

Abstract
Antiproliferative activity of twenty-nine (E)-4-aryl-4-oxo-2-butenoic acid amides against three human tumor cell lines (HeLa, FemX, and K562) is reported. Compounds showed antiproliferative activity in one-digit micromolar to submicromolar concentrations. The most active derivatives toward all the cell lines tested bear alkyl substituents on the aroyl moiety of the molecules. Fourteen compounds showed tubulin assembly inhibition at concentrations <20 μM. The most potent inhibitor of tubulin assembly was unsubstituted compound 1, with IC50 = 2.9 μM. Compound 23 had an oral LD50 in vivo of 45 mg/kg in mice. Cell cycle analysis on K562 cells showed that compounds 1, 2 and 23 caused accumulation of cells in the G2/M phase, but inhibition of microtubule polymerization is not the principal mode of action of the compounds. Nevertheless, they may be useful leads for the design of a new class of antitubulin agents.
Keywords: Antitumor agents, Structure–activity relationships, 4-aryl-4-oxo-2-butenoic acid amides, Inhibition of tubulin polymerization
1. Introduction
Chalcones, or 1,3-diaryl-2-propen-1-ones (I, Fig. 1), are considered as the intermediate precursors to all flavonoid compounds (II, Fig. 1). Diverse biological activities have been attributed to these compounds, in particular antioxidative [1], antimalarial [2], anti-leishmanial [3], antiinflamatory [4], and anticancer [5] properties. Numerous SAR studies of their anticancer activities have been described [6–9]. Chalcones that originate in plants have –OH and MeO– substitution on rings A and B, due to biosynthetic pathways [10]. Health benefits of natural chalcones can be ascribed to their redox properties and their ability to scavenge radical oxygen species [11]. Along with scavenging of free radicals, biological activity of chalcones can be ascribed to their ability to act as Michael acceptors with nucleophilic moieties, especially with the thiol groups of biological targets. So far, synthetic chalcones that exert mainly –OH, MeO–, and halogen substituents [12]. Alkyl substitution, especially on ring A, occurs relatively infrequently.
Fig 1.
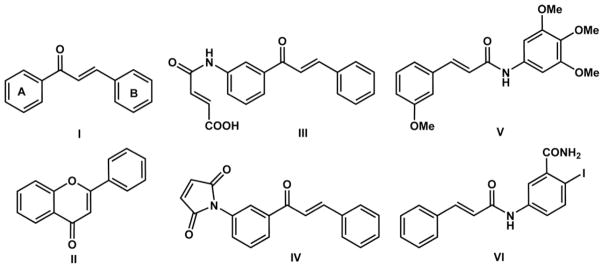
Structures of some compounds with the ketovinyl moiety that have antiproliferative activity: (I) chalcones, (II) flavones, (III) 4-aminochalcone maleamic acid, (IV) 4-aminochalcone maleimides, (V) phenylcinnamides, (VI) cinnamoyl anthranilates.
Chalcones and structurally similar compounds exert their anti-proliferative activity through various mechanisms. For example, recently described chalcone-based compounds that are proteosome inhibitors probably act as covalent binders, involving their ketovinyl moiety [13]. It has been postulated that the presence of a 4-amino function on ring A of the chalcone structure, protonated at physiological pH, increases their reactivity and thus their anti-proliferative activity [14,15]. It has also been confirmed that N-alkylmaleimides exert antiproliferative activity by inhibition of topoisomerase I [16,17]. This led to the design of the 4-aminochalcone maleamic acids (III) and imides (IV) [18]. Most of these derivatives exert antiproliferative activity in the low micromolar range. Moreover, it was shown that phenylcinnamides (V) bind to tubulin, causing inhibition of its polymerization and thereby altering the tubulin–microtubule equilibrium [19]. Structurally similar cinnamoyl anthranilates (VI) also act as antitubulin agents [20]. In addition, another type of molecule containing the ketovinyl moiety, aroylacrylic acids (C, Scheme 1), exert antiproliferative activity toward HeLa cells [21a] and other human dedifferentiated cell lines [21b].
Scheme 1.
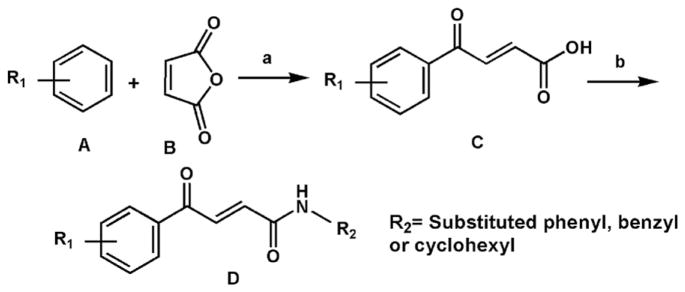
Synthetic pathway to compounds 1–29 (Table 1). Reagents: (a) AlCl3 in CH2Cl2; (b) POCl3, dry THF. For compounds 8 and 17, cyclohexylamine was used to obtain the corresponding aroylacrylic acid amides (D). Benzylamine was used to obtain derivatives 9, 10, 15 and 17. For the rest of the compounds, aniline or substituted anilines were used.
This rationale led us to design chalcone–aroylacrylic acid chimeras, by incorporation of the amidic moiety between the α,β-unsaturated carbonyl moiety and the B ring of chalcones. We have synthesized twenty-nine aroylacrylic acid amides, tested their inhibitory potency on three tumor cell lines, and examined their effects on tubulin polymerization.
2. Results and discussion
2.1. Chemistry
The synthetic pathway to 1–29 is shown in Scheme 1. Friedel– Crafts acylation of the commercially available substituted benzenes (A) with maleic acid anhydride (B) yielded aroylacrylic acids (C) [22]. Subsequently, the acids were converted to acid chlorides in dry THF by phosphorous oxychloride and then in situ reacted with equimolar amounts of aromatic or alicyclic amines to give the corresponding aroylacrylic acid amides (D). The coupling constants of vinylic protons (3JHH) of c.a. 15 Hz proved the E configuration of the double bond in all compounds (see characterization data within Section 5.1 Experimental/Chemistry). Along with this, the NOESY spectrum of compound 11 also proved the E configuration of the double bond. We chose this derivative due to the presence of para-substituted aromatic rings, having a simple AA′BB′ 1H NMR pattern. Distance between ortho- and meta-protons on the aroyl ring was used as reference. The –CH=CH– – NOESY integral appears less than 1% of the chosen reference (Figure S1 in Supplementary material), which also proved E geometry of the double bond. The same compound having Z geometry, as obtained by optimization on the semiempirical level of theory, showed significantly shorter distance of –CH=CH– protons, as expected (Figure S2 in Supplementary material). Molecular geometry, as obtained by single crystal X-ray analysis (unpublished results), of derivative 12 also proved the E geometry of the double bond (Figure S3 in Supplementary material). NMR spectra of representative compounds 7, 8, 9 and 23 are shown in Supplementary material, Figure S4. During evaluation of the purity of compounds by HPLC analysis, we observed a trend between the retention times (RT) and virtual log P values (r = 0.900, n = 29). This finding will be examined in detail separately.
2.2. Antiproliferative activities of 1–29
Previously, we showed that aroylacrylic acids exert anti-proliferative activity toward human tumor cell lines [21] at micromolar concentrations. The aim of this study was to investigate the effects of replacement of the carboxyl group with a cyclo-alkylamide or arylamide moiety. The presence of two aromatic rings bonded directly to each other or separated by one to four carbon atoms, so that they are spatially close but not coplanar, is a significant structural attribute of a number of anticancer compounds, especially antitubulin agents [23].
We tested the antiproliferative activity of 1–29 on HeLa (human cervix carcinoma), FemX (human melanoma) and K562 (human chronic myelogenous leukemia) cells. The results are shown in Table 1. Most of the compounds showed antiproliferative activity at lower concentrations as compared with similarly substituted aroylacrylic acids [21].
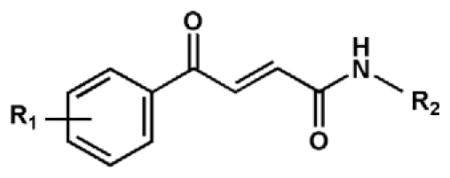
Table 1.
Antiproliferative activity of the synthesized compounds (1–29) toward cancer cell lines.
| Comp. no. |
|
IC50 (μM) | |||
|---|---|---|---|---|---|
|
|
|
||||
| R1 | R2 | HeLa | FemX | K562 | |
| 1 | H | Ph- | 4.91 3 0.99 | 2.20 3 0.50 | 5.30 3 0.08 |
| 2 | 2,5-di-Me | Ph- | 0.74 3 0.13 | 0.62 3 0.13 | 0.34 3 0.08 |
| 3 | 3,4-di-Me | Ph- | 2.95 3 0.27 | 1.84 3 0.65 | 0.72 3 0.19 |
| 4 | 3,4-di-Me | 3,5-di-OMe-Ph- | 2.89 3 0.24 | 1.17 3 0.22 | 0.45 3 0.01 |
| 5 | 3,4-di-Me | 4-i-Pr-Ph- | 2.84 3 0.15 | 1.79 3 0.01 | 1.12 3 0.15 |
| 6 | 4-i-Pr | Ph- | 3.09 3 1.78 | 0.64 3 0.13 | 0.48 3 0.02 |
| 7 | 4-i-Pr | 3,5-di-OMe-Ph- | 1.98 3 0.24 | 0.75 3 0.21 | 0.45 3 0.08 |
| 8 | 4-i-Pr | Ch- | 1.38 3 0.65 | 0.81 3 0.03 | 0.55 3 0.08 |
| 9 | 2,5-di-Me | Bn- | 2.24 3 0.79 | 1.48 3 0.54 | 1.19 3 0.42 |
| 10 | 4-i-Pr | Bn- | 1.37 3 0.11 | 0.49 3 0.21 | 0.34 3 0.01 |
| 11 | 4-i-Pr | 4-OMe-Ph- | 2.92 3 0.04 | 1.06 3 0.07 | 0.68 3 0.16 |
| 12 | 2,4-di-i-Pr | Ph- | 0.68 3 0.13 | 0.59 3 0.15 | 0.38 3 0.10 |
| 13 | 2,4-di-i-Pr | 3,5-di-OMe-Ph- | 0.83 3 0.61 | 0.95 3 0.06 | 0.71 3 0.18 |
| 14 | 2,4-di-i-Pr | 4-i-Pr-Ph | 0.75 3 0.29 | 0.78 3 0.06 | 0.39 3 0.06 |
| 15 | 2,4-di-i-Pr | Bn- | 1.52 3 0.39 | 1.01 3 0.09 | 0.73 3 0.07 |
| 16 | β-tetralinyl | 3,5-di-OMe-Ph- | 2.67 3 0.82 | 1.26 3 0.29 | 0.70 3 0.11 |
| 17 | β-tetralinyl | Ch- | 2.14 3 0.30 | 0.99 3 0.05 | 0.52 3 0.13 |
| 18 | β-tetralinyl | 4-i-Pr-Ph- | 1.80 3 0.17 | 1.02 3 0.09 | 0.41 3 0.04 |
| 19 | β-tetralinyl | Bn- | 1.63 3 0.44 | 0.80 3 0.15 | 0.36 3 0.01 |
| 20 | β-tetralinyl | Ph- | 2.11 3 0.09 | 0.73 3 0.16 | 0.52 3 0.06 |
| 21 | 4-n-Bu | Ph- | 1.06 3 0.30 | 0.62 3 0.06 | 0.45 3 0.02 |
| 22 | 2,3,5,6-tetra-Me | 3,5-di-OMe-Ph- | 0.86 3 0.53 | 0.65 3 0.15 | 0.55 3 0.05 |
| 23 | 4-F | Ph- | 2.99 3 0.65 | 0.63 3 0.32 | 0.55 3 0.07 |
| 24 | 4-Cl | Ph- | 2.91 3 0.29 | 2.84 3 0.37 | 0.61 3 0.07 |
| 25 | 3,4-di-Cl | Ph- | 2.80 3 0.07 | 2.30 3 0.28 | 0.51 3 0.10 |
| 26 | 4-Br | Ph- | 2.29 3 0.98 | 2.30 3 0.33 | 0.40 3 0.12 |
| 27 | 4-OMe | Ph- | 3.50 3 0.54 | 2.34 3 0.48 | 1.31 3 0.25 |
| 28 | 4-OMe | 3,5-di-OMe-Ph- | 2.55 3 0.35 | 0.52 3 0.09 | 0.38 3 0.03 |
| 29 | 3,5-di-Me-4-OMe | 3,5-di-OMe-Ph- | 1.36 3 0.38 | 0.76 3 0.33 | 0.82 3 0.35 |
| Cisplatina | / | / | 4.4 3 0.30 | 4.7 3 0.30 | 5.7 3 0.30 |
Adopted from Reference [25].
