation steps at 650 g and 5400g. JC-1 dye (5 µM, Molecular Probes, Grand Island, NY) or MitoSox dye (10 µM, Invitrogen, Grand Island, NY) was added to mitochondrial pellets (1 mg/ml). Membrane potential and ROS were detected by fluorescence excitation/emission spectra of 490/590 nm and 485/520 nm respectively.
CYP2E1 and proteasomal activity
CYP2E1 activity of microsomal protein was measured via hydroxylation of p-nitrophenol as previously described (14). Proteasomal activity of liver homogenates were assayed for chymotrypsin-like (CT-L) activity, and trypsin-like (T-L) activity, as previously described (15).
Ketone body measurement
Serum 3-hydroxybutyrate (BOH) was measured using EnzyChrom Ketone body assay kit (BioAssay Systems, Hayward, CA). Absorbance was measured at 340 nm.
Statistics
Statistical analysis was performed using the student’s t test. Differences in values were considered significant at P<0.05.
Results
CD1d−/− mice are significantly more susceptible to AILI than WT mice
Female WT and CD1d−/− mice were i.p. injected with APAP (385 mg/kg). CD1d−/− mice displayed significantly greater serum ALT levels than WT mice at 8 h and 24 h post-APAP challenge (Supplemental Figure 2S). Moreover, a significant decrease in survival was also observed in CD1d−/− mice compared to WT starting at 8 h post-APAP challenge. Only 25% of CD1d−/− mice survived at 24 h, whereas all the WT mice survived (Figure 1A). When a lower dose of APAP (350 mg/kg) was administered, marked increases in serum alanine transaminase (ALT) levels were observed in CD1d−/− mice compared to WT at 24 and 48 h post-APAP challenge (Figure 1B). Blinded histopathological evaluation of H&E stained liver tissue samples was performed. Histological analysis revealed more dramatic liver injury in CD1d−/− mice compared to WT mice 48 h post-APAP challenge (Figure 1E, F). To determine whether increased susceptibility of CD1d−/− mice to AILI is gender specific, we further compared the susceptibilities of male WT and CD1d−/− mice to AILI. Similar to female mice, a decrease in survival was observed in male CD1d−/− mice compared to WT starting at 8 h with no mice surviving at 48 h post-APAP challenge (235 mg/kg, Figure 1C). At this dose, CD1d−/− mice displayed significantly greater serum ALT levels at 8 h compared to WT mice; however, there were not enough mice that survived to measure ALT at 24 h and 48 h post-APAP challenge (Supplemental Figure 2S). At a lower dose, in which both groups of mice survive, ALT levels were significantly higher in CD1d−/− mice compared to WT mice at 24 h and 48 h post- APAP challenge (230 mg/kg, Figure 1D).
Figure 1. Increased susceptibility of CD1d−/− mice to AILI.

A) Decreased survival of female CD1d−/− mice compared to WT mice after APAP-challenge (385 mg/kg). B) Serum ALT levels, measured by using a colorimetric assay (Teco Diagnostics, Anaheim, CA), were increased in female CD1d−/− mice compared to WT mice at 24 h and 48 h post-APAP challenge (350 mg/kg ). C) Decreased survival of male CD1d−/− mice compared to WT mice after APAP-challenge (235 mg/kg APAP). D) Increased serum ALT levels in male CD1d−/− mice compared to WT mice at 24 h and 4 8h post-APAP challenge (230 mg/kg APAP). E) Photomicrograph (100× and 400X final magnification) of H&E-stained liver sections from female WT and CD1d−/− mice following 48 h APAP. CV, central vein. F) Necrosis, inflammatory infiltration and sinusoidal dilation scores from WT and CD1d−/− mice following 48 h APAP. The individual necrosis (A) and cellular infiltration (B) sinusoidal dilation (C) scores from each mouse strain (3 mice per group) were determined. Results in panel A-D represent mean ± SEM of 10 mice per group. Results in panel F represent average ± SE of 3 mice per group. *p < 0.05 versus WT mice. Data shown are representative of two independent experiments.
Increased APAP-protein adduct formation in CD1d−/− mice compared to WT mice
The mechanism of AILI involves APAP biotransformation into NAPQI, which depletes GSH in the liver. Upon GSH depletion, NAPQI binds to hepatocellular proteins, forming APAP-protein adducts (16). To assess whether differential amounts of APAP-protein adducts are formed in WT and CD1d−/− mice following APAP-challenge, female WT and CD1d−/− mice were treated for 2 h with APAP (350 mg/kg). The levels of hepatic protein-adducts were significantly increased in CD1d−/− mice compared to WT mice (Figure 2A,C). Studies have shown adduct formation in the mitochondria is essential in APAP-toxicity, as this leads to the induction of mitochondrial ROS formation and mitochondrial permeability transition (17). Therefore, mitochondria were isolated following 2 h APAP-challenge to measure APAP-protein adducts. The levels of mitochondrial protein-adducts were significantly higher in CD1d−/− than WT mice (Figure 2B,D).
Figure 2. Increased APAP-protein adducts in CD1d−/− mice compared to WT mice following APAP-challenge.

Female CD1d−/− and WT mice were starved overnight prior to i.p. injected with APAP (350 mg/kg). After 2 h, mice were sacrificed and livers harvested. A) APAP-protein adducts in liver homogenates and B) mitochondrial fractions were probed using anti-APAP antisera (1:5000; gift from Dr. Lance Pohl, National Institutes of Health, Bethesda, MD). Quantification of APAP-protein adducts in C) liver homogenates and D) mitochondrial fractions by densitometric analysis. Results represent mean ± SEM of 3 mice per group. * p < 0.05 versus WT mice. Data shown are representative of three independent experiments.
