

ABSTRACT
Bloodstream spread is a critical step in the pathogenesis of many viruses. However, mechanisms that promote viremia are not well understood. Reoviruses are neurotropic viruses that disseminate hematogenously to the central nervous system. Junctional adhesion molecule A (JAM-A) is a tight junction protein that serves as a receptor for reovirus. JAM-A is required for establishment of viremia in infected newborn mice and viral spread to sites of secondary replication. To determine how viruses gain access to the circulatory system, we examined reovirus infection of polarized human brain microvascular endothelial cells (HBMECs). Reovirus productively infects polarized HBMECs, but infection does not alter tight junction integrity. Apical infection of polarized HBMECs is more efficient than basolateral infection, which is attributable to viral engagement of sialic acid and JAM-A. Viral release occurs exclusively from the apical surface via a mechanism that is not associated with lysis or apoptosis of infected cells. These data suggest that infection of endothelial cells routes reovirus apically into the bloodstream for systemic dissemination in the host. Understanding how viruses invade the bloodstream may aid in the development of therapeutics that block this step in viral pathogenesis.
IMPORTANCE Bloodstream spread of viruses within infected hosts is a critical but poorly understood step in viral disease. Reoviruses first enter the host through the oral or respiratory route and infect cells in the central nervous system. Spread of reoviruses to the brain occurs by blood or nerves, which makes reoviruses useful models for studies of systemic viral dissemination. In this study, we examined how reoviruses infect endothelial cells, which form the walls of blood vessels. We found that reovirus infection of endothelial cells allows the virus to enter blood vessels and serves as a means for the virus to reach high titers in the circulation. Understanding how reovirus is routed through endothelial cells may aid in the design of antiviral drugs that target this important step in systemic viral infections.
IMPORTANCE
Bloodstream spread of viruses within infected hosts is a critical but poorly understood step in viral disease. Reoviruses first enter the host through the oral or respiratory route and infect cells in the central nervous system. Spread of reoviruses to the brain occurs by blood or nerves, which makes reoviruses useful models for studies of systemic viral dissemination. In this study, we examined how reoviruses infect endothelial cells, which form the walls of blood vessels. We found that reovirus infection of endothelial cells allows the virus to enter blood vessels and serves as a means for the virus to reach high titers in the circulation. Understanding how reovirus is routed through endothelial cells may aid in the design of antiviral drugs that target this important step in systemic viral infections.
Introduction
Bloodstream dissemination within an infected host is required for the pathogenesis of many viruses. In particular, many neurotropic viruses use the circulation to invade the central nervous system (CNS) from a distant site of primary replication. Regardless of the site of entry into the host, viruses that disseminate hematogenously must first traverse an endothelial barrier and egress from the circulation. Although viremia is a well-established dissemination process, precise mechanisms of viral entry into or exit from the bloodstream are not well understood.
Mammalian orthoreoviruses (reoviruses) are neurotropic viruses that disseminate hematogenously to the CNS, where they display serotype-specific patterns of tropism for neural cells. Serotype 1 reoviruses spread strictly by the bloodstream and infect ependymal cells within the CNS, causing nonlethal hydrocephalus (1–3). In contrast, serotype 3 reoviruses spread neurally and hematogenously, infect neurons within the CNS, and cause fatal encephalitis (1, 4, 5). These serotype-specific differences in neuropathogenesis segregate with the viral S1 gene (2, 3), which encodes attachment protein σ1 and nonstructural protein σ1s (6–8). Both S1 gene products play key roles in reovirus pathogenesis (4, 5, 9–11), with σ1 targeting reovirus to specific host cells (12–14) and σ1s contributing to lymphatic and bloodstream spread (5, 10).
Reoviruses engage two known cellular receptors, oligosaccharides terminating in sialic acid and junctional adhesion molecule A (JAM-A), via attachment protein σ1 by using an adhesion-strengthening mechanism (15). Virions are first tethered to the cell surface by low-affinity binding to the relatively more abundant sialic acid, followed by high-affinity interactions with JAM-A (15). JAM-A is a member of the immunoglobulin superfamily and is expressed in epithelial and endothelial cells, where it functions in the formation and maintenance of tight junctions (TJs) (16–18). JAM-A also is expressed on the surface of hematopoietic cells and platelets, where it facilitates leukocyte extravasation and platelet activation, respectively (16, 19, 20). In mice, the capacity of reovirus to bind sialic acid enhances neurovirulence (9, 21) and allows infection of bile duct epithelial cells, producing a disease that mimics biliary atresia in human infants (9). In contrast, the capacity of reovirus to bind JAM-A is required for the establishment of viremia and dissemination to sites of secondary replication through the blood (4). The function of sialic acid and JAM-A in reovirus infection of polarized endothelial cells is not known.
In this study, we examined reovirus infection of polarized endothelial cells to better understand mechanisms of viral entry into and egress from the bloodstream. We found that reovirus productively infects polarized endothelial cells from both apical and basolateral routes of adsorption. Infection was more efficient after adsorption from the apical surface, a property attributable to the binding of sialic acid and JAM-A. Interestingly, reovirus was released exclusively from the apical surface in a noncytolytic manner. These studies provide a new understanding of how viruses infect polarized endothelial cells and identify the endothelium as an important mediator of viral pathogenesis.
RESULTS
Reovirus infection of polarized endothelial cells is more efficient from the apical surface.
To determine whether reovirus productively infects polarized endothelial cells (see Fig. S1 in the supplemental material), we adsorbed either the apical or the basolateral surface of polarized human brain microvascular endothelial cells (HBMECs) with strain T3SA+, a virus that efficiently binds sialic acid and JAM-A (15, 22). The viral titer in cell lysates increased over time, regardless of the route of adsorption (Fig. 1A). Following apical adsorption, the viral titer peaked at 24 h postinfection, with the yield reaching approximately 1,000-fold over the input. In contrast, following basolateral adsorption, viral replication was delayed, with yields of 5-fold at 24 h and 100-fold at 48 h postinfection. These data indicate that reovirus infection of polarized HBMECs by either the apical or the basolateral entry route is productive, but apical adsorption results in more efficient replication and increased viral yields.
FIG 1 .

Reovirus infection of polarized HBMECs is more efficient following adsorption from the apical surface. Polarized HBMECs were adsorbed either apically (white bars) or basolaterally (black bars) with reovirus T3SA+ at an MOI of 10 PFU per cell. (A) Transwell inserts were excised at 0, 24, and 48 h postinfection, and viral titers in cell lysates were determined by plaque assay. A representative experiment of three performed, with each experiment conducted in duplicate, is shown. Error bars indicate the range of data for the duplicates. (B) HBMECs were incubated for 20 to 24 h and harvested by trypsinization. Cells were permeabilized and stained with Alexa Fluor-conjugated, reovirus-specific antiserum. The percentage of infected cells was determined by flow cytometry. A representative experiment of three performed, with each experiment conducted in duplicate, is shown. Error bars indicate the range of data for the duplicates. (C) HBMECs were removed immediately after adsorption and stained with Alexa Fluor-conjugated, reovirus-specific antiserum. MFI was determined by flow cytometry. A representative experiment of three performed, with each experiment conducted in duplicate, is shown. Error bars indicate the range of data for the duplicates. **, P < 0.005.
Because we observed higher peak titers in polarized HBMECs after apical adsorption, we sought to determine whether initiation of reovirus infection is more efficient when cells are infected apically than when they are infected basolaterally. Polarized HBMECs were adsorbed with virus by the apical or basolateral route, and the percentage of reovirus antigen-positive cells was quantified by flow cytometry. Apical adsorption resulted in approximately 10-fold more infected cells than did basolateral adsorption (Fig. 1B). As a control, apical or basolateral adsorption of nonpolarized L929 fibroblast cells cultivated on Transwell inserts yielded equivalent numbers of infected cells (Fig. 1B).