HeLa cells were the least sensitive toward the newly synthesized compounds, with IC50 values in the one-digit micromolar range for the majority of compounds. Compounds having alkyl substituents at positions 2 and 5 or at 2 and 4 on the aroyl phenyl ring (2, 12, 13, 14, and 22) exerted potency at submicromolar concentrations on the HeLa cells. A similar relationship between aroyl substitution and antiproliferative activity was observed for aroylacrylic acids [21]. The influence of structural variations of the amidic moiety on antiproliferative activity could not be interpreted in a straightforward manner, but we observed that more ovoid molecules, having larger apolar surface areas (ASA), displayed higher potency. Trends between ASA and an oval shape of the molecules and their potency toward HeLa cells are shown in Supplementary material, Figure S5a and b. We did not observe such a trend with the estimated virtual log P values [24].
The majority of alkyl-substituted derivatives with unsubstituted phenyl groups at the amidic portion of the molecule exerted sub-micromolar activities toward the FemX cell line. The least active were unsubstituted 1 and derivatives with halogen or methoxy substituents (23–27) at the aroyl moiety. These compounds had IC50 values above 2 μM. Generally, the presence of a 4-i-Pr substituent at the phenylamidic moiety of the molecules (5, 14, 18) slightly decreased antiproliferative activity in comparison with their unsubstituted counterparts. In most instances, 3,5-dimethoxy substitution on the amidic portion of the molecule had an unfavorable influence on antiproliferative activity. We did not observe other specific structural features that affected the antiproliferative potency of 1–29 toward FemX cells.
K562 cells were the most sensitive toward the newly synthesized compounds. Most compounds had IC50 values in the narrow range of 0.34–0.82 μM. The exceptions were the unsubstituted 1 and compounds 5 and 27.
The majority of the compounds contain phenylamide or 3,5-di-MeO-phenylamide moieties. It should be noted that among the phenylamide derivatives (1–3, 6, 12, 20, 21, 23, 24–27), compounds that have a more lipophilic aroyl moiety exerted a more pronounced antiproliferative activity toward all cell lines tested. We observed the same trend for 3,5-di-MeO-phenylamide derivatives (4, 7, 13, 16, 22, 28, 29) toward HeLa cells. Among the 3,5-di-MeO-phenylamide derivatives, the less lipophilic derivatives exerted a more pronounced antiproliferative activity toward FemX and K562 cell lines (see for example 28 vs. 13). Due to the narrow range of IC50’s toward FemX and K562 cells, this conclusion should be taken with caution.
A number of the compounds had submicromolar activity against all three cell lines examined. These were compounds 2, 12, 13, 14 and 22.
2.3. Inhibition of tubulin polymerization by 1–29 and cell cycle analysis
Turning to molecular mechanism of action, we first considered tubulin as a potential target, based on the structural similarity of 1– 29 with the known tubulin inhibitory activity of phenylcinnamides (V) [19] and cinnamoyl anthranilates (VI) [20] (Fig. 1). Compounds 1–29 were compared with combretastatin A-4 as a reference compound for inhibition of tubulin assembly (Table 2). Combretastatin A-4 was generously provided by Dr. George R. Pettit, Arizona State University.
Table 2.
Inhibition of tubulin polymerization and colchicine binding by 1–29.
| Comp. No. | Tubulin polymerization inhibition
|
Inhibition of colchicine binding (% ± SD)
|
|
|---|---|---|---|
| IC50 (μM ± SD) | 50 μM | 5 μM | |
| 1 | 2.9 ± 0.3 | 39 ± 5 | 24 ± 0.8 |
| 2 | 5.2 ± 0.2 | 38 ± 1 | 9.3 ± 3 |
| 3 | 6.5 ± 0.08 | 14 ± 5 | 7.0 ± 4 |
| 4 | >20 | / | / |
| 5 | >20 | / | / |
| 6 | 9.7 ± 1 | 25 ± 2 | 3.1 ± 3 |
| 7 | >20 | / | / |
| 8 | 11 ± 2 | 14 ± 0.8 | 9.6 ± 5 |
| 9 | 8 ± 0.08 | 19 ± 4 | 12 ± 0.01 |
| 10 | >20 | / | / |
| 11 | >20 | / | / |
| 12 | >20 | / | / |
| 13 | >20 | / | / |
| 14 | >20 | / | / |
| 15 | >20 | / | / |
| 16 | >20 | / | / |
| 17 | 11 ± 0.07 | 13 ± 2 | 8.9 ± 2 |
| 18 | >20 | / | / |
| 19 | >20 | / | / |
| 20 | 18 ± 0.9 | / | / |
| 21 | 11 ± 0.1 | 20 ± 5 | 2.7 ± 4 |
| 22 | >20 | / | / |
| 23 | 3.5 ± 0.3 | 31 ± 4 | 11 ± 3 |
| 24 | 11 ± 0.6 | 19 ± 0.8 | 2.8 ± 1 |
| 25 | 10 ± 2 | 20 ± 5 | 0.3 ± 2 |
| 26 | 8.6 ± 0.5 | 28 ± 4 | 6.2 ± 3 |
| 27 | 7.8 ± 1 | 24 ± 2 | 5.9 ± 5 |
| 28 | >20 | / | / |
| 29 | >20 | / | / |
| Combretastatin A-4 | 1.2 ± 0.007 | / | 99 ± 0.7 |
Fourteen compounds inhibited tubulin polymerization with IC50 values below 20 μM. The unsubstituted derivative 1 had the lowest IC50 value (2.9 μM), while the other effective inhibitors all had small substituents on the aroyl moiety and no substituent on the phenylamide portion of the molecule. Compounds substituted on the phenylamide moiety (4-i-Pr, 3,5-di-MeO-) showed only weak inhibitory activity in the polymerization assay or were inactive. The halogen and methoxy substituted derivatives (23–27) that exerted inferior antiproliferative activities, as compared with other compounds, were relatively active as inhibitors of tubulin polymerization. Four (compounds 12, 13, 14 and 22) of the five best antiproliferative agents were inactive as antitubulin agents. Therefore, the SAR of 1–29 for the inhibition of tubulin polymerization was quite different from the SAR observed in the cancer cell line screen.
Thirteen compounds that showed activity as inhibitors of tubulin polymerization were tested as inhibitors of colchicine binding, at 5 and at 50 μM (Table 2). All tested compounds showed some, albeit modest, inhibition of colchicine binding. The greatest inhibition of colchicine binding was observed with compounds 1, 2 and 23, which were also the three best inhibitors of tubulin polymerization. It is possible that these derivatives exert antitubulin activity by binding at the same site as colchicine, which was also observed with structurally similar antitubulin chalcones [26].
Molecules that alter tubulin assembly in cells invariably cause alteration of cell cycle distribution, with preferential G2/M blockade. We therefore examined the mechanism of action of four of the newly synthesized derivatives (Table 3 and Figure S6 in Supplementary material) by cytofluorimetric analysis, using propidium iodide to label the DNA of K562 cells. The effects of compounds 1, 2 and 23, which inhibited tubulin polymerization with IC50 values below 5 μM, were examined, as well as the effect of compound 14. The latter compound exerted significant anti-proliferative potency toward all cells tested and bears bulky alkyl substituents both on the aroyl phenyl and the phenylamido moieties. Colchicine was used as the reference compound. Compound 1 caused an increase in the proportion of cells in the G2/M phase, without dose dependence. In contrast, compounds 2 and 23 caused an increase in the proportion of G2/M cells in a dose-dependent manner, as compared with the control. Compound 14 did not cause an increase in the proportion of G2/M cells, even at 8 times the IC50 value. The increase in the proportion of cells in the G2/M phase caused by compounds 1, 2 and 23 was less than the effect of colchicine or of structurally similar compounds with an antitubulin mechanism of action (see, for example, reference [20]). Thus, it seems likely that there is another, more dominant molecular mechanism of action for our compounds through which they exert their antiproliferative activity. Our compounds are powerful Michael acceptors, acting as unsaturated ketones (kinetic study of the addition of the model thiol and amino compounds on representative congeners will be described elsewhere), so they probably also act as alkylating agents.
Table 3.
Cell cycle analysis.
| Compound no. | Concentration | G0/G1 (%) | S (%) | G2/M (%) |
|---|---|---|---|---|
| Control | 39.55 | 26.17 | 12.95 | |
| 1 | 2 × IC50 | 35.93 | 39.18 | 19.84 |
| 4 × IC50 | 40.24 | 35.26 | 18.32 | |
| 8 × IC50 | 40.56 | 36.54 | 17.30 | |
| 2 | 2 × IC50 | 42.92 | 27.44 | 12.57 |
| 4 × IC50 | 40.94 | 32.37 | 17.72 | |
| 8 × IC50 | 37.48 | 37.32 | 19.04 | |
| 23 | 2 × IC50 | 44.38 | 24.74 | 14.67 |
| 4 × IC50 | 41.80 | 29.01 | 16.41 | |
| 8 × IC50 | 37.70 | 37.56 | 19.13 | |
| 14 Colchicine | 8 × IC50 | 40.76 | 24.52 | 12.30 |
| 2 μM | 21.12 | 16.86 | 21.75 | |
| 4 μM | 24.03 | 17.78 | 24.14 | |
| 8 μM | 23.71 | 18.02 | 23.58 |
2.4. In vivo acute toxicity
Compound 23 was chosen for an in vivo acute toxicity test. This compound exerted both significant antiproliferative potency toward all cell lines tested and showed significant inhibition of tubulin polymerization. Compound 23 is also the only fluoro-substituted derivative in the set. The fluoro substituent at position 4 of the aroyl phenyl ring makes it, probably, more stable toward metabolic degradation by cytochromes (see, for example, [27]). Due to insufficient water solubility and the absence of ionizable moieties amenable to prepare the salt, the compound was dissolved in DMSO and given orally to mice. The LD50 value was 45 mg/kg. Further structural optimization is required to obtain less toxic compounds.
3. Molecular modeling
All compounds exert significant antiproliferative potency in low micromolar to submicromolar concentrations, spanning a narrow range of IC50 values. A narrow range of activity values usually does not produce statistically significant correlations, so we did not perform a QSAR study for data on the antiproliferative potency of 1–29. Among 1–29, roughly, compounds having a solvent-assessable area <620 Å2 and a volume <305 Å3 inhibited tubulin polymerization with IC50 values <20 μM (Table S7 in Supplementary material). The majority of tubulin assembly inhibitors that target the colchicine site, reported thus far, resemble colchicine or combretastatin A-4, bearing two or three MeO/OH substituents on the aromatic rings, which are connected by a two-to four-carbon spacer [23,28]. Within 1–29, the unsubstituted compound or compounds bearing a small substituent at position 4 of the aryl moiety are the most potent inhibitors of tubulin polymerization and of colchicine binding. Also, an unsubstituted phenyl of the amidic moiety is preferred in our compound series. Therefore, in our modeling study, among compounds cocrystallized with tubulin, we chose the minimally substituted compounds E70, from the PDB entry 3HKC, and N16, from the PDB entry 3HKD [29] (structures in Figure S8, Supplementary material) in their tubulin-bound, cocrystallized conformations, for comparison with compounds 1–3, 6, 8, 9, 17, 21, 20, and 23–27. The shape and the pharmacophoric pattern of the compounds were compared using the ROCS program [30]. We found a trend between pharmacophoric similarity with E70 of the set studied (OpenEye Color force field/ Explicit Meals-Dean [31], Tanimoto scoring) and the percentage of inhibition of colchicine binding at 50 μM (r = 0.89), with two outliers (17 and 24) (Figure S9 in Supplementary material). It should be mentioned that pharmacophoric similarity between templates and compounds studied did not exceed 0.23 for E70 or 0.35 N16, as obtained with the Tanimoto function. Other statistically significant correlations or trends were not observed. Despite this, compounds 1, 2, and 23 were always highly ranked using a combined scoring function that includes both the shape and the pharmacophoric similarity (Tanimoto combo), having a score >0.80 for E70, and >1.00 for N16 (Figure S10 in Supplementary material). The Tanimoto combo score for all compounds studied exceed 0.62 for E70, and 0.95 for N16.
