

Abstract
Increases in S-nitrosylation and inactivation of the neuroprotective ubiquitin E3 ligase, parkin, in the brains of patients with Parkinson’s Disease (PD) are thought to be pathogenic and suggest a possible mechanism linking parkin to sporadic PD. Here we demonstrate that physiologic modification of parkin by hydrogen sulfide (H2S), termed sulfhydration, enhances its catalytic activity. Sulfhydration sites are identified by mass spectrometry analysis and investigated by site directed mutagenesis. Parkin sulfhydration is markedly depleted in the brains of patients with PD, suggesting that this loss may be pathologic. This implies that H2S donors may be therapeutic.
INTRODUCTION
Parkin is an E3 ubiquitin ligase that ubiquitinates diverse substrates1. Mutations in parkin which disrupt its catalytic activity are the most common cause of autosomal recessive Parkinson’s Disease (PD), indicating that loss of parkin is neurotoxic, while its enhancement is neuroprotective1,2. Parkin may also participate in the pathophysiology of the much more common sporadic form of PD based on interactions with nitric oxide (NO). Dawson and associates3 and Lipton and colleagues4 reported that parkin is S-nitrosylated with greatly increased nitrosylation in brains of patients with PD. Nitrosylation of parkin inhibits its E3 ligase and neuroprotective activities, implying that the increased nitrosylation of parkin in PD is pathogenic. Recently, hydrogen sulfide (H2S) has been appreciated as a gasotransmitter comparable to NO and CO5–8. H2S signals primarily by attaching to SH groups of cysteines in proteins, a process termed sulfhydration9. Sulfhydration generally occurs on the same cysteines as nitrosylation so that the two processes may be reciprocal. This prompted us to explore relative roles of sulfhydration and nitrosylation in the function of parkin.
We report that parkin is physiologically sulfhydrated and that, whereas nitrosylation inactivates parkin, sulfhydration stimulates its activity. We identify major declines of parkin sulfhydration in the corpus striatum of PD patients. We also show that H2S donors are cytoprotective in parkin-related models of neurotoxicity. Thus, diminished sulfhydration of parkin may be pathogenic in PD and selective H2S donors may be therapeutic.
RESULTS
Parkin is physiologically sulfhydrated
The biotin switch method we developed10 for monitoring nitrosylation can be modified to identify sulfhydration9. Utilizing the modified biotin switch assay, we demonstrate sulfhydration of overexpressed parkin in response to treatment with the H2S donor NaHS (Fig. 1a). Recently we have developed improved methodology for monitoring sulfhydration employing fluorescently labeled derivatives of maleimide11. Virtues of the maleimide procedure include its greater specificity, the ability to quantify results readily, and the capacity of the technique to monitor nitrosylation and sulfhydration of the same samples11. Utilizing the maleimide procedure, we establish that under basal conditions parkin is robustly sulfhydrated in whole brain of mice and the striatum of rats (Fig. 1b).
Figure 1. Parkin is physiologically sulfhydrated.

(a) Parkin expressed in HEK293 cells is sulfhydrated by the H2S donor NaHS as detected by the modified biotin switch method. (b) Endogenous parkin is basally sulfhydrated in both mouse brain and rat striatum as detected by the maleimide assay in which loss of red fluorescence signal following DTT treatment indicates sulfhydration of the protein. (c) Sulfhydration of myc-parkin overexpressed in HEK293 cells is enhanced almost 20 fold upon overexpression of GST-CBS, one of the principle H2S producers in the brain. n=3, P<0.01 via one-way ANOVA. (d) Endogenous parkin sulfhydration in SH-SY5Y cells is enhanced over 5 fold upon treatment with 100 μM GYY4137, a hydrogen sulfide donor. n=3, P<0.01 via one-way ANOVA. (e) Endogenous parkin sulfhydration in SH-SY5Y cells is enhanced over 8 fold by overexpression of GST-CBS. n=3, P<0.01 via one-way ANOVA. All data expressed as mean ± s.e.m.