Increased mitochondrial ROS and dysfunction in CD1d−/− mice than WT mice
Mitochondrial ROS induction has been demonstrated following APAP-challenge (17). In agreement with these findings we observed a significant increase in mitochondrial superoxide following 1 h APAP-challenge in WT mice. Importantly, CD1d−/− mice exhibited significantly higher superoxide levels in the mitochondria compared to WT mice (Figure 3A). Interestingly, we also observed a significant increase in superoxide levels following 16 h starvation of CD1d−/− mice, but not in WT mice (Figure 3A). Associated with the increase in mitochondrial ROS, was a significant decrease in mitochondrial membrane potential in CD1d−/− mice compared with WT mice following starvation, and 1 and 2 h after APAP-challenge (Figure 3B). These data indicate that CD1d−/− mice are uniquely susceptible to mitochondrial oxidative stress and dysfunction following starvation and APAP-challenge.
Figure 3. Increased mitochondrial dysfunction and ROS generation in CD1d−/− mice compared to WT after starvation and APAP-challenge.
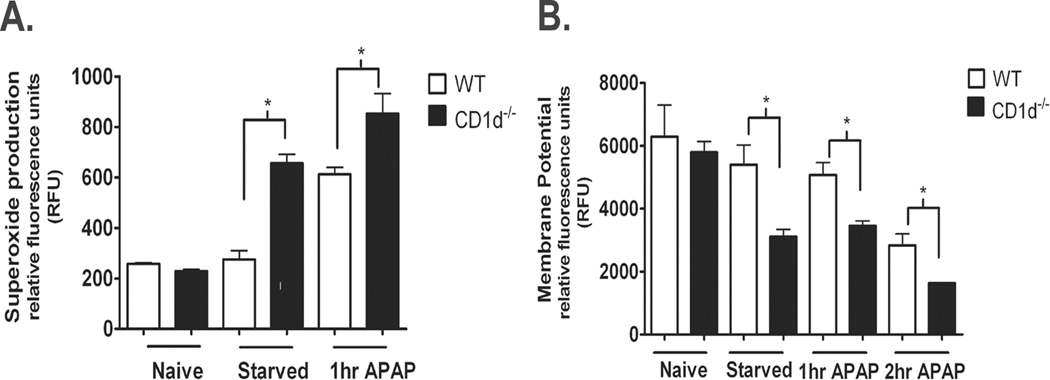
Female WT and CD1d−/− mice were starved overnight for 16 h prior to i.p. injected with APAP (350 mg/kg). After 0, 1, or 2 h of APAP treatment, mice were sacrificed and mitochondria isolated. A) Mitochondrial polarization was detected by fluorometric analysis using JC-1 cationic dye. B) Mitochondrial ROS was detected by using MitoSOX. Results represent mean ± SEM of 3 mice per group. * p < 0.05 versus WT mice. Data shown are representative of two independent experiments.
Similar GSH levels in WT and CD1d−/− mice prior to and following APAP-challenge
Covalent binding of NAPQI by GSH represents an important defense mechanism against APAP toxicity. To assess whether GSH levels were innately different between WT and CD1d−/− mice, liver GSH levels in naïve mice were measured, the data showed similar levels in WT and CD1d−/− mice. Starvation of mice for 16 h caused a similar reduction in GSH levels (approximately 50%) in WT and CD1d−/− mice (Figure 4). Following APAP challenge, GSH levels in WT and CD1d−/− mice decreased to the lowest level at 2 h, and began to rebound at 8 and 16 h. GSH levels were lower in CD1d−/− than WT mice at 8 h post-APAP challenge, perhaps due to the enhanced hepatotoxicity in these mice.
Figure 4. Comparison of GSH levels between WT and CD1d−/− mice.

GSH levels in liver homogenates were determined at 0 (before treatment), 2, 8 and 19 h following treatment of WT and CD1d−/− mice with APAP (350 mg/kg) by HPLC analysis. Results represent mean ± SEM of 10 mice per group. *p < 0.05 versus WT mice. Data shown are representative of two independent experiments.
CYP2E1 protein expression and activity are higher in CD1d−/− mice compared with WT following starvation
CYP2E1 is the major metabolizing enzyme in the biotransformation of APAP into NAPQI (3). Therefore, we compared the expression levels of CYP2E1 in WT and CD1d−/− mice. No difference in CYP2E1 protein levels between naïve WT and CD1d−/− mice was observed. However, following 16 h starvation, CYP2E1 protein levels were significantly higher in CD1d−/− mice compared to WT (Figure 5 A, B and D). While immunoblot analysis did not show changes in CYP2E1 protein levels in WT mice upon starvation, immunohistochemical staining revealed a significant increase in CYP2E1 expression in WT mice; however, starvation-induced up-regulation of CYP2E1 was markedly greater in CD1d−/− mice. CYP2E1 activity was higher in CD1d−/− mice than WT following 16 h starvation (Figure 5C). To confirm that the increased susceptibility of CD1d−/− mice was due to starvation-induced up-regulation of CYP2E1, we treated naïve non-starved female WT and CD1d−/− mice with APAP (700mg/kg). The results show no difference in serum ALT levels between WT and CD1d−/− mice. (Supplemental Figure 1S).
Figure 5. Increased CYP2E1 protein expression and activity in CD1d−/− mice compared to WT upon starvation.