To determine whether differences in infectivity are attributable to differences in virus binding, we assessed virus attachment to polarized HBMECs following apical or basolateral adsorption. In concordance with the infectivity data, approximately 10-fold more virus was bound to HBMECs following apical adsorption than following basolateral adsorption (Fig. 1C). As anticipated, virus bound equivalently to L929 fibroblasts following adsorption either apically or basolaterally (Fig. 1C). Together, these data suggest that reovirus binds more efficiently to the apical surface of polarized HBMECs, which results in increased infectivity and replication.
Sialic acid and JAM-A are required for reovirus infection of polarized endothelial cells.
To determine whether differences in the infectivity of polarized HBMECs after apical or basolateral adsorption are attributable to differences in receptor engagement, we used mutant reovirus strains impaired in the capacity to bind either sialic acid or JAM-A. Single amino acid mutations in the σ1 attachment protein can dramatically diminish binding to these receptors (15, 23). Polarized HBMECs were adsorbed apically or basolaterally with wild-type or mutant reovirus strains, and the percentage of infected cells was quantified at 24 h postinfection. There were significantly more infected cells following apical adsorption with wild-type strain type 3 Dearing (rsT3D) than after apical adsorption with mutant strain rsT3D-σ1R202W, which is deficient in sialic acid binding (21, 23), or mutant strain rsT3D-σ1G381A, which is deficient in JAM-A binding (24) (Fig. 2A). Treatment of polarized HBMECs with neuraminidase (to remove cell surface sialic acid) and JAM-A-specific antibody prior to apical virus adsorption significantly decreased infection by rsT3D. Similarly, neuraminidase and JAM-A-specific antibody pretreatment substantially decreased infection of polarized HBMECs by rsT3D-σ1G381A and rsT3D-σ1R202W, respectively (Fig. 2A). Concordantly, rsT3D bound more efficiently to the apical surface of polarized HBMECs than did the mutant virus strains, and virtually all virus binding was abolished by neuraminidase or JAM-A-specific antibody pretreatment (Fig. 2C). We observed a similar trend after basolateral adsorption in that diminished receptor engagement by mutant viruses or blockade of receptor engagement with inhibitors significantly decreased the percentages of virus-infected and virus-bound cells (Fig. 2B and D). However, the overall percentage of infected cells and levels of virus binding after basolateral adsorption were substantially lower than those following apical adsorption, which diminishes the magnitude of the observed differences (note the different y axis scales in Fig. 2C and D). Reovirus mutant rsT3D-σ1R202W bound to the basolateral surface of HBMECs equivalently to wild-type rsT3D but infected significantly fewer cells, suggesting that sialic acid engagement may enhance reovirus replication at a postattachment step following basolateral adsorption of polarized endothelial cells. These data suggest that infection of polarized endothelial cells is dependent on virus binding to sialylated glycans and JAM-A on the apical and basolateral surfaces of polarized endothelial cells, but binding to the apical surface is more efficient.
FIG 2 .
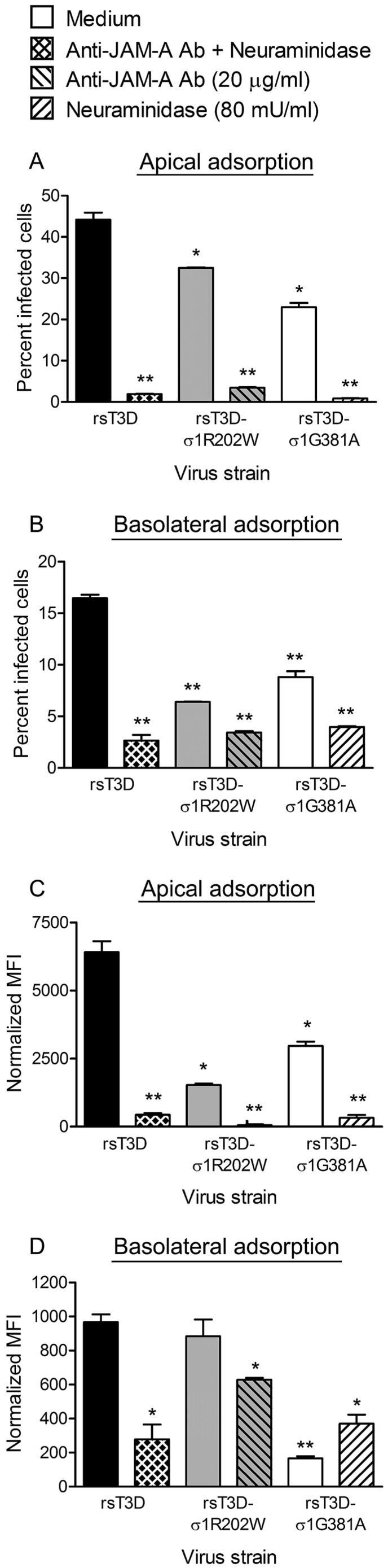
JAM-A and sialic acid are required for reovirus infection of polarized HBMECs. Polarized HBMECs were adsorbed either apically (A, C) or basolaterally (B, D) at an MOI of 10 PFU per cell with reovirus strain rsT3D, rsT3D-σ1R202W, or rsT3D-σ1G381A in the presence or absence of anti-JAM-A antibody (Ab; 20 µg/ml) or A. ureafaciens neuraminidase (80 mU/ml). (A, B) Cells were incubated for 20 to 24 h, removed from Transwell inserts with trypsin, permeabilized, and incubated with Alexa Fluor-conjugated, reovirus-specific antiserum. The percentage of infected cells was determined by flow cytometry. A representative experiment of two performed, with each experiment conducted in duplicate, is shown. Error bars indicate the range of data for the duplicates. (C, D) Cells were harvested from Transwell inserts immediately after adsorption and stained with Alexa Fluor-conjugated, reovirus-specific antiserum. MFI was quantified by flow cytometry. Note that different y axis scales are used for apical and basolateral adsorption. A representative experiment of two performed, with each experiment conducted in duplicate, is shown. Error bars indicate the range of data for the duplicates. *, P < 0.05; **, P < 0.005.
To determine whether increased binding of reovirus to the apical surface of polarized HBMECs is attributable to enhanced receptor expression, we examined the distribution of JAM-A on polarized HBMECs by confocal microscopy. Polarized HBMEC monolayers were stained with antibodies specific for TJ protein claudin-1, as well as JAM-A (Fig. 3A). Substantially more JAM-A staining was detected at the apical surface of the polarized cell monolayer (Fig. 3B), including nonjunction sites that lack detectable claudin-1 staining (Fig. 3A). Confocal micrographs of apical portions of cells showed a stippled pattern of JAM-A expression. In equatorial sections of cells, JAM-A was distributed at the cell periphery, presumably in contact with JAM-A on adjacent cells. In these images, TJ puncta marked by claudin-1 and JAM-A colocalization are clearly visible (Fig. 3A, white asterisks). At the basolateral surface, the JAM-A signal was diminished in intensity and diffusely localized compared with JAM-A staining at the apical surface (Fig. 3A and B). Increased distribution of JAM-A to the apical surface of polarized HBMECs may allow reovirus to bind and infect these cells more efficiently by this route.
FIG 3 .

Polarized HBMECs express JAM-A predominantly at the apical surface. (A) Polarized HBMECs were stained for JAM-A (green), claudin-1 (red), and nuclei (blue) and imaged by confocal microscopy. Shown are images of the apical, equatorial, and basolateral regions of a single representative z stack. Colocalization of TJ proteins is indicated by white asterisks. The scale bar indicates 10 µm. Enlarged images of the white-boxed areas are shown in the bottom panels. Cell images were captured with a Zeiss LSM 510 Meta laser-scanning confocal microscope with a 63×/1.40 Plan-Apochromat objective lens. (B) JAM-A channel MFI of apical and basolateral sections of individual cells (n = 5) was quantified. Error bars indicate standard deviations. *, P < 0.05.
