

ABSTRACT
Chlamydia psittaci is an obligate intracellular bacterium. Interest in Chlamydia stems from its high degree of virulence as an intestinal and pulmonary pathogen across a broad range of animals, including humans. C. psittaci human pulmonary infections, referred to as psittacosis, can be life-threatening, which is why the organism was developed as a bioweapon in the 20th century and is listed as a CDC biothreat agent. One remarkable recent result from comparative genomics is the finding of frequent homologous recombination across the genome of the sexually transmitted and trachoma pathogen Chlamydia trachomatis. We sought to determine if similar evolutionary dynamics occurred in C. psittaci. We analyzed 20 C. psittaci genomes from diverse strains representing the nine known serotypes of the organism as well as infections in a range of birds and mammals, including humans. Genome annotation revealed a core genome in all strains of 911 genes. Our analyses showed that C. psittaci has a history of frequently switching hosts and undergoing recombination more often than C. trachomatis. Evolutionary history reconstructions showed genome-wide homologous recombination and evidence of whole-plasmid exchange. Tracking the origins of recombinant segments revealed that some strains have imported DNA from as-yet-unsampled or -unsequenced C. psittaci lineages or other Chlamydiaceae species. Three ancestral populations of C. psittaci were predicted, explaining the current population structure. Molecular clock analysis found that certain strains are part of a clonal epidemic expansion likely introduced into North America by South American bird traders, suggesting that psittacosis is a recently emerged disease originating in New World parrots.
IMPORTANCE
Chlamydia psittaci is classified as a CDC biothreat agent based on its association with life-threatening lung disease, termed psittacosis, in humans. Because of the recent remarkable findings of frequent recombination across the genome of the human sexually transmitted and ocular trachoma pathogen Chlamydia trachomatis, we sought to determine if similar evolutionary dynamics occur in C. psittaci. Twenty C. psittaci genomes were analyzed from diverse strains that may play a pathogenic role in human disease. Evolution of the strains revealed genome-wide recombination occurring at a higher rate than for C. trachomatis. Certain strains were discovered to be part of a recent epidemic clonal expansion originating in South America. These strains may have been introduced into the United States from South American bird traders, suggesting that psittacosis is a recently emerged disease originating in New World parrots. Our analyses indicate that C. psittaci strains have a history of frequently switching hosts and undergoing recombination.
Introduction
The obligate intracellular bacterial family Chlamydiaceae is comprised of the genus Chlamydia, which includes nine species (1). Chlamydiaceae are globally important because they are responsible for a diversity of severe and debilitating diseases in livestock and humans. Collectively, these diseases impose a huge economic burden.
Chlamydiaceae are ancient pathogens that date back 50 to 250 million years ago (2, 3). They have spread throughout the animal kingdom, yet continue to give rise to an array of chlamydial species and strains that infect various avian and mammalian hosts. Chlamydiaceae appear to regularly jump host species but have maintained a small core genome for many millions of years (4). Other groups of intracellular bacteria either show evidence of recent emergence from more generalist lineages (5, 6) with ongoing genome reduction and frequent horizontal gene transfer or, at the other extreme, exist as slowly decaying endosymbionts (e.g., Buchnera spp. [7]). Somehow, Chlamydiaceae have avoided both of these paths. Most genomes sequenced to date have conserved gene order (synteny) over a very long time, with few pseudogenes and a general absence of disruptive mobile elements, except for the transposable tetracycline element identified in Chlamydia suis strains (8). Yet there is evidence of gene reduction, positive selection, and recombination (3, 9–15). It appears that speciation and strain emergence can be mediated by horizontal gene transfer or niche-specific genes that circulate among Chlamydiaceae (3, 8, 14, 16).
Chlamydia psittaci can be a particularly virulent pathogen in feral birds and domesticated poultry as well as cattle, pigs, sheep, and horses (17–21), where the pathogen is responsible for ocular, respiratory, intestinal, and arthritic diseases as well as abortion. In these hosts, clinical presentations and outcomes, however, range from asymptomatic colonization to death. This bacterium is also the causative agent of mild to life-threatening pulmonary and systemic diseases in humans (22–30). Humans are customarily thought of as an uncommon secondary host, although a recent study found widespread psittacosis among poultry farmers (27), suggesting that zoonotic infections had previously been greatly underestimated. A lack of epidemiological surveys and inadequate diagnostics may contribute to this underestimation.
Because of its infectiousness as an aerosol agent, C. psittaci was developed as a bioweapon by the United States during the first half of the 20th century, resulting in its subsequent classification as a biothreat agent by the Centers for Disease Control and Prevention (CDC). Virulent outbreak strains from San Francisco, CA (31), and Louisiana (32), where mortality rates were 42 to 63% in humans were studied as part of development efforts. The bacterium is also believed to have been weaponized by the Soviet Union’s offensive program (33).
Despite being responsible as an important global infectious disease and being a CDC class B select agent, C. psittaci has been relatively understudied. The diversity of hosts infected by C. psittaci and the range of diseases, even within the same host species, suggest that pathogenesis is linked to host-pathogen interactions that may in part be defined by variations in C. psittaci genomics. In order to better understand the virulence and host specificity of these strains, we expanded on recent genomics projects (3, 34), characterizing 20 diverse avian and mammalian strains representing the nine known serological variants of C. psittaci.
RESULTS
Whole-genome phylogeny reveals recent worldwide expansion of a pathogenic C. psittaci clone.
We analyzed 20 C. psittaci genomes (Table 1), including eight previously published strains (35–38) and 12 strains that we sequenced in this study using Roche 454 pyrosequencing technology. The 12 strains were chosen based on serotype diversity and distribution among different animal hosts, including humans. The redundancy of sequence coverage was 10- to 34-fold. All strains contained an approximate 1.1-Mb genome and a conserved plasmid of ~8 kb, except for strain GR9(GD), in which the plasmid was absent.
TABLE 1.
C. psittaci strain characteristics and accession numbers
| Strain | Serologic variant |
Source | Associated disease, event, or source |
Year of isolation/ country |
RefSeq ID/ SRA Seq ID |
No. of protein- coding genes |
No. of genes >90 nt in length |
Coverage (fold)a |
|---|---|---|---|---|---|---|---|---|
| 8DC60 | Human | Psittacosis | 2008/Germany | NC_017290.1 | 1,043 | 924 | NA | |
| RD1 | Mixed culture with C. trachomatis strain L2b |
Possible lab contamination |
2010/United Kingdom |
FQ482149.1 | 1,041 | 915 | NA | |
| 6BC/30 | A | Parrot | Ornithosis | 1930/Maryland, USA | NC_017287.1 | 1,037 | 920 | NA |
| 6BC/41 | A | Parakeet | Ornithosis | 1941/CA | NC_015470.1 | 1,031 | 915 | NA |
| C19/98 | Sheep | Afterbirth | 1998/Germany | NC_017291.1 | 1,033 | 916 | NA | |
| Cal10 | Ferret | Meningopneumonitis | 1934/USA | AEZD00000000 | 1,051 | 924 | NA | |
| DC15 | Cow | Abortion | 2002/Germany | NC_017292.1 | 1,044 | 919 | NA | |
| DC11 | Pig | Conjunctivitis | 2001/Germany | NC_017289.1 | 1,043 | 918 | NA | |
| CP3 | B | Pigeon | Systemic | 1958/CA | SRA061582 | 1,058 | 930 | 20 |
| 6BC/83 | A | Parakeet | Ornithosis | 1983/CA | SRA061584 | 1,297 | 1,055 | 19 |
| GR9(GD) | C | Duck | Systemic | 1960/Germany | SRA061587 | 1,052 | 932 | 25 |
| CT1 | C | Turkey (acquired from sparrows) |
Systemic | 1954/CA | SRA061680 | 1,063 | 920 | 18 |
| WC | WC | Cow | Hemorrhagic enteritis |
1963/CA | SRA061579 | 1,237 | 1,023 | 14 |
| VS225 | F | Orange-fronted parakeet |
Systemic | 1991/Tex. | SRA061577 | 1,053 | 926 | 25 |
| FalTex | D | Turkey | Systemic | 1980/Tex. | SRA061585 | 1,332 | 1,046 | 28 |
| NJ1 | D | Turkey | Systemic | 1954/NJ | SRA061578 | 1,364 | 1,064 | 21 |
| Borg | D | Human lung | Psittacosis | 1944/LA | SRA061576 | 1,159 | 984 | 34 |
| MN | E | Human throat washings |
Influenza-like illness |
1936/CA | SRA061581 | 1,386 | 1,086 | 11 |
| M56 | M56 | Muskrat | Systemic | 1961/Canada | SRA061583 | 1,200 | 989 | 11 |
| RTH | G | Red-tailed hawk | Systemic | 2003/CA | SRA061571 | 1,306 | 1,038 | 16 |
NA, not available because these genomes were not generated in this study.