To further substantiate the nature of parkin sulfhydration, we show that overexpression of cystathionine beta synthase (CBS), an H2S biosynthetic enzyme, increases parkin sulfhydration more than 20-fold (Fig. 1c, Supplementary Fig. S1a). The neuronal cell line SH-SY5Y displays endogenous sulfhydration of parkin which is increased more than 5-fold by treatment with the H2S donor GYY413712 (Fig. 1d, Supplementary Fig. S1b). Moreover, overexpressing CBS in SH-SY5Y cells increases parkin sulfhydration more than 8-fold (Fig. 1e, Supplementary Fig. S1c). There appears to be negligible basal nitrosylation of parkin present in brain tissue or cell lines, which is unchanged by treatment with H2S donors or generating enzymes (Supplementary Fig. S1d–g).
H2S enhances parkin E3 ligase activity via sulfhydration
Nitrosylation of parkin decreases its ubiquitination activity both exerted upon itself and on other substrates3,4. By contrast, in HEK293 cells the H2S donor GYY4137 markedly augments parkin autoubiquitination whereas “old” GYY4137 (GYY4137 exposed to air overnight to eliminate any H2S donating capacity), fails to influence ubiquitination (Fig. 2a). Treatment with GYY4137 results in an increase in parkin E3 ligase activity over time consistent with the activation of parkin via sulfhydration (Supplementary Fig. S2a). The stimulation by GYY4137 of parkin’s ubiquitination capacity applies also to proteins that are implicated in the pathogenicity of the disease. AIMP2 expression is elevated in human postmortem brain from both sporadic and familial PD, consistent with the notion that parkin is inactivated in PD and that AIMP2 is neurotoxic13,14. GYY4137 substantially increases ubiquitination of AIMP2 by parkin (Fig. 2b) as well as ubiquitination by parkin of synphilin-1, another parkin substrate implicated in Parkinson’s Disease (Fig. 2c).
Figure 2. Hydrogen sulfide enhances parkin E3 ligase activity via sulfhydration.

(a) Parkin E3 ligase activity in HEK293 cells is augmented by GYY4137 in a dose dependent manner, but not by “old” GYY4137, which was exposed to air overnight in PBS, and is subsequently unable to donate H2S. (b) Parkin activity, measured by target ubiquitination of AIMP2, is stimulated by GYY4137 (100 μM) and by GST-CBS. (c) Ubiquitination by parkin of synphilin-1 is enhanced by treatment with GYY4137 or GST-CBS. (d) Parkin E3 ligase activity in vitro is increased by the addition of 100 μM NaHS and decreased by GSNO. DTT, a reducing agent, returns activity to baseline by reversing sulfhydration or nitrosylation. (e) MPTP influences parkin sulfhydration. Parkin sulfhydration was determined by the maleimide technique in WT, nNOS−/−, and iNOS−/−mice injected with saline or MPTP and sacrificed at 2 h, 4 h, 24 h, and 48 h after MPTP injection. Following MPTP treatment, parkin sulfhydration in WT mice increases by almost 2 fold at 4 h before returning to baseline. Sulfhydration levels are substantially greater at 2–4 h in iNOS and nNOS mice (n=3 mice for each data point). Statistical significance is as noted **P<0.01 and *P<0.05 by ANOVA analysis with Tukey HSD post-hoc test. All data expressed as mean ± s.e.m.
To ascertain whether the influence of H2S upon ubiquitination is exerted in a direct fashion, we conducted experiments in vitro comparing actions of the NO donor GSNO and the H2S donor NaHS (Fig. 2d). As reported previously, GSNO substantially diminishes parkin’s autoubiquitination,3 whereas such autoubiquitination is markedly augmented by treatment with NaHS.
The contrasting actions of H2S and NO upon parkin’s ubiquitination activity suggest that the two gasotransmitters may exert reciprocal actions in the pathophysiology of PD. Sulfhydration and nitrosylation typically take place upon the same cysteines in proteins. This suggests that reciprocity between sulfhydration and nitrosylation of parkin impacts pathogenic features of PD. To examine this possibility, we monitored sulfhydration of parkin in brains of mice treated with the neurotoxin MPTP, which selectively damages dopamine neurons and is often employed as a model for PD (Fig. 2e, Supplementary Fig. S3a–c). We note increased sulfhydration of parkin 2–4 hours following MPTP treatment. In brains of mice with targeted deletion of inducible NO synthase (iNOS) or neuronal synthase (nNOS), in which parkin nitrosylation is lost,3 its sulfhydration is increased 2.5–3 fold. This implies that sulfhydration and nitrosylation of parkin occur reciprocally, presumably on the same cysteines.