A) CYP2E1 protein levels in female naïve and starved WT and CD1d−/− mice were detected in liver homogenates by immunoblot analysis with anti-CYP2E1 antibody (1:3000, Millipore, Billerica, MA). B) Quantification of CYP2E1 protein levels by densitometric analysis. C) CYP2E1 activity levels in female starved WT and CD1d−/− mice. D) Photomicrograph (400X final magnification) of CYP2E1 (1:1000) immunohistochemical staining of liver sections from naïve and starved female WT and CD1d−/− mice. CV, central vein. Results represent mean ± SEM of 3 mice per group. * p < 0.05 versus WT mice. Data shown are representative of three independent experiments.
Similar Cyp2e1 mRNA and proteasomal degradation but significantly higher ketone body levels in CD1d−/− mice compared to WT mice
CYP2E1 regulation occurs at both transcriptional and post-translational levels (18, 19). To explore the mechanism of the increased CYP2E1 protein expression in CD1d−/− mice following starvation, we examined hepatic Cyp2e1 mRNA levels. Real time PCR analysis of naïve and starved WT and CD1d−/− mice demonstrated that starvation did not up-regulate Cyp2e1 mRNA and that the mRNA levels are similar between WT and CD1d−/− mice (Figure 6A). Furthermore, proteasome peptidase activities, measured by ChT-L and T-L activity analyses, were similar between WT and CD1d−/− mice following starvation (Figure 6B,C), suggesting differential proteasomal degradation cannot explain the increased expression level in CD1d−/− mice. Post-translational stabilization mediated by substrate binding is another possible mechanism accounting for the increased protein expression of CYP2E1 (20). BOH represents a main ketone bodies produced in the liver, that is freely converted into acetoacetate and broken down into acetone, two molecules reported to stabilize CYP2E1 post-translationally (18, 21). Our data demonstrated that naïve WT and CD1d−/− mice exhibited similar levels of BOH in serum. Starvation of mice increased BOH levels in, WT and CD1d−/− mice. A significantly greater elevation of BOH levels was observed in CD1d−/− mice compared to WT mice (Figure 6D).
Figure 6. Transcriptional and post-translational regulation of CYP2E1 in WT and CD1d−/− mice.

A) Quantification of liver Cyp2e1 mRNA expression by real time PCR. B) Quantification of chymotypsin-like (ChT-L) proteasomal degradation activities in WT and CD1d−/− mice following 16 h starvation. C) Quantification of typsin-like (T-L) proteasomal degradation activities in WT and CD1d−/− mice following 16 h starvation. D) Quantification of serum 3-hydroxybutyric acid (BOH) in naïve and starved WT and CD1d−/− mice. Results represent mean ± SEM of 5–10 mice per group. Data shown are representative of two independent experiments.
Increased susceptibility of Jα18−/− mice to AILI
To determine whether the increased susceptibility of CD1d−/− mice to AILI was due to NKT cell depletion but not an unexpected effect of CD1d deletion, we examined another strain of NKT cell-deficient mice, i.e. Jα18−/− mice. Female WT and Jα18−/− mice were injected with APAP (350 mg/kg). A marked increase in serum ALT levels was observed in Jα18−/− mice compared to WT at 8, 24 and 48 h post-APAP challenge (Figure 7A). Similar to female mice, male Jα18−/− mice developed a greater degree of injury compared to WT mice (Figure 7B). Significantly higher APAP-protein adducts and a significant decrease in mitochondrial membrane potential were observed in Jα18−/− mice following 1 h APAP-challenge compared to WT (Figure 7 C,D). A trending decrease in membrane potential was observed following starvation in Jα18−/− compared to WT mice (data not shown). Similar to CD1d−/− mice, starvation resulted in a significant increase in CYP2E1 protein levels in Jα18−/− mice than WT (Figure 7E). Lastly, starvation caused greater elevations of serum BOH levels in Jα18−/− mice compared to WT (Figure 7F). These data confirm that it was the deletion of NKT cells that rendered the mice more susceptible to AILI.
Figure 7. Increased susceptibility of Jα18−/− mice to AILI.

A) Increased serum ALT levels in female Jα18−/− mice compared to WT mice at 8, 24 and 48 h post-APAP challenge (350 mg/kg APAP). B) Increased serum ALT levels in male Jα18−/− mice compared to WT mice at 24 h post-APAP challenge (235mg/kg APAP). Female WT and Jα18−/− mice were starved overnight prior to i.p. injection with APAP (350 mg/kg APAP). After 1 h, mice were sacrificed and livers harvested. C) APAP-protein adducts were detected in liver homogenates. After 1 h of APAP treatment, mice were sacrificed and mitochondria isolated. D) Mitochondrial polarization was detected by fluorometric analysis using JC-1 cationic dye. E) CYP2E1 protein levels in female WT and Jα18−/− mice following 16 h starvation were detected in liver homogenates by immunoblot analysis with anti-CYP2E1 antibody. F) Quantification of serum BOH in starved WT and Jα18−/− mice. Results in panels A, B and F represent mean ± SEM of 10 mice per group. * p < 0.05. Results in panel D represent 3 mice per group. * p < 0.05 versus WT mice. Data shown are representative of two independent experiments.
Discussion
Our data demonstrate that NKT cell-deficient mice are more susceptible to AILI than WT mice. This is due in part to starvation-induced up-regulation of CYP2E1 protein expression and activity, which is associated with marked increases in hepatic APAP-protein adduct formation. Starvation also caused greater elevations of ketone bodies in NKT cell-deficient mice, which may account for the increase in CYP2E1 protein levels.
Upon activation, NKT cells rapidly produce cytokines, such as IL-4 and IFN-γ (9). Many studies have shown both protective and pathological functions of these cytokines in liver disease models (22, 23). Based on these findings, we examined whether differential production of these cytokines between WT and CD1d−/− mice may explain the increased susceptibility of NKT cell-deficient mice to AILI. However, message levels of a number of cytokines were similar in liver tissues and isolated liver mononuclear cells (in which NKT cells are enriched) from APAP-treated WT and CD1d−/− mice (data not shown). These results suggest APAP treatment does not trigger NKT cells to produce protective cytokines.
