

Abstract
Aortic aneurysm is common, accounting for 1–2% of all deaths in industrialized countries. Early theories of the causes of human aneurysm mostly focused on inherited or acquired defects in components of the extracellular matrix in the aorta. Although several mutations in the genes encoding extracellular matrix proteins have been recognized, more recent discoveries have shown important perturbations in cytokine signalling cascades and intracellular components of the smooth muscle contractile apparatus. The modelling of single-gene heritable aneurysm disorders in mice has shown unexpected involvement of the transforming growth factor-β cytokine pathway in aortic aneurysm, highlighting the potential for new therapeutic strategies.
Aneurysm or arterial enlargement is the gross phenotype manifesting progressive organ failure of large arteries, including the aorta. Aortic aneurysm is usually not inherently dangerous, but enlarged arteries show a predisposition for tear (known as dissection), with high mortality rates. Clinicians recognize two predominant spatial distributions of aortic aneurysms in patient groups. The most common form, abdominal aortic aneurysm (AAA), is typically associated with advanced age and atherosclerosis, with attendant risk factors such as hypercholesterolaemia, hypertension and/or diabetes. These lesions are pathologically characterized by atheromata, invasion of inflammatory cells, destructive extracellular matrix remodelling, and depletion and dysfunction of vascular smooth muscle cells. Although it is clear that genetic determinants influence the expression of AAA, there has been no description of a single major gene or locus effect that is sufficient to cause isolated abdominal aneurysm (that is, in the absence of evidence for a more systemic vasculopathy). The weight of evidence therefore indicates that AAA is a complex disorder that integrates the influence of predisposing genes with lifestyle-associated risk factors — analogous to coronary artery disease.
The second common site for aneurysm is the thoracic ascending aorta. Unlike AAA, thoracic aortic aneurysms (TAAs) occur in all age groups, are more highly associated with hereditary influences and do not show obligate association with cardiovascular risk factors. Pathologically, inherited forms of TAA typically show destructive matrix remodelling with elastin fragmentation, proliferation of vascular smooth muscle cells and a less prominent inflammatory component without atheromata. Many presentations of TAA show classic Mendelian inheritance with high or complete penetrance, suggesting the major contribution of a single gene. Familial TAA can be subdivided into syndromic presentations that show prominent features of a systemic connective tissue disorder (such as Marfan and Loeys–Dietz syndrome) and non-syndromic presentations (such as bicommissural aortic valve with TAA, and isolated familial TAA). Both the syndromic and non-syndromic groups include many disorders in which disease predominates in the very proximal ascending thoracic aorta (Fig. 1). In fact, there are only rare exceptions in which the ascending aorta is infrequently involved — such as vascular Ehlers–Danlos syndrome. Table 1 includes a list of human and mouse single-gene disorders associated with aneurysm predisposition.
Figure 1. Sites of TAA in transforming growth factor-β vasculopathy syndromes.

Multidetector computer tomographic reconstruction images are shown. a, The thoracic aorta of a 9-year-old patient with Loeys-Dietz syndrome (LDS) is shown after emergent repair of a proximal descending (Stanford type B) aortic dissection, with a large unrepaired aortic root aneurysm (yellow arrowhead). The red arrowhead denotes the dacron tube graft. b, The thoracic aorta of a 16-year-old patient with LDS is shown after surgical repair of an aortic root aneurysm (yellow arrowhead), with a discrete fusiform aneurysm of the proximal descending aorta (red arrowhead). Bar = 3 cm.
Table 1.
Human hereditary aneurysm conditions and mouse models of aneurysm
| Gene (protein) | Human aneurysmal syndrome | Animal-model phenotype | Pathway |
|---|---|---|---|
|
Extracellular matrix protein | |||
| FBN1 (fibrillin-1) | MFS; highly penetrant ascending aortic aneurysm | KO: perinatal lethal, pulmonary hypoplasia, arteriopathy; hypomorphic: arteriopathy, aneurysm, dissection and systemic manifestations of MFS | TGF-β54,55 |
| EFEMP2 (fibulin-4) | Cutis laxa with aneurysm; ascending aortic aneurysm and tortuosity | KO: ascending aortic aneurysm, defective elastogenesis, perinatal lethality | TGF-β3,13 |
| ELN (elastin) | Cutis laxa with aneurysm; low penetrance ascending aortic aneurysm and dissection | Haploinsufficient: obstructive arterial disease with increased VSMC proliferation, increased lamellae number; KO: accentuated phenotype | Unknown1,56 |
| COL1A1 (collagen α-1(I)) | Osteogenesis imperfecta; extremely rare aortic aneurysm; EDS, type 7A; dissection of medium-sized arteries | KO: adult-onset aortic aneurysm and dissection | Collagen metabolism57,58 |
| COL1A2 (collagen α-2(I)) | Osteogenesis imperfecta; extremely rare aortic aneurysm; EDS, cardiac valvular dystrophy type 7B; borderline aortic root enlargement with aortic regurgitation | Homozygous LOF: decreased body weight, bony abnormalities, no arterial phenotype reported | Collagen metabolism59 |
| COL3A1 (collagen α-1(III)) | EDS, type 4; frequent arterial dissection with infrequent aneurysm | KO: frequent neonatal mortality, aortic rupture, intestinal rupture | Collagen metabolism60,61 |
| COL4A1 (collagen α-1(IV)) | Hereditary angiopathy, nephropathy, aneurysms and muscle cramps; infrequent aneurysms | KO: embryonic lethal (E10.5–11.5), basement membrane failure | Collagen metabolism62,63 |
| COL4A5 (collagen α-5(IV)) | X-linked Alport syndrome; ascending aortic and abdominal aneurysms and dissections | Nonsense mutation: no overt aortic disease noted | Collagen metabolism64 |
| LOX (lysyl oxidase) | No human phenotype described | KO: low penetrance aortic aneurysm, perinatal lethality | Collagen metabolism; TGF-β15 |
| PLOD1 (lysyl hydroxylase 1) | EDS, type 6; rare aneurysm | KO: spontaneous aneurysm and dissection, gait abnormalities | Collagen metabolism65,66 |
| PLOD3 (lysyl hydroxlase 3) | Bone fragility with contractures, arterial rupture and deafness; frequent medium-sized arterial aneurysms | KO: embryonic lethal (E9.5) and basement membrane fracture | Collagen metabolism67,68 |
|
Transmembrane protein | |||
| TGFBR1 (TGF-β receptor type 1) | LDS; highly penetrant root and diffuse large and medium arterial aneurysms | KO: midgestational death with yolk sac defects; M318R heterozygous knock-in: aortic root and diffuse aneurysm (D. Loch, unpublished observations) | TGF-β8,69 |
| TGFBR2 (TGF-β receptor type 2) | LDS; highly penetrant root and diffuse large and medium arterial aneurysms; familial thoracic aortic aneurysms and dissections; highly penetrant root and medium arterial aneurysms | KO: defects in haematopoesis and vasculogenesis, embryonic lethal (E10.5); Tgfbr2max; impaired elastogenesis, decreased lysyl oxidase in aorta; G357W heterozygous knock-in: aortic root and diffuse aneurysm (D. Loch, unpublished observations) | TGF-β8,19,70 |
| ENG (endoglin) | Hereditary haemorrhagic telangiectasia; incompletely penetrant aortic and medium-sized arterial aneurysms | KO: defective vasculogenesis, embryonic lethal (E10); haploinsufficient: haemorrhagic telangiectasia causing strokes, fatal haemorrhage and heart failure | TGF-β superfamily71,72 |
| ACVRL1 (activin receptor-like kinase I) | Hereditary haemorrhagic telangiectasia; incompletely penetrant aortic and medium-sized arterial aneurysms | KO: defective vasculogenesis, embryonic lethal, excessive fusion of capillary plexuses; haploinsufficient: haemorrhagic telangiectasia | TGF-β superfamily73,74 |
| SLC2A10 (glucose transporter type 10) | Arterial tortuosity syndrome; diffuse arterial tortuosity, stenoses, aneurysms | Homozygote missense: arterial thickening with increased elastin deposition, elastin fractures at advanced age | TGF-β12,75 |
| NOTCH1 (NOTCH1) | Bicuspid valve with ascending aortic aneurysm | KO: embryonic lethal (E9.5), required for somite segmentation, defects in angiogenesis | NOTCH1–JAGGED176,77 |
| JAG1 (JAGGED1) | Alagille syndrome; intracranial aneurysms, coarctation of the aorta, aortic aneurysm | KO: embryonic lethal (E9.5) with diffuse haemorrhages | NOTCH1–JAGGED178,79 |
| GJA1 (connexin-43) | Hypoplastic left heart syndrome (HLHS) | Nonsense mutation (W45X): coronary artery aneurysms | Unknown83 |
| PKD1 (polycystin-1) | Polycystic kidney disease with intracranial aneurysms | KO: embryonic lethal (E14.5) with polycystic kidneys; hypomorphic expression: adult-onset aortic aneurysm and dissection | mTOR80,81 |
| PKD2 (polycystin-2) | Polycystic kidney disease with intracranial aneurysms | KO: defects in cardiac septation and left–right axis determination, kidney and pancreatic cysts | Unknown82 |