Parkin sulfhydration enhances it protective functions
To ascertain the pathophysiologic relevance of reciprocal nitrosylation/sulfhydration of parkin, we sought to identify the sites of sulfhydration. High resolution ESI-MS-MS technique was implemented, which can differentiate sulfhydration from sulfinic acid oxidation of the cysteine residues. Mass spectrometric analysis reveals five sites of parkin sulfhydration at cysteines 59, 95, 182, 212 and 377 (Fig. 3a, Supplementary Fig. S4). At least one of these sites, cysteine 95, has been identified as being nitrosylated (Harry Ischiropoulos, personal communication). In order to determine the relative importance of these sites in mediating regulation of parkin ubiquitination activity by H2S, we performed systematic mutations of the various cysteines and assessed their activities (Fig. 3b). Parkin with C212S or C377S mutations fails to express or is unstable and could not be evaluated. Enhanced ubiquitination activity of parkin in response to GYY4137 is abolished with C95S mutations, while substantial diminution of the enhancement of ubiquitination is evident with C59S and C182S mutations.
Figure 3. Parkin sulfhydration at specific cysteine residues enhances its protective functions.

(a) Mass spectrometry detects probable parkin cysteines sulfhydrated by H2S donor NaHS. (b) C59S, C95S, and C182S mutations all substantially reduce parkin activity stimulation by H2S in HEK293 cells. (c) Cell death as monitored by trypan blue exclusion assay in tet-repressible AIMP2 expressing PC12 cells is prevented by parkin overexpression and by GYY4137, whose protective effect is not evident in the absence of parkin, with parkin-C95S or with the T240R catalytically inactive parkin mutant n=3–6 with *P<0.01 by one-way ANOVA. All data expressed as mean ± s.e.m.
To determine whether sulfhydration of parkin regulates pathogenic events associated with PD, we monitored cell death using trypan blue exclusion as well as MTT assays in PC12 cells overexpressing AIMP2, whose ubiquitination and destruction are elicited by parkin in cellular models of PD15 (Fig. 3c, Supplementary Fig. S5a). Overexpression of AIMP2 triples cell death, while parkin overexpression reverses this cytotoxicity. GYY4137 markedly reduces cell death in the parkin treated cells but not in those overexpressing AIMP2 in the absence of parkin. The selective action of GYY4137 indicates that its cytoprotective effects reflect modifications of parkin rather than some generalized antioxidant influence. This conclusion is supported by GYY4137’s lack of cytoprotective influence in cells overexpressing AIMP2 along with catalytically inactive parkin-T240R.
Further evidence that GYY4137 protects by enhancing parkin sulfhydration comes from experiments in which the cytoprotective action of GYY4137 is lost in cells overexpressing parkin-C95S, which is not activated by sulfhydration. Furthermore, we utilized the MPP+ model of PD in PC12 cells and SH-SY5Y cells in which H2S donors provide significant protection against MPP+ toxicity and inhibition of the H2S producing enzymes results in enhanced toxicity of MPP+ which is relieved by administration of H2S donors (Supplementary Fig. S5b,c). In order to resolve whether the anti-oxidant properties of H2S releasing agents were partially responsible for the neuroprotective effects seen in the AIMP2 overexpressing cells, we determined ROS levels in these cells overexpressing vector or various parkin mutants (Supplementary Fig. 6). The various parkin cysteine-serine mutants exhibited increased ROS levels as described previously16. However, there was not a significant difference in the GYY4137 treated samples at the concentrations that were employed suggesting that this is not the primary mechanism of protection seen in the experiments described in Fig. 3c.
Decreased sulfhydration and increased nitrosylation in PD
The pronounced cytoprotective action of parkin sulfhydration as well as the reciprocal relationship of parkin’s nitrosylation and sulfhydration suggest that alterations of sulfhydration participate in the pathophysiology of PD. Accordingly, we monitored sulfhydration and nitrosylation in the striatum of PD patients (Fig. 4, Supplementary Fig. 7). We confirm the increase of parkin nitrosylation in PD brain,3 and also observe a 60% decrease in parkin sulfhydration in patient brain.
Figure 4. Parkin sulfhydration is decreased in PD brain while nitrosylation is increased in PD.