It is established that APAP metabolism to NAPQI and its covalent modification of liver proteins are essential in triggering hepatocyte damage (16). Because a series of downstream events, such as mitochondrial dysfunction, ATP depletion, and DNA damage, take place prior to ALT release, there is a delay between NAPQI generation and increase of serum ALT levels. Compared with WT mice, CD1d−/− mice had significantly higher levels of APAP-protein adducts as early as 2 h post-APAP (Fig. 2); however, depending on the dose of APAP a significantly higher ALT level was not observed until 8 h (Supplemental Figure 2) or 24 h following APAP challenge (Figure 1).
GSH plays a pivotal role in AILI through scavenging NAPQI. It has been demonstrated that mice deficient in both IL-10 and IL-4 (IL-10/4−/− mice) are more susceptible to AILI compared to WT mice. This appears to be due to lower GSH levels in IL-I0/4−/− mice before, and more dramatically after APAP-challenge (24). We observed no differences in total GSH levels in the liver of naïve or starved WT and CD1d−/− mice (Figure 4). Although there appears to be a slight delay in GSH rebound in the CD1d−/− mice at 8 h, the GSH levels are similar in WT and CD1d−/− mice at 19 h after APAP treatment. Further, we did not observe significant differences in expression and holoenzyme formation of glutamate cysteine ligase, the rate-limiting enzyme for GSH synthesis (data not shown). These data suggest the increased susceptibility of NKT-deficient mice to AILI compared with WT mice is not caused by differential levels of GSH stores or synthesis.
While CYP2E1 protein expression is similar in naïve WT and NKT cell-deficient mice, it is significantly higher in CD1d−/− and Jα18−/− mice than WT mice upon starvation (Figure 5A, B, D, 7E). CYP2E1 activity is induced under a variety of physiological and pathological conditions, including chronic alcohol consumption, nonalcoholic steatohepatitis, and diabetes (20). CYP2E1 protein levels, but not mRNA levels, have been shown to increase 2- to 8-fold following treatment with ethanol, acetone, pyrazole and isoniazid. In pathological conditions, such as diabetes, and obesity CYP2E1 levels have been observed to increase 3- to 8-fold at both the mRNA and protein levels (20). The elevation of CYP2E1 following these conditions has been attributed to changes in metabolism, specifically, the increase of ketone bodies during these states (25). Our data demonstrated no significant difference in transcriptional activation in starved WT and CD1d−/− mice (Figure 6A). WT and CD1d−/− mice displayed similar amounts of proteasomal activity following starvation (Figure 6B, C), indicating that a change in overall proteasomal function was not responsible for the increased CYP2E1 protein and activity. CYP2E1 substrates, such as acetone, pyrazole and ethanol have been reported to enhance CYP2E1 protein expression through increasing protein stability (26). Studies of in vivo protein labeling in rats revealed a biphasic turnover of CYP2E1 at 7 and 32 h. Acetone treatment resulted in the loss of the 7 h degradation of CYP2E1, a process termed “substrate-induced stabilization” (18). Computational modeling of a predicted cytosolic domain of CYP2E1 identified a potential ubiquitylation site, which may also serve as a site for substrate interaction. This finding provides a possible mechanism for the ability of substrate to bind and shield the enzyme from proteasomal degradation (27). Additional CYP enzymes have been shown to be regulated by substrate-induced stabilization. For example, CYP3A protein is stabilized by troleandomycin (28).
Ketone bodies are produced primarily in the liver and serve as a source of energy during starvation. Our data demonstrated following 16 h starvation, NKT cell-deficient mice produced significantly higher amounts of BOH than WT mice (Figure 6D and 7F). The correlation of increased ketone bodies to induction of CYP2E1 is supported by many reports. In a rat model of streptozocin-induced hyperketonemia, increased CYP2E1 protein expression and activity were observed (29). Diabetic rats with severe ketosis, consisting of high BOH in plasma, were found to have significantly higher CYP2E1 than non-diabetic control mice (25). Furthermore, treatment of cultured mouse hepatocytes with acetoacetate stabilizes CYP2E1 protein expression in vitro (21). Acetone has also been implicated in the induction of CYP2E1 activity. When administered to rats in the drinking water, acetone induced CYP2E1 2-fold higher compared to control (18). Aside from the up-regulation of CYP2E1, starvation of CD1d−/− mice significantly affected the mitochondria, as evident by increased ROS production and decreased mitochondrial viability (Figure 3). This may be attributable to starvation-induced elevation in ketone bodies, as they are known to be produced in the mitochondrial matrix of hepatocytes and have been shown to induce mitochondrial ROS and dysfunction (30). The elevation of ketone bodies (acetoacetate) has been associated with decreased glutathione (GSH) levels in diabetic patients as well as, in vitro cell culture models (31). Since GSH is a potent ROS scavenger, reduction in GSH levels is important in causing mitochondrial dysfunction. The mitochondrial impairment was dramatically worsened in CD1d−/− and Jα18−/− than WT mice upon APAP challenge, which likely contributes to increased susceptibility of CD1d−/− mice to AILI (Figure 3B, 8D).
Figure 8. Proposed mechanism for the increased susceptibility of NKT cell deficient mice to APAP-induced liver injury.