4. Conclusion
(E)-4-aryl-4-oxo-2-butenoic acid amides exert antiproliferative activity toward three human tumor cell lines (HeLa, FemX, and K562) with one-digit micromolar to submicromolar IC50 values. All derivatives had antiproliferative activity that was significantly greater than that of their precursors, aroylacrylic acids. The replacement of the carboxyl group of aroylacrylic acids with an arylamide or cycloalkylamide moiety proved beneficial for anti-proliferative activity. The most active derivatives toward all three cell lines tested were compounds with alkyl substituents on the aroyl moiety. Although fourteen compounds inhibited tubulin assembly with IC50 values <20 μM, cell cycle analysis of compound-treated K562 cells showed that only a few compounds caused accumulation of cells in the G2/M phase. The mode of action competitive to tubulin polymerization inhibition obviously exists. Nevertheless, the newly synthesized compounds may represent good leads for the design of a new class of antitubulin agents with the SAR data we obtained. The most potent inhibitor of tubulin polymerization was the compound unsubstituted on both the aroyl and the arylamido moieties, having an IC50 = 2.9 μM. Our data further indicate that bulkier substituents on both the aroyl and amidic moieties of the molecules impose steric hindrance for the binding of this class of compounds to tubulin. Structural modification to further test this hypothesis is underway.
5. Experimental
5.1. Chemistry
All chemicals were purchased from Fluka, Aldrich or Merck, having >98% purity, and were used as received. Melting points were determined in open capillary tubes on an SMP-10 Stuart apparatus and are uncorrected. ESI-MS spectra were recorded on an Agilent Technologies 6210-1210 TOF-LC-ESI-MS instrument in positive mode. For HPLC analysis the Agilent HPLC system 1200 (equipped with vacuum degasser, binary pump, autosampler, thermostatted column compartment and DAD UV detector) with Zorbax Extend C18 column (RRHT, 50 × 4.6 mm, 1.8 μm) was used. Compounds were dissolved in MeOH (0.5 mg/mL), sample volume was 0.2 μL, column temperature was 40 °C. The UV detector was set on 280 and 310 nm. Mobile phase A was 0.2% HCOOH in H2O, mobile phase B was MeCN. Gradient mode: 0–2 min 40% A: 60% B; 2–10 min 60% B – 100% B. All compounds had >99.5% purity. 1H and 13C NMR spectra were recorded in DMSO-d6 or CDCl3 on a Bruker AVANCE 500/125 MHz or a Varian Gemini2000 200/ 50 MHz instrument. NOESY spectrum of compound 11 was recorded on a Bruker AVANCE 500 instrument. Chemical shifts are reported in parts per million (ppm) relative to tetramethylsilane (TMS), and spin multiplicities are given as follows: s (singlet), d (doublet), t (triplet), h (heptet), m (multiplet) or br (broad).
5.2. General procedure for synthesis of compounds is given under 2.1 Chemistry
5.2.1. (E)-4-oxo-4-phenyl-2-butenoic acid phenylamide (1)
C16H13NO2, starting from (E)-4-phenyl-4-oxo-2-butenoic acid (0.017 mol) and a corresponding amount of aniline, 1.70 g of 1 was obtained, 39.81% yield, light yellow solid, m.p. = 143–145 °C decomposition, (AcOEt). 1H NMR (500 MHz, DMSO-d6) δ = 7.15 (t, J = 7.63 Hz, 1H, amido p-phenyl), 7.36–7.32 (multiplet, 3H, including ~7.33 (d, J = 14.35 Hz, 1H, C(O)–CH=CH–) and 7.35 (t. J = 7.32 Hz, 2H, aroyl m-phenyl)), 7.50 (t, J = 7.93 Hz, 2H, amido m-phenyl), 7.62 (t, J = 7.32 Hz, 1H, aroyl p-phenyl), 7.67 (d, J = 7.94 Hz, 2H, amido o-phenyl), 8.04 (d, J = 7.02 Hz, 2H, aroyl o-phenyl), 8.11 (d, 14.95 Hz, 1H, C(O)–CH=CH–), 8.45 (s,b, 1H, NH–C(O)). 13C NMR (125 MHz, DMSO-d6) δ = 120.19, 125.03, 128.92, 129.10, 133.79, 134.01, 136.21, 136.79, 137.65, 162.10, 190.05. HR MS (ESI): 252.1003 (M + 1), Calc. 252.1025. HPLC purity >99.5%; RT 3.447 min.
5.2.2. (E)-4-(2,5-dimethylphenyl)-4-oxo-2-butenoic acid phenylamide (2)
C18H17NO2, starting from (E)-4-(2,5-dimethylphenyl)-4-oxo-2-butenoic acid (0.017 mol) and a corresponding amount of aniline, as described above, 2.56 g of 6 was obtained, 70.52% yield, yellow solid, m.p. = 159–160 °C, (AcOEt). 1H NMR (500 MHz, DMSO-d6) δ: 2.34 (s, 3H, m-CH3), 2.50 (s, 3H, o-CH3), 6.97 (d, J = 15.11 Hz, 1H, C(O)–CH=CH–), 7.11 (t, J1,2 = 7.16 Hz, 1H, amido p-phenyl), 7.24 (d, J = 7.95, 1H, aroyl p-phenyl), 7.30 (doublet like peaks, 1H, aroyl m-phenyl), 7.35 (t, J1,2 = 8.71 Hz, 2H, amido m-phenyl), 7.43 (d, J = 15.11 Hz, 1H, C(O)–CH=CH–), 7.46 (s, 1H, aroyl o-phenyl), 7.69 (d, J = 8.75 Hz, 2H, amido o-phenyl), 10.54 (1H, s, NH–C(O)). 13C NMR (125 MHz, DMSO-d6) δ = 19.65, 20.36, 119.38, 124.07, 128.88, 129.23, 131.40, 132.10, 133.91, 135.10, 136.39, 137.22, 138.65, 161.80, 193.75. HR MS (ESI): 280.1338 (M + 1), Calc. 280.1338. HPLC purity >99.5%; RT 4.696 min.
5.2.3. (E)-4-(3,4-dimethylphenyl)-4-oxo-2-butenoic acid phenylamide (3)
C18H17NO2, starting from (E)-4-(3,4-dimethylphenyl)-4-oxo-2-butenoic acid (0.013 mol) and a corresponding amount of aniline, 2.00 g of 3 was obtained, 55.10% yield, yellow solid, m.p. = 163– 165 °C, (AcOEt). 1H NMR (500 MHz, DMSO-d6) δ = 2.32 (s, 6H, m,p-CH3), 7.12 (t, J1,2 = 7.30 Hz, 1H, amido p-phenyl), 7.22 (d, J = 15.11 Hz, 1H, C(O)–CH=CH–), 7.37 (m, 3H, amido m-phenyl and aroyl m-phenyl), 7.73 (d, J = 7.30 Hz, 2H, amido o-phenyl), 7.80 (d, J = 8.32 Hz, 1H, aroyl o-phenyl), 7.85 (s, 1H, aroyl o-phenyl), 7.92 (d, 1H, J = 15.11 Hz, C(O)–CH=CH–), 10.60 (s, 1H, NH–C(O)). 13C NMR (125 MHz, DMSO-d6) δ = 19.28,19.66,119.44,124.04,128.49,128.90, 129.59, 130.08, 132.08, 133.00, 134.48, 136.27, 137.22, 138.75, 143.37, 162.00, 189.05. HR MS (ESI): 280.1335 (M + 1), Calc. 280.1338. HPLC purity >99.5%; RT 4.618 min.
5.2.4. (E)-4-(3,4-dimethylphenyl)-4-oxo-2-butenoic acid (3,5-dimethoxyphenyl)amide (4)
C20H21NO4, starting from (E)-4-(3,4-dimethylphenyl)-4-oxo-2-butenoic acid (0.0114 mol) and a corresponding amount of 3,5-dimethoxyaniline, 2.32 g of 4 was obtained, 59.95% yield, yellow solid, m.p. = 162–164 °C, decomposition (AcOEt). 1H NMR (500 MHz, CDCl3) δ = 2.31 (s, 3H, p-CH3), 2.34 (s, 3H, m-CH3), 3.77 (s, 6H, –OCH3), 6.26 (t, J = 2.35 Hz, 1H, amido p-phenyl), 7.01 (doublet like peak, 2H, aroyl o-phenyl), 7.25 (d, J = 7.70 Hz, 1H, aroyl m-phenyl), 7.39 (d, J = 14.96 Hz, 1H, C(O)–CH=CH–), 7.81 (d, J = 7.89 Hz, 1H, aroyl o-phenyl), 7.84 (s, 1H, aroyl o-phenyl), 8.12 (d, J = 14.75 Hz, 1H, C(O)–CH=CH–), 8.85 (sb, 1H, NH–C(O)). 13C NMR (125 MHz, CDCl3) δ = 19.68, 20.18, 55.39, 97.51, 98.48, 126.79, 130.09,130.20, 133.94, 134.79, 134.79, 135.95, 137.53, 139.70,144.05, 161.06,162.48,189.79. ESI-MS HR: 340.1544 (M + 1), Calc. 340.1549. HPLC purity >99.5%; RT 4.802 min.
5.2.5. (E)-4-(3,4-dimethylphenyl)-4-oxo-2-butenoic acid (4-isopropylphenyl)amide (5)
C21H23NO2, starting from (E)-4-(3,4-dimethylphenyl)-4-oxo-2-butenoic acid (0.0076 mol) and a corresponding amount of 4-isopropylaniline, 1.65 g of 5 was obtained, 67.62% yield, yellow solid, m.p. = 129–130 °C, decomposition (AcOEt). 1H NMR (500 MHz, CDCl3) δ = 1.24 (d, J = 6.83 Hz, 6H, i-PrCH3), 2.29 (s, 3H, p-CH3), 2.33 (s, 3H, m-CH3), 2.89 (h, J = 6.82 Hz, 1H, i-PrCH), 7.20 (d, J = 8.58 Hz, 2H, amido m-phenyl), 7.24 (d, J = 7.88 Hz, 1H, aroyl m-phenyl), 7.45 (d, J = 14.87 Hz, 1H, C(O)–CH=CH–), 7.64 (d, J = 8.58 Hz, 2H, amido o-phenyl), 7.88 (d, J = 7.88 Hz, 1H, aroyl o-phenyl), 7.83 (s, 1H, aroyl o-phenyl), 8.13 (d, J = 14.87 Hz, 1H, C(O)– CH=CH–), 8.91 (sb, 1H, NH–C(O)). 13C NMR (125 MHz, CDCl3) δ = 19.95, 20.12, 23.95, 33.63, 120.36, 126.79, 126.90, 130.05, 130.13, 133.47, 133.47, 134.77, 135.61, 136.60, 137.39, 143.91 145.61, 162.27, 189.88. ESI-MS HR: 322.1814 (M + 1), Calc. 321.1729. HPLC purity >99.5%; RT 6.703 min.
5.2.6. (E)-4-(4-isopropylphenyl)-4-oxo-2-butenoic acid phenylamide (6)
C19H19NO2, starting from (E)-4-(4-isopropylphenyl)-4-oxo-2-butenoic acid (0.017 mol) and a corresponding amount of aniline, 2.15 g of 6 was obtained, 42.99% yield, yellow solid, m.p. = 132– 136 °C, decomposition (AcOEt). 1H NMR (500 MHz, CDCl3) δ = 1.29 (d, J = 6.94 Hz, 6H, i-PrCH3), 2.99 (m, 1H, i-PrCH), 7.16 (t, J1,2 = 7.38 Hz, 1H, amido p-phenyl), 7.33–7.37 (overlapping multiplets, 5H, amido m-phenyl, aroyl m-phenyl and C(O)–CH=CH–), 7.69 (d, J = 7.73 Hz, 2H, amido o-phenyl); 8.00 (d, J = 8.23 Hz, 2H, aroyl o-phenyl), 8.13 (d, J = 14.97 Hz, 1H, C(O)–CH=CH–), 8.50 (s, 1H, NH–C(O)). 13C NMR (125 MHz, DMSO-d6) δ = 23.57, 34.36, 120.20, 124.97, 127.07, 129.09, 129.28, 133.94, 134.67, 135.90, 137.74, 155.89, 162.25, 189.56. HR MS (ESI): 294.1493 (M + H), Calc. 294.1494. HPLC purity >99.5%; RT 5.464 min.