Reovirus is released apically from infected polarized endothelial cells.
We next determined whether progeny virus is released apically or basolaterally from infected polarized endothelial cells. Polarized HBMECs were adsorbed apically or basolaterally with virus, and titers within the apical and basolateral compartments were quantified at various intervals by plaque assay. After apical adsorption, the viral titer in the apical compartment increased more than 30-fold at 24 h and more than 3,000-fold at 48 h (Fig. 4A). Interestingly, no virus was detected in the basolateral compartment at any time point tested (Fig. 4A). After basolateral adsorption, virus was detected in the basolateral compartment at all of the intervals tested (Fig. 4B). However, titers did not increase over time, suggesting that infectious virus in this compartment is most likely residual virus from the inoculum. The viral titer within the apical compartment was detected at 24 h postinfection and increased approximately 100,000-fold by 48 h postinfection (Fig. 4B). Therefore, regardless of the route of adsorption, reovirus egress from polarized endothelial cells occurs from the apical surface.
FIG 4 .

Reovirus release from polarized HBMECs occurs from the apical surface. Polarized HBMECs were adsorbed either apically (A) or basolaterally (B) with reovirus T3SA+ at an MOI of 10 PFU per cell. Cells were washed, fresh medium was added to the apical and basolateral compartments, and cells were incubated for the times shown. Viral titers in the medium from the apical (white bars) and basolateral (black bars) compartments were determined by plaque assay. A representative experiment of three performed, with each experiment conducted in duplicate, is shown. Error bars indicate the range of data for the duplicates.
Reovirus infection does not alter endothelial cell TJ integrity.
To determine whether reovirus infection alters the integrity of TJs in the polarized monolayer, we quantified the transendothelial electrical resistance (TEER) at both early and late times postadsorption. After adsorption with a multiplicity of infection (MOI) of 1,000 PFU per cell, no significant alteration in TEER was observed in the 2-h postinfection interval (Fig. 5A). Similarly, after adsorption with an MOI of 10 PFU per cell, no significant alteration in TEER was observed at 1 or 2 days postinfection (Fig. 5B). We conclude from these data that reovirus does not alter the function of endothelial TJs during infection.
FIG 5 .

Reovirus infection of polarized HBMECs does not disrupt TJs. Polarized HBMECs were mock infected (closed circle, solid line) or adsorbed either apically (closed circle, dashed line) or basolaterally (open circle, dotted line) with reovirus T3SA+ at an MOI of 1,000 PFU per cell (A) or 10 PFU per cell (B). Cells were washed, fresh medium was added to the apical and basolateral compartments, and TEER was determined at the times shown. A representative experiment of two (A) or three (B) performed, with each experiment conducted in duplicate, is shown. Error bars indicate the range of data for the duplicates. TEER from the various samples was compared by one-way ANOVA. Student’s t test was used to evaluate differences between mock-infected and apically infected (A) or mock-infected and basolaterally infected (B) samples. No differences were statistically significant.
Reovirus egress from polarized HBMECs occurs noncytolytically.
To determine whether reovirus egress from infected polarized HMBECs is associated with cell lysis, we assessed cell viability with trypan blue. Polarized HBMECs or confluent L929 cells cultured on Transwell inserts were adsorbed apically or basolaterally at an MOI of 10 PFU per cell, and cell viability was quantified at 24 h postinfection. Levels of HBMEC lysis were lower than the background levels of lysis in mock-treated HBMECs after either apical or basolateral virus adsorption (Fig. 6A). In contrast, more than half of the population of infected L929 cells was lysed at 24 h postinfection. These data suggest that reovirus infection of polarized HBMECs does not compromise cell viability.
FIG 6 .
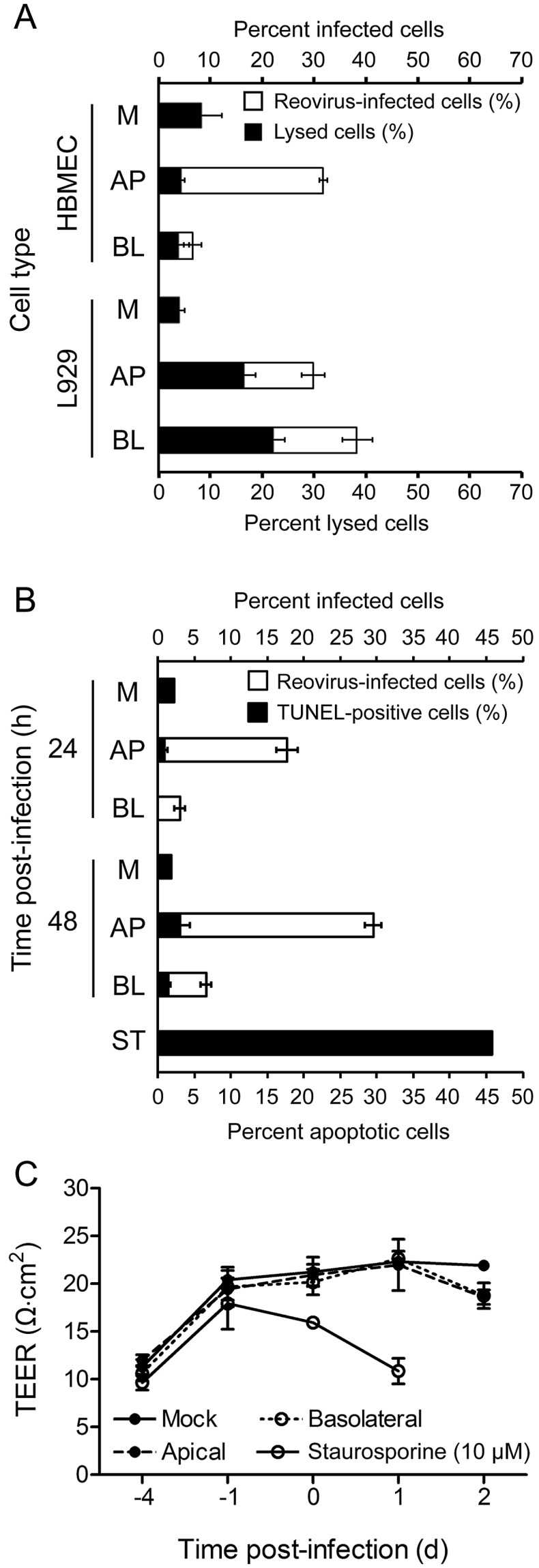
Reovirus infection of polarized HBMECs is noncytolytic. (A) Polarized HBMECs or confluent L929 cells cultured on Transwell inserts were mock infected (M) or adsorbed either apically (AP) or basolaterally (BL) with reovirus T3SA+ at an MOI of 10 PFU per cell. Cells were washed, fresh medium was added to the apical and basolateral compartments, and cells were incubated at 37°C for 20 to 24 h. Cells were harvested and incubated with trypan blue or permeabilized and stained for reovirus antigen with Alexa Fluor-conjugated, reovirus-specific antiserum. The percentage of infected cells (white bars) and the percentage of lysed cells (black bars) are shown in a stacked-column graph. A representative experiment of two performed, with each experiment conducted in duplicate, is shown. Error bars indicate the range of data for the duplicates. (B, C) Polarized HBMECs were mock infected (M) or adsorbed either apically (AP) or basolaterally (BL) with reovirus T3SA+ at an MOI of 100 PFU per cell. Cells were incubated at 37°C and harvested at 24 or 48 h postinfection. As a control for apoptosis, staurosporine (ST, 10 µM) was added to the medium in the apical and basolateral compartments of uninfected cells, which were incubated for 18 h. (B) Cells were stained for reovirus antigen with Alexa Fluor-conjugated, reovirus-specific antiserum and for apoptosis by the TUNEL technique. The percentage of infected cells (white bars) and the percentage of TUNEL-positive cells (black bars) within the population of infected cells are shown in a stacked-column graph. A representative experiment of three performed, with each experiment conducted in duplicate, is shown. Error bars indicate the range of data for the duplicates. (C) TEER was recorded for each sample at the time of cell harvest. A representative experiment of three performed, with each experiment conducted in duplicate, is shown. Error bars indicate the range of data for the duplicates.