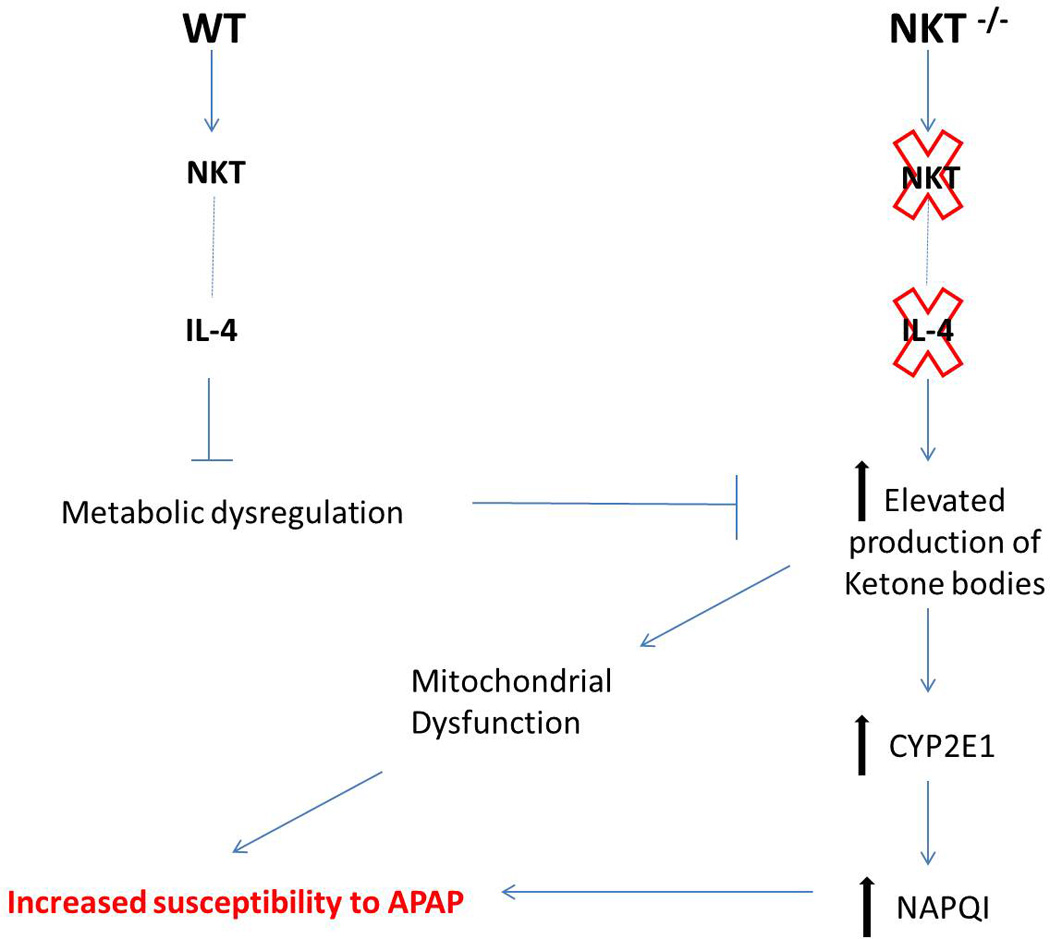
We hypothesize that the intrinsic IL-4 production by NKT cells may be critical in maintaining metabolic homeostasis. Thus, inhibiting the elevation of ketone bodies that are known modulators of CYP2E1 stability. Elevated CYP2E1 can lead to elevated NAPQI metabolite and overall increased susceptibility to AILI.
The increased ketone body production in NKT cell deficient mice suggests an underlying role of NKT cells in metabolism. Several lines of evidence support a link between NKT cells and metabolism. Patients with abetalipoproteinemia, a rare mendelian disorder characterized by a lack of functional microsomal triglyceride transfer protein, also exhibits reduced number of NKT cells and impaired functionality of these cells (32). In murine models of obesity (ob/ob mice), NKT cells are decreased in number (33). Upon adoptive transfer of NKT to ob/ob mice, a significant reduction in liver steatosis was observed, coinciding with marked improvement in glucose sensitivity (34). Furthermore, stimulation and expansion of NKT cell populations by means of norepinephrine or glucocerebroside injection has been shown to decrease the size and fat accumulation in the liver, and decrease overall hepatic injury (35).
The mechanisms by which NKT cells regulate metabolism during conditions of energy deficit or oversupply remain largely unknown despite several recent studies on this topic (36, 37). We hypothesize that the intrinsic IL-4 production by NKT cells may be critical in maintaining metabolic homeostasis. A recent report suggests that IL4 activation of STAT 6 in hepatocytes can regulate fatty acid oxidation by the suppression of PPAR-α (38). It is also reported that IL-4 increases thermogenic gene expression, fatty acid mobilization and energy expenditure, by means of stimulating alternatively activated macrophages (39). Another study demonstrate that IL-4 produced by eosinophils in the adipose tissues is important in protecting mice from high fat diet-induce obesity (40). It is our plan for future studies to examine the role of endogenous IL-4 production by NKT cells in metabolic regulation, which will require the use of IL-4-reporter mice.