5.2.7. (E)-4-(4-isopropylphenyl)-4-oxo-2-butenoic acid (3,5-dimethoxyphenyl)amide (7)
C21H23NO4, starting from (E)-4-(4-isopropylphenyl)-4-oxo-2-butenoic acid (0.0114 mol) and a corresponding amount of 3,5-dimethoxyaniline, 1.70 g of 7 was obtained, 41.44% yield, yellow solid, m.p. = 132–134 °C, (AcOEt). 1H NMR (500 MHz, CDCl3) δ = 1.27 (d, J = 6.84 Hz, 6H, i-PrCH3), 2.98 (h, J = 2.98 Hz, 1H, i-PrCH), 3.76 (s, 6H, –OCH3), 6.97 (d, J = 2.14 Hz, 2H, amido o-phenyl), 7.35 (d, J = 8.12 Hz, 2H, aroyl m-phenyl), 7.39 (d, J = 14.96 Hz, 1H, C(O)– CH=CH–), 8.00 (d, J = 8.33 Hz, 2H, aroyl o-phenyl), 8.13 (d, J = 14.75 Hz, 2H, C(O)–CH=CH–), 8.74 (sb, 1H, NH–C(O)). 13C NMR (125 MHz, CDCl3) δ = 23.55, 34.34, 55.36, 97.60, 98.39, 127.07, 129.26, 133.83, 134.69, 136.03, 139.56, 155.88, 161.03, 162.36, 189.61. ESI-MS HR: 354.1709 (M + H), Calc. 354.1705. HPLC purity >99.5%; RT 5.616 min.
5.2.8. (E)-4-(4-isopropylphenyl)-4-oxo-2-butenoic acid cyclohexylamide (8)
C19H25NO2, starting from (E)-4-(4-isopropylphenyl)-4-oxo-2-butenoic acid (0.0092 mol) and a corresponding amount of cyclohexylamine, 0.65 g of 8 was obtained, 23.78% yield, yellow solid, m.p. = 136–138 °C, (AcOEt); 1H NMR (500 MHz, CDCl3) δ = 1.16– 1.25 (m, 3H, cyclohexyl-CH2–), 1.27 (d, J = 7.0 Hz, 6H, cyclohexyl-CH2–), 1.36-1.44 (m, 2H, cyclohexyl-CH2–), 1.62–1.67 (m, 1H, cyclohexyl-CH2–), 1.72–1.76 (m, 2H, cyclohexyl-CH2–), 1.97–2.00 (m, 2H, cyclohexyl-CH2), 2.96 (h, J = 6.5 Hz, 1H, i-PrCH3), 3.93 (m, 1H, cyclohexyl-CH), 6.56 (d, J = 8 Hz, 1H, NH–C(O)), 7.10 (d, J = 15.00 Hz, 1H, C(O)–CH=CH–), 7.34 (d, J = 8.5 Hz, 2H, aroyl m-phenyl), 7.97 (d, J = 8.5 Hz, 2H, aroyl o-phenyl), 7.98 (d, J = 15.50 Hz, 1H, C(O)–CH=CH–). 13C NMR (125 MHz, CDCl3) δ = 23.53, 24.76, 25.46, 32.88, 34.26, 48.68, 126.87, 129.11, 132.78, 134.76, 135.88, 155.43, 163.17, 189.54. ESI-MS HR: 300.1958 (M + 1), Calc. 300.1964. HPLC purity >99.5%; RT 5.440 min.
5.2.9. (E)-4-(2,5-dimethylphenyl)-4-oxo-2-butenoic acid benzylamide (9)
C19H19NO2, starting from (E)-4-(2,5-dimethylphenyl)-4-oxo-2-butenoic acid (0.0078 mol) and a corresponding amount of benzylamine, as described above,1.04 g of 9 was obtained, 45.41% yield, yellow solid, m.p. =142–144 °C, (AcOEt). 1H NMR (500 MHz, CDCl3) δ = 2.33 (s, 3H, m-CH3), 2.38 (s, 3H, o-CH3), 4.52 (d, J = 5.92 Hz, 2H, benzyl-CH2–), 6.80 (sb, 1H, benzylamide-NH), 6.87 (d, J = 15.36 Hz, 1H, C(O)–CH=CH–), 7.12 (d, J = 7.84 Hz, 1H, aroyl p-phenyl), 7.20 (dd, J = 7.68 Hz, 1H, aroyl m-phenyl), 7.24–7.30 (m, 5H, benzyl-CH), 7.40 (d, J = 1.12 Hz, 1H, aroyl o-phenyl), 7.65 (d, J = 15.20 Hz, 1H, C(O)–CH=CH–). 13C NMR (125 MHz, CDCl3) δ = 20.31, 20.74, 43.94, 127.60, 127.79, 128.68, 129.74, 131.68, 132.58, 134.97, 135.25, 135.51, 136.71, 137.01, 137.48, 164.07, 194.09. ESI-MS HR: 294.1490 (M + 1) 100%, Calc. 294.1494. HPLC purity >99.5%; RT 4.123 min.
5.2.10. (E)-4-(4-isopropylphenyl)-4-oxo-2-butenoic acid benzylamide (10)
C20H21NO2, starting from (E)-4-(4-isopropylphenyl)-4-oxo-2-butenoic acid (0.0092 mol) and a corresponding amount of benzylamine, 0.94 g of 10 was obtained, 33.29% yield, yellow solid, m.p. = 125–127 °C, decomposition (AcOEt). 1H NMR (200 MHz, DMSO-d6) δ = 1.27 (d, J = 7.30 Hz, 6H, i-PrCH3), 2.96 (h, J = 6.74 Hz, 1H, i-PrCH), 4.57 (d, J = 5.62 Hz, 2H, benzyl-CH2–), 7.23–7.53 (overlapped m, 9H, benzyl-CH–, aroyl m-phenyl, C(O)–CH=CH–, and benzylamide-NH), 7.83 (d, J = 6.74 Hz, 2H, aroyl o-phenyl), 8.03 (d, J = 15.16 Hz, 1H, C(O)–CH=CH–). 13C NMR (50 MHz, DMSO-d6) δ = 23.47, 34.20, 43.94, 95.68, 126.89, 127.51, 127.83, 128.63, 129.11, 132.97, 134.52, 135.46, 137.57, 155.53, 164.28, 189.48. ESI-MS HR: 308.1647 (M + 1), Calc. 308.1651. HPLC purity >99.5%; RT 4.849 min.
5.2.11. (E)-4-(4-isopropylphenyl)-4-oxo-2-butenoic acid (4-methoxyphenyl)amide (11)
C20H21NO3, starting from (E)-4-(4-isopropylphenyl)-4-oxo-2-butenoic acid (0.0078 mol) and a corresponding amount of 3,5-dimethoxyaniline, 0.71 g of 11 was obtained, 28.09% yield, yellow solid, m.p. = 142–144 °C, decomposition (AcOEt). 1H NMR (500 MHz, CDCl3) δ = 1.27 (d, J = 6.71 Hz, 6H, i-PrCH3), 2.97 (h, J = 6.72 Hz, 1H, i-PrCH), 3.78 (s, 3H, –OCH3), 6.86 (d, J = 9.06 Hz, 2H, amido m-phenyl), 7.33 (d, J = 8.39 Hz, 2H, aroyl m-phenyl), 7.41 (d, J = 15.10 Hz, 1H, C(O)–CH=CH–), 7.62 (d, J = 8.06 Hz, 2H, amido o-phenyl), 7.97 (d, J = 8.39 Hz, 2H, aroyl o-phenyl), 8.11 (d, J = 14.77 Hz, 1H, C(O)–CH=CH–), 8.90 (sb, 1H, NH–C(O)). 13C NMR (125 MHz, CDCl3) δ = 23.54, 34.31, 55.40, 114.16, 121.93, 127.00, 129.23, 131.02, 133.28, 134.69, 136.30, 155.76, 155.77, 162.15, 189.66. ESI-MS HR: 324.1599 (M + 1), Calc. 324.1600. HPLC purity >99.5%; RT 5.129 min.
5.2.12. (E)-4-(2,4-diisopropylphenyl)-4-oxo-2-butenoic acid phenylamide (12)
C22H25NO2, starting from (E)-4-(2,4-diisopropylphenyl)-4-oxo-2-butenoic acid (0.013 mol) and corresponding amount of aniline, 2.06 g of 12 was obtained, 45.87% yield, yellow solid, m.p. = 122– 124 °C, (AcOEt). 1H NMR (500 MHz, DMSO-d6) δ = 1.20 (d, J = 6.86 Hz, 6H, p-i-PrCH3), 1.24 (d, J = 6.75 Hz, 6H, o-i-PrCH3); 2.96 (m, 1H, p-i-PrCH), 3.19 (m, 1H, o-i-PrCH); 6.91 (d, J = 14.93 Hz, 1H, C(O)–CH=CH–), 7.10 (t, J1,2 = 6.97 Hz, 1H, amido p-phenyl), 7.21, 7.22 (doublet like peaks, 1H, aroyl m-phenyl), 7.30 (d, J = 14.93 Hz, 1H, C(O)–CH=CH–), 7.34 (t, J1,2 = 8.30 Hz, 2H, amido m-phenyl), 7.37 (br, 1H, aroyl m-phenyl), 7.42 (d, J = 7.94 Hz, 1H, aroyl o-phenyl), 7.67 (d, J = 8.63 Hz, 2H, amido o-phenyl), 10.54 (s, 1H, NH– C(O)). 13C NMR (125 MHz, DMSO-d6) δ = 23.63, 23.90, 29.23, 33.57, 119.28, 123.39, 124.08, 128.88, 134.95, 137.40, 137.57, 136.63, 147.58, 151.82, 161.72, 196.11. HR MS (ESI): 336.1975 (M + 1), Calc. 336.1964. HPLC purity >99.5%; RT 7.254 min.
5.2.13. (E)-4-(2,4-diisopropylphenyl)-4-oxo-2-butenoic acid (3,5-dimethoxyphenyl)amide (13)
C24H29NO4, starting from (E)-4-(2,4-diisopropylphenyl)-4-oxo-2-butenoic acid (0.0096 mol) and a corresponding amount of 3,5-dimethoxyaniline, 1.90 g of 13 was obtained, 50.00% yield, yellow solid, m.p. = 120–122 °C, decomposition (AcOEt). 1H NMR (500 MHz, CDCl3) δ = 1.23 (d, J = 6.72 Hz, 6H, p-i-PrCH3), 1.27 (d, J = 6.88 Hz, 6H, o-i-PrCH3), 2.94 (h, J = 6.72 Hz, 1H, p-i-PrCH), 3.42 (h, J = 6.72 Hz, 1H, o-i-PrCH), 3.73 (s, 6H, –OCH3), 6.26 (s, 1H, amido p-phenyl), 6.86 (s, 2H, amido o-phenyl), 7.98 (d, J = 15.03 Hz, 1H, C(O)–CH=CH–), 7.11 (d, J = 7.67 Hz, 1H, aroyl m-phenyl), 7.28 (s, 1H, aroyl m-phenyl), 7.43 (d, J = 7.84 Hz, 1H, aroyl o-phenyl), 7.65 (d, J = 15.20 Hz, 1H, C(O)–CH=CH–), 8.11 (sb, 1H, NH–C(O)). 13C NMR (125 MHz, CDCl3) δ = 23.71, 24.14, 29.58, 34.36, 55.53, 97.55, 98.26, 123.45, 124.82, 128.87, 135.00,135.64, 136.12,139.28, 148.93, 153.09, 161.02, 162.18, 194.99. ESI-MS HR: 396.2169 (M + 1), Calc. 396.2175. HPLC purity >99.5%; RT 7.327 min.