|
Cytoplasmic protein | |||
| SMAD3 (SMAD family member 3) | LDS; aortic aneurysm with osteoarthritis | KO: metastatic colorectal cancer | TGF-β11 |
| ACTA2 (α-smooth muscle actin) | Familial aortic aneurysm with livedo reticularis and iris flocculi | KO: viable offspring with normal lifespan and impaired vascular contractility | IGF-1, Ang II38,84 |
| MYH11 (smooth muscle myosin) | Familial aortic aneurysm with patent ductus arteriosus | KO: neonatal lethality, urinary retention, dilated cardiomyopathy | IGF-1, Ang II36,37,85 |
| FLNA (filamin-A) | Periventricular nodular heterotopia with EDS features; ascending aortic aneurysm and valvular dystrophy | KO: neonatal lethality, persistent truncus arteriosus, endothelial cell–cell contact defects | Unknown40,86 |
| NF1 (neurofibromin-1) | Neurofibromatosis; medium-sized arterial aneurysm and stenosis | KO: enlarged head, pale liver, cardiac malformations | Ras–MEK–ERK30 |
| PTPN11 (protein-tyrosine phosphatase 2C) | Noonan and LEOPARD syndromes; coronary artery aneurysms and rare ascending aortic aneurysm | KO: embryos die at preimplantation; missense mutation (D61G): cardiac defects, defective valvulogenesis, skeletal anomalies, myeloproliferative disorder | Ras–MEK–ERK29,87,88 |
| NPHP3 (nephrocystin-3) | Nephronophthisis | KO: low penetrance intracranial aneurysms | Unknown89 |
| NOS3 (nitric oxide synthase 3) | Refractory hypertension | KO: abnormal aortic development with bicuspid aortic valve; in combination with Apoe−/−, mice show abdominal arterial aneurysm and dissections | Nitric oxide90,91 |
| TSC2 (tuberin) | Tuberous sclerosis; diffuse thoracoabdominal aneurysms | Heterozygous KO: increased proliferation of VSMCs after injury | mTOR92 |
| GAA (lysosomal α-glucosidase) | Acid maltase deficiency, adult onset; intracranial aneurysms | KO: lysosomal accumulation in heart, aorta, skeletal muscle | Unknown93 |
| S100A12 (S100A12) | No human phenotype; increased S100A12 protein expression in human MYH11-mutation aneurysmal tissues | Sm22α promoter–S100A12 transgenic mouse: vascular smooth muscle disarray, elastin fragmentation, thoracic aneurysm | IL-6, TGF-β39 |
|
Nuclear protein | |||
| MED12 (mediator complex subunit 12) | Lujan–Fryns syndrome; extremely rare aneurysm | Hypomorphic mutants: embryonic lethal (E10), defects in neural tube closure, somatogenesis, heart formation | WNT–β-catenin, WNT–PCP94 |
| KLF15 (Krüppel-like factor 15) | No human phenotype; Krüppel-like factor 15 downregulated in human abdominal aortic aneurysm | KO: aortic aneurysm and cardiomyopathy | TSP-1, p53, TGF-β95 |
| KLF2 (Krüppel-like factor 2) | No human phenotype | KO: embryonic aortic aneurysm and dissection | Unknown96 |
|
Chromosomal anomaly | |||
| 45, X | Turner syndrome; bicuspid aortic valve, coarctation of the aorta, ascending aneurysm | XO mice (with a single X chromosome): no phenotypic heart disease | Unknown97 |
|
Chemical model | |||
| No human phenotype | Ang-II-infusion model | Ang II, MCP-1, IL-6, TGF-β98 | |
| No human phenotype | Elastase-infusion model | Unknown99 | |
| No human phenotype | Periarterial calcium application | JNK1 (ref. 100) | |
E10.5, embryonic day 10.5; EDS, Ehlers–Danlos syndrome; KO, knockout; LOF, loss of function; mTOR, mammalian target of rapamycin; PCP, planar cell polarity. PTPN11 is also known as SHP2.
There has been disproportionate historical focus on elastic fibres in pathogenetic models of inherited TAA. This derives from the near-uniform histological observation of reduced elastin content and elastic fibre fragmentation in the aortic media (the middle aortic layer), known as cystic medial necrosis. However, many elastin-deficiency states do not associate with aneurysm as a prominent phenotype. Although aortic aneurysm is an extremely rare manifestation of cutis laxa syndromes caused by mutations in the elastin gene1, it is not observed in mice or humans with dominant and recessive forms of cutis laxa caused by deficiency of fibulin-5 (ref. 2), a crucial mediator of elastogenesis. By contrast, aneurysmal disease, including prominent involvement of the ascending aorta, is highly penetrant in inherited cutis laxa caused by fibulin-4 deficiency3. Humans and mice with a fibrillin-1 deficiency show failed elastic fibre homeostasis and highly penetrant aortic root aneurysms in the context of Marfan syndrome (MFS) (see ref. 4 and references therein). Pseudoxanthoma elasticum caused by ABCC6 deficiency in humans also shows postnatally acquired elastic fibre fragmentation in the aorta, but does not typically show aneurysm5. These observations raise the important question of what molecular events, besides elastin-related issues, are common to the fibrillin-1- and fibulin-4-deficiency states but not recapitulated by fibulin-5 deficiency. The focus on the aortic media and elastic fibres may have been a distraction, and further insight could come from considering other distinguishing features of the proximal ascending aorta, including developmental cellular ontology. The discovery that the genes mutated in MFS and vascular Ehlers–Danlos syndrome (FBN1 and COL3A1, respectively) encode extracellular matrix elements (fibrillin-1 and collagen α-1(III)) led to the generation of pathogenetic models that singularly invoke inherent structural weakness of the tissues. Such a view boded poorly for the development of productive medical treatment strategies, requiring a means to alter the structural composition of inherently weak tissues. Fortunately, for both patients and researchers, the story of hereditary aneurysm has turned out to be much more complex and potentially permissive for therapeutic intervention.
Over the past couple of decades, the genes responsible for heritable aortic aneurysm have been identified at an accelerating pace. Gene identification has allowed the creation of mouse models of inherited aortic aneurysm, providing the first opportunity to temporally and comprehensively interrogate the pathogenic sequence of aneurysm, extending from predisposition to clinical consequence, in an experimental context that mimics the physiological complexity of the human system. This combination of human molecular genetics and animal modelling has shown the involvement of diverse cytokine pathways, prominently the role of the transforming growth factor-β (TGF-β) pathway in aortic aneurysm. This review will focus on the mechanisms of failure of large vessel wall homeostasis that challenge or inform historical perspectives, will attempt to integrate emerging models of disease, and will discuss the remaining challenges and opportunities in aneurysm research.
Marfan syndrome provides a link between aneurysm and TGF-β
A shift in thinking regarding the pathogenesis of aortic aneurysm occurred during the study of MFS (ref. 4 and references therein). Although many of the clinical manifestations of the disorder could be caused by simple tissue weakness imposed by fibrillin-1 deficiency (such as aortic aneurysm, eye lens dislocation and emphysema), others were not so easily reconciled (bone overgrowth, craniofacial alterations and myxomatous valve disease). Insight came from studying lung disease in fibrillin-1-deficient mice. Contrary to expectation, mouse models of MFS did not show destructive and inflammatory emphysema. Instead, they showed primary failure of distal alveolar septation during late embryogenesis and the perinatal period6.
Mechanistic hypotheses built on the observation that fibrillin proteins show marked homology to latent TGF-β-binding proteins (LTBPs). Most TGF-β is secreted from cells in the context of a large latent complex (LLC) that includes the mature cytokine, a dimer of its processed amino-terminal propeptide (latency-associated peptide (LAP)) and one of three LTBP isoforms (LTBP-1, LTBP-3 or LTBP-4). LTBPs target the TGF-β LLC to binding partners such as fibronectin and microfibrils composed of fibrillin-1 — an event that is thought to regulate TGF-β bioavailability and activity by controlling access to, or the efficiency of, TGF-β activators. One hypothesis was that failed or improper matrix sequestration of the LLC owing to fibrillin-1 deficiency could lead to promiscuous TGF-β activation. In keeping with this concept, the developing lungs of fibrillin-1-deficient mice showed decreased LLC levels but raised levels of free TGF-β and increased TGF-β signalling, as demonstrated by nuclear translocation and phosphorylation of receptor-activated SMAD proteins 2 and 3 (pSMAD2/3). Notably, distal alveolar septation could be restored in fibrillin-1-deficient mice by the administration of a pan-specific anti-TGF-β neutralizing antibody6. Other manifestations of MFS, including myxomatous mitral valve disease, skeletal myopathy and, most importantly, aortic root aneurysm, were associated with increased TGF-β signalling in mice models of MFS, and were attenuated or prevented by TGF-β antagonism with a neutralizing anti-TGF-β antibody in mice in vivo4,7.