A total of 911 core genes were found to be present among all 20 strains, about 90% of the number of genes present in each genome. This high proportion of core genes, typical for a Chlamydiaceae genome (4), was likely to be slightly underestimated because whole-genome shotgun assemblies constructed using 454 sequencing technology are known to contain a low rate of false-positive frameshifts into genes due to aberrant homopolymer base calling (for example, see the article by Zwick et al. [39]). The whole-genome phylogeny was inferred from both the concatenated core proteome and the whole-genome alignment (Fig. 1; also, see Fig. S1 in the supplemental material). It showed that 10 strains formed a clade, referred to as the 6BC Clade 1, with very little genetic diversity, suggesting recent epidemic spread. Of the 32,484 variant positions in the core genome region (1,114,643 bp), only 482 were polymorphic within this clade. The 6BC Clade 1, which includes the canonical laboratory strain 6BC/30 originating in 1930 from a parrot (35), contains strains isolated from three avian species (pigeon, parrot, and parakeet) and five mammalian species (ferret, pig, cow, sheep, and human) corresponding to serotypes A and B (Table 1). The strains MN, WC, and VS225 (serotypes E, WC, and F, respectively) are the most closely related to the 6BC Clade but do not form a distinct clade themselves. We identified two other clades (Fig. 1). Clade 2 contains strains FalTex, Borg, and NJ1, which are all serotype D and derived from avian hosts. Clade 3 comprises strains GR9 and CT1, which are both serotype C and from avian origins. In addition, strains M56, isolated from a muskrat, and RTH, isolated from a red-tailed hawk, were particularly diverged from the rest of the strains.
FIG 1 .
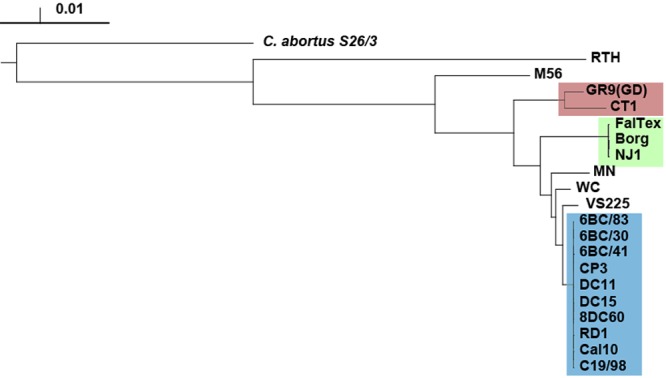
Whole-genome phylogeny of C. psittaci. The tree was constructed using a neighbor-joining algorithm based on whole-genome alignment (see Materials and Methods). The 6BC Clade 1 is highlighted in blue, and Clades 2 and 3 are in green and red, respectively. All branches were supported by 100% confidence in 100 bootstrap samplings except for some branches internal to the 6BC Clade (not shown) and the Borg/NJ1/FalTex branch. The scale is in substitutions per position.
We used the BEAST (40) and ClonalFrame (41) programs to infer the dated evolutionary history of C. psittaci. Both programs (see Fig. S2A and B in the supplemental material) yielded the same tree topology as the neighbor-joining analysis, although there were some differences between the two methods regarding the estimates of nucleotide substitution rates and divergence times.
The reconstructions suggested a high rate of evolution in C. psittaci. BEAST estimated a mean rate of 1.682 × 10−4 substitution per year per site, with a 95% credibility interval ranging from 2.97 × 10−5 to 2.805 × 10−4 and an estimated date for the most recent common ancestor (MRCA) of 1770. ClonalFrame estimated the mutation rate to be 1.74 × 10−5 per site per year, with a 95% credibility interval ranging from 1.71 × 10−5 to 1.76 × 10−5, and dated the MRCA at 1521 with a 95% credibility interval ranging from 1514 to 1527. The date of the MRCA of the 6BC Clade 1 was 1916 according to BEAST and 1930 according to ClonalFrame. ClonalFrame differs from BEAST in the fact that recombination events are identified, and the dating is based only on the remaining mutation events. In contrast, the BEAST method estimated a substitution rate, which could be caused by either mutation or recombination, and this explains why it is higher than the mutation rate estimated by ClonalFrame. C. psittaci had a substitution rate greater than that previously reported for other bacterial species, including Staphylococcus aureus (3 × 10−6) (42), Streptococcus pneumoniae (1.57 × 10−6) (43), and Vibrio cholerae (8 × 10−7) (44), but similar to the estimated mutation rate of Helicobacter pylori (1.9 × 10−5) (45).
It should be noted that the strains in our data set have been cultivated in vitro for an unknown number of passages. While there are few studies that address selection and drift in in vitro culture systems, Labiran et al. (46) found no genetic changes after 8 to 72 passages for nine gene loci in Chlamydia trachomatis, although whole-genome sequencing was not performed. Similar studies for C. psittaci have not been undertaken, although we think it unlikely that in vitro systems have introduced relaxed selection and drift.
The plasticity zone (PZ) of the C. psittaci chromosome.
The 20 C. psittaci genomes were very similar in gene content, with very few significant insertions and deletions (indels) of DNA that distinguished strains (see Fig. S1 in the supplemental material). The exception to the otherwise very similar genomes is the PZ, a region near the predicted replication termination region on the main chromosome that is a nexus for indels of genes in Chlamydiaceae (9) (Fig. 2). The 6BC Clade 1 and the three closest relatives, strains MN, WC, and VS225, and the FalTex/Borg/NJ1 Clade 2, had an intact large toxin/adhesin (tox) gene as well as a three-gene guaAB-add cluster, encoding IMP dehydrogenase, guanosine monophosphate synthase, and adenine deaminase, respectively. However, the remaining four C. psittaci strains (Clade 3 strains GC6 and CT1 and strains M56 and RTH) had degraded or missing guaAB-add clusters, and had multiple deletions and frameshifts in the tox gene. The most closely related species, Chlamydia abortus, had missing or degraded tox and guaAB-add loci (47) (Fig. 2).
FIG 2 .
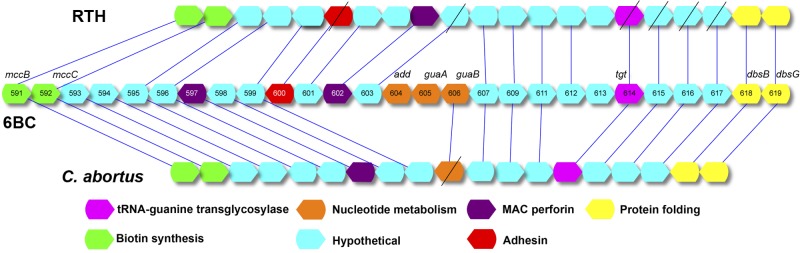
Comparison of the plasticity zones (PZs) of C. psittaci 6BC and RTH and C. abortus S26/3. The structure in the center shows the order of genes in the PZ of 6BC, with the numbers referring to the locus tag prefix (see RefSeq accession NC_017287.1). Some small genes (<150 nucleotides) were omitted in order to make the diagram less complex. The genes are color coded by function and matched to best hits in the other genomes. Genes with diagonal slashes are predicted to have loss-of-function frameshifts.