5.2.14. (E)-4-(2,4-diisopropylphenyl)-4-oxo-2-butenoic acid (4-isopropylphenyl)amide (14)
C25H31NO2, starting from (E)-4-(2,4-diisopropylphenyl)-4-oxo-2-butenoic acid (0.0096 mol) and a corresponding amount of 4-isopropylaniline, 2.30 g of 14 was obtained, 63.53% yield, yellow solid, m.p. = 119–121 °C, decomposition (AcOEt). 1H NMR (500 MHz, DMSO-d6) δ = 1.23 (d, J = 6.50 Hz, 6H, aroyl o-i-PrCH3), 1.17–1.20 (m, 12 H, aroyl p-i-PrCH3, amido p-i-PrCH3), 2.96 (h, J = 6.31 Hz, 1H, aroyl p-i-PrCH3), 2.84 (h, J = 6.31 Hz, 1H, amido p-i-PrCH3), 3.18 (h, J = 6.31 Hz, 1H, aroyl o-i-Pr–CH), 6.90 (d, J = 15.49 Hz, 1H, C(O)–CH=CH–), 7.19–7.21 (m, 3H, aroyl m-phenyl, amido m-phenyl), 7.29 (d, J = 15.49 Hz, 1H, C(O)–CH=CH–), 7.37 (s, 1H, aroyl m-phenyl), 7.41 (d, J = 7.65 Hz, 1H, aroyl o-phenyl), 7.59 (d, J = 7.27 Hz, 2H, amido o-phenyl), 10.5 (sb, 1H, NH–C(O)). 13C NMR (125 MHz, CDCl3) δ = 23.62, 23.85, 23.89, 29.23, 32.89, 33.57, 119.43, 123.36, 124.44, 126.57, 128.34, 134.99, 136.42, 137.19, 137.72, 144.20, 147.51, 151.76, 161.46, 196.12. ESI-MS HR: 378.2421 (M + 1), Calc. 378.2433. HPLC purity >99.5%; RT 7.511 min.
5.2.15. (E)-4-(2,4-diisopropylphenyl)-4-oxo-2-butenoic acid benzylamide (15)
C23H27NO2, starting from (E)-4-(2,4-diisopropylphenyl)-4-oxo-2-butenoic acid (0.0077 mol) and a corresponding amount of benzylamine, 0.92 g of 15 was obtained, 35.11% yield, yellow solid, m.p. = 125–127 °C, (AcOEt). 1H NMR (500 MHz, CDCl3) δ = 1.22 (d, J = 6.85 Hz, 6H, p-i-PrCH3), 1.26 (d, J = 6.85 Hz, 6H, o-i-PrCH3), 2.93 (h, J = 7.00 Hz, 1H, p-i-PrCH), 3.37 (h, J = 6.85 Hz, 1H, o-i-PrCH), 4.52 (d, J = 5.79 Hz, 2H, benzyl-CH2–), 6.48 (m,1H, benzyl-p-CH), 6.75 (d, J = 15.37 Hz, 1H, C(O)–CH=CH–), 7.09 (d, J = 8.06 Hz, 1H, aroyl m-phenyl), 7.26–7.30 (m, 6H, benzyl-CH–, aroyl m-phenyl and benzyl-NH–), 7.38 (d, J = 8.06 Hz, 1H, aroyl o-phenyl), 7.53 (d, J = 15.37 Hz, 1H, C(O)–CH=CH–). 13C NMR (125 MHz, CDCl3) δ = 23.70, 24.10, 29.49, 34.33, 44.01, 123.32, 124.72, 127.69, 127.87, 128.74, 134.90, 135.04, 137.45, 137.75, 148.79, 152.80, 164.02, 195.06. ESI-MS HR: 350.2114 (M + 1), Calc. 350.2120. HPLC purity >99.5%; RT 6.590 min.
5.2.16. (E)-4-oxo-4-(5,6,7,8-tetrahydronaphthalenyl)-2-butenoic acid (3,5-dimethoxyphenyl)-amide (16)
C22H23NO4, starting from (E)-4-(5,6,7,8-tetrahydronaphtalenyl)-4-oxo-2-butenoic acid (0.0065 mol) and a corresponding amount of 3,5-dimethoxyaniline, 1.48 g of 16 was obtained, 62.40% yield, yellow solid, m.p. = 172–174 °C, decomposition (AcOEt). 1H NMR (500 MHz, CDCl3) δ = 1.83 (m, 4H, tetralinoyl-CH2–), 2.82 (m, 4H, tetralinoyl-CH2–), 3.78 (m, 6H, –OCH3), 6.29 (s, 1H, amido p-phenyl), 6.95 (m, 2H, amido o-phenyl), 7.17 (d, J = 8.50 Hz, 1H, aroyl m-phenyl), 7.30 (d, J = 14.72 Hz, 1H, C(O)–CH=CH–), 7.79 (m, 2H, aroyl o-phenyl), 8.12 (d, J = 15.05 Hz, 1H, C(O)–CH=CH–), 8.37 (sb, 1H, NH–C(O)). 13C NMR (125 MHz, CDCl3) δ = 22.71, 22.86, 29.26, 29.78, 55.39, 97.51, 98.37, 125.98, 129.74, 129.99, 134.17, 134.32, 135.57, 136.02, 139.47, 144.64, 161.08, 162.33, 189.63. ESI-MS HR: 366.1710 (M + 1), Calc. 366.1705. HPLC purity >99.5%; RT 5.906 min.
5.2.17. (E)-4-oxo-4-(5,6,7,8-tetrahydronaphthalenyl)-2-butenoic acid cyclohexylamide (17)
C20H25NO2, starting from (E)-4-(5,6,7,8-tetrahydronaphthalenyl)-4-oxo-2-butenoic acid (0.0078 mol) and a corresponding amount of cyclohexylamine, 1.06 g of 17 was obtained, 43.46% yield, yellow solid, m.p. = 166–168 °C, decomposition (AcOEt). 1H NMR (500 MHz, CDCl3) δ = 1.16–1.29 (m, 3H, cyclohexyl-CH2–), 1.39–1.43 (m, 2H, cyclohexyl-CH2–), 1.64 (m, 1H, cyclohexyl-CH2–), 1.75 (m, 2H, cyclohexyl-CH2–), 1.82 (sb, 4H, tetralinoyl-CH2–), 2.00 (m, 2H, cyclohexyl-CH2-), 2.81 (sb, 4H, tetralinoyl-CH2–), 3.93 (m, 1H, cyclohexyl-CH–), 6.66 (d, J = 8.10 Hz, 1H, amide-NH–), 7.13 (d, J = 15.15 Hz, 1H, C(O)–CH=CH-), 7.17 (d, J = 8.56 Hz, 1H, aroyl m-phenyl), 7.75 (m, 2H, aroyl o-phenyl), 7.97 (d, J = 14.95 Hz, 1H, C(O)–CH=CH–). 13C NMR (125 MHz, CDCl3) δ = 22.66, 22.81, 24.77, 25.46, 29.44, 29.66, 32.89, 48.67, 125.86, 129.52, 129.78, 132.79, 134.38, 135.79, 137.71, 144.16, 163.25, 189.74. ESI-MS HR: 312.1956 (M + 1), Calc. 312.1964. HPLC purity >99.5%; RT 5.692 min.
5.2.18. (E)-4-oxo-4-(5,6,7,8-tetrahydronaphthalenyl)-2-butenoic acid (4-isopropylphenyl)amide (18)
C23H25NO2, starting from (E)-4-(5,6,7,8-tetrahydronaphthalenyl)-4-oxo-2-butenoic acid (0.0096 mol) and a corresponding amount of 4-isopropylaniline, 1.76 g of 18 was obtained, 52.54% yield, yellow solid, m.p. = 146–148 °C, decomposition (AcOEt). 1H NMR (500 MHz, CDCl3) δ = 1.23 (d, J = 6.99 Hz, 6H, i-PrCH3),1.81 (m, 4H, tetralinoyl-CH2-), 2.79 (db, 4H, tetralinoyl-CH2-), 2.88 (h, J = 6.99 Hz, 1H, i-PrCH), 7.15 (d, J = 8.43 Hz, 2H, amido m-phenyl), 7.19 (d, J = 8.43 Hz, 1H, aroyl m-phenyl), 7.47 (d, J = 14.81 Hz, 1H, C(O)–CH=CH–), 7.65 (d, J = 8.43 Hz, 2H, amido o-phenyl), 7.76 (m, 2H, aroyl o-phenyl), 8.13 (d, J = 14.81 Hz, 1H, C(O)–CH=CH–), 9.06 (sb, 1H, NH–C(O)). 13C NMR (125 MHz, CDCl3) δ = 22.60, 22.80, 23.94, 29.93, 29.71, 33.60, 120.35, 125.98, 126.85, 129.64, 129.92, 133.43, 134.35,135.65, 136.93, 137.86,144.49, 145.53, 162.23, 189.92. ESI-MS HR: 348.1957 (M + 1), Calc. 348.1964. HPLC purity >99.5%; RT 7.832 min.
5.2.19. (E)-4-oxo-4-(5,6,7,8-tetrahydronaphthalen-2-yl)-2-butenoic acid benzylamide (19)
C21H21NO2, starting from (E)-4-(5,6,7,8-tetrahydronaphtalenyl)-4-oxo-2-butenoic acid (0.0078 mol) and a corresponding amount of benzylamine, 1.03 g of 19 was obtained, 41.20% yield, yellow solid, m.p. = 148–150 °C, decomposition (AcOEt). 1H NMR (500 MHz, CDCl3) δ = 1.81 (sb, 4H, tetralinoyl-CH2–), 2.80 (mb, 4H, tetralinoyl-CH2–), 4.57 (d, J = 5.94 Hz, 2H, benzylamine-CH2–), 6.87 (m, 1H, benzylamine–CH–), 7.09 (d, J = 15.07 Hz, 1H, C(O)–CH=CH–), 7.13 (d, J = 8.06 Hz, 1H, aroyl m-phenyl), 7.27 (m, 1H, benzyl–NH–), 7.31 (m, 4H, benzylamine-CH–), 7.65 (d, J = 7.91 Hz, 1H, aroyl o-phenyl), 7.70 (sb, 1H, aroyl o-phenyl), 8.00 (d, J = 14.92 Hz, 1H, C(O)–CH=CH–). 13C NMR (125 MHz, CDCl3) δ = 22.69, 22.83, 29.26, 29.71, 44.02, 125.85, 127.62, 127.83, 128.73, 129.62, 129.86, 133.55, 134.25, 134.79, 137.59, 137.77, 144.29, 164.19, 189.53. ESI-MS HR: 320.1649 (M + 1), Calc. 320.1651. HPLC purity >99.5%; RT 5.082 min.
5.2.20. (E)-4-(5,6,7,8-tetrahydronaphtalenyl)-4-oxo-2-butenoic acid phenylamide (20)
C20H19NO2, starting from (E)-4-(5,6,7,8-tetrahydronaphthalenyl)-4-oxo-2-butenoic acid (0.017 mol) and a corresponding amount of aniline, 3.10 g of 20 was obtained, 78.08% yield, yellow solid, m.p. = 135–167 °C, decomposition (AcOEt). 1H NMR (500 MHz, CDCl3) δ = 1.82 (m, 4H, tetralinoyl-CH2–), 2.82 (m, 4H, tetralinoyl-CH2–), 7.14–7.19 (overlapping peaks, 2H, amido p-phenyl and aroyl m-phenyl), 7.36 (t, J1,2 = 8.32 Hz, 2H, amido m-phenyl), 7.41 (d, J = 14.94 Hz, 1H, C(O)–CH=CH–), 7.71 (d, J = 8.63 Hz, 2H, amido o-phenyl), 7.77 (s, 1H, aroyl m-phenyl), 7.78 (s, 1H, aroyl o-phenyl), 8.14 (d, J = 14.94 Hz, 1H, C(O)–CH=CH–), 8.68 (s, 1H, NH–C(O)). 13C NMR (125 MHz, CDCl3) δ = 22.69, 22.84, 29.30, 29.77, 120.20, 124.91, 126.02, 129.06, 129.73, 129.97, 133.90, 134.34, 135.94, 137.85, 137.97, 144.66, 162.32, 189.85. HR MS (ESI): 306.1491 (M + 1), Calc. 306.1494. HPLC purity >99.5%; RT 5.756 min.