TGF-β receptor mutations
Perhaps the most direct evidence for a major role of TGF-β in aneurysm pathogenesis came from the finding that mutations in the TGFBR1 and TGFBR2 genes — which encode the TGF-β receptor subunits TGFR-1 (also known as ALK-5) and TGFR-2, respectively — result in aneurysm conditions with undeniable phenotypic overlap with MFS, a notable example of which is Loeys–Dietz syndrome (LDS)8,9 (Table 1). Similar to MFS, patients with LDS typically show skeletal involvement including long fingers, chest-wall deformity and scoliosis. Other shared features include widening of the dural sac, skin stretch marks and mitral valve prolapse. Patients with LDS typically do not show lens dislocation, and can show many discriminating systemic features such as widely spaced eyes (hypertelorism), cleft palate or bifid uvula, cervical spine malformation or instability, osteoporosis and club foot deformity8,10. Most importantly, patients with LDS show highly penetrant arterial tortuosity (an elongation of an artery resulting in a twisted course) and a strong predisposition for aneurysm and dissection throughout the arterial tree. Vascular disease in patients with LDS is more aggressive than in those with MFS, with rupture at a younger age (as young as 6 months) and at smaller aortic dimensions10.
Nearly all patients with LDS are heterozygous for missense substitutions mutations in the kinase domain of TGFR-1 or TGFR-2. Recombinant expression of mutant receptors in cells naive for the corresponding receptor subunit failed to support TGF-β signalling, leading to the hypothesis that haploinsufficiency was the relevant mechanism9. If this was the case, however, the identification of early nonsense mutations that elicit messenger RNA clearance through nonsense-mediated mRNA decay or whole allele deletions might be expected. Instead, the skewed mutational repertoire seems to manifest selection for receptor variants that traffic to the cell surface and bind ligand but lack the ability to propagate signal. Co-transfection studies in human cells using equimolar concentrations of normal and mutant receptors and experiments with heterozygous patient cells has shown apparent preservation of the signalling potential of wild-type receptor subunits, excluding a conventional dominant-negative mechanism8,9. Notably, patient vascular tissue obtained at surgery or autopsy has consistently shown paradoxically enhanced TGF-β signalling, demonstrated by nuclear accumulation of pSMAD2 in vascular smooth muscle cells (VSMCs) and increased output of TGF-β-driven gene products such as collagens and connective tissue growth factor (CTGF)8,10. The architectural changes seen in the aortic wall of patients with LDS are highly reminiscent of those seen in MFS and other inherited forms of aortic root aneurysm. The mechanisms underlying this paradoxical effect remain unknown, but potentially include altered receptor trafficking, impaired autoregulation of TGF-β signalling, alternative signalling cascades or non-autonomous cellular events (see ‘Cancer biology as a guide to aneurysm’). Heterozygous loss-of-function mutations in SMAD3 recapitulate both the LDS phenotype and paradoxical enhancement of TGF-β signalling in the aortic wall11.
Other TGF-β links to aneurysm
Several other aneurysmal disorders have been linked to TGF-β signalling4. High TGF-β signalling, as assessed by nuclear pSMAD2 accumulation, has been observed in surgical samples from patients with diverse aneurysm conditions such as isolated familial TAA and bicommissural aortic valve with TAA4. Patients with autosomal-recessive arterial tortuosity syndrome show diffuse and severe arterial tortuosity that is often associated with vascular stenoses, segmental vascular hypoplasia and arterial aneurysms, caused by loss-of-function mutations in the SLC2A10 (also known as GLUT10) gene that encodes the integral membrane protein glucose transporter type 10 (ref. 12). Vascular tissue from patients with arterial tortuosity syndrome shows the same signature of high TGF-β signalling seen in tissue from patients with MFS and LDS. Although the mechanism is poorly understood, cultured fibroblasts from patients with arterial tortuosity syndrome show impaired expression of the proteoglycan decorin — an antagonist of TGF-β superfamily signalling12. Deficiency of the extracellular protein fibulin-4 causes autosomal recessive cutis laxa in association with arterial tortuosity and aortic aneurysm in both humans and mice3,13. Curiously, fibulin-5 deficiency causes cutis laxa with arterial tortuosity but not aneurysm. Both fibulin-4 and fibulin-5 bind to fibrillin-1 and elastin, and both deficiency states are associated with profound failure of elastogenesis — the probable cause of arterial tortuosity. Fibulin-5 mainly promotes elastin fibre assembly by the recruitment of tropoelastin to microfibrils14. By contrast, fibulin-4 is needed for the recruitment of lysyl oxidase (LOX), a copper-dependent enzyme that catalyzes cross-linking of elastin molecules14. In keeping with these findings, LOX-deficient mice show severely disrupted aortic laminae, arterial tortuosity and low penetrance aneurysm15. Mice and humans deficient in fibulin-4 show increased TGF-β signalling in the vessel wall, which may be directly related to the ability of LOX to enzymatically inhibit TGF-β16. In this light, it seems that the aneurysm phenotype specific to fibulin-4 deficiency manifests a loss of a function other than elastin cross-linking, plausibly including TGF-β repression.
Despite several lines of evidence invoking high TGF-β signalling in aneurysm, conflicting observations exist. For example, high vascular TGF-β signalling was shown in Emilin1-deficient mice in association with a diffusely small vascular system and hypertension in juvenile animals17. The developmental timing and vascular distribution of high TGF-β signalling were not assessed, and there was no comprehensive analysis for aneurysms17. Another study has shown that lineage-specific ablation of TGF-β signalling in VSMCs in the ascending aorta results in perturbations of vascular morphogenesis in fetal mice, including persistent truncus arteriosus, impaired elastogenesis and apparent vessel widening that was equated with aneurysm18; similar impairment of elastogenesis was seen only in the descending aorta of mice with global VSMC deletion of Tgfbr2 (ref. 19). There is further indirect evidence that low TGF-β signalling may also be involved in developmental presentations of aneurysm in MFS. An emerging view is that the fibrillin proteins have a dichotomous role in TGF-β regulation. Fibrillin-2 was shown to concentrate TGF-β ligands (prominently BMP7), and this is required to support morphogenetic events at sites of intended function. In the developing autopod, fibrillin-2-deficient mice show BMP7 deficiency and recapitulate the syndactyly phenotype observed in BMP7-targeted mice20. Mice deficient in both fibrillin-1 and fibrillin-2 show persistent truncus arteriosus, which has been historically associated with loss of TGF-β signalling (F. Ramirez, personal communication).
Downstream of TGF-β
Little is known about the precise pathogenetic sequence downstream of TGF-β that is involved in aneurysm progression. Enhancement of matrix metalloproteinase (MMP) activity is frequently invoked. Such a model is both theoretically appealing and experimentally validated. Evidence includes high MMP expression and activity in many natural and experimentally induced presentations of aneurysm, and the ability of MMP inhibitors (such as doxycycline) to attenuate aneurysm progression, including in mouse models of MFS21. While TGF-β has been associated with reduced expression and activity of multiple MMPs in many tissues and contexts, it has been shown to specifically induce MMP2 and MMP9 expression — the MMPs that are most closely associated with aneurysm conditions such as MFS22,23.
Most studies of TGF-β-related disease states have focused on ‘canonical’ (SMAD-dependent) signalling cascades, with a more historic than empirical basis for such an emphasis. More recently, it has been shown that ligand-activated TGF-β receptors can stimulate signalling through non-canonical pathways, including the phosphatidylinositol-3-OH kinase (PI(3)K)/AKT, Rho-associated protein kinase (ROCK) and mitogen-activated protein kinase (MAPK) cascades24 (Fig. 2). Although mouse and human models of MFS demonstrate upregulation of canonical signalling7, its importance is not clear. There is emerging evidence that MAPKs may have a role in aneurysm. For example, activation of p38 has been observed in the aorta of young mice homozygous for a hypomorphic fibillin-1 (Fbn1) allele25, and we have observed TGF-β- and angiotensin II type 1 receptor (AT1R)-dependent activation of extracellular-signal regulated kinases (ERK1 and ERK2) in the aorta of fibrillin-1-deficient mice and abrogation of pathologic aortic root growth upon treatment with a specific ERK inhibitor26,27. ERK1 upregulation has also been observed in fibulin-4 deficiency in mice and humans4,13, perhaps providing a link between the loss of function of fibulin-4 and fibrillin-1.
Figure 2. The TGF-β- and angiotensin II-signalling pathways.

Signalling cascades of TGF-β and angiotensin II have been implicated in the pathogenesis of aneurysm. Fibrillin-1, the major component of extracellular microfibrils, binds and sequesters the large latent complex (LLC) of TGF-β. After TGF-β activation (release), ligand binds to the TGF-β receptor and activates both canonical (grey) and non-canonical (blue) signalling cascades. The extensive cross-talk between the TGF-β and angiotensin II type 1 receptor (AT1R) signalling pathways is indicated. Key terminal events in the pathogenesis of aneurysm may include MMP-mediated proteolysis, CTGF-mediated epithelial-to-mesenchymal transition and tissue remodelling, or IL-6- and MCP-1-mediated inflammation. Proteins indicated in purple have been directly implicated in human hereditary aneurysmal disease (see Table 1). α-SMA, α-smooth muscle actin; MEK1, MAP kinase kinase 1; MLCK, myosin light chain kinase; MLCP, myosin light chain phosphatase; p190 RhoGAP, Rho GTPase-activating protein 5; SHP2, protein-tyrosine phosphatase 2C; TAK1, mitogen-activated protein kinase kinase kinase 7 (also known as TAK1); MEK1, MAP kinase kinase 1; MLCK, myosin light chain kinase; MLCP, myosin light chain phosphatase; p190 RhoGAP, Rho GTPase-activating protein 5; SHP2, protein tyrosine phosphatase 2C; α-SMA, α-smooth muscle actin.