Reovirus is capable of inducing apoptosis in many types of cultured cells (25–28) and in the CNS of infected mice (1–3, 29). Although polarized HBMECs remain intact after reovirus infection, we wondered whether reovirus egress from polarized HBMEC monolayers might occur via apoptosis. To test this hypothesis, we adsorbed polarized HBMECs apically or basolaterally at an MOI of 100 PFU per cell and quantified levels of apoptosis at 24 and 48 h postinfection by using terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) staining. At 24 h postinfection, 17.7% of the cells were infected after apical adsorption but apoptosis was detectable in only 0.9% of those cells (Fig. 6B). At 24 h after basolateral adsorption, 3.0% of the cells were infected but apoptosis was not detected in those cells (Fig. 6B). At 48 h after apical adsorption, 29.5% of the cells were infected with reovirus, with only 3.0% showing evidence of apoptosis (Fig. 6B). After basolateral adsorption, 6.6% of the cells were infected with reovirus, yet only 1.4% of those cells were apoptotic (Fig. 6B). As a positive control, treatment of polarized HBMECs with staurosporine resulted in ~50% of the cells displaying evidence of apoptosis with a concomitant decrease in TEER (Fig. 6B and C), suggesting that the low levels of apoptosis in reovirus-infected cells are not attributable to an inherent block to apoptosis in HBMECs. These data suggest that reovirus egress from polarized HBMECs occurs without inducing apoptosis.
DISCUSSION
Many viruses cause disease in infected hosts after bloodstream spread from an initial site of infection to a distant target site. Reoviruses are neurotropic viruses that first replicate within the small intestine and disseminate systemically via the blood, nerves, and lymphatics. Reovirus penetration of the endothelium to invade the bloodstream may occur within the intestine or lymph nodes to allow the establishment of primary viremia. To investigate reovirus infection of the endothelium, we cultured HBMECs on Transwell membranes until polarization was achieved (see Fig. S1 in the supplemental material). Although reoviruses use TJ protein JAM-A as a receptor, TEER was not altered immediately following reovirus adsorption (Fig. 5), suggesting that TJ integrity remains intact after infection. Adsorption of polarized endothelial cells either apically or basolaterally with reovirus resulted in productive infection (Fig. 1; see Fig. S2 in the supplemental material). Interestingly, reovirus strain T3D replicated more efficiently than strain type 1 Lang (T1L) in polarized endothelial cells (compare Fig. 1; see Fig. S2). This discrepancy might be due to differences in the cell surface expression of the sialylated glycans used by the different reovirus serotypes or cell-intrinsic properties of endothelial cells that confer serotype-dependent differences in reovirus susceptibility. Regardless of the serotype, replication was more efficient when reovirus was adsorbed to the endothelial cell apical surface (Fig. 1; see Fig. S2), and significantly more reovirus antigen-positive cells were detected following adsorption by this route (Fig. 1B; see Fig. S2). The observed increase in infectivity and replication after apical adsorption was most likely due to increased virus binding to the apical surface (Fig. 1C). The number of cells bound by virus was actually higher than the number of cells productively infected. This finding suggests that not all viral particles bound to the cell surface complete an infectious cycle, a phenomenon observed in other cell lines (30–32). Reovirus infection of polarized endothelial cells by either the apical or the basolateral route requires the engagement of sialylated glycans and JAM-A (Fig. 2). Consistent with these findings, substantially more JAM-A is distributed to the apical than to the basolateral surface of polarized HBMECs (Fig. 3). Subconfluent, nonpolarized HBMECs are substantially more susceptible to reovirus infection than are polarized HBMECs (data not shown), presumably because of higher levels of JAM-A on the cell surface and the absence of a restriction of JAM-A expression to TJs.
Regardless of the route of adsorption, reovirus egress from infected polarized HBMECs occurs solely from the apical surface (Fig. 4; see Fig. S2 in the supplemental material). Similarly, reovirus infection of polarized human airway epithelial cells results in apical release of progeny virions (33). Although TEER did not change appreciably over a time course of reovirus infection of HBMECs (Fig. 5), we questioned whether infected cells are extruded from the monolayer in a manner analogous to epithelial cell turnover (34). If they are, we would expect TEER to be maintained despite the detection of an increased number of nonviable cells over time. To test this hypothesis, we used trypan blue staining to determine whether polarized HBMECs infected with reovirus are lysed. Compared with infected L929 cells, which display substantial cytopathic effect after reovirus infection (28) (Fig. 6A), polarized HBMECs infected with reovirus apically or basolaterally do not undergo cell lysis (Fig. 6A), despite the presence of high viral titers in cells and supernatants (Fig. 1 and 4). Sonication of supernatants harvested from the apical surface of polarized HBMECs did not lead to an increased viral titer, suggesting that released virus was not trapped within extruded cells or membrane-bound vesicles (data not shown). Apical or basolateral adsorption of polarized HBMECs with reovirus led to an increase in reovirus antigen-positive cells, but the number of apoptotic cells did not increase above that in mock-treated samples (Fig. 6B). Additionally, levels of apoptosis in reovirus-infected HBMECs were lower than in mock-infected cells by the complementary acridine orange and annexin V staining assays (see Fig. S3 in the supplemental material). We conclude from these data that regardless of the route of entry, reovirus release occurs from the apical surface in a manner that maintains cell viability. Because infection of polarized endothelial cells is noncytolytic, clearance of reovirus from an infected host may require cytotoxic T lymphocyte-mediated immunity in addition to neutralizing antibodies (35–39).
Virus infection of endothelial cells may serve as an additional mechanism to produce and maintain high levels of viremia. For example, dengue virus infection of endothelial cells leads to high-titer viremia by inducing endothelial cell apoptosis, resulting in endothelial barrier dysfunction and vascular leakage (40). Murine cytomegalovirus primarily infects hepatocytes, but virus produced from infected hepatic endothelial cells is responsible for dissemination to other organs (41, 42). Similarly, reovirus may use the endothelium as a means to amplify to high titers in the bloodstream (Fig. 7). Reovirus infection from the basolateral route is not efficient, but progeny viral particles are efficiently transported to and released from the apical surface of polarized endothelial cells. Once released, progeny virions have access to the apical surface of adjacent endothelial cells and can enter those cells efficiently. This cycle may serve as a mechanism to generate high titers of virus in the bloodstream, which are observed during reovirus infection (4, 10, 21). Sialylated glycans and JAM-A are required for the infection of endothelial cells by both the apical and basolateral routes, which may account for the markedly diminished viremia in reovirus-infected JAM-A-deficient mice (4).
FIG 7 .

Model of reovirus infection of the endothelium. A cross-sectional schematic of a blood vessel is shown. The blood vessel is lined with endothelial cells that are linked via TJs (black bars). Following reovirus infection of endothelial cells from the basolateral surface (step 1), virus is routed apically (or luminally) into the bloodstream (step 2). Once within the bloodstream, virus is capable of infecting endothelial cells from the apical surface (step 3). Reovirus binding to JAM-A, found mostly within TJs, and sialic acid at the apical surface may account for the increased efficiency of infection. After reovirus infects cells from the apical surface, progeny virions are routed apically into the bloodstream. The efficiency of apical infection may allow for endothelial amplification of reovirus (step 4), resulting in higher levels of viremia within an infected host.