5.2.21. (E)-4-(4-n-butylphenyl)-4-oxo-2-butenoic acid phenylamide (21)
C20H21NO2, starting from (E)-4-(4-n-buthylphenyl)-4-oxo-2-butenoic acid (0.013 mol) and a corresponding amount of aniline, 1.29 g of 21 was obtained, 32.25% yield, yellow solid, m.p. = 133– 135 °C, (AcOEt). 1H NMR (500 MHz, DMSO-d6) δ = 0.91 (t, J1,2 = 7.07 Hz, 3H, CH3CH2),1.32 (sp, J1,2 = 7.48 Hz, J1,3 = 14.95 Hz, 2H, CH3CH2CH2), 1.59 (m, J1,2 = 7.47 Hz, J1,3 = 14.95 Hz, 2H, CH3CH2CH2), 2.68 (t, J1,2 = 7.47 Hz, 2H, CH3CH2CH2CH2), 7.12 (t, J1,2 = 7.08 Hz, 1H, amido p-phenyl), 7.21 (d, J = 15.33 Hz, 1H, C(O)–CH=CH–), 7.37 (t, J1,2 = 7.47 Hz, 2H, amido m-phenyl), 7.42 (d, J = 7.87 Hz, 2H, aroyl m-phenyl), 7.73 (d, J = 7.47 Hz, 2H, amido o-phenyl), 7.89 (d, J = 15.33 Hz, 1H, C(O)–CH=CH–), 7.99 (d, J = 8.26 Hz, 2H, aroyl o-phenyl), 10.59 (s, 1H, NH–C(O)). 13C NMR (125 MHz, DMSO-d6) δ = 13.70, 21.71, 32.65, 34.81, 119.41, 124.02, 128.88, 149.11, 161.92, 189.06. HR MS (ESI): 308.1656 (M + 1), Calc. 308.1651. HPLC purity > 99.5%; RT 6.572 min.
5.2.22. (E)-4-(2,3,5,6-tetramethylphenyl)-4-oxo-2-butenoic acid (3,5-dimethoxyphenyl)amide (22)
C22H25NO4, starting from (E)-4-(2,3,5,6-tetramethylphenyl)-4-oxo-2-butenoic acid (0.0094 mol) and a corresponding amount of 3,5-dimethoxyaniline, 2.50 g of 22 was obtained, 72.46% yield, yellow solid, m.p. =198–200 °C, (AcOEt). 1H NMR (200 MHz, CDCl3) δ = 2.03 (s, 6H, o- and m-CH3), 2.19 (s, 6H, m- and p-CH3), 3.67 (s, 6H, –OCH3), 6.23 (t, J1,2 = 2.16 Hz, 1H, amido p-phenyl), 6.80 (d, J = 15.41 Hz, 1H, C(O)–CH=CH–), 6.79 (s,1H, amido o-phenyl), 6.80 (s, 1H, amido o-phenyl), 6.99 (s, 1H, aroyl o-phenyl), 7.31 (d, J = 15.41 Hz, 1H, C(O)–CH=CH–), 8.38 (s, 1H, NH–C(O)). 13C NMR (50 MHz, CDCl3) δ = 16.19, 19.36, 55.17, 97.45, 97.99, 129.35, 132.31, 134.52,137.32,138.52,139.20,139.52,160.92,162.03, 201.92. ESI-MS HR: 368.1856 (M + 1), Calc. 368.1862. HPLC purity >99.5%; RT 5.551 min.
5.2.23. (E)-4-(4-fluorophenyl)-4-oxo-2-butenoic acid phenylamide (23)
C16H12FNO2, starting from (E)-4-(4-fluorophenyl)-4-oxo-2-butenoic acid (0.013 mol) and a corresponding amount of aniline, 1.19 g of 23 was obtained, 34.00% yield, yellow solid, m.p. = 163– 173 °C, (AcOEt). 1H NMR (500 MHz, DMSO-d6) δ = 7.13 (t, J1,2 = 7.54 Hz, 1H, amido p-phenyl), 7.23 (d, J = 15.38 Hz, 1H, C(O)– CH=CH–), 7.37 (t, J1,2 = 7.57 Hz, 2H, amido m-phenyl), 7.42 (t, J1,2 = 7.42 Hz, 2H, aroyl m-phenyl), 7.73 (d, J = 7.66 Hz, 2H, amido o-phenyl), 7.92 (d, J = 15.32 Hz, 1H, C(O)–CH=CH–), 8.17 (m, J1,2 = 5.55 Hz, J1,3 = 9.07 Hz; 2H, aroyl o-phenyl), 10.60 (s, 1H, NH–C(O)). 13C NMR (125 MHz, DMSO-d6) δ = 115.52, 116.23, 119.41, 124.08, 128,90, 131.82, 131.89, 132.73, 133.28, 133.30, 136.75, 138.66, 161.89, 166.37, 164.36, 188.22. HR MS (ESI): 270.0935 (M + 1), Calc. 270.0930. HPLC purity >99.5%; RT 3.610 min.
5.2.24. (E)-4-(4-chlorophenyl)-4-oxo-2-butenoic acid phenylamide (24)
C16H12ClNO2, starting from (E)-4-(4-chlorophenyl)-4-oxo-2-butenoic acid (0.017 mol) and a corresponding amount of aniline, 2.30 g of 24 was obtained, 47.42% yield, yellow solid, m.p. = 181– 183 °C, decomposition (AcOEt). 1H NMR (500 MHz, DMSO-d6) δ = 7.12 (t, J1,2 = 7.26 Hz, 1H, amido p-phenyl), 7.22 (d, J = 15.29 Hz, 1H, C(O)–CH=CH–), 7.37 (t, J1,2 = 7.58 Hz, 2H, amido m-phenyl), 7.72 (d, J = 7.44 Hz, 2H, amido o-phenyl), 7.80 (d, J = 8.42 Hz, 2H, aroyl m-phenyl), 7.87 (d, J = 15.29 Hz, 1H, C(O)–CH=CH–), 8.00 (d, J = 8.42 Hz, 2H, aroyl o-phenyl), 10.60 (s, 1H, NH–C(O)). 13C NMR (125 MHz, DMSO-d6) δ = 119.42, 124.09, 128.00, 128.01, 130.73, 132.08, 132.61, 135.55, 137.00, 138.66, 161.76, 188.91. HR MS (ESI): 286.0634 (M + 1), Calc. 286.0635. HPLC purity >99.5%; RT 4.387 min.
5.2.25. (E)-4-(3,4-dichlorophenyl)-4-oxo-2-butenoic acid phenylamide (25)
C16H11Cl2NO2, starting from (E)-4-(3,4-dichlorophenyl)-4-oxo-2-butenoic acid (0.013 mol) and a corresponding amount of aniline, 2.80 g of 25 was obtained, 67.31% yield, yellow solid, m.p. = 187– 194 °C, decomposition (AcOEt). 1H NMR (500 MHz, DMSO-d6) δ: 7.12 (t, J1,2 = 7.35 Hz, 1H, amido p-phenyl), 7.24 (d, J = 15.30 Hz, 1H, C(O)–CH=CH–), 7.37 (t, J1,2 = 7.65 Hz, 2H, amido m-phenyl), 7.72 (d, J = 7.65 Hz, 2H, amido o-phenyl), 7.84 (d, J = 8.46 Hz,1H, aroyl m-phenyl), 7.87 (d, J = 15.32 Hz, 1H, C(O)–CH=CH–), 8.03 (dd, 1H, J1,2 = 2.12 Hz, J1,3 = 8.48 Hz, aroyl o-phenyl), 8.24 (s, 1H, aroyl o-phenyl), 10.61 (s, 1H, NH–C(O)). 13C NMR (500 MHz, DMSO-d6) δ = 119.43, 124.11, 128.77, 128.90, 130.49, 131.29, 132.08, 132.35, 136.61, 136.69, 137.45, 136.64, 161.67, 187.85. HR MS (ESI): 320.0241 (M + 1), Calc. 320.0245. HPLC purity >99.5%; RT 5.37 min.
5.2.26. (E)-4-(4-bromophenyl)-4-oxo-2-butenoic acid phenylamide (26)
C16H12BrNO2, starting from (E)-4-(4-bromophenyl)-4-oxo-2-butenoic acid (0.013 mol) and a corresponding amount of aniline, as described above, 2.60 g of 26 was obtained, 60.60% yield, yellow solid, m.p. = 187–194 °C, decomposition (AcOEt). 1H NMR (500 MHz, DMSO-d6) δ = 7.12 (t, J1,2 = 7.55 Hz, 1H, amido p-phenyl); 7.23 (d, J = 14.37, 1H, C(O)–CH=CH–); 7.37 (t, J1,2 = 7.85 Hz, 2H, amido m-phenyl); 7.73 (d, J = 7.60, 2H, amido o-phenyl); 7.82 (d, J = 8.74, 2H, aroyl m-phenyl); 7.88 (d, J = 15.26, 1H, C(O)–CH=CH– ); 8.00 (d, J = 8.77 Hz, 2H, aroyl o-phenyl); 10.60 (s, 1H, NH–C(O)). 13C NMR (125 MHz, DMSO-d6) δ = 119.41, 124.08, 127.99, 128.90, 130.72, 132.07, 132.60, 135.54, 136.99, 138.65, 161.75, 188.90. HR MS (ESI): 330.0120/332.0102 (M + 1), Calc. 330.0124/332.0105. HPLC purity >99.5%; RT 4.626 min.
5.2.27. (E)-4-(4-methoxyphenyl)-4-oxo-2-butenoic acid phenylamide (27)
C17H15NO3, starting from (E)-4-(4-methoxyphenyl)-4-oxo-2-butenoic acid (0.017 mol) and a corresponding amount of aniline, 2.50 g of 27 was obtained, 52.30% yield, yellow solid, m.p. = 157– 161 °C, decomposition (AcOEt). 1H NMR (500 MHz, DMSO-d6) δ = 3.88 (s, 3H, p-OCH3), 6.95 (d, J = 9.03, 2H, aroyl m-phenyl), 7.15 (t, J1,2 = 7.35 Hz, 1H, amido p-phenyl), 7.35 (t, J1,2 = 8.16 Hz, 2H, amido m-phenyl), 7.42 (d, J = 14.81 Hz, 1H, C(O)–CH=CH–), 7.70 (d, J = 8.18, 2H, amido o-phenyl), 8.04 (d, J = 9.03 Hz, 2H, aroyl o-phenyl), 8.13 (d, J = 14.81 Hz, 1H, C(O)–CH=CH–), 8.87 (1H, s, NH–C(O)). 13C NMR (125 MHz, DMSO-d6) δ: 55.57, 114.18, 120.25, 124.87, 129.04, 128.85, 131.46, 133.69, 135.84, 137.88, 162.43, 164.40, 188.36. HR MS (ESI): 282.1133 (M + 1), Calc. 282.1134. HPLC purity >99.5%; RT 3.360 min.
5.2.28. (E)-4-(4-methoxyphenyl)-4-oxo-2-butenoic acid (3,5-dimethoxyphenyl)amide (28)
C19H19NO5, starting from (E)-4-(4-methoxyphenyl)-4-oxo-2-butenoic acid (0.0076 mol) and a corresponding amount of 3,5-dimethoxyaniline, 0.81 g of 28 was obtained, 31.40% yield, yellow solid, m.p. = 141–143 °C, decomposition (AcOEt). 1H NMR 500 MHz (CDCl3) δ = 3.76 (s, 6H, –OCH3), 3.90 (s, 3H, –OCH3), 6.29 (sb, 1H, amido p-phenyl), 6.94 (m, 2H, amido o-phenyl), 6.98 (d, J = 8.96 Hz, 2H, aroyl m-phenyl), 7.29 (d, J = 14.72 Hz, 1H, C(O)–CH=CH–), 8.07 (d, J = 8.64 Hz, 2H, aroyl o-phenyl), 8.11 (d, J = 15.04 Hz, 1H, C(O)– CH=CH–), 8.33 (sb, 1H, NH–C(O)). 13C NMR (125 MHz, CDCl3) δ = 55.41, 55.60, 97.53, 98.37, 114.21, 129.86, 131.44, 134.05, 135.35, 139.44, 161.09, 162.36, 164.41, 188.05. ESI-MS HR: 342.1338 (M + 1), Calc. 342.1341. HPLC purity >99.5%; RT 3.500 min.