In conclusion, our data demonstrate that NKT cells protect mice from AILI because genetic deletion of these cells causes significantly higher ketone body production upon starvation. The increased ketone bodies lead to mitochondrial stress even before APAP challenge, rendering the mice more susceptible to another insult. The increased ketone bodies also stabilize CYP2E1 protein, resulting in a marked increase of APAP bioactivation to generate the hepatotoxic metabolite, which causes liver injury (Figure 8). We found that message levels of a number of cytokines were similar in liver tissues and liver mononuclear cells (in which NKT cells are enriched) isolated from APAP-treated WT and CD1d−/− mice (data not shown). These results suggest that APAP treatment does not trigger NKT cells to produce protective cytokines. Our data do not support an active protective role for NKT cells, but rather, that the lack of NKT cells renders mice more susceptible to AILI. This is the first study to examine the specific role of NKT cells in AILI. The findings provide further insights into the underlying mechanisms of drug-induced liver injury, as well as, other liver conditions in which CYP2E1-mediated ROS generation plays an important pathological role (41). Aside from genetic conditions, such as abetalipoproteinemia, lipid antigens, bacterial and viral pathogens have been demonstrated to activate NKT cells, which leads to decreased cell number (42). Under such situations, NKT cell deficiency may result in increased susceptibility to metabolic stress, as well as hepatotoxin-induced liver injury.
Supplementary Material
Naïve non-starved female WT and CD1d−/− mice were i.p injected with APAP (700mg/kg). Serum ALT levels were measured at 8 and 24 hr post-APAP challenge. Results are presented as mean ± SE of 5 mice per group.
A) Male CD1d−/− mice and WT mice were treated with APAP (235 mg/kg), and serum levels of ALT were measured at 8 and 24 h. *p< 0.05 compared with WT mice. n=1 (panel A) due to death of CD1d−/− mice between 8 and 24hr after APAP-challenge. B) Female CD1d−/− mice and WT mice were treated with APAP (385 mg/kg) and serum levels of ALT were measured at 8 and 24 h. *p<0.05 compared with WT mice.
Acknowledgements
Funding:
Work was supported by F31DK082269 (B.V.M) and RO1ES012914 (C.J.)
We wish to thank Dr. Lance Pohl for the generous gift of anti-APAP antibody, Dr. Laurent Gapin for the generous gift of Jα18−/− mice. We thank Dr. Chris Franklin and Dr. Don Backos for assistance with GCL western blot analysis. We thank Casey Trambly for conducting the proteasome and CYP2E1 activity assays and Dr. James Galligan for assistance in CYP2E1 IHC. Special thanks to Dr. Sean Colgan for the generous use of HPLC instrumentation and Brittelle Bowers and Adrianne Burgess for technical assistance with HPLC setup.
Abbreviations
- APAP
Acetaminophen
- AILI
APAP-induced liver injury
- CYP2E1
cytochrome P450 2E1
- NAPQI
N-acetyl-p-benzoquinone imine
- GSH
glutathione
- NK
natural killer
- NKT
cells with T cell receptor
- ALT
alanine transaminase
- IFN
interferon
- TNF
tumor necrosis factor
- IL
interleukin
- i.p.
intraperitoneal
- ROS
reactive oxygen species
- BOH
3-hydroxybutyrate
Reference List
- 1.Kaplowitz N. Drug-induced liver disorders: implications for drug development and regulation. Drug Saf. 2001;24(7):483–490. doi: 10.2165/00002018-200124070-00001. [DOI] [PubMed] [Google Scholar]
- 2.Raucy JL, Lasker JM, Lieber CS, Black M. Acetaminophen activation by human liver cytochromes P450IIE1 and P450IA2. Arch Biochem Biophys. 1989 Jun;271(2):270–283. doi: 10.1016/0003-9861(89)90278-6. [DOI] [PubMed] [Google Scholar]
- 3.Lee SS, Buters JT, Pineau T, Fernandez-Salguero P, Gonzalez FJ. Role of CYP2E1 in the hepatotoxicity of acetaminophen. J Biol Chem. 1996 May 17;271(20):12063–12067. doi: 10.1074/jbc.271.20.12063. [DOI] [PubMed] [Google Scholar]
- 4.Masubuchi Y, Bourdi M, Reilly TP, Graf ML, George JW, Pohl LR. Role of interleukin-6 in hepatic heat shock protein expression and protection against acetaminophen-induced liver disease. Biochem Biophys Res Commun. 2003 Apr 25;304(1):207–212. doi: 10.1016/s0006-291x(03)00572-2. [DOI] [PubMed] [Google Scholar]
- 5.Bourdi M, Masubuchi Y, Reilly TP, Amouzadeh HR, Martin JL, George JW, et al. Protection against acetaminophen-induced liver injury and lethality by interleukin 10: role of inducible nitric oxide synthase. Hepatology. 2002 Feb;35(2):289–298. doi: 10.1053/jhep.2002.30956. [DOI] [PubMed] [Google Scholar]
- 6.Ju C, Reilly TP, Bourdi M, Radonovich MF, Brady JN, George JW, et al. Protective role of Kupffer cells in acetaminophen-induced hepatic injury in mice. Chem Res Toxicol. 2002 Dec;15(12):1504–1513. doi: 10.1021/tx0255976. [DOI] [PubMed] [Google Scholar]