Infusion of angiotensin II (AngII) or the application of CaCl2 promotes AAA in mice. Antagonism of c-Jun N-terminal protein kinase 1 (JNK1) signalling has been shown to attenuate disease progression in an AAA-induced mouse model28. This occurred in association with reduced MMP2 and MMP9 activity and enhanced LOX expression. ERK activity is known to be instrumental in diverse aspects of vascular pathology, including VSMC proliferation and migration, and has been linked to TGF-β-mediated MMP upregulation and epithelial-to-mesenchymal transition. Finally, low-penetrance aneurysm has been observed in human conditions known to positively modify Ras signalling, a major upstream activator of ERK. These include gain-of-function mutations in PTPN11, which cause Noonan syndrome, and loss-of-function mutations in NF1, encoding a Ras GTPase-activating protein, in neurofibromatosis type 1 (refs 29, 30) (Table 1 and Fig. 2).
Angiotensin II and aneurysm
Aortic aneurysm and dissection can be modelled through the infusion of AngII in mice deficient for apolipoprotein E (Apoe−/ −) (ref. 4 and references therein), or with higher doses in aged wild-type mice31. Aneurysm formation occurs with high penetrance in the suprarenal abdominal aorta; the ascending aorta can also be involved with lower frequency and severity. Aneurysm has been shown to be independent of hypercholesterolaemia and hypertension in this model, but requires intact AT1R signalling, innate immunity and MMP activity (ref. 4 and references therein).
The increased expression of monocyte chemoattractant protein-1 (MCP-1), its receptor (CCR2) and interleukin-6 (IL-6) have been demonstrated in models of AngII-induced aneurysm, and CCR2 signalling has been demonstrated to contribute to IL-6 expression. AngII infusion in mice was shown to associate with adventitial accumulation of CCR2+ macrophages specifically at the sites of aneurysm formation and most prominently at the sites of dissection31. Mice lacking CCR2 protein showed reduced macrophage accumulation, decreased IL-6 and MCP-1 expression, and protection from dissection in response to AngII infusion. In vitro modelling has demonstrated that monocytes co-cultured with adventitial fibroblasts upregulate IL-6 and MCP-1, and show enhanced differentiation to macrophages. Although the activity of a fibroblast-derived paracrine factor was suggested, there was no speculation about its identity. The potent anti-inflammatory cytokine TGF-β is a promising candidate. TGF-β is known to induce the expression of both IL-6 and MCP-1 in many cell types, including fibroblasts and VSMCs, and can positively regulate monocyte recruitment and macrophage differentiation4. Increased MCP-1 expression was also proposed as a determinant of disease in response to JNK signalling, with JNK suppressing the expression of LOX that normally negatively regulates MCP-1 (ref. 32). It is notable that although the loss of CCR2 or IL-6 expression prevented early dissection in response to acute AngII infusion in mice, it did not preclude dissection after chronic infusion. Increased adventitial IL-6 expression was observed in human aneurysms, but only at sites of dissection. In this light, it seems that the described IL-6 and MCP-1 amplification loop contributes to, but is not required for, AngII-induced aneurysm and dissection in mice, and more work needs to be done to determine its contribution to disease pathogenesis in humans.
Attempts to integrate TGF-β signalling into the pathogenesis of AngII-induced aneurysm models are frustrated by limited and contradictory empirical knowledge. As previously mentioned, AngII signalling through AT1R has the capacity to enhance TGF-β signalling by inducing the expression of ligands, receptors and activators. It has also been reported that AngII can activate the intracellular SMAD signalling cascade in VSMCs, in a TGF-β-independent manner33 (Fig. 2). AngII can also regulate MAPK signalling cascades independently of TGF-β, with the suggestion that signalling through its different receptor subtypes (AT1R and AT2R) can have varying and even opposing effects (Fig. 2). Conversely, a recent study has shown that the resistance to AngII-induced aneurysm in normocholesterolaemic C57BL/6 mice is disrupted by systemic treatment with a neutralizing anti-TGF-β antibody34. Again, these lesions were inflammatory in nature, and the incidence of aneurysm and dissection was greatly attenuated after monocyte depletion. These observations indicate that TGF-β may be protective specifically in the setting of acute and intense inflammation. Another study has shown that neutralizing anti-TGF-β antibody provided significant protection from AngII-induced inflammatory aneurysms after targeted silencing of CXCL10, a known chemoattractant for monocytes and macrophages35. Taken together, these data suggest that TGF-β has biphasic and discordant roles in the pathogenesis of mouse AngII-induced aneurysm, and that TGF-β antagonism can be protective in a context-dependent manner.
Smooth muscle cytoskeletal elements and aneurysm
Studies with MFS mouse models have shown that phenotypic changes in aortic VSMCs preceded elastolysis and gross medial remodelling22. These included adoption of a general ‘synthetic’ (as opposed to ‘contractile’) character, and morphological changes consistent with cytoskeletal rearrangement. More recent work identifying genes for isolated familial TAA has directly implicated perturbation of the contractile apparatus in the pathogenesis of aneurysm. Heterozygous mutations in MYH11, which encodes myosin heavy chain 11, cause non-syndromic ascending aortic aneurysm in association with patent ductus arteriosis and rare incidence of bicuspid aortic valve36,37. By contrast, heterozygous mutations in ACTA2, which encodes α-smooth muscle actin, cause a vascular disorder involving high, but incomplete, penetrance of ascending aortic aneurysm and dissection, with lower incidence of descending aortic aneurysm and dissection, patent ductus arteriosis and bicuspid aortic valve38. Many patients show a purplish discoloration of the skin in a network pattern caused by altered tone in deep dermal capillaries (livedo reticularis) and pigmented cysts of the iris (iris flocculi). VSMCs isolated from patients with MYH11 mutations showed high proliferative rates and upregulation of insulin-like growth factor 1 (IGF-1) signalling37 and components of the AngII signalling cascade. Although it has been reported that isolated patient cells do not show evidence of increased TGF-β signalling, the data and experimental conditions were not provided. This is particularly important, because the high TGF-β signalling seen in the aorta of humans and/or mice with MFS or LDS is not recapitulated in cultured VSMCs, suggesting the necessity of tissue-specified contexts. More recently, a study found that aortic tissue from a patient with MYH11 mutations showed high VSMC expression of the calcium-binding protein S100A12, an event previously linked to high TGF-β expression and signalling in mice39.
In addition to myosin heavy chain 11 and α-smooth muscle actin, a third cytoskeletal (but non-contractile) protein has been implicated in aortic aneurysm. The large actin-binding protein filamin-A shows altered expression or function in the neurological condition periventricular nodular heterotopia40. Encoded by the FLNA gene on the X chromosome, filamin-A has a diverse repertoire of binding partners, making the delineation of a specific causal pathway difficult. Moreover, only a small subset of filamin-A-deficient women have been reported to show a predisposition for aneurysm, generally in association with other systemic connective-tissue findings. Mutations in FLNA have independently been implicated in myxomatous valvular dystrophy, a phenotype commonly seen in syndromic aneurysm conditions such as MFS and LDS. Filamin-A can function as a positive regulator of TGF-β signalling through modulation of RhoA and SMAD protein trafficking, but also contributes to negative regulation of ERKs41,42 (Fig. 2).
Many aspects of TGF-β signalling have links to the cytoskeleton, prominently including trafficking and activity of TGF-β receptors and signalling effectors (reviewed in ref. 24). Filamin-A can bind to receptor-activated SMAD proteins, including SMAD2, and filamin-A-deficient melanoma cells show impaired TGF-β signalling compared with cells transfected with a filamin-A-encoding vector42. The force generated by cellular contraction against a resistance imposed by the neighbouring matrix has been shown to contribute positively to TGF-β activation in an integrin-dependent manner43. Acute disruption of the actin cytoskeleton in human mesangial cells using cytochalasin D, for example, has been shown to reduce SMAD2 phosphorylation and TGF-β-induced α-1(I) but not α-1(IV) collagen expression44. Other studies have shown that cytoskeletal disruption can induce the expression of TGF-β-driven gene products such as plasminogen activator inhibitor 1 (PAI-1) and connective tissue growth factor in VSMCs in a TGF-β-ligand- and TGF-β-receptor-independent manner, and that these effects are at least partly mediated by activation of the ROCK and/or MAPK signalling cascades45. It remains impossible to determine the chronic consequences of such manipulations in the context of a healthy or diseased tissue.
Prospects for aneurysm treatment
Standard medical therapy for aortic aneurysm has revolved around blood pressure control to limit aortic wall stress. The implication of TGF-β signalling in the pathogenesis of aortic aneurysm suggested an opportunity for more specific therapy. Blockade of AngII signalling through AT1R had previously been shown to limit TGF-β signalling and fibrosis in rodent models of chronic kidney disease. Indeed, in a mouse model of MFS, the AT1R blocker losartan prevented progressive aortic aneurysm7. Potential mechanisms for aneurysm treatment include the prevention of AT1R-induced expression of TGF-β ligands, receptors and activators such as thrombospondin-1 (TSP-1) or MMPs. Prenatal initiation of losartan treatment in MFS mouse models resulted in full normalization of aortic root size, aortic root growth rate and aortic wall architecture. Importantly, postnatal initiation of therapy in the context of established aneurysmal dilatation and medial degeneration also achieved full suppression of aortic root growth and productive remodelling of the aortic wall, with decreased elastin fragmentation and matrix deposition. Several observations pointed to TGF-β antagonism as the relevant mechanism. First, the protection achieved by losartan correlated with reduced nuclear accumulation of pSMAD2 and reduced expression of TGF-β-driven gene products such as plasminogen activator inhibitor-1 (PAI-1) and CTGF. Second, other agents with comparable blood-pressure-lowering effects that did not alter TGF-β signalling were associated with a small decline in aortic root growth rates when compared with losartan, and had no effect on aortic wall architecture. Third, losartan limited the growth of the aortic root, which showed pathological dilatation and increased TGF-β signalling, but had no effect on the growth of other aortic segments, which showed neither. Losartan also limited aortic root growth in a subset of children with severe and rapidly progressive MFS46. Several large and randomized clinical trials of losartan in MFS are under way47.