How reovirus exits the bloodstream is not clear from our study. Because JAM-A is present on the surface of hematopoietic cells, it is possible that reovirus-infected hematopoietic cells transport the virus from the bloodstream to sites of secondary replication, including the CNS. It also is possible that cells adjacent to blood vessels become infected as a consequence of infection of the endothelium. Epstein-Barr virus (EBV) binding to B cells leads to conjugate formation between B cells and epithelial cells, resulting in EBV entry into epithelial cells (43, 44). Blood vessels in the brain closely appose pericytes and astrocytes, and reovirus infection of endothelial cells may induce modifications of these cells, resulting in invasion of the CNS.
Bloodstream spread is an important step in the pathogenesis of many viral diseases, but the mechanisms used by viruses to gain entry into the bloodstream are not well understood. Our work describes how viral infection of endothelial cells may allow access to and amplification within the circulation. We show that reovirus productively infects polarized endothelial cells by both the apical and basolateral routes. Infection after apical adsorption is more efficient than basolateral adsorption because of increased utilization of sialic acid and JAM-A at the apical surface. Reovirus release from polarized endothelial cells occurs exclusively from the apical surface in a manner that maintains TJ integrity and cell viability. Since TJ proteins are used as receptors by a diverse array of viruses, including adenovirus (45), feline calicivirus (46), hepatitis C virus (47, 48), and several picornaviruses (45, 49), our findings may provide a more general understanding of how viruses establish viremia for bloodstream spread. Moreover, the apical release mechanism employed by reovirus may be similarly generalizable, providing a potential new target for a host-specific, broad-spectrum antiviral therapeutic.
MATERIALS AND METHODS
Cells, viruses, enzymes, and antibodies.
Spinner-adapted murine L929 fibroblast cells were grown in either suspension or monolayer cultures as previously described (10, 50). HBMECs (51, 52) were grown in RPMI 1640 medium (Mediatech) supplemented to contain 10% fetal bovine serum, 10% NuSerum (BD Biosciences), nonessential amino acids (Sigma), 1 mM sodium pyruvate, MEM Vitamins (Mediatech), 2 mM l-glutamine, 100 U/ml penicillin, 100 µg/ml streptomycin, and 25 ng/ml amphotericin B. HBMECs and L929 cells were cultured on collagen-coated Transwell inserts (6.5-mm diameter, 0.4-µm pores; Costar) for 7 days prior to infection or imaging experiments.
Reovirus strain T1L is a laboratory stock. Strain T3SA+ was generated as previously described (15). Recombinant viruses rsT3D, rsT3D-σ1R202W, and rsT3D-σ1G381A were generated by plasmid-based reverse genetics (21, 24). Virus was purified as previously described (53). Viral titers were determined by plaque assay with L929 cells (37).
The immunoglobulin G (IgG) fraction of a rabbit antiserum raised against strains T1L and T3D (31) was purified by protein A-Sepharose as previously described (9, 15). Reovirus-specific IgG was conjugated to Alexa Fluor 647 with an APEX antibody labeling kit (Invitrogen). JAM-A-specific monoclonal antibody J10.4 (provided by Charles Parkos, Emory University) and claudin-1-specific antibody ab15098 (Abcam) were used in confocal microscopy imaging experiments. Alexa Fluor-conjugated antibodies (Invitrogen) were used as secondary antibodies.
TEER measurements.
TEER across polarized HBMEC monolayers was quantified at 3 and 6 days postseeding, on the day of infection, and at various intervals postinfection with an EVOM voltohmmeter and an EndOhm-6 cup electrode (World Precision Instruments). TEER readings for test samples were normalized by subtracting the TEER of blank collagen-coated Transwell inserts. The data are presented as unit area resistance (Ω·cm2) (54).
Virus assays.
Polarized HBMECs cultivated on Transwell inserts were adsorbed with virus apically or basolaterally at an MOI of 10 PFU per cell. For apical adsorption, 30 µl of virus inoculum was added to the apical compartment. For basolateral adsorption, the Transwell insert was inverted in a sterile dish and 30 µl of virus inoculum was added to the basolateral surface. In some experiments, cells were treated with medium, anti-JAM-A antibody (20 µg/ml), or Arthrobacter ureafaciens neuraminidase (80 mU/ml; MP Biomedicals) prior to virus adsorption. After adsorption of virus at room temperature for 1 h, cells were washed twice with phosphate-buffered saline (PBS) and 200 µl of medium was added to the apical compartment and 1 ml of medium was added to the basolateral compartment. For viral release assays, medium from the apical or basolateral compartment was collected at various intervals and viral titers in medium from each compartment were determined by plaque assay with L929 cells (37). For viral replication assays, Transwell membrane inserts were removed from Transwell inserts with a scalpel, submerged in 500 µl of medium, and subjected to two cycles of freezing and thawing. Viral titers in cell lysates were determined by plaque assay with L929 cells (37).
For infectivity studies, cells were incubated at 37°C for 20 to 24 h, harvested with 0.05% trypsin-EDTA (Invitrogen) at room temperature, and quenched with medium collected from the apical compartment of the respective sample. Cells were stained with Alexa Fluor-conjugated, reovirus-specific antiserum as previously described (50). The percentage of reovirus antigen-positive cells was determined by flow cytometry. For binding studies, cells were detached from the Transwell insert immediately after adsorption with Cellstripper (Mediatech) at 37°C for 5 min and stained with Alexa Fluor-conjugated, reovirus-specific antiserum as previously described (50). The mean fluorescence intensity (MFI) of each sample was determined by flow cytometry. All cell staining was quantified with FlowJo software (Tree Star).
Cell imaging.
Polarized HBMECs were fixed in 100% methanol at −20°C for 5 min. Cells were blocked in PBS containing 5% bovine serum albumin at room temperature for 30 min. Cells were stained with antibodies specific for JAM-A (1:1,000) and claudin-1 (1:100) as previously described (50, 55). After staining, Transwell membranes containing cells were excised with a scalpel. Membranes were placed onto glass slides, and glass coverslips (#1.5; Thermo Scientific) were mounted with Aqua-Poly/Mount mounting medium (Polysciences, Inc.). Cell images were captured with a Zeiss LSM 510 Meta laser-scanning confocal microscope with a 63×/1.40 Plan-Apochromat objective lens. A standard threshold pixel intensity was used for all images, and the pinhole size used was the same for all fluorophores. Images represent a single section or a series of sections from within a z stack and were adjusted for brightness and contrast to the same extent. The MFI of pixels from apical and basolateral sections of cells (n = 5) was quantified with ImageJ software (NIH).
Trypan blue exclusion assay.
HBMECs and L929 cells were cultured on Transwell inserts until polarized and confluent, respectively. Virus was adsorbed apically or basolaterally at an MOI of 10 PFU per cell, and cells were incubated at 37°C for 20 to 24 h. After incubation, cells were harvested with trypsin-EDTA, quenched with medium collected from the apical compartment, and washed once with PBS. A small aliquot (20 µl) of cells was removed for analysis of cell lysis. An equal volume of trypan blue (0.4% [wt/vol] in PBS; Mediatech) was added to cells, which were then incubated at room temperature for 3 min. Lysed and intact cells were enumerated using a hemocytometer with bright-field microscopy. The percentage of reovirus-infected cells in the remainder of each sample was quantified by flow cytometry.
TUNEL assay.
Polarized HBMECs were adsorbed with virus at an MOI of 100 PFU per cell, washed twice with PBS, and incubated at 37°C for 24 or 48 h. Cells were removed from the Transwell insert with trypsin-EDTA, quenched with medium collected from the apical compartment, washed once with PBS, and assayed for the percentage of apoptotic cells by the TUNEL technique (APO-BrdU TUNEL assay kit; Invitrogen) according to the manufacturer’s instructions. After TUNEL staining, cells were stained with Alexa Fluor-conjugated, reovirus-specific antiserum (1:1,000) at 4°C for 30 min, washed, and pelleted. The samples were resuspended in 0.5 ml propidium iodide-containing buffer. Stained cells were analyzed for apoptosis and the presence of reovirus antigen by flow cytometry. See Text S1 in the supplemental material for the additional methods used.