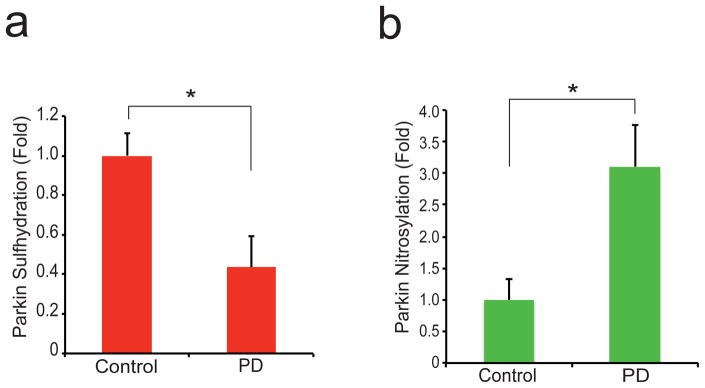
(a) The maleimide technique demonstrates that parkin sulfhydration is depleted in the striatum of PD patients. (b) Nitrosylation of parkin is increased in PD patient brain. P<0.02 by one-way ANOVA (n=7 control and n=6 PD samples for (a)); P<0.01 by one-way ANOVA (n=5 each control and PD for (b)). All data expressed as mean ± s.e.m.
DISCUSSION
In the present study, we have demonstrated that parkin is physiologically sulfhydrated, a process that enhances its ubiquitination activity. This contrasts markedly with nitrosylation, which decreases such activity. Sulfhydration and nitrosylation of parkin appear to be reciprocal events. This may be related to the difference in chemical reactivity between the two modifications. Nitrosylated cysteines will present a distinctly different chemical group to the local environment than will sulfhydration. In patient striatum, we demonstrate major decreases in parkin sulfhydration, which are reciprocal to the increases in nitrosylation.
What might be the comparative roles of parkin nitrosylation and sulfhydration in the pathophysiology of PD? One possibility is that the cell stress of PD leads to increased generation of NO, which accounts for the augmented nitrosylation of parkin in patient brain. Cell stressors do lead to activation of both iNOS and nNOS, and increased S-nitrosylation of several proteins, such as parkin, protein-disulfide isomerase, and XIAP, has been speculated to impact PD17–19. However, increased NO formation in PD has not been directly demonstrated, and measurements of H2S generation in PD are lacking. Nitrosylation and sulfhydration are sensitive and specific reflections of the presence of ambient NO and H2S respectively, implying that altered nitrosylation/sulfhydration in PD brain reflects changes in levels of the two gasotransmitters.
Our experiments also establish cytoprotective actions of H2S donors, which appear to reflect sulfhydration of parkin. Mutation of C95, a principal site of parkin sulfhydration, largely prevents the protective influences of H2S donors indicating that the donors act via parkin sulfhydration to enhance its ubiquitination activity. C182 and C59 also appear to contribute to influences of H2S donors upon parkin. We could not evaluate effects of H2S on C212 and C377, as they did not express or were unstable in our cell lines. Interestingly, C95 occurs in human but not rodent parkin, while the other sulfhydrated cysteines are conserved in rodent and human species.
The beneficial effects of H2S donors in PD models may have therapeutic implications. H2S donors have already been noted to be beneficial in rodent models of PD20–22, and an H2S donating variant of L-DOPA has shown promising effects in cellular models of PD23. These influences had been speculated to reflect general antioxidant and anti-inflammatory actions of H2S. However, numerous studies have failed to reveal a beneficial effect of antioxidants in PD24,25. Our findings provide a specific molecular mechanism whereby H2S therapy may benefit PD and can explain the ineffectiveness of generalized antioxidant treatment. Modifications of parkin and other interactors of parkin may offer promise in the therapy of PD15,26,27 so that H2S donors selectively targeted to parkin may provide notable benefit.
Materials and Methods
Generation of plasmids
The full-length parkin cDNA was cloned into pRK5-myc and pCMV-FLAG (Stratagene) vectors between the SalI and NotI. Full-length cDNAs of synphilin-1 and AIMP2 were cloned into pRK5-myc vector and pCMV-FLAG respectively between the SalI and NotI sites. The cDNA of ubiquitin was cloned into pRK5-HA vector between the SalI and NotI sites. The generation of C-S mutants was done using the pRK5-myc parkin construct and site-directed mutagenesis. The integrity of the constructs was confirmed by sequencing.