5.2.29. (E)-4-(4-methoxy-3,5-dimethylphenyl)-4-oxo-2-butenoic acid (3,5-dimethoxyphenyl)-amide (29)
C21H23NO5, starting from (E)-4-(4-methoxy-3,5-dimethyl-phenyl)-4-oxo-2-butenoic acid (0.0076 mol) and a corresponding amount of 3,5-dimethoxyaniline, as described above, 0.93 g of 29 was obtained, 33.02% yield, yellow solid, m.p. = 160–162 °C, decomposition (AcOEt). 1H NMR 500 MHz (CDCl3) δ = 2.19 (s, 3H, –CH3), 2.54 (s, 3H, –CH3), 5.70 (s, 6H, –OCH3), 3.88 (s, 3H, –OCH3), 6.25 (s, 1H, amido p-phenyl), 6.67 (s, 1H, aroyl o-phenyl), 6.91 (s, 2H, amido o-phenyl), 7.12 (d, J = 14.88 Hz, 1H, C(O)–CH=CH–), 7.58 (s, 1H, aroyl o-phenyl), 7.89 (d, J = 14.88 Hz, 1H, C(O)–CH=CH–), 8.55 (s, 1H, NH–C(O)). 13C NMR (125 MHz, CDCl3) δ = 15.77, 21.94, 55.30, 55.42, 97.46, 98.28, 113.33, 123.97, 128.92, 132.95, 134.61, 137.32, 139.53, 140.25, 160.77, 160.95, 162.62, 191.61. ESI-MS HR (solvent MeOH/CH2Cl2, positive mode): 370.1649 [M + H]+ 100%, (Calc. Mass 369.1576). HPLC purity >99.5%; RT 4.863 min.
5.3. Biological assays
5.3.1. Cytotoxicity assays
5.3.1.1. Preparation of compound solutions
Stock solutions of the investigated compounds were made in dimethyl sulfoxide (Fluka Chemie AG Buchs, Switzerland) at a concentration of 20 mM, filtered through Millipore filters (0.22 μm) before use and afterwards diluted to various working concentrations with RPMI-1640 cell culture medium (Sigma Chemical Co. St Louis, MO) supplemented with 3 mmol/L L-glutamine, 100 μg/mL streptomycin, 100 IU/mL penicillin, 10% heat inactivated fetal bovine serum (FBS – Sigma Chemical Co.), and 25 mM Hepes, adjusted to pH 7.2 with a bicarbonate solution.
5.3.1.2. Treatment of cells
Cells were seeded into 96-well microtiter plates, 2000 cells in 0.1 mL of culture medium per well. After 20 h, to wells with the cells, five different concentrations of compounds 1–29 were applied, except to the control wells, in which the cells were grown in medium only. All concentrations were set up in triplicate. Culture medium with corresponding concentrations of investigated agent, but without cells, was used as a blank, also in triplicate.
5.3.1.3. Determination of FemX, HeLa, and K562 cell survival
Cell survival was determined by the MTT test, according to the method of Mosmann [32] as modified by Ohno and Abe [33], 72 h after compound addition. Briefly, 20 μL of MTT solution (5 mg/mL in phosphate buffered saline) was added to each well. Cells were incubated for 4 h at 37 °C in a humidified atmosphere with 5% CO2. Then, 100 μL of 10% SDS was added to each well. Absorbance was measured at 570 nm. Cell survival was measured/quantified using absorbance at 570 nm of a sample with cells grown in the presence of various concentrations of agent divided by the absorbance of the control sample (the absorbance of cells grown only in nutrient medium). Absorbance of blank was always subtracted from absorbance of a corresponding sample with cells. All experimentally obtained IC50 data, reported in Table 1, are means of at least three measurements done in triplicate.
5.3.1.4. Cell cycle determination
Aliquots of 2.5 × 105 control cells or cells treated with investigated compounds for 24 h (concentrations corresponded to 2 × IC50, 4 × IC50, and 8 × IC50 values) were fixed in 70% ethanol for 1 h on ice, then at −20 °C for at least 1 week. The cells were then collected by centrifugation. The pellets were treated with RNase (100 μg/mL) at 37 °C for 30 min and then incubated with propidium iodide (40 μg/mL) for at least 30 min. DNA content and cell cycle distribution were analyzed using a Becton Dickinson FAC-Scan flow cytometer. Flow cytometric analysis was performed using a CellQuestR (Becton Dickinson, San Jose, CA, USA) on a minimum of 10,000 cells per sample [34].
5.3.2. Inhibition of tubulin polymerization and colchicine binding
The tubulin assembly and [3H]colchicine binding assays were performed with electrophoretically homogenous bovine brain tubulin [35]. The assembly assay [36] was performed with 10 μM (1.0 mg/mL) tubulin, 0.8 M monosodium glutamate (pH 6.6 with HCl in a 2.0 M stock solution), 0.4 mM GTP, and 4% (v/v) dimethyl sulfoxide (as the compound solvent). Tubulin and varying compound concentrations were preincubated without GTP for 15 min at 30 °C, samples were placed on ice, and GTP was added. The samples were transferred to 0 °C cuvettes in Beckman DU7400 and DU7500 recording spectrophotometers equipped with electronic temperature controllers. After baselines were established at 350 nm, the temperature was jumped to 30 °C (less than 1 min), and sample turbidity was followed for 20 min. The IC50 was the compound concentration that reduced the turbidity reading at 20 min by 50%, relative to a control reaction mixture without compound.
The colchicine binding assay was described in detail by Verdier-Pinard et al. [37]. The reaction mixtures contained a tubulin stabilizing buffer system [37] and 1.0 μM (0.1 mg/mL) tubulin, 5.0 μM [3H]colchicine (from Perkin–Elmer, Boston, MA), and compounds at 5.0 or 60 μM in a final dimethyl sulfoxide concentration of 5% (v/ v). The 0.1 mL reaction mixtures were incubated for 10 min at 37 °C, at which time the reaction is about 40–50% complete in the absence of the compounds. The reactions were stopped with 3 mL of ice water, and each diluted reaction mixture was filtered through a stack of two DEAE-cellulose filters from Whatman. The amount of radiolabeled colchicine bound to the tubulin adsorbed to the filters was determined in a scintillation counter.
5.3.3. Acute toxicity
5.3.3.1. Animals
Male albino mice (NMRI Hann) weighing 18–22 g, obtained from Vivarium–Galenika a.d. Belgrade, Serbia, selected by random sampling, were used in the study. They were housed under standard animal conditions, at 23 ± 2 °C with 50% relative air humidity. The animals had free access to food (standard pallet diet) and water. This study was performed following the National (‘The Law on the Experimental Animal Treatment’, Republic of Serbia), and European (Directive 2010/63/EU; European convention for the protection of vertebrate animals used for experimental and other scientific purposes) regulations and standards. The acute toxicity tests were performed in accordance with OECD-423 guidelines. Experiments were approved by the Animal Ethics Committee-Galenika a.d. (Permission No 3/12).
5.3.3.2. Acute toxicity studies
20 mg of compound 23 was dissolved in 10 mL of DMSO. The tested substance (dose level of 10 mg/ kg body weight) was given to mice by gavage. If mortality was not observed, the procedure was repeated with higher doses of 20, 30, 40, or 50 mg/kg body weight. If mortality was observed in 2 or 3, among 6 animals, then the dose administered was assigned as a toxic dose. If mortality was observed in one animal, the substance was applied again in the same dose, to confirm the toxic dose. Following the administration of the tested substance, the animals were followed for signs of toxicity for 72 h. Observation was continued for 14 days. The toxic dose was confirmed as 45 mg/kg body weight for the oral route of administration.
5.4. Molecular modeling
The shape and the pharmacophoric pattern of compounds 1–3, 6, 8, 9, 17, 21, 20, and 23–27 were compared with the ligands cocrystallized with tubulin. As templates for comparison we used compounds E70, from the PDB entry 3HKC [29], and N16, from the PDB entry 3HKD [29], in their cocrystallized conformations. 2D structures of templates are shown in Figure S7 in Supplementary material. Compounds E70 (N-{2-[(4-hydroxyphenyl)amino]pyridin-3-yl}-4-methoxy-benzenesulfonamide) and N16 ((3Z, 5S)-5-benzyl-3-[1-(phenylamino)ethylidene]pyrrolidine-2,4-dione) were extracted from the PDB files and hydrogens added in VegaZZ 2.4.0 [38]. Structures were saved as mol2 files. Up to 100 conformations of compounds 1–3, 6, 8, 9, 17, 21, 20, and 23–27 were generated, from SMILES notation, in the OMEGA program [39], by using MMFF94s force field [40] (both to build initial structures and for search of the conformational space). To obtain 100 conformations per compound having four rotatable bonds in their backbone, rmsd options in OMEGA was set to 0.1. The set of conformers of each compound was saved as mol2 files. The ROCS program was used for the shape and for the pharmacophoric pattern comparison. The Tanimoto cut-off was set to −1. The 100 random starts and the explicit Meals-Dean force field, with modifications by OpenEye Inc., were used. The 3D dependent molecular properties of 1–29 (solvent-assessable area, polar surface area, apolar surface area, volume, virtual log P and ovality) were calculated for the minimum-energy conformation of each molecule, as obtained by OMEGA and MMFF94s force field, and by using a 1.4 Å probe (water), or 0 for volume, in VegaZZ. Only molecular properties that show some regularity in comparisons with antiproliferative or antitubulin activity are shown in Table S1 in Supplementary material. Structures of the E and the Z isomer of compound 11 were optimized in MOPAC2009 [41], using PM6 semiempirical MO method [42], with eigenvector-following optimizer to root-mean-square gradient >0.01 kcal/mol/Å.
Supplementary Material
Acknowledgments
This work was supported by the Ministry of Education and Science of the Republic of Serbia Grant 172035. Authors gratefully acknowledge Mr Milka Jadranin and Dr Nenad Milosavič for help with HPLC analysis.
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.ejmech.2013.01.006.
References
- 1.Vaya J, Belinsky PA, Aviram M. Antioxidant constituents from licorice roots: isolation, structure elucidation and antioxidative capacity toward LDL oxidation. Free Radic Biol Med. 1997;23:302–313. doi: 10.1016/s0891-5849(97)00089-0. [DOI] [PubMed] [Google Scholar]
- 2.Chen M, Christensen SB, Zhai L, Rasmunssen MH, Theander TG, Frokajaer S, Steffensen B, Davidson J, Kharazmi A. The novel oxygenated chalcone, 2,4-dimethoxy-4′-butoxychalcone, exhibits potent activity against human malaria parasite Plasmodium falciparum in vitro and rodent parasites Plasmodium berghei and Plasmodium yoelii in vivo. J Infect Dis. 1997;176:1327–1333. doi: 10.1086/514129. [DOI] [PubMed] [Google Scholar]
- 3.Nielsen SF, Christensen SB, Cruciani G, Kharazmi A, Liljefors T. Anti-leishmanial chalcones: statistical design, synthesis, and three-dimensional quantitative structure–activity relationship analysis. J Med Chem. 1998;41:4819–4832. doi: 10.1021/jm980410m. [DOI] [PubMed] [Google Scholar]
- 4.Kim YH, Kim J, Park H, Kim HP. Anti-inflammatory activity of the synthetic chalcone derivatives: inhibition of inducible nitric oxide synthase-catalyzed nitric oxide production from lipopolysaccharide-treated RAW 264.7 cells. Biol Pharm Bull. 2007;30:1450–1455. doi: 10.1248/bpb.30.1450. [DOI] [PubMed] [Google Scholar]
- 5.Boumendjel A, Ronot X, Boutonnat J. Chalcones derivatives acting as cell cycle blockers: potential anticancer drugs? Curr Drug Targets. 2009;10:363–371. doi: 10.2174/138945009787846416. [DOI] [PubMed] [Google Scholar]
- 6.Cabrera M, Simoens M, Falchi G, Lavaggi ML, Piro OE, Castellano EE, Vidal AL, Azqueta A, Monge A, Lopez de Cerain A, Sagrera G, Seoane G, Cerecetto H, Gonzalez M. Synthetic chalcones, flavanones, and flavones as antitumoral agents: biological evaluation and structure–activity relationships. Bioorg Med Chem. 2007;15:3356–3367. doi: 10.1016/j.bmc.2007.03.031. [DOI] [PubMed] [Google Scholar]
- 7.Nakamura C, Kawasaki N, Miyataka H, Jayachandran E, Kim IH, Kirk KL, Taguchi T, Takeuchi Y, Horie H, Satoha T. Synthesis and biological activities of fluorinated chalcone derivatives. Bioorg Med Chem. 2002;10:699–706. doi: 10.1016/s0968-0896(01)00319-4. [DOI] [PubMed] [Google Scholar]
- 8.Modzelewska A, Pettit C, Achanta G, Davidson NE, Huang P, Khan SR. Anticancer activities of novel chalcone and bis-chalcone derivatives. Bioorg Med Chem. 2006;14:3491–3495. doi: 10.1016/j.bmc.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 9.Liu X, Go M. Antiproliferative properties of piperidinylchalcones. Bioorg Med Chem. 2006;14:153–163. doi: 10.1016/j.bmc.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 10.Halbwirth H. The creation and physiological relevance of divergent hydroxylation patterns in the flavonoid pathway. Int J Mol Sci. 2010;11:595–621. doi: 10.3390/ijms11020595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Snijman PW, Joubert E, Ferreira D, Li X-C, Ding Y, Green IR, Gelderblom WCA. Antioxidant activity of the dihydrochalcones aspalathin and nothofagin and their corresponding flavones in relation to other rooibos (Aspalathus linearis) flavonoids, epigallocatechin gallate, and trolox. J Agric Food Chem. 2009;57:6678–6684. doi: 10.1021/jf901417k. [DOI] [PubMed] [Google Scholar]
- 12.See for example: Katsori A-M, Hadjipavlou-Litina D. Chalcones in cancer: understanding their role in terms of QSAR. Curr Med Chem. 2009;16:1062–1081. doi: 10.2174/092986709787581798.Batovski DI, Parushev SP. An update on the anticancer effects of chalcones. Int J Curr Chem. 2010;1:217–236.Boumendjel A, Boccard J, Carrupt P-A, Nicolle E, Blanc M, Geze A, Choisnard L, Wouessidjewe D, Matera E-L, Dumontet C. Antimitotic and antiproliferative activities of chalcones: forward structure–activity relationship. J Med Chem. 2008;51:2307–2310. doi: 10.1021/jm0708331.