Genome-wide recombination across the C. psittaci genome.
The impact of recombination on C. psittaci genomes was quantified by applying the ClonalFrame algorithm to whole-genome sequences to estimate the number of recombination events (Fig. 3). ClonalFrame estimates two values, namely, ρ/θ and r/m; the former measures the frequency of occurrence of recombination relative to mutation, while the latter measures how important the effect of recombination is in genetic diversification relative to mutation. ClonalFrame estimated ρ/θ to be around 0.06 (with a 95% credibility interval of 0.054 to 0.065), indicating that recombination happened about twenty times less frequently than mutation (Table 2). The value for r/m was estimated to be around 3.19 (with a 95% credibility interval of 2.9217 to 3.4496), meaning that even though recombination was less frequent than mutation, each recombination event affected several nucleotides at a time so that, in total, about three times more substitutions were introduced via recombination than via mutation (Table 2). In C. trachomatis, ρ/θ and r/m were estimated to be around 0.14 and 1.4, respectively (3), which are lower than the corresponding values for C. psittaci. Recombination therefore happened less frequently in C. psittaci than in C. trachomatis but has had a bigger effect on genomic diversification, probably because of a higher diversity in C. psittaci. Similar to C. trachomatis, the detected recombination events were spread across the genome (3, 13–15, 48).
FIG 3 .

Results of the ClonalFrame analysis on an alignment of the 20 C. psittaci genomes. The inferred clonal genealogy is shown on the left. Each branch of the tree corresponds to a row of the heat map, which is horizontally aligned with it according to the core MAUVE whole-genome alignment. Each row of the heat map shows the posterior probability of recombination estimated by ClonalFrame on the corresponding branch (y axis) and along the positions of the alignment (x axis).
TABLE 2 .
ClonalFrame analysis
| Clade or group | No. of: |
ρ/θa | r/mb | |||
|---|---|---|---|---|---|---|
| Genomes | Mutation events |
Recombination events |
Substitutions introduced by recombination |
|||
| GR9/CT1 Clade 3 | 2 | 495 | 30 | 5,720 | 0.06 | 11.56 |
| FalTex/Borg/NJ1 Clade 2 | 3 | 109 | 10 | 48 | 0.09 | 0.44 |
| MN, WC, VS22 | 10 | 3,812 | 131 | 3,682 | 0.03 | 0.97 |
| 6BC Clade 1 | 10 | 482 | 41 | 954 | 0.09 | 1.98 |
| All | 20 | 32,486 | 1,514 | 77,170 | 0.05 | 2.38 |
Relative rate of recombination and mutation.
Relative effect of recombination and mutation.
Recombination was found to affect segments with a length of 1,116 bp on average (with a 95% credibility interval of 1,032 to 1,203 bp). The estimated mean track length of recombined fragments in C. trachomatis was 357 bp (3). Other bacteria, such as Bacillus cereus, have recombinant segments similar in size to those in C. psittaci (49). Table S1 in the supplemental material lists all the clade-specific recombinant genes identified by the ClonalFrame analysis.
Strikingly, there was limited evidence of recombination throughout the 6BC Clade 1 (Fig. 3), consistent with recent clonal expansion from a single original strain. Only strain 6BC/83, originating from a parakeet, appears to have acquired DNA sequences, and the acquisitions occurred 18 times with an average length of 44 bp. In order to understand whether C. psittaci strains undergo recombination with other C. psittaci strains or from an external source, we extracted all the recombinant segments and tracked their origin using BLASTN (see Materials and Methods) (Fig. 4). For the 6BC Clade 1, only six recombinant segments had an identity to other C. psittaci strains of <95%, whereas the remaining segments were highly similar to other C. psittaci strains. Of the six recombinant segments that had an identity of <95%, the smallest and the largest segments were, respectively, 163 and 762 bp. Similarly, the BLASTN control experiment performed by randomly selecting genomic regions within the boundaries of the length of the predicted recombinant segments showed that all the segments have >95% similarity to other C. psittaci strains. In the 6BC Clade 1, most of the recombination occurred with other C. psittaci strains rather than from an external source. However, within the GR9 and CT1 Clade 3, 60% of recombinant segments were <95% identical to other C. psittaci strains, indicating that those imports came from an external source. At the same time, the control experiment performed for the GR9 and CT1 clade using randomly selected regions in the genome showed statistically significantly higher identity to other C. psittaci strains (P < 0.001) compared to recombinant segments. For strains RTH and M56, 54% and 57%, respectively, of the recombinant segments were <95% identical to other C. psittaci strains, whereas the control experiment showed 80% of the random sequences to be highly similar to other C. psittaci strains (P < 0.001). For strains MN, WC, and VS22, the majority of the recombined segments were highly similar to other C. psittaci strains, thereby ruling out the possibility of less external influx of DNA into these strains. These data indicate that there is a wide range of variability among the C. psittaci strains with importation of DNA segments either from another Chlamydia species or from other as-yet-unsequenced clades of C. psittaci.
FIG 4 .
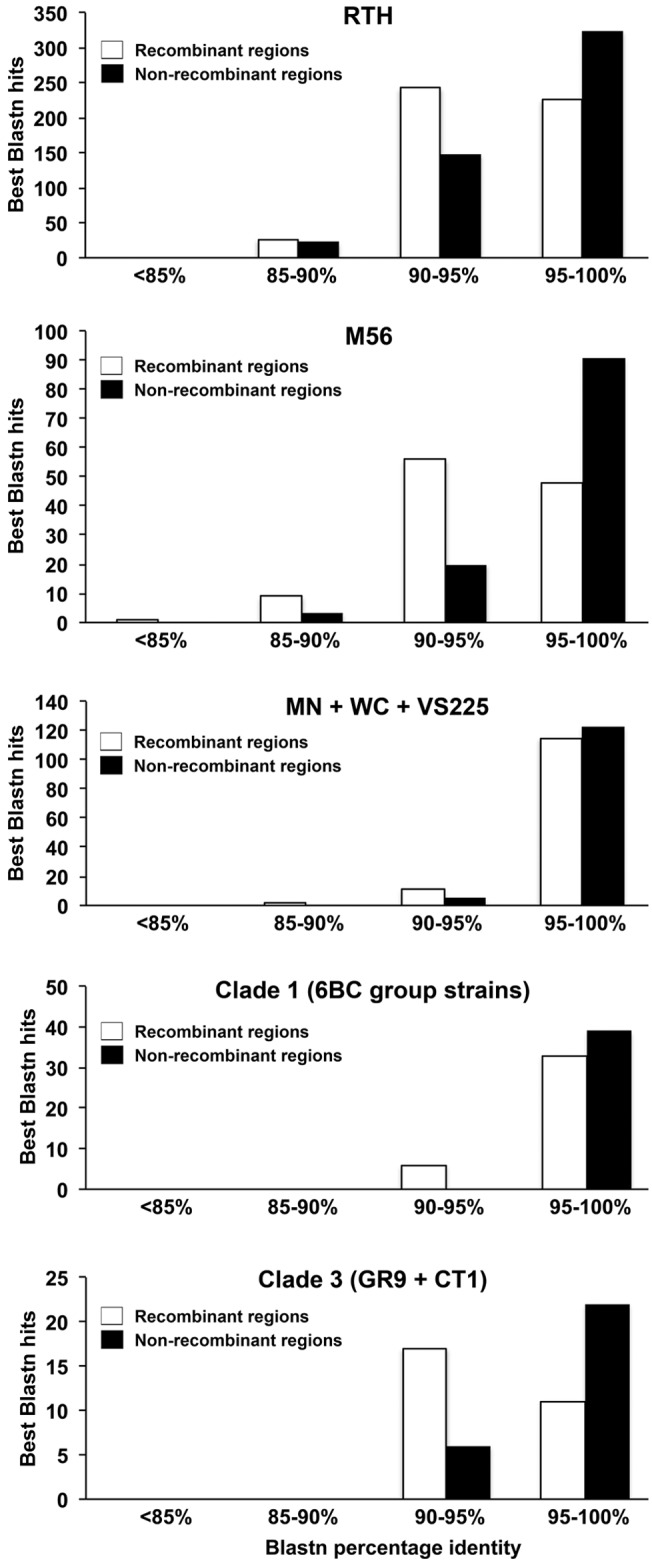
BLAST analysis of origins of recombinant fragments. Distribution of the best BLASTN hits of the putative recombinant segments identified by ClonalFrame and nonrecombinant regions extracted randomly (within the range of the size of recombinant segments) categorized based on 4 ranges of percent identity. For each of the clades/strains, both the recombinant segments and the nonrecombinant segments were searched against a BLAST database containing all the C. psittaci genomes minus the strain/clade that was affected by the import using BLASTN. Clade 2 (FalTex, Borg, and NJ1) contained a minimal number of recombinant segments, which were shorter to perform, and, therefore, BLASTN analysis was not included here.