Statistical analysis.
Experiments were performed in duplicate and repeated at least twice. Representative results of single experiments are shown. Mean values were compared with an unpaired Student's t test or one-way analysis of variance (ANOVA) (GraphPad Prism). Error bars denote the range of data or standard deviation. P values of <0.05 were considered statistically significant.
SUPPLEMENTAL MATERIAL
Supplemental methods used in this study. Download
Barrier properties of polarized HBMECs. (A) HBMECs (solid line) and L929 cells (dashed line) cultivated on Transwell inserts were monitored for TEER daily for 12 days. The data are presented as unit area resistance, where normalized TEER is multiplied by the area of the Transwell insert (Ω·cm2). A representative experiment of two performed, with each experiment conducted in duplicate, is shown. Error bars indicate the range of data for the duplicates. (B) Fluorescein isothiocyanate (FITC)-labeled dextrans were added to the apical compartment of HBMECs cultivated on Transwell inserts at 1, 4, or 7 days postseeding. Prior to the addition of dextrans, TEER (Ω·cm2) was determined and is presented in parentheses above each bar. The percent permeability was determined with the following equation: Permeability (%) = [FITC-dextran]basolateral/([FITC-dextran]basolateral + [FITC-dextran]apical) × 100. On day 7, 2.5 mM EDTA was added to the apical and basolateral compartments as a control to disrupt TJs. A representative experiment of three performed, with each experiment conducted in duplicate, is shown. Error bars indicate the range of data for the duplicates. (C) HBMECs cultured on Transwell inserts for 7 days were stained for TJ proteins claudin-1 (red) and JAM-A (green) and nuclei (blue). At the bottom of the merged image, blue staining shows the Transwell membrane. Representative images of the cell monolayer in the x-z plane are shown. White asterisks indicate colocalization of TJ proteins. Cell images were captured with a Zeiss LSM 510 Meta laser-scanning confocal microscope with a 63×/1.40 Plan-Apochromat objective lens. Download
T1L infection of polarized HBMECs is more efficient by the apical route. Polarized HBMECs were adsorbed either apically or basolaterally with reovirus T1L at an MOI of 10 PFU per cell. After adsorption of virus, cells were incubated for various intervals. (A) Transwell inserts were excised at 0, 24, and 48 h postinfection, and viral titers in cell lysates were determined by plaque assay. A representative experiment of two performed, with each experiment conducted in duplicate, is shown. Error bars indicate the range of data for the duplicates. (B) HBMECs were incubated for 20 to 24 h and harvested by trypsinization. Cells were permeabilized and stained with Alexa Fluor-conjugated, reovirus-specific antiserum, and the percentage of infected cells was determined by flow cytometry. A representative experiment of two performed, with each experiment conducted in duplicate, is shown. Error bars indicate the range of data for the duplicates. (C, D) After adsorption of polarized HBMECs with virus from either the apical (C) or the basolateral (D) surface, medium from the apical (white bars) and basolateral (black bars) compartments was harvested at various intervals and viral titers in the medium were determined by plaque assay. A representative experiment of three performed, with each experiment conducted in duplicate, is shown. Error bars indicate the range of data for the duplicates. Download
Reovirus release from polarized HBMECs occurs noncytolytically. Polarized HBMECs were mock infected (M) or adsorbed either apically (AP) or basolaterally (BL) with reovirus T3SA+ at an MOI of 100 PFU per cell. Cells were incubated at 37°C and harvested at 24 h postinfection. As a control for apoptosis, staurosporine (ST, 10 µM) was added to the medium in the apical and basolateral compartments of uninfected cells, which were incubated for 18 h. (a) Cells were harvested, washed, and stained with acridine orange dye. The apoptotic cells were enumerated under bright-field microscopy. A representative experiment of three performed, with each experiment conducted in duplicate, is shown. Error bars indicate the range of data for the duplicates. (b) Cells were harvested and stained either for apoptosis with Alexa Fluor-conjugated antibody specific for annexin V or for reovirus antigen with Alexa Fluor-conjugated, reovirus-specific antiserum. The percentage of infected cells (in parentheses above the respective bars) and the percentage of annexin V-positive cells are shown. A representative experiment of three performed, with each experiment conducted in duplicate, is shown. Error bars indicate the range of data for the duplicates. Download
ACKNOWLEDGMENTS
We thank members of the Dermody laboratory for many useful discussions and Alison Ashbrook, Jennifer Konopka, and Jennifer Stencel for critical review of the manuscript.
This research was supported by Public Health Service awards T32 GM007347 (C.M.L.), F31 NS074596 (C.M.L.), T32 HL07751 (B.A.M.), F32 A1801082 (B.A.M.), and R37 AI38296 (T.S.D.) and the Elizabeth B. Lamb Center for Pediatric Research. Additional support was provided by the Vanderbilt Institute for Clinical and Translational Research (UL1 RR024975), the Vanderbilt-Ingram Cancer Center (CA68485), the Vanderbilt Flow Cytometry Shared Resource (DK058404), and the Vanderbilt Cell Imaging Shared Resource (DK20593).
Footnotes
Citation Lai CM, Mainou BA, Kim KS, Dermody TS. 2013. Directional release of reovirus from the apical surface of polarized endothelial cells. mBio 4(2):e00049-13. doi:10.1128/mBio.00049-13.