- 13.Bazzaro M, Anchoori RK, Mudiam MKR, Issaenko O, Kumar S, Karanam B, Lin Z, Isaksson Vogel R, Gavioli R, Destro F, Ferretti V, Roden RBS, Khan SR. α,β-unsaturated carbonyl system of chalcone-based derivatives is responsible for broad inhibition of proteasomal activity and preferential killing of human papilloma virus (HPV) positive cervical cancer cells. J Med Chem. 2011;54:449–456. doi: 10.1021/jm100589p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dimmock JR, Jha A, Zello GA, Allen TM, Santos CL, Balzarini J, De Clercq E, Manavathu EK, Stables JP. Cytotoxic 4′-aminochalcones and related compounds. Pharmazie. 2003;58:227–232. [PubMed] [Google Scholar]
- 15.Robinson T-P, Hubbard RB, Ehlers TJ, Arbiser JL, Goldsmith DJ, Bowen JP. Synthesis and biological evaluation of aromatic enones related to curcumin. Bioorg Med Chem. 2005;13:4007–4013. doi: 10.1016/j.bmc.2005.03.054. [DOI] [PubMed] [Google Scholar]
- 16.Yellaturu CR, Manjula B, Neeli I, Rao GN. N-ethylmaleimide inhibits platelet-derived growth factor BB-stimulated Akt phosphorylation via activation of protein phosphatase 2A. J Biol Chem. 2002;227:40148–40155. doi: 10.1074/jbc.M206376200. [DOI] [PubMed] [Google Scholar]
- 17.Jensen LH, Renodon-Corniere A, Wessel I, Langer SW, Søkilde B, Carstensen EV, Sehested M, Jensen PB. Maleimide is a potent inhibitor of topoisomerase II in vitro and in vivo: a new mode of catalytic inhibition. Mol Pharmacol. 2002;61:1235–1243. doi: 10.1124/mol.61.5.1235. [DOI] [PubMed] [Google Scholar]
- 18.Jha A, Mukherjee C, Rolle AJ, De Clercq E, Balzarini J, Stablesc JP. Cytostatic activity of novel 4′-aminochalcone-based imides. Bioorg Med Chem Lett. 2007;17:4545–4550. doi: 10.1016/j.bmcl.2007.05.094. [DOI] [PubMed] [Google Scholar]
- 19.Leslie BJ, Holaday CR, Nguyen T, Hergenrother PJ. Phenylcinnamides as novel antimitotic agents. J Med Chem. 2010;53:3964–3972. doi: 10.1021/jm901805m. [DOI] [PubMed] [Google Scholar]
- 20.Raffa D, Maggio B, Plescia F, Cascioferro S, Plescia S, Raimondi MV, Daidone G, Tolomeo M, Grimaudo S, Di Cristina A, Pipitone RM, Bai R, Hamel E. Synthesis, antiproliferative activity, and mechanism of action of a series of 2-{[(2E)-3-phenylprop-2-enoyl]amino}benzamides. Eur J Med Chem. 2011;46:2786–2796. doi: 10.1016/j.ejmech.2011.03.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.(a) Juraniá Z, Stevovič Lj, Drakulič B, Stanojkovič T, Radulovič S, Juranič I. Substituted (E)-β-(benzoyl)acrylic acids suppressed survival of neoplastic human HeLa cells. J Serb Chem Soc. 1999;64:505–512. [Google Scholar]; (b) Drakulič BJ, Stanojkovič TP, Žižak ŽS, Dabovič MM. Antiproliferative activity of aroylacrylic acids. Structure–activity study based on molecular interaction fields. Eur J Med Chem. 2011;46:3265–3273. doi: 10.1016/j.ejmech.2011.04.043. [DOI] [PubMed] [Google Scholar]
- 22.Papa D, Schwenk E, Villani F, Klingsberg E. β-aroylacrylic acids. J Am Chem Soc. 1948;70:3356–3360. doi: 10.1021/ja01190a041. [DOI] [PubMed] [Google Scholar]
- 23.Massarotti A, Coluccia A, Silvestri R, Sorba G. The tubulin colchicine domain: a molecular modeling perspective. Chem Med Chem. 2012;7:33–42. doi: 10.1002/cmdc.201100361. [DOI] [PubMed] [Google Scholar]
- 24.Gaillard P, Carrupt PA, Testa B, Boudon A. Molecular lipophilicity potential, a tool in 3D QSAR: method and applications. J Comp Aided Mol Des. 1994;8:83–96. doi: 10.1007/BF00119860. [DOI] [PubMed] [Google Scholar]
- 25.Gómez-Ruiz S, Ceballos-Torresa J, Prashara S, Fajardoa M, Žižak Ž, Juranič ZD, Kaluđerovič GN. One ligand different metal complexes: biological studies of titanium(IV), tin(IV) and gallium(III) derivatives with the 2,6-dimethoxypyridine-3-carboxylato ligand. J Organomet Chem. 2011;696:3206–3213. [Google Scholar]
- 26.Peyrot V, Leynadier D, Sarrazin M, Briand C, Rodriquez A, Nieto JM, Andreu JM. Interaction of tubulin and cellular microtubules with the new antitumor drug MDL 27048. A powerful and reversible microtubule inhibitor. J Biol Chem. 1989;264:21296–21301. [PubMed] [Google Scholar]
- 27.(a) Cruciani G, Carosati E, De Boeck B, Ethirajulu K, Mackie C, Howe T, Vianello R. MetaSite: understanding metabolism in human cytochromes from the perspective of the chemist. J Med Chem. 2005;48:6970–6979. doi: 10.1021/jm050529c. [DOI] [PubMed] [Google Scholar]; (b) Ahlström MM, Ridderström M, Zamora I. CYP2C9 structure–metabolism relationships: substrates, inhibitors, and metabolites. J Med Chem. 2007;50:5382–5391. doi: 10.1021/jm070745g. [DOI] [PubMed] [Google Scholar]
- 28.Nguyen TL, McGrath C, Hermone AR, Burnett JC, Zaharevitz DW, Day BW, Wipf P, Hamel E, Gussio R. A common pharmacophore for a diverse set of colchicine site inhibitors using a structure-based approach. J Med Chem. 2005;48:6107–6116. doi: 10.1021/jm050502t. [DOI] [PubMed] [Google Scholar]
- 29.Dorleans A, Gigant B, Ravelli RBG, Mailliet P, Mikol V, Knossow M. Variations in the colchicine-binding domain provide insight into the structural switch of tubulin. Proc Natl Acad Sci U S A. 2009;106:13775–13779. doi: 10.1073/pnas.0904223106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grant JA, Gallardo MA, Pickup BT. A fast method of molecular shape comparison. A simple application of a Gaussian description of molecular shape. J Comp Chem. 1996;17:1653–1666. ROSC v3.1.1, http://www.eyesopen.com/ [Google Scholar]
- 31.Mills JEJ, Dean PM. Three-dimensional hydrogen-bond geometry and probability information from a crystal survey. J Comput Aided Mol Des. 1996;10:607–622. doi: 10.1007/BF00134183. [DOI] [PubMed] [Google Scholar]
- 32.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Meth. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 33.Ohno M, Abe TJ. Rapid colorimetric assay for the quantification of leukemia inhibitory factor (LIF) and interleukin-6 (IL-6) J Immunol Meth. 1991;145:199–203. doi: 10.1016/0022-1759(91)90327-c. [DOI] [PubMed] [Google Scholar]
- 34.Clothier RH. The FRAME cytotoxicity test. Methods Mol Biol. 1995;43:109–118. doi: 10.1385/0-89603-282-5:109. [DOI] [PubMed] [Google Scholar]
- 35.Hamel E, Lin CM. Separation of active tubulin and microtubule-associated proteins by ultracentrifugation and isolation of a component causing the formation of microtubule bundles. Biochemistry. 1984;23:4173–4184. doi: 10.1021/bi00313a026. [DOI] [PubMed] [Google Scholar]
- 36.Hamel E. Evaluation of antimitotic agents by quantitative comparisons of their effects on the polymerization of purified tubulin. Cell Biochem Biophys. 2003;38:1–21. doi: 10.1385/CBB:38:1:1. [DOI] [PubMed] [Google Scholar]
- 37.Verdier-Pinard P, Lai J-Y, Yoo H-D, Yu J, Marquez B, Nagle DG, Nambu M, White JD, Falck JR, Gerwick WH, Day BW, Hamel E. Structure–activity analysis of the interaction of curacin A, the potent colchicine site antimitotic agent, with tubulin and effects of analogs on the growth on MCF-7 breast cancer cells. Mol Pharmacol. 1998;53:62–76. doi: 10.1124/mol.53.1.62. [DOI] [PubMed] [Google Scholar]
- 38.Pedretti A, Villa L, Vistoli G. Vega – an open platform to develop chemo-bioinformatics applications, using plug-in architecture and script programming. J Comput Aided Mol Des. 2004;18:167–173. doi: 10.1023/b:jcam.0000035186.90683.f2. http://www.ddl.unimi.it. [DOI] [PubMed] [Google Scholar]
- 39.(a) Boström J. Reproducing the conformations of protein bound ligands: a critical evaluation of several popular conformational tools. J Comput Aided Mol Des. 2002;15:1137–1152. doi: 10.1023/a:1015930826903. [DOI] [PubMed] [Google Scholar]; (b) Boström J, Greenwood JR, Gottfries J. Assessing the performance of OMEGA with respect to retrieving bioactive conformations. J Mol Graph Model. 2003;21:449–462. doi: 10.1016/s1093-3263(02)00204-8. OMEGA v 2.2.1, www.eyesopen.com. [DOI] [PubMed] [Google Scholar]
- 40.Halgren TA. MMFF VI. MMFF94s option for energy minimization studies. J Comp Chem. 1999;20:720–729. doi: 10.1002/(SICI)1096-987X(199905)20:7<720::AID-JCC7>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 41.James J, Stewart P, editors. MOPAC 2009. Stewart Computational Chemistry; Colorado Springs, CO, USA: HTTP://OpenMOPAC.net. [Google Scholar]
- 42.Stewart JJP. Optimization of parameters for semiempirical methods V: modification of NDDO approximations and application to 70 elements. J Mol Model. 2007;13:1173–1213. doi: 10.1007/s00894-007-0233-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.