For all the putative DNA recombinant imports, BLASTN analysis was performed against the NCBI’s nucleotide database containing all nucleotide sequences. There were 110 recombinant DNA sequences identified that had C. abortus as the significant best hit along with a single additional hit to Chlamydia caviae. These sequences were also subjected to a BLAST search against the local database of our 20 C. psittaci strains, which was not yet present in the BLAST nucleotide database at the time of this analysis. Of the 110 sequences, 53 maintained the best hit to C. abortus, while the best hit for the remainder was to C. psittaci. Of the 53, three sequences had 100% identity to C. abortus. The remaining had <100% identity to C. abortus.
Given the frequent exchange of DNA by homologous recombination predicted above, we examined the possible population structure of C. psittaci using STRUCTURE (50, 51). The number of ancestral populations (K) needed to explain the current population structure was estimated to be 3 (see Fig. S3 in the supplemental material). The proportion of ancestry from each of these ancestral populations is shown in color for each strain in Fig. 5. The strains fell into several groups based on the major ancestral source of genetic material of each strain, mostly following the phylogeny of the clonal regions of the genome (Fig. 1). Group 1 consisted of the 10 strains from the 6BC Clade 1 strains, and these genomes were entirely from one ancestral population (blue in Fig. 5). Group 2 comprised the strains GR9, CT1, WC, and VS225, which all had roughly 90% of material originating from one ancestral population (Fig. 5, blue) and smaller proportions from the other two (green and red). Group 3 consisted of strains NJ1, FalTex, and Borg, which showed almost equal proportions of two ancestral populations (Fig. 5, blue and green). Group 4 was made of strains MN and M56, which showed a mixture from the three ancestral populations. Finally, strain RTH consisted of almost entirely a single ancestral population (Fig. 5, red). There was no correlation between the ancestral population and the type of animal (mammal or bird) from which the strain was isolated, except for group 3.
FIG 5 .

Bar plot representing the result from the STRUCTURE linkage model. Each vertical line represents one of the 20 C. psittaci strains. The y axis shows the proportion of ancestry from each of the three ancestral populations. The tree at the top represents a complete linkage clustering of the strains based on their proportions of ancestry from each population and are represented by different groups as noted. The lettering of each strain is color coded to identify clades as in Fig. 1.
We found no evidence of recombination of loci between the sequences of the C. psittaci virulence plasmids using ClonalFrame analysis. However, the phylogeny of the whole plasmids reveals that the MN plasmid clustered among the plasmids of the 6BC Clade 1, suggesting a possible recent exchange event involving these strains (see Fig. S5 in the supplemental material).
DISCUSSION
Recent comparative genomic studies have revealed that there is a relatively high rate of homologous recombination in the archetypical infectious agent C. trachomatis despite the ecological barriers to cell-to-cell contact posed by its being an obligate intracellular pathogen (3, 13–15). Based on the most comprehensive analysis of the broadest diversity of C. psittaci genomes to date, we found evidence that recombination plays as much of a role in C. psittaci as in C. trachomatis.
Some of the patterns of microevolution among the 20 diverse C. psittaci strains echo those recently discovered in C. trachomatis. There also are some very stark and interesting differences. Both organisms undergo mostly intraspecific homologous recombination at various sites across the genome. However, the effect of recombination was higher in C. psittaci than in C. trachomatis (mean r/m of 3.2 versus 1.4). It is notable that there is no strong association between the host animal species and diseases for the 6BC Clade 1, in contradistinction to the other lineages, where strain host source, disease, and serotype are linked (Clades 2 and 3) (Table 1).
There was a wide range of variability of DNA importation among the 20 C. psittaci strains, either from an external source or from other C. psittaci strains. For the 110 putative DNA recombinant imports identified, C. abortus was the best hit, although there were additional hits to C. caviae and C. psittaci strains. When these sequences were subjected to a BLAST search against our database of 20 C. psittaci genomes, 53 were still best hits to C. abortus, with the remainder being best hits to C. psittaci; 50 of these had <100% identity to C. abortus, suggesting that the origin is not necessarily importation from C. abortus but may be importation from as-yet-unsequenced/unsampled C. psittaci strains or possibly other as-yet-unknown Chlamydiaceae strains. The conclusion drawn from this is that there is extensive diversity still to be sampled at the whole-genome level in the species C. psittaci.
Within each species, a recently emerged epidemic clonal strain has appeared: the LGV clade in C. trachomatis (15) and the 6BC Clade 1 in C. psittaci, which we describe here. The LGV strains are known to be more invasive, readily infecting intact epithelia and spreading via regional lymphatics, and have a shorter developmental cycle than non-LGV urogenital C. trachomatis strains. The latter may explain their rapid transmission and epidemic spread. 6BC strains are similar in both infection and developmental characteristics to the LGV strains. Our findings are consistent with the clinical knowledge of these strains, suggesting that they are more virulent than the remainder of the C. psittaci strains.
The presence in the PZ of the tox and guaAB-add loci in contrast to other Chlamydiaceae species may reflect adaptation of C. psittaci strains to diverse epithelial tissues in the lung and intestinal tract. It is important to understand whether 6BC and related strains have gained the tox and guaAB-add functions recently or whether these genes have instead been selectively lost by C. abortus and outlying C. psittaci strains. The 6BC Clade 1 and its three closest relatives may have gained these genes through one horizontal gene transfer event or alternatively deleted them from the PZ on at least three independent occasions. Acquisition of the tox gene by a clinical C. trachomatis L2 strain from a C. trachomatis D strain resulted in a recombinant that caused severe localized infection in the rectum of a patient without evidence of dissemination via regional lymphatics (14). The latter dissemination often results in what is referred to as the lymphatic syndrome and is typical of LGV infections. That study suggested that the tox gene may have restricted systemic disease due to localized cellular toxicity. However, if the patient had not sought out medical treatment, the course of the disease might have resembled the systemic disease seen with C. psittaci animal infections in their respective hosts.
The guaAB-add cluster represents accessory virulence genes present in Chlamydia pneumoniae (albeit with a frameshift in guaA), Chlamydia muridarum, Chlamydia felis, and some C. psittaci genomes (the 6BC Clade 1 and its three closest relatives) but absent in C. trachomatis or C. abortus. These genes play a role in salvaging biosynthesis of purine nucleotides necessary for the growth of the bacterium. Their presence intact in a subset of C. psittaci strains presumably reflects adaptation to a niche with biochemical restriction not encountered by other strains. In this regard, there may be a parallel with the finding that the guaAB genes in the Lyme disease pathogen Borrelia burgdorferi (horizontally acquired by the bacterium on a plasmid) are essential for survival in the infectious phase of the tick-blood cycle (52). While the 6BC strains are known to cause severe disease in humans, it is curious that other strains that cause systemic disease did not acquire these genes. It is a fascinating parallel that the C. pneumoniae strains that infect humans contain guaBA-add homologs but very closely related C. pneumoniae strains that infect koalas lack these genes (53).