Modified biotin switch assay
A modified version of the modified biotin switch assay as described in9 was used. In brief, overexpressed myc-parkin was transfected into HEK293 cells with polyfect (Qiagen) for 24 hours. Cells were treated with 100 μM NaHS (Sigma) as indicated and harvested in HEN buffer with 1% triton and spun down at 14,000 r.p.m. for 15 min. The supernatant is then added to lysis buffer plus 2% SDS and 10 mM NEM for 1 h at 37°C while shaking. Blocked proteins are acetone precipitated with acetone (−20°C) to remove free NEM followed by 1 mM DTT treatment for 1 hour at 25°C while shaking. Another round of acetone precipitation was performed to remove the DTT and reduced S-NEM from sulfhydrated residues followed by treatment with 200 μM biotin-NEM for 1 hour at 25°C followed by precipitation with neutravidin (Thermo Scientific) beads and subsequent analysis via western blot with anti-myc antibody.
Maleimide Assay
Performed as described in11. In brief: cells or tissue were lysed in 20 mM Tris-HCl pH 7.5, 0.1% triton, 100 mM NaCl and immunoprecipitated with parkin antibody (Cell Signal) or appropriate antibody for 16 h at 4°C followed by washing 5 X with lysis buffer plus 300 mM NaCl (wash buffer). Beads were then incubated with 5 μM Red-Maleimide for 2 h at 4°C while rotating followed by 3 X wash with wash buffer. Beads were then divided equally between two tubes with one tube receiving 1 mM DTT and the other buffer and both rotated for 2 h at 4°C followed by 3 X wash with wash buffer. 50 μL of 2X LDS (Invitrogen) was then added, the beads boiled for 2 min, and proteins separated on SDS-PAGE and visualized on the LiCor fluorescent scanner. If the 2-color maleimide assay was used, an additional labeling step was included: after incubation with Red-Maleimide and subsequent wash, 1 mM ascorbate was added for 2 h followed by treatment with Green-Maleimide for 2 h and subsequent wash. Beads were then split equally again and treated with DTT as described above. Western blot analysis with the appropriate antibody was performed to ensure that any differences between bead allocations between tubes were taken into account for the experiment.
In vitro ubiquitination assay
Reactions were performed in a 20 μl mixture containing 50 mM Hepes, pH 7.5, 1 mM MgCl2, 1 mM ATP, 1 mM biotin-ubiquitin (BostonBiochem), 110 ng of E1 (BostonBiochem), 900 ng of UbcH7 (BostonBiochem), His6-Parkin (1.5 μg) that had been treated with either 100 μM of GSNO or NaHS (as indicated) for 20 min prior to the addition of buffer or 1 mM DTT (as indicated). After 30 min this was added to the main reaction which was carried out in darkness and devoid of any reducing agents such as DTT at 37 °C (except where specified). After 1 h, the reactions were terminated with an equal volume of 2X SDS sample buffer and the products were subject to Western-blot analysis with anti-ubiquitin antibody (Cell Signaling).
Ubiquitination assay
HEK293 cells were transfected with 4 μg of plasmids. After 24 h, the cells were treated with the MG132 (Sigma) followed by the selected drugs (as indicated) for the specified time course (3–9 h). If cells were transfected with CBS or CSE, 500 μM of L-Cysteine was added to the media as a supplement for H2S generation. After 2–9 h all samples were harvested together to ensure that all samples were treated by MG132 for equal time and only the time treated with H2S varied. The cells were harvested by washing with cold PBS and then lysed with immunoprecipitation buffer (25 mM Hepes, pH 7.5, 100 mM NaCl, 0.5% triton X-1000, 1mM EDTA, Roche Complete Protease Inhibitor Tablet). The lysates were then sonicated at 4°C for 10 sec and rotated for 15 min at 4°C followed by centrifugation at 14,000 r.p.m. for 15 min. The supernatants were combined with 30 μL EZ-View anti-myc or anti-FLAG (Sigma) beads overnight at 4°C. The beads were pelleted and washed 5 times using immunoprecipitation buffer with 500 mM NaCl. The precipitates were resolved on SDS-PAGE gel and subjected to Western-blot analysis with antibodies against myc or HA (Roche). Bands were visualized with chemiluminescence (Pierce).