Embracing paradox
Despite recent advances in understanding aneurysm pathogenesis, many paradoxes remain to be reconciled. For example, it is not clear why defects in structural or regulatory matrix elements, signalling molecules or contractile proteins culminate in focal aneurysms rather than a diffusely fragile and dilated arterial tree. If haemodynamic stress is the answer, it remains unclear why lesions occur on both the high- and low-pressure (that is, the root of the pulmonary artery) sides of the circulation. Another paradox is why conditions such as MFS and LDS show a high predisposition for focal aneurysm and primary dissection in the periductal region of the proximal descending thoracic aorta, In such conditions, it is not known why therapies aimed at blunting TGF-β signalling at the level of ligand bioavailability (such as an anti-TGF-β neutralizing antibody or losartan) or events far downstream of TGF-β (such as MMP inhibition with doxycycline) make things better, whereas events or manipulations that target the intracellular signalling cascade routinely make things worse (such as TGFBR1 or TGFBR2 mutations in LDS or introduction of Smad4 haploinsufficiency in fibrillin-1-deficient mice 26,27). The paradoxical increase in TGF-β signalling observed in patients with LDS could be explained if receptor-mediated activation of the canonical and non-canonical cascades rely on separable activities of the receptor complex. It has been suggested that distinct kinase activities underlie the phosphorylation of SMADs and the adaptor protein ShcA, the proximal event in the ERK cascade48. Furthermore, TGF-β-mediated activation of other MAPKs has been linked to receptor-mediated ubiquitination of the ubiquitin ligase TRAF6 (ref. 49) (Fig. 2). If abnormal receptor complexes in LDS have relative preservation of non-canonical signalling but feedback regulation is disproportionately governed by canonical signalling, then compensatory mechanisms, such as increased ligand expression and/or activation, would drive excessive non-canonical TGF-β signalling in a cell-autonomous manner (Fig. 3a).
Figure 3. TGF-β signalling in heritable aneurysm syndromes.
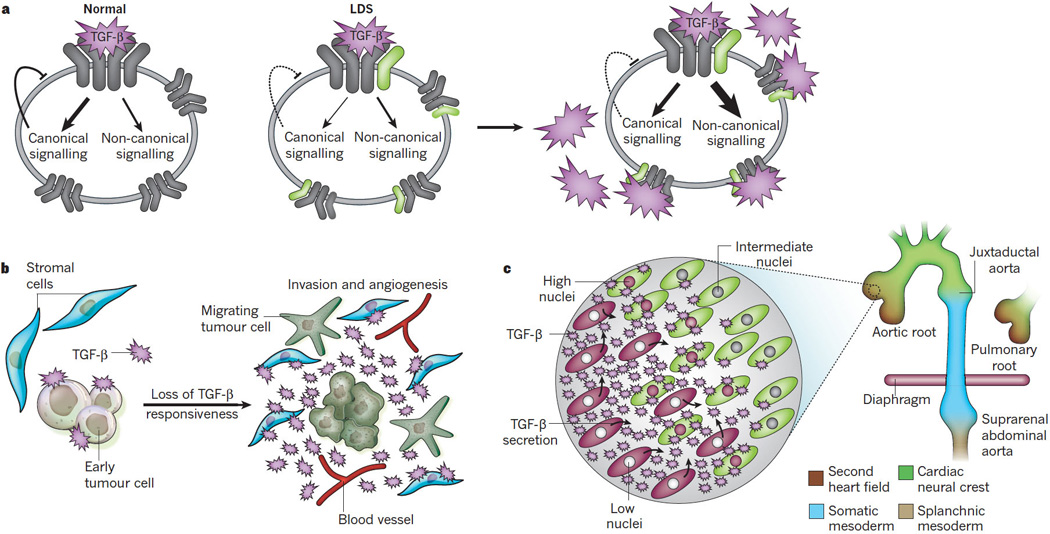
The proposed mechanisms for the amplification of TGF-β signalling in conditions such as LDS are shown. a, Potential cell-autonomous mechanism of upregulation of TGF-β signalling in LDS. TGF-β ligand usually stimulates canonical and non-canonical pathways (left), with canonical signalling providing feedback inhibition. In LDS (right), TGF-β receptor kinase domain mutations (depicted in green) may cause a selective decrease in canonical signalling and thus in feedback inhibition. Cell-autonomous compensation (that is, increased ligand expression and activation) to maintain canonical signalling would result in excessive activation of non-canonical signalling cascades. b, The TGF-β cancer paradox. During the progression of tumorigenesis, tumour cells often lose TGF-β responsiveness as a method of escaping TGF-β-mediated cell-cycle arrest. A lack of feedback inhibition results in upregulation of TGF-β expression by tumour cells and excessive activation of neighbouring signalling-competent stromal cells, which promotes angiogenesis and tumour invasion. c, Potential non-cell-autonomous mechanism of upregulation of TGF-β signalling in vascular disease. Sites of developmental field boundaries correspond anatomically to sites of predisposition for aneurysm in TGF-β vasculopathy syndromes (the aortic and pulmonary roots, the juxtaductal aorta and the suprarenal abdominal aorta). Inset shows cellular events thought to occur at the transition between second heart field (brown)- and cardiac neural crest (green)-derived VSMCs. A relative perturbation of TGF-β signalling would have a disproportionate effect on the more vulnerable lineage (second heart field), resulting in increased ligand expression and excessive TGF-β signalling by adjacent cells of a different lineage (cardiac neural crest) with relative preservation of signalling potential.
Outlook: Cancer biology as a guide to aneurysm?
In many respects, there are intriguing parallels to be drawn to the TGF-β cancer paradox. TGF-β can function as a tumour suppressor, with prominent roles in the maintenance of cellular differentiation and induction of cell-cycle arrest and apoptosis. During early tumorigenesis, many tumour types lose responsiveness to TGF-β through biallelic loss-of-function mutations in genes that encode the TGF-β receptors or intracellular mediators of signalling (ref. 4 and references therein). Attenuation or loss of tumour responsiveness to TGF-β leads to increased signalling by the neighbouring signalling-competent stroma, owing to increased TGF-β ligand expression (Fig. 3b). Consequences include impaired tumour surveillance owing to inhibition of adaptive immunity, acceleration of tumour growth because of enhanced angiogenesis, tumour invasion and metastasis, enhancement of innate immunity (mediated, at least in part, through MCP-1), and/or stimulation of epithelial-to-mesenchymal transition. TGF-β signalling can be further amplified in the tumour microenvironment through enhanced ligand expression by recruited inflammatory cells or enhanced TGF-β activation as a consequence of increased expression of activators such as MMPs. This sequence of events is not simply a function of the cell types per se, but more crucially the interface between cell types with a mismatch in their intrinsic capacity to support TGF-β signalling50.
Could the cancer paradox inform the pathogenesis of aneurysm? The first potential link comes from a consideration of cellular ontology in the vascular system. VSMCs at the base of the aorta and pulmonary artery derive from specialized cardiogenic mesoderm termed the second heart field (Fig. 3c), a finding with possible implications in MFS51. The ascending aorta is a chimaera between cells derived from the second heart field and the ectodermal cardiac neural crest (CNC), whereas the CNC is the sole origin of VSMCs in the more distal ascending aorta and the transverse arch. There is an abrupt transition to somatic mesoderm-derived cells in the proximal descending thoracic (juxtaductal) aorta and a contribution of splanchnic mesoderm to the descending aorta beginning just below the diaphragm52. Thus, although there is not a common origin of VSMCs at frequent sites of predisposition for aneurysm, an interaction between cells of divergent origin exists at each location. Lineage-specific differences in intrinsic TGF-β signalling capacity are addressed in a study comparing the performance of ectoderm-derived cells from the aortic arch and mesoderm-derived cells from the abdominal aorta of chick embryos53. After stimulation with TGF-β1, ectoderm-derived VSMCs showed increased DNA synthesis and robust transcriptional activation of a TGF-β reporter allele, whereas mesoderm-derived VSMCs showed little reporter activation and growth inhibition. TGF-β responsiveness correlated with differences in the glycosylation status of TGFR-2. The authors concluded that different SMC populations within a common vessel wall respond in lineage-dependent ways to growth factors that govern developmental events and that might participate in vascular disease in later life53. It seems plausible that cells of one lineage, such as mesodermal cells in the aortic root and descending thoracic aorta, would be more sensitive to a perturbation of TGF-β signalling (e.g. due to failure to concentrate cytokine in the setting of fibrillin-1 deficiency or altered signalling capacity with heterozygous loss-of-function TGF-β receptor mutations). Loss of TGF-β feedback would initiate compensatory events such as increased TGF-β ligand expression, which could, in turn, stimulate neighbouring cells that can tolerate the primary insult better owing to improved reserves in their inherent signalling capacity (such as CNC-derived VSMCs). Indeed, inspection of the aortic wall from aneurysm tissue obtained at surgery shows an apparent binary status of medial VSMCs for TGF-β signalling (nuclear pSMAD2), with neighbouring cells showing either strong or absent activity (Fig. 4). Such a model not only accommodates but also mandates a mechanism for impaired TGF-β signalling, and would explain how compensatory events could lead to functional overshoot. It would also explain why aneurysms occur at the margins of the CNC developmental field, but not typically in its middle (Figs 1 and 3c). In such a model, manipulations that limit TGF-β ligand bioavailability or block terminal pathogenetic events would be effective at preventing aneurysm, whereas those that accentuate the signalling imbalance would prove detrimental. Despite challenges, the effort to refine mechanistic understanding is justified by the high probability that these insights will derive treatment strategies for aneurysms and perhaps other clinical states associated with impaired vessel wall homeostasis.