REFERENCES
- 1. Tyler KL, McPhee DA, Fields BN. 1986. Distinct pathways of viral spread in the host determined by reovirus S1 gene segment. Science 233:770–774 [DOI] [PubMed] [Google Scholar]
- 2. Weiner HL, Drayna D, Averill DR, Jr, Fields BN. 1977. Molecular basis of reovirus virulence: role of the S1 gene. Proc. Natl. Acad. Sci. U. S. A. 74:5744–5748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Weiner HL, Powers ML, Fields BN. 1980. Absolute linkage of virulence and central nervous system cell tropism of reoviruses to viral hemagglutinin. J. Infect. Dis. 141:609–616 [DOI] [PubMed] [Google Scholar]
- 4. Antar AR, Konopka JL, Campbell JA, Henry RA, Perdigoto AL, Carter BD, Pozzi A, Abel TW, Dermody TS. 2009. Junctional adhesion molecule-A is required for hematogenous dissemination of reovirus. Cell Host Microbe 5:59–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Boehme KW, Frierson JM, Konopka JL, Kobayashi T, Dermody TS. 2011. The reovirus sigma1s protein is a determinant of hematogenous but not neural virus dissemination in mice. J. Virol. 85:11781–11790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jacobs BL, Atwater JA, Munemitsu SM, Samuel CE. 1985. Biosynthesis of reovirus-specified polypeptides: the s1 mRNA synthesized in vivo is structurally and functionally indistinguishable from in vitro-synthesized s1 mRNA and encodes two polypeptides, sigma 1a and sigma 1bNS. Virology 147:9–18 [DOI] [PubMed] [Google Scholar]
- 7. Jacobs BL, Samuel CE. 1985. Biosynthesis of reovirus-specified polypeptides: the reovirus S1 mRNA encodes two primary translation products. Virology 143:63–74 [DOI] [PubMed] [Google Scholar]
- 8. Sarkar G, Pelletier J, Bassel-Duby R, Jayasuriya A, Fields BN, Sonenberg N. 1985. Identification of a new polypeptide coded by reovirus gene S1. J. Virol. 54:720–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Barton ES, Youree BE, Ebert DH, Forrest JC, Connolly JL, Valyi-Nagy T, Washington K, Wetzel JD, Dermody TS. 2003. Utilization of sialic acid as a coreceptor is required for reovirus-induced biliary disease. J. Clin. Invest. 111:1823–1833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Boehme KW, Guglielmi KM, Dermody TS. 2009. Reovirus nonstructural protein sigma1s is required for establishment of viremia and systemic dissemination. Proc. Natl. Acad. Sci. U. S. A. 106:19986–19991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kaye KM, Spriggs DR, Bassel-Duby R, Fields BN, Tyler KL. 1986. Genetic basis for altered pathogenesis of an immune-selected antigenic variant of reovirus type 3 (Dearing). J. Virol. 59:90–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tardieu M, Weiner HL. 1982. Viral receptors on isolated murine and human ependymal cells. Science 215:419–421 [DOI] [PubMed] [Google Scholar]
- 13. Dichter MA, Weiner HL. 1984. Infection of neuronal cell cultures with reovirus mimics in vitro patterns of neurotropism. Ann. Neurol. 16:603–610 [DOI] [PubMed] [Google Scholar]
- 14. Tardieu M, Powers ML, Hafler DA, Hauser SL, Weiner HL. 1984. Autoimmunity following viral infection: demonstration of monoclonal antibodies against normal tissue following infection of mice with reovirus and demonstration of shared antigenicity between virus and lymphocytes. Eur. J. Immunol. 14:561–565 [DOI] [PubMed] [Google Scholar]
- 15. Barton ES, Connolly JL, Forrest JC, Chappell JD, Dermody TS. 2001. Utilization of sialic acid as a coreceptor enhances reovirus attachment by multistep adhesion strengthening. J. Biol. Chem. 276:2200–2211 [DOI] [PubMed] [Google Scholar]
- 16. Bazzoni G. 2003. The JAM family of junctional adhesion molecules. Curr. Opin. Cell Biol. 15:525–530 [DOI] [PubMed] [Google Scholar]
- 17. Martìn-Padura I, Lostaglio S, Schneemann M, Williams L, Romano M, Fruscella P, Panzeri C, Stoppacciaro A, Ruco L, Villa A, Simmons D, Dejana E. 1998. Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J. Cell Biol. 142:117–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Woodfin A, Reichel CA, Khandoga A, Corada M, Voisin MB, Scheiermann C, Haskard DO, Dejana E, Krombach F, Nourshargh S. 2007. JAM-A mediates neutrophil transmigration in a stimulus-specific manner in vivo: evidence for sequential roles for JAM-A and PECAM-1 in neutrophil transmigration. Blood 110:1848–1856 [DOI] [PubMed] [Google Scholar]
- 19. Sobocka MB, Sobocki T, Banerjee P, Weiss C, Rushbrook JI, Norin AJ, Hartwig J, Salifu MO, Markell MS, Babinska A, Ehrlich YH, Kornecki E. 2000. Cloning of the human platelet F11 receptor: a cell adhesion molecule member of the immunoglobulin superfamily involved in platelet aggregation. Blood 95:2600–2609 [PubMed] [Google Scholar]
- 20. Ostermann G, Weber KS, Zernecke A, Schröder A, Weber C. 2002. Jam-1 is a ligand of the beta(2) integrin LFA-1 involved in transendothelial migration of leukocytes. Nat. Immunol. 3:151–158 [DOI] [PubMed] [Google Scholar]
- 21. Frierson JM, Pruijssers AJ, Konopka JL, Reiter DM, Abel TW, Stehle T, Dermody TS. 2012. Utilization of sialylated glycans as coreceptors enhances the neurovirulence of serotype 3 reovirus. J. Virol. 86:13164–13173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Barton ES, Forrest JC, Connolly JL, Chappell JD, Liu Y, Schnell FJ, Nusrat A, Parkos CA, Dermody TS. 2001. Junction adhesion molecule is a receptor for reovirus. Cell 104:441–451 [DOI] [PubMed] [Google Scholar]
- 23. Reiter DM, Frierson JM, Halvorson EE, Kobayashi T, Dermody TS, Stehle T. 2011. Crystal structure of reovirus attachment protein σ1 in complex with sialylated oligosaccharides. PLoS Pathog. 7:e1002166 http://doi.10.1371/journal.ppat.1002166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kirchner E, Guglielmi KM, Strauss HM, Dermody TS, Stehle T. 2008. Structure of reovirus sigma1 in complex with its receptor junctional adhesion molecule-A. PLoS Pathog. 4:e1000235 http://doi.10.1371/journal.ppat.1000235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Connolly JL, Rodgers SE, Clarke P, Ballard DW, Kerr LD, Tyler KL, Dermody TS. 2000. Reovirus-induced apoptosis requires activation of transcription factor NF-kappaB. J. Virol. 74:2981–2989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Connolly JL, Barton ES, Dermody TS. 2001. Reovirus binding to cell surface sialic acid potentiates virus-induced apoptosis. J. Virol. 75:4029–4039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rodgers SE, Barton ES, Oberhaus SM, Pike B, Gibson CA, Tyler KL, Dermody TS. 1997. Reovirus-induced apoptosis of MDCK cells is not linked to viral yield and is blocked by Bcl-2. J. Virol. 71:2540–2546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tyler KL, Squier MK, Rodgers SE, Schneider BE, Oberhaus SM, Grdina TA, Cohen JJ, Dermody TS. 1995. Differences in the capacity of reovirus strains to induce apoptosis are determined by the viral attachment protein sigma 1. J. Virol. 69:6972–6979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Morrison LA, Sidman RL, Fields BN. 1991. Direct spread of reovirus from the intestinal lumen to the central nervous system through vagal autonomic nerve fibers. Proc. Natl. Acad. Sci. U. S. A. 88:3852–3856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dermody TS, Nibert ML, Wetzel JD, Tong X, Fields BN. 1993. Cells and viruses with mutations affecting viral entry are selected during persistent infections of L cells with mammalian reoviruses. J. Virol. 67:2055–2063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wetzel JD, Chappell JD, Fogo AB, Dermody TS. 1997. Efficiency of viral entry determines the capacity of murine erythroleukemia cells to support persistent infections by mammalian reoviruses. J. Virol. 71:299–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rubin DH, Wetzel JD, Williams WV, Cohen JA, Dworkin C, Dermody TS. 1992. Binding of type 3 reovirus by a domain of the sigma 1 protein important for hemagglutination leads to infection of murine erythroleukemia cells. J. Clin. Invest. 90:2536–2542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Excoffon KJ, Guglielmi KM, Wetzel JD, Gansemer ND, Campbell JA, Dermody TS, Zabner J. 2008. Reovirus preferentially infects the basolateral surface and is released from the apical surface of polarized human respiratory epithelial cells. J. Infect. Dis. 197:1189–1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gu Y, Rosenblatt J. 2012. New emerging roles for epithelial cell extrusion. Curr. Opin. Cell Biol. 24:865–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Barkon ML, Haller BL, Virgin HW. 1996. Circulating immunoglobulin G can play a critical role in clearance of intestinal reovirus infection. J. Virol. 70:1109–1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. George A, Kost SI, Witzleben CL, Cebra JJ, Rubin DH. 1990. Reovirus-induced liver disease in severe combined immunodeficient (SCID) mice. A model for the study of viral infection, pathogenesis, and clearance. J. Exp. Med. 171:929–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Virgin HW. IV, Bassel-Duby R, Fields BN, Tyler KL. 1988. Antibody protects against lethal infection with the neurally spreading reovirus type 3 (Dearing). J. Virol. 62:4594–4604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Virgin HW, Tyler KL. 1991. Role of immune cells in protection against and control of reovirus infection in neonatal mice. J. Virol. 65:5157–5164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tyler KL, Mann MA, Fields BN, Virgin HW., IV 1993. Protective anti-reovirus monoclonal antibodies and their effects on viral pathogenesis. J. Virol. 67:3446–3453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dalrymple NA, Mackow ER. 2012. Roles for endothelial cells in dengue virus infection. Adv. Virol. 2012:840654 PubMed; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sacher T, Podlech J, Mohr CA, Jordan S, Ruzsics Z, Reddehase MJ, Koszinowski UH. 2008. The major virus-producing cell type during murine cytomegalovirus infection, the hepatocyte, is not the source of virus dissemination in the host. Cell Host Microbe 3:263–272 [DOI] [PubMed] [Google Scholar]