- 7.Ishida Y, Kondo T, Kimura A, Tsuneyama K, Takayasu T, Mukaida N. Opposite roles of neutrophils and macrophages in the pathogenesis of acetaminophen-induced acute liver injury. Eur J Immunol. 2006 Apr;36(4):1028–1038. doi: 10.1002/eji.200535261. [DOI] [PubMed] [Google Scholar]
- 8.Ishida Y, Kondo T, Tsuneyama K, Lu P, Takayasu T, Mukaida N. The pathogenic roles of tumor necrosis factor receptor p55 in acetaminophen-induced liver injury in mice. J Leukoc Biol. 2004 Jan;75(1):59–67. doi: 10.1189/jlb.0403152. [DOI] [PubMed] [Google Scholar]
- 9.Godfrey DI, Hammond KJ, Poulton LD, Smyth MJ, Baxter AG. NKT cells: facts, functions and fallacies. Immunol Today. 2000 Nov;21(11):573–583. doi: 10.1016/s0167-5699(00)01735-7. [DOI] [PubMed] [Google Scholar]
- 10.Van Kaer L. NKT cells: T lymphocytes with innate effector functions. Curr Opin Immunol. 2007 Jun;19(3):354–364. doi: 10.1016/j.coi.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 11.Liu ZX, Govindarajan S, Kaplowitz N. Innate immune system plays a critical role in determining the progression and severity of acetaminophen hepatotoxicity. Gastroenterology. 2004 Dec;127(6):1760–1774. doi: 10.1053/j.gastro.2004.08.053. [DOI] [PubMed] [Google Scholar]
- 12.Masson MJ, Carpenter LD, Graf ML, Pohl LR. Pathogenic role of natural killer T and natural killer cells in acetaminophen-induced liver injury in mice is dependent on the presence of dimethyl sulfoxide. Hepatology. 2008 Sep;48(3):889–897. doi: 10.1002/hep.22400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cui J, Shin T, Kawano T, Sato H, Kondo E, Toura I, et al. Requirement for Valpha14 NKT cells in IL-12-mediated rejection of tumors. Science. 1997 Nov 28;278(5343):1623–1626. doi: 10.1126/science.278.5343.1623. [DOI] [PubMed] [Google Scholar]
- 14.Chen Q, Cederbaum AI. Cytotoxicity and apoptosis produced by cytochrome P450 2E1 in Hep G2 cells. Mol Pharmacol. 1998 Apr;53(4):638–648. doi: 10.1124/mol.53.4.638. [DOI] [PubMed] [Google Scholar]
- 15.Osna NA, White RL, Todero S, McVicker BL, Thiele GM, Clemens DL, et al. Ethanol-induced oxidative stress suppresses generation of peptides for antigen presentation by hepatoma cells. Hepatology. 2007 Jan;45(1):53–61. doi: 10.1002/hep.21442. [DOI] [PubMed] [Google Scholar]
- 16.Pumford NR, Halmes NC, Hinson JA. Covalent binding of xenobiotics to specific proteins in the liver. Drug Metab Rev. 1997 Feb;29(1–2):39–57. doi: 10.3109/03602539709037572. [DOI] [PubMed] [Google Scholar]
- 17.Hanawa N, Shinohara M, Saberi B, Gaarde WA, Han D, Kaplowitz N. Role of JNK translocation to mitochondria leading to inhibition of mitochondria bioenergetics in acetaminophen-induced liver injury. J Biol Chem. 2008 May 16;283(20):13565–13577. doi: 10.1074/jbc.M708916200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gonzalez FJ. The 2006 Bernard B. Brodie Award Lecture. Cyp2e1. Drug Metab Dispos. 2007 Jan;35(1):1–8. doi: 10.1124/dmd.106.012492. [DOI] [PubMed] [Google Scholar]
- 19.Lieber CS. Cytochrome P-4502E1: its physiological and pathological role. Physiol Rev. 1997 Apr;77(2):517–544. doi: 10.1152/physrev.1997.77.2.517. [DOI] [PubMed] [Google Scholar]
- 20.Novak RF, Woodcroft KJ. The alcohol-inducible form of cytochrome P450 (CYP 2E1): role in toxicology and regulation of expression. Arch Pharm Res. 2000 Aug;23(4):267–282. doi: 10.1007/BF02975435. [DOI] [PubMed] [Google Scholar]
- 21.Abdelmegeed MA, Carruthers NJ, Woodcroft KJ, Kim SK, Novak RF. Acetoacetate induces CYP2E1 protein and suppresses CYP2E1 mRNA in primary cultured rat hepatocytes. J Pharmacol Exp Ther. 2005 Oct;315(1):203–213. doi: 10.1124/jpet.105.084608. [DOI] [PubMed] [Google Scholar]
- 22.Ishida Y, Kondo T, Ohshima T, Fujiwara H, Iwakura Y, Mukaida N. A pivotal involvement of IFN-gamma in the pathogenesis of acetaminophen-induced acute liver injury. FASEB J. 2002 Aug;16(10):1227–1236. doi: 10.1096/fj.02-0046com. [DOI] [PubMed] [Google Scholar]
- 23.Ryan PM, Bourdi M, Korrapati MC, Proctor WR, Vasquez RA, Yee SB, et al. Endogenous interleukin-4 regulates glutathione synthesis following acetaminophen-induced liver injury in mice. Chem Res Toxicol. 2012 Jan 13;25(1):83–93. doi: 10.1021/tx2003992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bourdi M, Eiras DP, Holt MP, Webster MR, Reilly TP, Welch KD, et al. Role of IL-6 in an IL-10 and IL-4 double knockout mouse model uniquely susceptible to acetaminophen-induced liver injury. Chem Res Toxicol. 2007 Feb;20(2):208–216. doi: 10.1021/tx060228l. [DOI] [PubMed] [Google Scholar]
- 25.Bellward GD, Chang T, Rodrigues B, McNeill JH, Maines S, Ryan DE, et al. Hepatic cytochrome P-450j induction in the spontaneously diabetic BB rat. Mol Pharmacol. 1988 Feb;33(2):140–143. [PubMed] [Google Scholar]
- 26.Song BJ, Veech RL, Park SS, Gelboin HV, Gonzalez FJ. Induction of rat hepatic N-nitrosodimethylamine demethylase by acetone is due to protein stabilization. J Biol Chem. 1989 Feb 25;264(6):3568–3572. [PubMed] [Google Scholar]