Figure 4. TGF-β signalling in LDS aorta.

Immunohistochemical staining for nuclear pSMAD2 (a marker of TGF-β signalling) in the aortic root of a control individual and a patient with LDS. An enlarged view of the LDS aorta is shown on the right. Although TGF-β signalling is markedly increased in LDS, two distinct cell populations are observed in the aortic media, with either absent (blue nuclei) or strongly positive (brown nuclei) activity. The low-power images represent a montage of images taken at 10× magnification to span the entire thickness of the aortic wall. The image on the right is shown at 60× magnification.
Acknowledgments
We would like to thank E. Arbustini for the use of the multidetector computer tomographic image in Fig. 1a. We acknowledge funding support by the National Heart, Lung, and Blood Institute, the Howard Hughes Medical Institute and the National Marfan Foundation and the William S. Smilow Center for Marfan Research.
Footnotes
The authors declare no competing financial interests.
References
- 1.Szabo Z, et al. Aortic aneurysmal disease and cutis laxa caused by defects in the elastin gene. J. Med. Genet. 2006;43:255–258. doi: 10.1136/jmg.2005.034157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Loeys B, et al. Homozygosity for a missense mutation in fibulin-5 (FBLN5) results in a severe form of cutis laxa. Hum. Mol. Genet. 2002;11:2113–2118. doi: 10.1093/hmg/11.18.2113. [DOI] [PubMed] [Google Scholar]
- 3.Dasouki M, et al. Compound heterozygous mutations in fibulin-4 causing neonatal lethal pulmonary artery occlusion, aortic aneurysm, arachnodactyly, and mild cutis laxa. Am. J. Med. Genet. A. 2007;143A:2635–2641. doi: 10.1002/ajmg.a.31980. [DOI] [PubMed] [Google Scholar]
- 4.Dietz HC. TGF-β in the pathogenesis and prevention of disease: a matter of aneurysmic proportions. J. Clin. Invest. 2010;120:403–407. doi: 10.1172/JCI42014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bergen AA, et al. Mutations in ABCC6 cause pseudoxanthoma elasticum. Nature Genet. 2000;25:228–231. doi: 10.1038/76109. [DOI] [PubMed] [Google Scholar]
- 6. Neptune ER, et al. Dysregulation of TGF-β activation contributes to pathogenesis in Marfan syndrome. Nature Genet. 2003;33:407–411. doi: 10.1038/ng1116. This study first implicated upregulation of TGF-β activity in the multisystem manifestations of Marfan syndrome.
- 7.Habashi JP, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312:117–121. doi: 10.1126/science.1124287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loeys BL, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nature Genet. 2005;37:275–281. doi: 10.1038/ng1511. [DOI] [PubMed] [Google Scholar]
- 9.Mizuguchi T, et al. Heterozygous TGFBR2 mutations in Marfan syndrome. Nature Genet. 2004;36:855–860. doi: 10.1038/ng1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Loeys BL, et al. Aneurysm syndromes caused by mutations in the TGF-β receptor. N. Engl. J. Med. 2006;355:788–798. doi: 10.1056/NEJMoa055695. [DOI] [PubMed] [Google Scholar]
- 11.van de Laar IM, et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nature Genet. 2010;43:121–126. doi: 10.1038/ng.744. [DOI] [PubMed] [Google Scholar]
- 12.Coucke PJ, et al. Mutations in the facilitative glucose transporter GLUT10 alter angiogenesis and cause arterial tortuosity syndrome. Nature Genet. 2006;38:452–457. doi: 10.1038/ng1764. [DOI] [PubMed] [Google Scholar]
- 13.Huang J, et al. Fibulin-4 deficiency results in ascending aortic aneurysms: a potential link between abnormal smooth muscle cell phenotype and aneurysm progression. Circ. Res. 2010;106:583–592. doi: 10.1161/CIRCRESAHA.109.207852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Horiguchi M, et al. Fibulin-4 conducts proper elastogenesis via interaction with cross-linking enzyme lysyl oxidase. Proc. Natl Acad. Sci. USA. 2009;106:19029–19034. doi: 10.1073/pnas.0908268106. This study demonstrates fibulin-4-dependent recruitment of lysyl oxidase to elastin.
- 15.Maki JM, et al. Inactivation of the lysyl oxidase gene Lox leads to aortic aneurysms, cardiovascular dysfunction, and perinatal death in mice. Circulation. 2002;106:2503–2509. doi: 10.1161/01.cir.0000038109.84500.1e. [DOI] [PubMed] [Google Scholar]
- 16. Atsawasuwan P, et al. Lysyl oxidase binds transforming growth factor-β and regulates its signaling via amine oxidase activity. J. Biol. Chem. 2008;283:34229–34240. doi: 10.1074/jbc.M803142200. This biochemical analysis demonstrates direct inactivation of TGF-β ligand by the enzymatic activity of lysyl oxidase.
- 17.Zacchigna L, et al. Emilin1 links TGF-β maturation to blood pressure homeostasis. Cell. 2006;124:929–942. doi: 10.1016/j.cell.2005.12.035. [DOI] [PubMed] [Google Scholar]
- 18.Choudhary B, et al. Absence of TGF-β signaling in embryonic vascular smooth muscle leads to reduced lysyl oxidase expression, impaired elastogenesis, and aneurysm. Genesis. 2009;47:115–121. doi: 10.1002/dvg.20466. [DOI] [PubMed] [Google Scholar]
- 19.Langlois D, et al. Conditional inactivation of TGF-β type II receptor in smooth muscle cells and epicardium causes lethal aortic and cardiac defects. Transgenic Res. 2010;19:1069–1082. doi: 10.1007/s11248-010-9379-4. [DOI] [PubMed] [Google Scholar]
- 20. Arteaga-Solis E, et al. Regulation of limb patterning by extracellular microfibrils. J. Cell Biol. 2001;154:275–281. doi: 10.1083/jcb.200105046. This study demonstrates that fibrillins can act as positive modulators of TGF-β superfamily function.
- 21.Chung AW, Yang HH, Radomski MW, van Breemen C. Long-term doxycycline is more effective than atenolol to prevent thoracic aortic aneurysm in Marfan syndrome through the inhibition of matrix metalloproteinase-2 and -9. Circ. Res. 2008;102:e73–e85. doi: 10.1161/CIRCRESAHA.108.174367. [DOI] [PubMed] [Google Scholar]
- 22.Bunton TE, et al. Phenotypic alteration of vascular smooth muscle cells precedes elastolysis in a mouse model of Marfan syndrome. Circ. Res. 2001;88:37–43. doi: 10.1161/01.res.88.1.37. [DOI] [PubMed] [Google Scholar]
- 23.Sakalihasan N, Delvenne P, Nusgens BV, Limet R, Lapière CM. Activated forms of MMP2 and MMP9 in abdominal aortic aneurysms. J. Vasc. Surg. 1996;24:127–133. doi: 10.1016/s0741-5214(96)70153-2. [DOI] [PubMed] [Google Scholar]
- 24.Moustakas A, Heldin C-H. The regulation of TGFβ signal transduction. Development. 2009;136:3699–3714. doi: 10.1242/dev.030338. [DOI] [PubMed] [Google Scholar]
- 25.Carta L, et al. p38 MAPK is an early determinant of promiscuous Smad2/3 signaling in the aortas of fibrillin-1 (Fbn1)-null mice. J. Biol. Chem. 2009;284:5630–5636. doi: 10.1074/jbc.M806962200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Holm T, et al. Noncanonical TGF-β Signaling Contributes to Aortic Aneurysm Progression in Marfan Syndrome Mice. Science. doi: 10.1126/science.1192149. (in Press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Habashi JP, et al. Angiotensin II Type 2 Receptor Signaling Attenuates Aortic Aneurysm in Mice through ERK Antagonism. Science. doi: 10.1126/science.1192152. (in Press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoshimura K, et al. Regression of abdominal aortic aneurysm by inhibition of c-Jun N-terminal kinase. Nature Med. 2005;11:1330–1338. doi: 10.1038/nm1335. [DOI] [PubMed] [Google Scholar]
- 29.Purnell R, Williams I, Von Oppell U, Wood A. Giant aneurysms of the sinuses of Valsalva and aortic regurgitation in a patient with Noonan's syndrome. Eur. J. Cardiothorac. Surg. 2005;28:346–348. doi: 10.1016/j.ejcts.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 30.Friedman JM, et al. Cardiovascular disease in neurofibromatosis 1: report of the NF1 Cardiovascular Task Force. Genet. Med. 2002;4:105–111. doi: 10.1097/00125817-200205000-00002. [DOI] [PubMed] [Google Scholar]
- 31. Tieu BC, et al. An adventitial IL-6/MCP1 amplification loop accelerates macrophage-mediated vascular inflammation leading to aortic dissection in mice. J. Clin. Invest. 2009;119:3637–3651. doi: 10.1172/JCI38308. This paper describes a major role for adventitial macrophage in aortic dissection in mice.