- 42. Sacher T, Andrassy J, Kalnins A, Dölken L, Jordan S, Podlech J, Ruzsics Z, Jauch KW, Reddehase MJ, Koszinowski UH. 2011. Shedding light on the elusive role of endothelial cells in cytomegalovirus dissemination. PLoS Pathog. 7:e1002366 http://doi.10.1371/journal.ppat.1002366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shannon-Lowe CD, Neuhierl B, Baldwin G, Rickinson AB, Delecluse HJ. 2006. Resting B cells as a transfer vehicle for Epstein-Barr virus infection of epithelial cells. Proc. Natl. Acad. Sci. U. S. A. 103:7065–7070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shannon-Lowe C, Rowe M. 2011. Epstein-Barr virus infection of polarized epithelial cells via the basolateral surface by memory B cell-mediated transfer infection. PLoS Pathog. 7:e1001338 http://doi.10.1371/journal.ppat.1001338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bergelson JM, Cunningham JA, Droguett G, Kurt-Jones EA, Krithivas A, Hong JS, Horwitz MS, Crowell RL, Finberg RW. 1997. Isolation of a common receptor for coxsackie B viruses and adenoviruses 2 and 5. Science 275:1320–1323 [DOI] [PubMed] [Google Scholar]
- 46. Makino A, Shimojima M, Miyazawa T, Kato K, Tohya Y, Akashi H. 2006. Junctional adhesion molecule 1 is a functional receptor for feline calicivirus. J. Virol. 80:4482–4490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Evans MJ, von Hahn T, Tscherne DM, Syder AJ, Panis M, Wölk B, Hatziioannou T, McKeating JA, Bieniasz PD, Rice CM. 2007. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 446:801–805 [DOI] [PubMed] [Google Scholar]
- 48. Ploss A, Evans MJ, Gaysinskaya VA, Panis M, You H, de Jong YP, Rice CM. 2009. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 457:882–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bewley MC, Springer K, Zhang YB, Freimuth P, Flanagan JM. 1999. Structural analysis of the mechanism of adenovirus binding to its human cellular receptor, CAR. Science 286:1579–1583 [DOI] [PubMed] [Google Scholar]
- 50. Mainou BA, Dermody TS. 2012. Transport to late endosomes is required for efficient reovirus infection. J. Virol. 86:8346–8358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Stins MF, Badger J, Sik Kim K. 2001. Bacterial invasion and transcytosis in transfected human brain microvascular endothelial cells. Microb. Pathog. 30:19–28 [DOI] [PubMed] [Google Scholar]
- 52. Stins MF, Gilles F, Kim KS. 1997. Selective expression of adhesion molecules on human brain microvascular endothelial cells. J. Neuroimmunol. 76:81–90 [DOI] [PubMed] [Google Scholar]
- 53. Furlong DB, Nibert ML, Fields BN. 1988. Sigma 1 protein of mammalian reoviruses extends from the surfaces of viral particles. J. Virol. 62:246–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Grab DJ, Perides G, Dumler JS, Kim KJ, Park J, Kim YV, Nikolskaia O, Choi KS, Stins MF, Kim KS. 2005. Borrelia burgdorferi, host-derived proteases, and the blood-brain barrier. Infect. Immun. 73:1014–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mainou BA, Dermody TS. 2011. Src kinase mediates productive endocytic sorting of reovirus during cell entry. J. Virol. 85:3203–3213 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental methods used in this study. Download
Barrier properties of polarized HBMECs. (A) HBMECs (solid line) and L929 cells (dashed line) cultivated on Transwell inserts were monitored for TEER daily for 12 days. The data are presented as unit area resistance, where normalized TEER is multiplied by the area of the Transwell insert (Ω·cm2). A representative experiment of two performed, with each experiment conducted in duplicate, is shown. Error bars indicate the range of data for the duplicates. (B) Fluorescein isothiocyanate (FITC)-labeled dextrans were added to the apical compartment of HBMECs cultivated on Transwell inserts at 1, 4, or 7 days postseeding. Prior to the addition of dextrans, TEER (Ω·cm2) was determined and is presented in parentheses above each bar. The percent permeability was determined with the following equation: Permeability (%) = [FITC-dextran]basolateral/([FITC-dextran]basolateral + [FITC-dextran]apical) × 100. On day 7, 2.5 mM EDTA was added to the apical and basolateral compartments as a control to disrupt TJs. A representative experiment of three performed, with each experiment conducted in duplicate, is shown. Error bars indicate the range of data for the duplicates. (C) HBMECs cultured on Transwell inserts for 7 days were stained for TJ proteins claudin-1 (red) and JAM-A (green) and nuclei (blue). At the bottom of the merged image, blue staining shows the Transwell membrane. Representative images of the cell monolayer in the x-z plane are shown. White asterisks indicate colocalization of TJ proteins. Cell images were captured with a Zeiss LSM 510 Meta laser-scanning confocal microscope with a 63×/1.40 Plan-Apochromat objective lens. Download
T1L infection of polarized HBMECs is more efficient by the apical route. Polarized HBMECs were adsorbed either apically or basolaterally with reovirus T1L at an MOI of 10 PFU per cell. After adsorption of virus, cells were incubated for various intervals. (A) Transwell inserts were excised at 0, 24, and 48 h postinfection, and viral titers in cell lysates were determined by plaque assay. A representative experiment of two performed, with each experiment conducted in duplicate, is shown. Error bars indicate the range of data for the duplicates. (B) HBMECs were incubated for 20 to 24 h and harvested by trypsinization. Cells were permeabilized and stained with Alexa Fluor-conjugated, reovirus-specific antiserum, and the percentage of infected cells was determined by flow cytometry. A representative experiment of two performed, with each experiment conducted in duplicate, is shown. Error bars indicate the range of data for the duplicates. (C, D) After adsorption of polarized HBMECs with virus from either the apical (C) or the basolateral (D) surface, medium from the apical (white bars) and basolateral (black bars) compartments was harvested at various intervals and viral titers in the medium were determined by plaque assay. A representative experiment of three performed, with each experiment conducted in duplicate, is shown. Error bars indicate the range of data for the duplicates. Download
Reovirus release from polarized HBMECs occurs noncytolytically. Polarized HBMECs were mock infected (M) or adsorbed either apically (AP) or basolaterally (BL) with reovirus T3SA+ at an MOI of 100 PFU per cell. Cells were incubated at 37°C and harvested at 24 h postinfection. As a control for apoptosis, staurosporine (ST, 10 µM) was added to the medium in the apical and basolateral compartments of uninfected cells, which were incubated for 18 h. (a) Cells were harvested, washed, and stained with acridine orange dye. The apoptotic cells were enumerated under bright-field microscopy. A representative experiment of three performed, with each experiment conducted in duplicate, is shown. Error bars indicate the range of data for the duplicates. (b) Cells were harvested and stained either for apoptosis with Alexa Fluor-conjugated antibody specific for annexin V or for reovirus antigen with Alexa Fluor-conjugated, reovirus-specific antiserum. The percentage of infected cells (in parentheses above the respective bars) and the percentage of annexin V-positive cells are shown. A representative experiment of three performed, with each experiment conducted in duplicate, is shown. Error bars indicate the range of data for the duplicates. Download