In the case of the tox locus, there is a very significant sequence difference between the genes of the 6BC Clade 1 and the non-6BC C. psittaci strains, suggesting that the tox genes have evolved through recombination with other Chlamydiaceae strains (Fig. S4E). The closest intact relative of the 6BC guaAB-add genes is found in C. felis, a species restricted to felines and causing conjunctivitis and pneumonitis. However, these genes do not share the same phylogeny as the core genomes, as evidenced by the C. muridarum guaAB-add locus, which is more conserved in C. felis than the latter is in Chlamydia pecorum (see Fig. S4 in the supplemental material). This indicates a history of mobile transfer between strains.
Using the North American strains, we estimated the date of the most recent common ancestor of the 20 C. psittaci genomes to be around the year 1500. In our evolutionary reconstruction, the 6BC strains have a common ancestor dating from about 1916 to 1930 depending on the method used. Based on available literature, the first mention of parrot-related diseases in humans was in 1879 in Switzerland and another occurred in 1892 in Paris, where the illness was associated with imported parrots from Buenos Aires (54, 55). Descriptions of parrots dying of psittacosis were published in 1893 (reviewed by Pospischil [56]). However, Old World birds have been kept domestically by literate societies for millennia, and cave paintings of psittacine birds in Brazil date back approximately 5,000 years, suggesting a long history of interaction with humans in South America. It is thus surprising that there are no ancient records of this highly visible disease in Eurasia or Africa. Our dating suggests that the 6BC lineage expansion may come from a single outbreak caused by an Argentinian parrot exporter in 1929 (57), which involved birds originally caught in Brazil. Although we still have limited data, our results are consistent with the possibility that psittacosis is a very recently emerged disease originating in New World parrots. The presence of strains causing human infections in North America from outside the 6BC lineage points to either parallel importation of multiple pathogenic C. psittaci or possibly native pre-Columbian pathogens. Additional sequencing of a global population of strains is required to help resolve the geographic origin of all C. psittaci strains.
MATERIALS AND METHODS
C. psittaci strains, clonal purification, and generation of genomic DNA.
The 20 strains of C. psittaci used in this study are summarized in Table 1. This included eight strains with previously published genomes. We sequenced 12 additional strains of animal origin, two of which were isolated from humans. The 20 strains represented all known serotypes of the organism from four different countries isolated between 1930 and 2010.
Each of the 12 new strains was individually propagated in McCoy or HeLa 229 cells and clonally purified; purity was confirmed by ompA sequencing of 10 clones of each strain as previously described (3, 14). Following density gradient centrifugation, each isolate was treated with DNase prior to purification of the DNA using a Roche High Pure kit as previously described (3, 14).
Genome sequencing.
Genomes were sequenced using GS-FLX (454 Life Sequencing Inc., Branford, CT). Libraries for sequencing were prepared from 1 to 5 µg of genomic DNA. De novo assembly and further processing of the genomes were performed as previously described (3, 39). The contigs were aligned against the previously sequenced C. psittaci 6BC genome using MUMmer (58) to create concatenated, ordered “pseudocontigs.” These pseudocontigs were considered the bacterial chromosome for each strain and were annotated using the ISGA bacterial annotation pipeline (59). All the published genomes were also reannotated using the same pipeline.
Whole-genome alignment and identification of core genes.
The 20 C. psittaci genomes were aligned using the MAUVE progressive alignment algorithm with default settings (60, 61). There were 95 locally collinear blocks (LCBs) of >500 bp, which was set as the minimum length of the core alignment blocks. LCBs for each genome were extracted from the output of the program and concatenated. We also performed a second MAUVE alignment by including the genome sequence of Chlamydia abortus (47) as an outgroup.
The complete predicted proteome from all the genomes annotated in this study along with the annotated protein sequences of the previously published genomes was searched against itself using BLASTP with an e-value cutoff of 1e-05. The best BLASTP scores were utilized for identifying the orthologous sequences using the OrthoMCL (62) algorithm as previously described (3, 13, 39). Core genes are defined as the protein-coding gene clusters that are shared by all C. psittaci strains. The program MUSCLE (63) was used with default settings to align the core genes, and each of the protein alignments was filtered by GBLOCKS (64) to remove gaps and highly divergent regions.
Phylogenetic reconstruction and temporal analysis.
We concatenated the alignments of all the core protein families and reconstructed a first phylogeny using the neighbor-joining algorithm implemented in PHYLIP. To evaluate statistical support, a majority rule-consensus tree of 100 bootstrap replicates was computed. We also used the concatenated MAUVE alignment to reconstruct phylogenies using both neighbor joining in PHYLIP and the coalescent-based ClonalFrame method (see below).
Bayesian analysis of evolutionary rates and divergence times was performed based on the MAUVE alignment using BEAST v1.7.2 with the GTR+G substitution model and with tip dates defined as the year of isolation (Table 1). Four independent runs were performed, each consisting of 145 million Markov chain Monte Carlo (MCMC) iterations, sampled every 1,000 generations and with the first 10% discarded as burn-in. These four runs were combined to provide robust estimates of posterior parameter distributions (calculated using the program Tracer v1.5). This BEAST analysis used the relaxed molecular clock model along with the Bayesian skyline demographic model.
ClonalFrame analysis.
ClonalFrame (41) version 1.2 was applied to the MAUVE alignment. Four independent runs were performed, each consisting of 20,000 MCMC iterations with the first half discarded as burn-in, and compared to unsure of good convergence and mixing properties. A key feature of ClonalFrame is that it detects genomic regions where inheritance did not follow the expected clonal genealogy, and we extracted the genomic position of these recombination events. These extracted recombination events were searched against a BLAST database containing all the C. psittaci genomes minus the strains that were affected by the import using BLASTN. The number of best BLASTN hits with the highest normalized BLASTN score along with the highest percent identity was compared to the same metrics obtained by extracting random regions (within the range of the size of recombinant segments) of the C. psittaci genome using chi square.
ClonalFrame also estimates the clonal genealogy as well as the number of mutations that occurred on each branch of the tree (41). Combining this information with the known dates of isolation for each genome, which spanned 80 years (1930 to 2010), allowed the simultaneous estimation of the mutation rate per year as well as the dates of existence of the nodes in the ClonalFrame tree. This inference was performed assuming the Kingman coalescent with the temporally offset leaves model (65) for the tree and a uniform prior for the mutation rate.
Structure analysis.
The linkage model of the Bayesian method STRUCTURE (50, 51) version 2.3 was used to analyze the population structure of C. psittaci. In order to estimate the value of the parameter K, representing the number of ancestral populations, four independent runs were performed for each value of K between 2 and 10. Each run consisted of 100,000 MCMC iterations, with the first half discarded as burn-in. Comparisons of the four independent runs with the same value of K confirmed that convergence and mixing were satisfactory. The optimal K value was found to be 3 using two different approaches: the posterior probability plateau method described in reference 66 and the second-derivatives likelihood method described in reference 67.
Nucleotide sequence accession numbers.
The genome and plasmid sequences have been deposited at NCBI under the accession numbers given in Table 1.