- 32.Onoda M, et al. Lysyl oxidase resolves inflammation by reducing monocyte chemoattractant protein-1 in abdominal aortic aneurysm. Atherosclerosis. 2010;208:366–369. doi: 10.1016/j.atherosclerosis.2009.07.036. [DOI] [PubMed] [Google Scholar]
- 33.Rodríguez-Vita J, et al. Angiotensin II activates the Smad pathway in vascular smooth muscle cells by a transforming growth factor-β-independent mechanism. Circulation. 2005;111:2509–2517. doi: 10.1161/01.CIR.0000165133.84978.E2. [DOI] [PubMed] [Google Scholar]
- 34.Wang Y, et al. TGF-β activity protects against inflammatory aortic aneurysm progression and complications in angiotensin II-infused mice. J. Clin. Invest. 2010;120:422–432. doi: 10.1172/JCI38136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.King VL, et al. Interferon-γ and the interferon-inducible chemokine CXCL10 protect against aneurysm formation and rupture. Circulation. 2009;119:426–435. doi: 10.1161/CIRCULATIONAHA.108.785949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhu L, et al. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nature Genet. 2006;38:343–349. doi: 10.1038/ng1721. This was the first study to implicate the smooth muscle contraction apparatus in thoracic aortic aneurysm.
- 37.Pannu H, et al. MYH11 mutations result in a distinct vascular pathology driven by insulin-like growth factor 1 and angiotensin II. Hum. Mol. Genet. 2007;16:2453–2462. doi: 10.1093/hmg/ddm201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo D-C, et al. Mutations in smooth muscle α-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nature Genet. 2007;39:1488–1493. doi: 10.1038/ng.2007.6. [DOI] [PubMed] [Google Scholar]
- 39.Hofmann Bowman M, et al. S100A12 mediates aortic wall remodeling and aortic aneurysm. Circ. Res. 2010;106:145–154. doi: 10.1161/CIRCRESAHA.109.209486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sheen VL, et al. Filamin A mutations cause periventricular heterotopia with Ehlers-Danlos syndrome. Neurology. 2005;64:254–262. doi: 10.1212/01.WNL.0000149512.79621.DF. [DOI] [PubMed] [Google Scholar]
- 41.Zhu T-N, et al. Filamin A-mediated down-regulation of the exchange factor Ras-GRF1 correlates with decreased matrix metalloproteinase-9 expression in human melanoma cells. J. Biol. Chem. 2007;282:14816–14826. doi: 10.1074/jbc.M611430200. [DOI] [PubMed] [Google Scholar]
- 42.Sasaki A, Masuda Y, Ohta Y, Ikeda K, Watanabe K. Filamin associates with Smads and regulates transforming growth factor-β signaling. J. Biol. Chem. 2001;276:17871–17877. doi: 10.1074/jbc.M008422200. [DOI] [PubMed] [Google Scholar]
- 43.Wipff P-J, Rifkin DB, Meister J-J, Hinz B. Myofibroblast contraction activates latent TGF-β1 from the extracellular matrix. J. Cell Biol. 2007;179:1311–1323. doi: 10.1083/jcb.200704042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hubchak SC, Runyan CE, Kreisberg JI, Schnaper HW. Cytoskeletal rearrangement and signal transduction in TGF-β1-stimulated mesangial cell collagen accumulation. J. Am. Soc. Nephrol. 2003;14:1969–1980. doi: 10.1097/01.asn.0000076079.02452.92. [DOI] [PubMed] [Google Scholar]
- 45.Samarakoon R, Higgins CE, Higgins SP, Higgins PJ. Differential requirement for MEK/ERK and SMAD signaling in PAI-1 and CTGF expression in response to microtubule disruption. Cell Signal. 2009;21:986–995. doi: 10.1016/j.cellsig.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brooke BS, et al. Angiotensin II blockade and aortic-root dilation in Marfan's syndrome. N. Engl. J. Med. 2008;358:2787–2795. doi: 10.1056/NEJMoa0706585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lacro RV, et al. Rationale and design of a randomized clinical trial of β-blocker therapy (atenolol) versus angiotensin II receptor blocker therapy (losartan) in individuals with Marfan syndrome. Am. Heart J. 2007;154:624–631. doi: 10.1016/j.ahj.2007.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee MK, et al. TGF-β activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 2007;26:3957–3967. doi: 10.1038/sj.emboj.7601818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yamashita M, et al. TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-β. Mol Cell. 2008;31:918–924. doi: 10.1016/j.molcel.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bachman KE, Park BH. Duel nature of TGF-β signaling: tumor suppressor vs. tumor promoter. Curr. Opin. Oncol. 2005;17:49–54. doi: 10.1097/01.cco.0000143682.45316.ae. [DOI] [PubMed] [Google Scholar]
- 51.Waldo KL, et al. Secondary heart field contributes myocardium and smooth muscle to the arterial pole of the developing heart. Dev. Biol. 2005;281:78–90. doi: 10.1016/j.ydbio.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 52.Majesky MW. Developmental basis of vascular smooth muscle diversity. Arterioscler. Thromb. Vasc. Biol. 2007;27:1248–1258. doi: 10.1161/ATVBAHA.107.141069. [DOI] [PubMed] [Google Scholar]
- 53. Topouzis S, Majesky MW. Smooth muscle lineage diversity in the chick embryo: two types of aortic smooth muscle cell differ in growth and receptor-mediated transcriptional responses to transforming growth factor-β. Dev. Biol. 1996;178:430–445. doi: 10.1006/dbio.1996.0229. This study first noted divergent TGF-β responsiveness in different aortic regions.
- 54.Carta L, et al. Fibrillins 1 and 2 perform partially overlapping functions during aortic development. J. Biol. Chem. 2006;281:8016–8023. doi: 10.1074/jbc.M511599200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dietz HC, et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352:337–339. doi: 10.1038/352337a0. [DOI] [PubMed] [Google Scholar]
- 56.Li DY, et al. Elastin is an essential determinant of arterial morphogenesis. Nature. 1998;393:276–280. doi: 10.1038/30522. [DOI] [PubMed] [Google Scholar]
- 57.Rahkonen O, et al. Mice with a deletion in the first intron of the Col1a1 gene develop age-dependent aortic dissection and rupture. Circ. Res. 2004;94:83–90. doi: 10.1161/01.RES.0000108263.74520.15. [DOI] [PubMed] [Google Scholar]
- 58.Malfait F, et al. Three arginine to cysteine substitutions in the pro-alpha (I)-collagen chain cause Ehlers-Danlos syndrome with a propensity to arterial rupture in early adulthood. Hum. Mutat. 2007;28:387–395. doi: 10.1002/humu.20455. [DOI] [PubMed] [Google Scholar]
- 59.Schwarze U, et al. Rare autosomal recessive cardiac valvular form of Ehlers-Danlos syndrome results from mutations in the COL1A2 gene that activate the nonsense-mediated RNA decay pathway. Am. J. Hum. Genet. 2004;74:917–930. doi: 10.1086/420794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu X, Wu H, Byrne M, Krane S, Jaenisch R. Type III collagen is crucial for collagen I fibrillogenesis and for normal cardiovascular development. Proc. Natl Acad. Sci. USA. 1997;94:1852–1856. doi: 10.1073/pnas.94.5.1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Superti-Furga A, Gugler E, Gitzelmann R, Steinmann B. Ehlers-Danlos syndrome type IV: a multi-exon deletion in one of the two COL3A1 alleles affecting structure, stability, and processing of type III procollagen. J. Biol. Chem. 1988;263:6226–6232. This was the first genetic study linking a single-gene disorder to aneurysmal disease.