- 27.Banerjee A, Kocarek TA, Novak RF. Identification of a ubiquitination-Target/Substrate-interaction domain of cytochrome P-450 (CYP) 2E1. Drug Metab Dispos. 2000 Feb;28(2):118–124. [PubMed] [Google Scholar]
- 28.Watkins PB, Wrighton SA, Schuetz EG, Molowa DT, Guzelian PS. Identification of glucocorticoid-inducible cytochromes P-450 in the intestinal mucosa of rats and man. J Clin Invest. 1987 Oct;80(4):1029–1036. doi: 10.1172/JCI113156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shimojo N, Ishizaki T, Imaoka S, Funae Y, Fujii S, Okuda K. Changes in amounts of cytochrome P450 isozymes and levels of catalytic activities in hepatic and renal microsomes of rats with streptozocin-induced diabetes. Biochem Pharmacol. 1993 Aug 17;46(4):621–627. doi: 10.1016/0006-2952(93)90547-a. [DOI] [PubMed] [Google Scholar]
- 30.Rains JL, Jain SK. Hyperketonemia decreases mitochondrial membrane potential and its normalization with chromium (III) supplementation in monocytes. Mol Cell Biochem. 2011 Mar;349(1–2):77–82. doi: 10.1007/s11010-010-0662-8. [DOI] [PubMed] [Google Scholar]
- 31.Jain SK, McVie R. Hyperketonemia can increase lipid peroxidation and lower glutathione levels in human erythrocytes in vitro and in type 1 diabetic patients. Diabetes. 1999 Sep;48(9):1850–1855. doi: 10.2337/diabetes.48.9.1850. [DOI] [PubMed] [Google Scholar]
- 32.Zeissig S, Dougan SK, Barral DC, Junker Y, Chen Z, Kaser A, et al. Primary deficiency of microsomal triglyceride transfer protein in human abetalipoproteinemia is associated with loss of CD1 function. J Clin Invest. 2010 Aug;120(8):2889–2899. doi: 10.1172/JCI42703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guebre-Xabier M, Yang S, Lin HZ, Schwenk R, Krzych U, Diehl AM. Altered hepatic lymphocyte subpopulations in obesity-related murine fatty livers: potential mechanism for sensitization to liver damage. Hepatology. 2000 Mar;31(3):633–640. doi: 10.1002/hep.510310313. [DOI] [PubMed] [Google Scholar]
- 34.Elinav E, Pappo O, Sklair-Levy M, Margalit M, Shibolet O, Gomori M, et al. Adoptive transfer of regulatory NKT lymphocytes ameliorates non-alcoholic steatohepatitis and glucose intolerance in ob/ob mice and is associated with intrahepatic CD8 trapping. J Pathol. 2006 May;209(1):121–128. doi: 10.1002/path.1950. [DOI] [PubMed] [Google Scholar]
- 35.Li Z, Oben JA, Yang S, Lin H, Stafford EA, Soloski MJ, et al. Norepinephrine regulates hepatic innate immune system in leptin-deficient mice with nonalcoholic steatohepatitis. Hepatology. 2004 Aug;40(2):434–441. doi: 10.1002/hep.20320. [DOI] [PubMed] [Google Scholar]
- 36.Miyagi T, Takehara T, Uemura A, Nishio K, Shimizu S, Kodama T, et al. Absence of invariant natural killer T cells deteriorates liver inflammation and fibrosis in mice fed high-fat diet. J Gastroenterol. 2010 Dec;45(12):1247–1254. doi: 10.1007/s00535-010-0272-y. [DOI] [PubMed] [Google Scholar]
- 37.Kotas ME, Lee HY, Gillum MP, Annicelli C, Guigni BA, Shulman GI, et al. Impact of CD1d deficiency on metabolism. PLoS One. 2011;6(9):e25478. doi: 10.1371/journal.pone.0025478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ricardo-Gonzalez RR, Red EA, Odegaard JI, Jouihan H, Morel CR, Heredia JE, et al. IL-4/STAT6 immune axis regulates peripheral nutrient metabolism and insulin sensitivity. Proc Natl Acad Sci U S A. 2010 Dec 28;107(52):22617–22622. doi: 10.1073/pnas.1009152108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nguyen KD, Qiu Y, Cui X, Goh YP, Mwangi J, David T, et al. Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature. 2011 Dec 1;480(7375):104–108. doi: 10.1038/nature10653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu D, Molofsky AB, Liang HE, Ricardo-Gonzalez RR, Jouihan HA, Bando JK, et al. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science. 2011 Apr 8;332(6026):243–247. doi: 10.1126/science.1201475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cederbaum AI, Lu Y, Wu D. Role of oxidative stress in alcohol-induced liver injury. Arch Toxicol. 2009 Jun;83(6):519–548. doi: 10.1007/s00204-009-0432-0. [DOI] [PubMed] [Google Scholar]
- 42.Kinjo Y, Tupin E, Wu D, Fujio M, Garcia-Navarro R, Benhnia MR, et al. Natural killer T cells recognize diacylglycerol antigens from pathogenic bacteria. Nat Immunol. 2006 Sep;7(9):978–986. doi: 10.1038/ni1380. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Naïve non-starved female WT and CD1d−/− mice were i.p injected with APAP (700mg/kg). Serum ALT levels were measured at 8 and 24 hr post-APAP challenge. Results are presented as mean ± SE of 5 mice per group.
A) Male CD1d−/− mice and WT mice were treated with APAP (235 mg/kg), and serum levels of ALT were measured at 8 and 24 h. *p< 0.05 compared with WT mice. n=1 (panel A) due to death of CD1d−/− mice between 8 and 24hr after APAP-challenge. B) Female CD1d−/− mice and WT mice were treated with APAP (385 mg/kg) and serum levels of ALT were measured at 8 and 24 h. *p<0.05 compared with WT mice.