SUPPLEMENTAL MATERIAL
Comparative mapping of C. psittaci genome data. Whole-genome shotgun contigs were mapped to the C. psittaci 6BC (35) genome using BLASTN. Matches with >90% DNA identity are shown in color; nonmatching regions are white. From the center, the strains mapped are 6BC/30, 6BC/41, CP3, DC11, DC15, 8DC6, RD1, Cal10, C19/8, VS22, WC, and MN (blue), FalTex, Borg, and NJ1 (yellow), GR9, CT1, and M56 (salmon), and RTH (red). The plasticity zone (PZ) is located at approximately the 7 o’clock position on the circle. The inner ring is the G+C skew: transitions from positive to negative and vice versa indicate the origin of replication and termination (A. C. Frank and J. R. Lobry, Gene 238:65–77, 1999). The figure was produced using BRIG software (N. F. Alikhan, N. K. Petty, N. L. Ben Zakour, S. A. Beatson, BMC Genomics 12:402, 2011). Download
(A) ClonalFrame-based Bayesian phylogenetic reconstruction of C. psittaci genomes. The analysis was performed by combining the estimated number of mutations that occurred on each branch inferred from the whole-genome core alignment of the 20 C. psittaci strains along with the known dates of isolation for each genome. ClonalFrame estimated the mutation rate to be 1.74 × 10−5 per site per year with a 95% credibility interval ranging from 1.71 × 10−5 to 1.76 × 10−5 and dated the MRCA at 1521 with 95% credibility interval ranging from 1514 to 1527. (B) Bayesian phylogenetic reconstruction of all C. psittaci genomes based on the program BEAST (41). Bayesian analysis of evolutionary rates and divergence times was performed based on the MAUVE alignment using BEAST v1.7.2 under the GTR+G substitution model and with tip dates defined as the year of isolation. We used the relaxed molecular clock model along with the Bayesian skyline demographic model. BEAST estimated a mean rate of 1.682 × 10−4 substitution per year per site, with a 95% credibility interval ranging from 2.97 × 10−5 to 2.805 × 10−4 and an estimated date for the MRCA of 1770. Download
Procedure of Evanno et al. (67) to determine the number of ancestral populations in C. psittaci using the data from STRUCTURE. (A) Plot of the estimated ln probability of the data across each of the K values (2 to 10) specified. (B) (Clockwise) (1) Plot of mean likelihood L(K) for each of the K values specified. (2) Plot of the mean difference between successive likelihood values of K; L′(K) = L(K) − L(K − 1) across each of the K values specified. (3) Plot of the mean difference between successive values of L′(K), where |L″(K)| = |L′(K + 1) − L′(K)|. (4) Plot of ΔK; ΔK = m|L″(K)|/s|L(K)|, where m is the mean of absolute values of L″(K) and s is the standard deviation of L(K). Download
Phylogeny of Gua-Add proteins in C. psittaci and other Chlamydiaceae species. All the trees were constructed using a maximum-likelihood algorithm by PhyML based on protein alignments. The evolutionary distances were computed using the JTT substitution model with 100 bootstrap sampling. (A) GuaA; (B) GuaB; (C) Add; (D) RecA (control). Download
Phylogeny of C. psittaci plasmids. Maximum-likelihood trees were constructed from aligned plasmid sequences using PHyML with 100 bootstrap sampling. Branches with >80% support are shown with diamonds. Download
Genes with evidence of recombination based on ClonalFrame analysis in each of the three clades.
ACKNOWLEDGMENTS
This study was supported by National Science Foundation/United States Department of Agriculture Microbial Genome Sequencing Program grant 2009-65109-05760 (to D.D.) and NIH/NIAID grant R01 AI098843 (to D.D.). Sequencing was provided by the Emory Genomics Center, which was partially funded by the Georgia Research Alliance and Atlanta Clinical & Translational Sciences Institute. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
We are indebted to Arthur Andersen for enabling his collection of animal chlamydial strains at the USDA to be moved and maintained in D.D.’s laboratory and to James Rothschild and Thomas Pham for excellent technical assistance.
Footnotes
Citation Read TD, Joseph SJ, Didelot X, Liang B, Patel L, Dean D. 2013. Comparative analysis of Chlamydia psittaci genomes reveals the recent emergence of a pathogenic lineage with a broad host range. mBio 4(2):e00604-12. doi:10.1128/mBio.00604-12
REFERENCES
- 1. Greub G. 2010. International Committee on Systematics of Prokaryotes. Subcommittee on the taxonomy of the Chlamydiae: minutes of the inaugural closed meeting, 21 March 2009, Little Rock, AR, USA. Int. J. Syst. Evol. Microbiol. 60:2691–2694 [DOI] [PubMed] [Google Scholar]
- 2. Horn M, Collingro A, Schmitz-Esser S, Beier CL, Purkhold U, Fartmann B, Brandt P, Nyakatura GJ, Droege M, Frishman D, Rattei T, Mewes HW, Wagner M. 2004. Illuminating the evolutionary history of Chlamydiae. Science 304:728–730 [DOI] [PubMed] [Google Scholar]
- 3. Joseph SJ, Didelot X, Rothschild J, de Vries HJ, Morré SA, Read TD, Dean D. 2012. Population genomics of Chlamydia trachomatis: insights on drift, selection, recombination, and population structure. Mol. Biol. Evol. 29:3933–3946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Collingro A, Tischler P, Weinmaier T, Penz T, Heinz E, Brunham RC, Read TD, Bavoil PM, Sachse K, Kahane S, Friedman MG, Rattei T, Myers GS, Horn M. 2011. Unity in variety–the pan-genome of the Chlamydiae. Mol. Biol. Evol. 28:3253–3270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Darby AC, Cho NH, Fuxelius HH, Westberg J, Andersson SG. 2007. Intracellular pathogens go extreme: genome evolution in the Rickettsiales. Trends Genet. 23:511–520 [DOI] [PubMed] [Google Scholar]
- 6. Sällström B, Andersson SG. 2005. Genome reduction in the alpha-proteobacteria. Curr. Opin. Microbiol. 8:579–585 [DOI] [PubMed] [Google Scholar]
- 7. Tamas I, Klasson L, Canbäck B, Näslund AK, Eriksson AS, Wernegreen JJ, Sandström JP, Moran NA, Andersson SG. 2002. 50 million years of genomic stasis in endosymbiotic bacteria. Science 296:2376–2379 [DOI] [PubMed] [Google Scholar]
- 8. Dugan J, Rockey DD, Jones L, Andersen AA. 2004. Tetracycline resistance in Chlamydia suis mediated by genomic islands inserted into the chlamydial inv-like gene. Antimicrob. Agents Chemother. 48:3989–3995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Read TD, Brunham RC, Shen C, Gill SR, Heidelberg JF, White O, Hickey EK, Peterson J, Utterback T, Berry K, Bass S, Linher K, Weidman J, Khouri H, Craven B, Bowman C, Dodson R, Gwinn M, Nelson W, DeBoy R, Kolonay J, McClarty G, Salzberg SL, Eisen J, Fraser CM. 2000. Genome sequences of Chlamydia trachomatis MoPn and Chlamydia pneumoniae AR39. Nucleic Acids Res. 28:1397–1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Millman KL, Tavaré S, Dean D. 2001. Recombination in the ompA gene but not the omcB gene of Chlamydia contributes to serovar-specific differences in tissue tropism, immune surveillance, and persistence of the organism. J. Bacteriol. 183:5997–6008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gomes JP, Bruno WJ, Nunes A, Santos N, Florindo C, Borrego MJ, Dean D. 2007. Evolution of Chlamydia trachomatis diversity occurs by widespread interstrain recombination involving hotspots. Genome Res. 17:50–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dean D, Bruno WJ, Wan R, Gomes JP, Devignot S, Mehari T, de Vries HJ, Morré SA, Myers G, Read TD, Spratt BG. 2009. Predicting phenotype and emerging strains among Chlamydia trachomatis infections. Emerg. Infect. Dis. 15:1385–1394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Joseph SJ, Didelot X, Gandhi K, Dean D, Read TD. 2011. Interplay of recombination and selection in the genomes of Chlamydia trachomatis. Biol. Direct 6:28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Somboonna N, Wan R, Ojcius DM, Pettengill MA, Joseph SJ, Chang A, Hsu R, Read TD, Dean D. 2011. Hypervirulent Chlamydia trachomatis clinical strain is a recombinant between lymphogranuloma venereum (L(2)) and D lineages. mBio 2:e00045-00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Harris SR, Clarke IN, Seth-Smith HM, Solomon AW, Cutcliffe LT, Marsh P, Skilton RJ, Holland MJ, Mabey D, Peeling RW, Lewis DA, Spratt BG, Unemo M, Persson K, Bjartling C, Brunham R, de Vries HJ, Morré SA, Speksnijder A, Bébéar CM, Clerc M, de Barbeyrac B, Parkhill J, Thomson NR. 2012. Whole-genome analysis of diverse Chlamydia trachomatis strains identifies phylogenetic relationships masked by current clinical typing. Nat. Genet. 