- 62.Plaisier E, et al. COL4A1 mutations and hereditary angiopathy, nephropathy, aneurysms, and muscle cramps. N. Engl. J. Med. 2007;357:2687–2695. doi: 10.1056/NEJMoa071906. [DOI] [PubMed] [Google Scholar]
- 63.Pöschl E, et al. Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development. 2004;131:1619–1628. doi: 10.1242/dev.01037. [DOI] [PubMed] [Google Scholar]
- 64.Kashtan CE, et al. Aortic abnormalities in males with Alport syndrome. Nephrol. Dial. Transplant. 2010;25:3554–3560. doi: 10.1093/ndt/gfq271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wenstrup RJ, Murad S, Pinnell SR. Ehlers-Danlos syndrome type VI: clinical manifestations of collagen lysyl hydroxylase deficiency. J. Pediatr. 1989;115:405–409. doi: 10.1016/s0022-3476(89)80839-x. [DOI] [PubMed] [Google Scholar]
- 66.Takaluoma K, et al. Tissue-specific changes in the hydroxylysine content and cross-links of collagens and alterations in fibril morphology in lysyl hydroxylase 1 knock-out mice. J. Biol. Chem. 2007;282:6588–6596. doi: 10.1074/jbc.M608830200. [DOI] [PubMed] [Google Scholar]
- 67.Salo AM, et al. A connective tissue disorder caused by mutations of the lysyl hydroxylase 3 gene. Am. J. Hum. Genet. 2008;83:495–503. doi: 10.1016/j.ajhg.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ruotsalainen H, et al. Glycosylation catalyzed by lysyl hydroxylase 3 is essential for basement membranes. J. Cell Sci. 2006;119:625–635. doi: 10.1242/jcs.02780. [DOI] [PubMed] [Google Scholar]
- 69.Larsson J, et al. Abnormal angiogenesis but intact hematopoietic potential in TGF-β type I receptor-deficient mice. EMBO J. 2001;20:1663–1673. doi: 10.1093/emboj/20.7.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Oshima M, Oshima H, Taketo MM. TGF-β receptor type II deficiency results in defects of yolk sac hematopoiesis and vasculogenesis. Dev. Biol. 1996;179:297–302. doi: 10.1006/dbio.1996.0259. [DOI] [PubMed] [Google Scholar]
- 71.Hsi DH, Ryan GF, Hellems SO, Cheeran DC, Sheils LA. Large aneurysms of the ascending aorta and major coronary arteries in a patient with hereditary hemorrhagic telangiectasia. Mayo Clin. Proc. 2003;78:774–776. doi: 10.4065/78.6.774. [DOI] [PubMed] [Google Scholar]
- 72.Arthur HM, et al. Endoglin, an ancillary TGFβ receptor, is required for extraembryonic angiogenesis and plays a key role in heart development. Dev. Biol. 2000;217:42–53. doi: 10.1006/dbio.1999.9534. [DOI] [PubMed] [Google Scholar]
- 73.Andersen ND, et al. Thoracic endografting in a patient with hereditary hemorrhagic telangiectasia presenting with a descending thoracic aneurysm. J. Vasc. Surg. 2010;51:468–470. doi: 10.1016/j.jvs.2009.08.058. [DOI] [PubMed] [Google Scholar]
- 74.Oh SP, et al. Activin receptor-like kinase 1 modulates transforming growth factor-β 1 signaling in the regulation of angiogenesis. Proc. Natl Acad. Sci. USA. 2000;97:2626–2631. doi: 10.1073/pnas.97.6.2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cheng C-H, et al. Mutations in the SLC2A10 gene cause arterial abnormalities in mice. Cardiovasc. Res. 2009;81:381–388. doi: 10.1093/cvr/cvn319. [DOI] [PubMed] [Google Scholar]
- 76.Garg V, et al. Mutations in NOTCH1 cause aortic valve disease. Nature. 2005;437:270–274. doi: 10.1038/nature03940. [DOI] [PubMed] [Google Scholar]
- 77.Conlon RA, Reaume AG, Rossant J. Notch1 is required for the coordinate segmentation of somites. Development. 1995;121:1533–1545. doi: 10.1242/dev.121.5.1533. [DOI] [PubMed] [Google Scholar]
- 78.Xue Y, et al. Embryonic lethality and vascular defects in mice lacking the Notch ligand Jagged1. Hum. Mol. Genet. 1999;8:723–730. doi: 10.1093/hmg/8.5.723. [DOI] [PubMed] [Google Scholar]
- 79.Kamath BM, et al. Vascular anomalies in Alagille syndrome: a significant cause of morbidity and mortality. Circulation. 2004;109:1354–1358. doi: 10.1161/01.CIR.0000121361.01862.A4. [DOI] [PubMed] [Google Scholar]
- 80.Distefano G, et al. Polycystin-1 regulates extracellular signal-regulated kinase-dependent phosphorylation of tuberin to control cell size through mTOR and its downstream effectors S6K and 4EBP1. Mol. Cell. Biol. 2009;29:2359–2371. doi: 10.1128/MCB.01259-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hassane S, et al. Pathogenic sequence for dissecting aneurysm formation in a hypomorphic polycystic kidney disease 1 mouse model. Arterioscler. Thromb. Vasc Biol. 2007;27:2177–2183. doi: 10.1161/ATVBAHA.107.149252. [DOI] [PubMed] [Google Scholar]
- 82.Wu G, et al. Somatic inactivation of Pkd2 results in polycystic kidney disease. Cell. 1998;93:177–188. doi: 10.1016/s0092-8674(00)81570-6. [DOI] [PubMed] [Google Scholar]
- 83.Yu Q, et al. ENU induced mutations causing congenital cardiovascular anomalies. Development. 2004;131:6211–6223. doi: 10.1242/dev.01543. [DOI] [PubMed] [Google Scholar]
- 84.Schildmeyer LA, et al. Impaired vascular contractility and blood pressure homeostasis in the smooth muscle α-actin null mouse. FASEB J. 2000;14:2213–2220. doi: 10.1096/fj.99-0927com. [DOI] [PubMed] [Google Scholar]
- 85.Morano I, et al. Smooth-muscle contraction without smooth-muscle myosin. Nature Cell Biol. 2000;2:371–375. doi: 10.1038/35014065. [DOI] [PubMed] [Google Scholar]
- 86.Feng Y, et al. Filamin A (FLNA) is required for cell-cell contact in vascular development and cardiac morphogenesis. Proc. Natl Acad. Sci. USA. 2006;103:19836–19841. doi: 10.1073/pnas.0609628104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Araki T, et al. Mouse model of Noonan syndrome reveals cell type- and gene dosage-dependent effects of Ptpn11 mutation. Nature Med. 2004;10:849–857. doi: 10.1038/nm1084. [DOI] [PubMed] [Google Scholar]
- 88.Iwasaki Y, et al. Coronary artery dilatation in LEOPARD syndrome. A child case and literature review. Congenit. Heart Dis. 2009;4:38–41. doi: 10.1111/j.1747-0803.2008.00243.x. [DOI] [PubMed] [Google Scholar]
- 89.Takahashi H, et al. A hereditary model of slowly progressive polycystic kidney disease in the mouse. J. Am. Soc. Nephrol. 1991;1:980–989. doi: 10.1681/ASN.V17980. [DOI] [PubMed] [Google Scholar]
- 90.Lee TC, Zhao YD, Courtman DW, Stewart DJ. Abnormal aortic valve development in mice lacking endothelial nitric oxide synthase. Circulation. 2000;101:2345–2348. doi: 10.1161/01.cir.101.20.2345. [DOI] [PubMed] [Google Scholar]
- 91.Kuhlencordt PJ, et al. Accelerated atherosclerosis, aortic aneurysm formation, and ischemic heart disease in apolipoprotein E/endothelial nitric oxide synthase double-knockout mice. Circulation. 2001;104:448–454. doi: 10.1161/hc2901.091399. [DOI] [PubMed] [Google Scholar]
- 92.Cao J, et al. Thoracic aortic disease in tuberous sclerosis complex: molecular pathogenesis and potential therapies in Tsc2+/− mice. Hum. Mol. Genet. 2010;19:1908–1920. doi: 10.1093/hmg/ddq066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Raben N, et al. Targeted disruption of the acid α-glucosidase gene in mice causes an illness with critical features of both infantile and adult human glycogen storage disease type II. J. Biol. Chem. 1998;273:19086–19092. doi: 10.1074/jbc.273.30.19086. [DOI] [PubMed] [Google Scholar]
- 94.Rocha PP, Scholze M, Bleiss W, Schrewe H. Med12 is essential for early mouse development and for canonical Wnt and Wnt/PCP signaling. Development. 2010;137:2723–2731. doi: 10.1242/dev.053660. [DOI] [PubMed] [Google Scholar]
- 95.Haldar SM, et al. Klf15 deficiency is a molecular link between heart failure and aortic aneurysm formation. Sci. Transl. Med. 2010;2:26ra26. doi: 10.1126/scitranslmed.3000502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kuo CT, et al. The LKLF transcription factor is required for normal tunica media formation and blood vessel stabilization during murine embryogenesis. Genes Dev. 1997;11:2996–3006. doi: 10.1101/gad.11.22.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lopez L, et al. Turner syndrome is an independent risk factor for aortic dilation in the young. Pediatrics. 2008;121:e1622–e1627. doi: 10.1542/peds.2007-2807. [DOI] [PubMed] [Google Scholar]
- 98. Daugherty A, Manning MW, Cassis LA. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J. Clin. Invest. 2000;105:1605–1612. doi: 10.1172/JCI7818. This study formed the basis for the AngII-infusion model of aneurysmal disease, the most widespread mouse model of aneurysm.
- 99.Anidjar S, et al. Elastase-induced experimental aneurysms in rats. Circulation. 1990;82:973–981. doi: 10.1161/01.cir.82.3.973. [DOI] [PubMed] [Google Scholar]
- 100.Chiou AC, Chiu B, Pearce WH. Murine aortic aneurysm produced by periarterial application of calcium chloride. J. Surg. Res. 2001;99:371–376. doi: 10.1006/jsre.2001.6207. [DOI] [PubMed] [Google Scholar]