44:413–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Read TD, Myers GS, Brunham RC, Nelson WC, Paulsen IT, Heidelberg J, Holtzapple E, Khouri H, Federova NB, Carty HA, Umayam LA, Haft DH, Peterson J, Beanan MJ, White O, Salzberg SL, Hsia RC, McClarty G, Rank RG, Bavoil PM, Fraser CM. 2003. Genome sequence of Chlamydophila caviae (Chlamydia psittaci GPIC): examining the role of niche-specific genes in the evolution of the Chlamydiaceae. Nucleic Acids Res. 31:2134–2147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Harkinezhad T, Geens T, Vanrompay D. 2009. Chlamydophila psittaci infections in birds: a review with emphasis on zoonotic consequences. Vet. Microbiol. 135:68–77 [DOI] [PubMed] [Google Scholar]
- 18. Longbottom D, Coulter LJ. 2003. Animal chlamydioses and zoonotic implications. J. Comp. Pathol. 128:217–244 [DOI] [PubMed] [Google Scholar]
- 19. Theegarten D, Sachse K, Mentrup B, Fey K, Hotzel H, Anhenn O. 2008. Chlamydophila spp. infection in horses with recurrent airway obstruction: similarities to human chronic obstructive disease. Respir. Res. 9:14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lenzko H, Moog U, Henning K, Lederbach R, Diller R, Menge C, Sachse K, Sprague LD. 2011. High frequency of chlamydial co-infections in clinically healthy sheep flocks. BMC Vet. Res. 7:29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Reinhold P, Sachse K, Kaltenboeck B. 2011. Chlamydiaceae in cattle: commensals, trigger organisms, or pathogens? Vet. J. 189:257–267 [DOI] [PubMed] [Google Scholar]
- 22. Chahota R, Ogawa H, Mitsuhashi Y, Ohya K, Yamaguchi T, Fukushi H. 2006. Genetic diversity and epizootiology of Chlamydophila psittaci prevalent among the captive and feral avian species based on VD2 region of ompA gene. Microbiol. Immunol. 50:663–678 [DOI] [PubMed] [Google Scholar]
- 23. Cunha BA. 2006. The atypical pneumonias: clinical diagnosis and importance. Clin. Microbiol. Infect. 12(Suppl 3):12–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Raso TdeF, Godoy SN, Milanelo L, de Souza CA, Matuschima ER, Araújo Júnior JP, Pinto AA. 2004. An outbreak of chlamydiosis in captive blue-fronted Amazon parrots (Amazona aestiva) in Brazil. J. Zoo. Wildl. Med 35:94–96 [DOI] [PubMed] [Google Scholar]
- 25. Smith KA, Bradley KK, Stobierski MG, Tengelsen LA, National Association of State Public Health Veterinarians 2005. Compendium of measures to control Chlamydophila psittaci (formerly Chlamydia psittaci) infection among humans (psittacosis) and pet birds, 2005. J. Am. Vet. Med. Assoc. 226:532–539 [DOI] [PubMed] [Google Scholar]
- 26. Toyokawa M, Kishimoto T, Cai Y, Ogawa M, Shiga S, Nishi I, Hosotsubo H, Horikawa M, Asari S. 2004. Severe Chlamydophila psittaci pneumonia rapidly diagnosed by detection of antigen in sputum with an immunochromatography assay. J. Infect. Chemother. 10:245–249 [DOI] [PubMed] [Google Scholar]
- 27. Verminnen K, Duquenne B, De Keukeleire D, Duim B, Pannekoek Y, Braeckman L, Vanrompay D. 2008. Evaluation of a Chlamydophila psittaci infection diagnostic platform for zoonotic risk assessment. J. Clin. Microbiol. 46:281–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Petrovay F, Balla E. 2008. Two fatal cases of psittacosis caused by Chlamydophila psittaci. J. Med. Microbiol. 57:1296–1298 [DOI] [PubMed] [Google Scholar]
- 29. Deschuyffeleer TP, Tyberghien LF, Dickx VL, Geens T, Saelen JM, Vanrompay DC, Braeckman LA. 2012. Risk assessment and management of Chlamydia psittaci in poultry processing plants. Ann. Occup. Hyg. 56:340–349 [DOI] [PubMed] [Google Scholar]
- 30. Heddema ER, van Hannen EJ, Duim B, de Jongh BM, Kaan JA, van Kessel R, Lumeij JT, Visser CE, Vandenbroucke-Grauls CM. 2006. An outbreak of psittacosis due to Chlamydophila psittaci genotype A in a veterinary teaching hospital. J. Med. Microbiol. 55:1571–1575 [DOI] [PubMed] [Google Scholar]
- 31. Eaton MD, Beck MD, Pearson HE. 1941. A virus from cases of atypical pneumonia: relation to the viruses of meningopneumonitis and psittacosis. J. Exp. Med. 73:641–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Olson BJ, Treuting WL. 1944. Epidemic of severe pneumonitis in bayou region of Louisiana, preliminary note on etiology. U.S. Public Health Rep. 69:1373–1374 [Google Scholar]
- 33. Leitenberg M. 2001. Biological weapons in the twentieth century: a review and analysis. Crit. Rev. Microbiol. 27:267–320 [DOI] [PubMed] [Google Scholar]
- 34. Voigt A, Schöfl G, Saluz HP. 2012. The Chlamydia psittaci genome: a comparative analysis of intracellular pathogens. PLoS One 7:e35097 http://dx.doi.org/10.1371/journal.pone.0035097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Grinblat-Huse V, Drabek EF, Creasy HH, Daugherty SC, Jones KM, Santana-Cruz I, Tallon LJ, Read TD, Hatch TP, Bavoil P, Myers GS. 2011. Genome sequences of the zoonotic pathogens Chlamydia psittaci 6 bc and Cal10. J. Bacteriol. 193:4039–4040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Voigt A, Schöfl G, Heidrich A, Sachse K, Saluz HP. 2011. Full-length de novo sequence of the Chlamydophila psittaci type strain 6BC. J. Bacteriol. 193:2662–2663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Seth-Smith HM, Harris SR, Rance R, West AP, Severin JA, Ossewaarde JM, Cutcliffe LT, Skilton RJ, Marsh P, Parkhill J, Clarke IN, Thomson NR. 2011. Genome sequence of the zoonotic pathogen Chlamydophila psittaci. J. Bacteriol. 193:1282–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schöfl G, Voigt A, Litsche K, Sachse K, Saluz HP. 2011. Complete genome sequences of four mammalian isolates of Chlamydophila psittaci. J. Bacteriol. 193:4258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zwick ME, Joseph SJ, Didelot X, Chen PE, Bishop-Lilly KA, Stewart AC, Willner K, Nolan N, Lentz S, Thomason MK, Sozhamannan S, Mateczun AJ, Du L, Read TD. 2012. Genomic characterization of the Bacillus cereus sensu lato species: backdrop to the evolution of Bacillus anthracis. Genome Res. 22:1512–1524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Drummond AJ, Suchard MA, Xie D, Rambaut A. 2012. Bayesian phylogenetics with BEAUti and the BEAST. Mol. Biol. Evol. 1 7:29:1969-1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Didelot X, Falush D. 2007. Inference of bacterial microevolution using multilocus sequence data. Genetics 175:1251–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Young BC, Golubchik T, Batty EM, Fung R, Larner-Svensson H, Votintseva AA, Miller RR, Godwin H, Knox K, Everitt RG, Iqbal Z, Rimmer AJ, Cule M, Ip CL, Didelot X, Harding RM, Donnelly P, Peto TE, Crook DW, Bowden R, Wilson DJ. 2012. Evolutionary dynamics of Staphylococcus aureus during progression from carriage to disease. Proc. Natl. Acad. Sci. U. S. A. 109:4550–4555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Croucher NJ, Harris SR, Fraser C, Quail MA, Burton J, van der Linden M, McGee L, von Gottberg A, Song JH, Ko KS, Pichon B, Baker S, Parry CM, Lambertsen LM, Shahinas D, Pillai DR, Mitchell TJ, Dougan G, Tomasz A, Klugman KP, Parkhill J, Hanage WP, Bentley SD. 2011. Rapid pneumococcal evolution in response to clinical interventions. Science 331:430–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mutreja A, Kim DW, Thomson NR, Connor TR, Lee JH, Kariuki S, Croucher NJ, Choi SY, Harris SR, Lebens M, Niyogi SK, Kim EJ, Ramamurthy T, Chun J, Wood JL, Clemens JD, Czerkinsky C, Nair GB, Holmgren J, Parkhill J, Dougan G. 2011. Evidence for several waves of global transmission in the seventh cholera pandemic. Nature 477:462–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kennemann L, Didelot X, Aebischer T, Kuhn S, Drescher B, Droege M, Reinhardt R, Correa P, Meyer TF, Josenhans C, Falush D, Suerbaum S. 2011. Helicobacter pylori genome evolution during human infection. Proc. Natl. Acad. Sci. U. S. A. 108:5033–5038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Labiran C, Clarke IN, Cutcliffe LT, Wang Y, Skilton RJ, Persson K, Bjartling C, Herrmann B, Christerson L, Marsh P. 2012. Genotyping markers used for multi locus VNTR analysis with ompA (MLVA-ompA) and multi sequence typing (MST) retain stability in Chlamydia trachomatis. Front. Cell. Infect. Microbiol. 2:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Thomson NR, Yeats C, Bell K, Holden MT, Bentley SD, Livingstone M, Cerdeño-Tárraga AM, Harris B, Doggett J, Ormond D, Mungall K, Clarke K, Feltwell T, Hance Z, Sanders M, Quail MA, Price C, Barrell BG, Parkhill J, Longbottom D. 2005. The Chlamydophila abortus genome sequence reveals an array of variable proteins that contribute to interspecies variation. Genome Res. 15:629–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Joseph SJ, Read TD. 2012. Genome-wide recombination in Chlamydia trachomatis. Nat. Genet. 44:364–366 [DOI] [PubMed] [Google Scholar]
- 49. Didelot X, Lawson D, Darling A, Falush D. 2010. Inference of homologous recombination in bacteria using whole-genome sequences. Genetics 186:1435–1449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pritchard JK, Stephens M, Donnelly P. 2000. Inference of population structure using multilocus genotype data. Genetics 155:945–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Falush D, Stephens M, Pritchard JK. 2003. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jewett MW, Lawrence KA, Bestor A, Byram R, Gherardini F, Rosa PA. 2009. GuaA and GuaB are essential for Borrelia burgdorferi survival in the tick-mouse infection cycle. J. Bacteriol. 191:6231–6241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Myers GS, Mathews SA, Eppinger M, Mitchell C, O’Brien KK, White OR, Benahmed F, Brunham RC, Read TD, Ravel J, Bavoil PM, Timms P. 2009. Evidence that human Chlamydia pneumoniae was zoonotically acquired. J. Bacteriol. 191:7225–7233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ritter J. 1880. Beitrag zur Frage des Pneumotyphus [Eine Hausepidemie in Uster (Schweiz) betreffend]. Dtsch. Arch. Klin. Med. 25:53–96 [Google Scholar]
- 55. Bedson SP, Western GT. 1930. Psittacosis—Ministry of Health report. Br. Med. J. 2:1018–1019 [PMC free article] [PubMed] [Google Scholar]
- 56. Pospischil A. 2009. From disease to etiology: historical aspects of Chlamydia-related diseases in animals and humans. Drugs Today (Barc.) 45(Suppl B):141–146 [PubMed] [Google Scholar]
- 57. Meyer K, Chaffee E, Hobby GL, Dawson MH. 1941. Hyaluronidases of bacterial and animal origin. J. Exp. Med. 73:309–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. 2004. Versatile and open software for comparing large genomes. Genome Biol. 5:R12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hemmerich C, Buechlein A, Podicheti R, Revanna KV, Dong Q. 2010. An Ergatis-based prokaryotic genome annotation web server. Bioinformatics 26:1122–1124 [DOI] [PubMed] [Google Scholar]
- 60. Darling AC, Mau B, Blattner FR, Perna NT. 2004. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14:1394–1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Darling AE, Mau B, Perna NT. 2010. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147 http://dx.doi.org/10.1371/journal.pone.0011147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Li L, Stoeckert CJ, Jr, Roos DS. 2003. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13:2178–2189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Castresana J. 2000. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17:540–552 [DOI] [PubMed] [Google Scholar]
- 65. Drummond AJ, Nicholls GK, Rodrigo AG, Solomon W. 2002. Estimating mutation parameters, population history and genealogy simultaneously from temporally spaced sequence data. Genetics 161:1307–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pritchard JK, Falush D. Documentation for STRUCTURE software, version 2.3. 2009. [Google Scholar]
- 67. Evanno G, Regnaut S, Goudet J. 2005. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14:2611–2620 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Comparative mapping of C. psittaci genome data. Whole-genome shotgun contigs were mapped to the C. psittaci 6BC (35) genome using BLASTN. Matches with >90% DNA identity are shown in color; nonmatching regions are white. From the center, the strains mapped are 6BC/30, 6BC/41, CP3, DC11, DC15, 8DC6, RD1, Cal10, C19/8, VS22, WC, and MN (blue), FalTex, Borg, and NJ1 (yellow), GR9, CT1, and M56 (salmon), and RTH (red). The plasticity zone (PZ) is located at approximately the 7 o’clock position on the circle. The inner ring is the G+C skew: transitions from positive to negative and vice versa indicate the origin of replication and termination (A. C. Frank and J. R. Lobry, Gene 238:65–77, 1999). The figure was produced using BRIG software (N. F. Alikhan, N. K. Petty, N. L. Ben Zakour, S. A. Beatson, BMC Genomics 12:402, 2011). Download
(A) ClonalFrame-based Bayesian phylogenetic reconstruction of C. psittaci genomes. The analysis was performed by combining the estimated number of mutations that occurred on each branch inferred from the whole-genome core alignment of the 20 C. psittaci strains along with the known dates of isolation for each genome. ClonalFrame estimated the mutation rate to be 1.74 × 10−5 per site per year with a 95% credibility interval ranging from 1.71 × 10−5 to 1.76 × 10−5 and dated the MRCA at 1521 with 95% credibility interval ranging from 1514 to 1527. (B) Bayesian phylogenetic reconstruction of all C. psittaci genomes based on the program BEAST (41). Bayesian analysis of evolutionary rates and divergence times was performed based on the MAUVE alignment using BEAST v1.7.2 under the GTR+G substitution model and with tip dates defined as the year of isolation. We used the relaxed molecular clock model along with the Bayesian skyline demographic model. BEAST estimated a mean rate of 1.682 × 10−4 substitution per year per site, with a 95% credibility interval ranging from 2.97 × 10−5 to 2.805 × 10−4 and an estimated date for the MRCA of 1770. Download
Procedure of Evanno et al. (67) to determine the number of ancestral populations in C. psittaci using the data from STRUCTURE. (A) Plot of the estimated ln probability of the data across each of the K values (2 to 10) specified. (B) (Clockwise) (1) Plot of mean likelihood L(K) for each of the K values specified. (2) Plot of the mean difference between successive likelihood values of K; L′(K) = L(K) − L(K − 1) across each of the K values specified. (3) Plot of the mean difference between successive values of L′(K), where |L″(K)| = |L′(K + 1) − L′(K)|. (4) Plot of ΔK; ΔK = m|L″(K)|/s|L(K)|, where m is the mean of absolute values of L″(K) and s is the standard deviation of L(K). Download
Phylogeny of Gua-Add proteins in C. psittaci and other Chlamydiaceae species. All the trees were constructed using a maximum-likelihood algorithm by PhyML based on protein alignments. The evolutionary distances were computed using the JTT substitution model with 100 bootstrap sampling. (A) GuaA; (B) GuaB; (C) Add; (D) RecA (control). Download
Phylogeny of C. psittaci plasmids. Maximum-likelihood trees were constructed from aligned plasmid sequences using PHyML with 100 bootstrap sampling. Branches with >80% support are shown with diamonds. Download
Genes with evidence of recombination based on ClonalFrame analysis in each of the three clades.