Animals and treatment
All experiments were approved and conformed to the guidelines set by the Institutional Animal Care Committee. Ten-week-old iNOS deficient mice and nNOS deficient mice (Jackson Laboratories) and their wild-type counterparts were used. Mice received four intraperitoneal injections of MPTP-HCl (20 mg/kg of free base; Sigma) in saline at 2 h intervals in 1 day, and were sacrificed at selected time points as indicated after the last injection. Control mice received saline only. The mouse brains were harvested and S-sulfhydration of parkin in the whole brain was determined by maleimide assay.
Human Tissue
Human brain tissue was obtained through the brain donation program of the Morris K. Udall Parkinson’s Disease Research Center at Johns Hopkins Medical Institutions (JHMI) according to HIPAA regulations. This research proposal involves anonymous autopsy material that lacks identifiers of gender, race, or ethnicity. The JHMI Joint Committee on Clinical Investigations decided that the studies in this proposal are exempt from Human Subjects Approval because of Federal Register 46.101 exemption number 4. Seven age-matched control brains, and six PD and/or DLBD brains were utilized for the detection of S-sulfhydration and S-nitrosylation of parkin by the maleimide assay.
AIMP2-inducible cell lines
As described previously, PC12 cells were grown in DMEM containing 10% horse serum, 5% No-Tet FBS in a 5% CO2 atmosphere15. Tet-off cells (Clontech) were used to create PC12 cell lines expressing inducible AIMP2 as described previously. Differentiation was initiated by the addition of 100 ng/mL NGF to the culture medium. NGF was replenished daily for differentiation.
Cell-viability analysis
AIMP2-inducible PC12 cells were plated in a six-well plate for viability. Cells were transfected with indicated plasmids using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Cells were incubated with 100 μM GYY4137 or vehicle after 2 d of induction and differentiation with NGF. To assess cell viability in the PC12 cell experiments, we used the trypan blue exclusion assay. Cells were resuspended in plating medium with trypan blue stain for 5 minutes. We counted the percentage of blue-stained cells among total cells using the Cell Countess cell counter to determine a percentage of cell death in the trypan blue exclusion assay described previously10. The MTT assay was also used to determine cell viability. Briefly, cells were incubated with 3,-4,5 dimethylthiazol-2,5 diphenyl tetrazolium bromide for 2 h after which the supernatant was removed and centrifuged to collect any floating cells. 1 mL of DMSO was used to lyse the remaining cells after which any pelleted cells were lysed with 1 mL of DMSO and added to the original well from which it was taken. This was incubated at RT for 20 minutes while shaking. Absorbance measurements were then taken at 570 nm and 630 nm to determine cell viability.
Statistical Analysis
All data are expressed as mean ± s.e.m. Statistical significance between sample sets was analyzed by ANOVA analysis with post-hoc test where appropriate.
Mass Spectral analysis
Purified His6-Parkin (BostonBiochem) was purified to remove any trace DTT with spin column followed by incubation in 50 mM HEPES, 100 mM NaCl buffer with 100 μM NaHS for 1 h. These samples were then digested with tripsin and run on high resolution tandem mass spectrometry array for analysis. Database searching: Tandem mass spectra were extracted, charge state deconvoluted and deisotoped by 1 version 3. All MS/MS samples were analyzed using Mascot (Matrix Science, London, UK; version Mascot) and Sequest. Mascot was set up to search the NCBInr_20080819 database (selected for Homo sapiens, 2, 133769 entries) assuming the digestion enzyme trypsin. Mascot was searched with a fragment ion mass tolerance of 0.050 Da and a parent ion tolerance of 15 ppm. Oxidation of methionine, persulfide of cysteine, sulphur dioxide of cysteine (sulfination), sulfitolysis of cysteine, N-ethylmaleimide on cysteines of cysteine, N-ethylmaleimide hydrolysis of cysteine, NEM+S (NEM modified sulfhydration) of cysteine and NEM+S+H2O of cysteine were specified in Mascot as variable modifications. Criteria for protein identification: Scaffold (version Scaffold_3.4.3, ProteomeSoftware Inc., Portland, OR) was used to validate MS/MS based peptide and protein identifications. Peptide identifications were accepted if they could be established at greater than 95.0% probability as specified by the Peptide Prophet algorithm28. Protein identifications were accepted if they could be established at greater than 95.0% probability and contained at least 2 identified peptides. Protein probabilities were assigned by the Protein Prophet algorithm.29 Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony.
Supplementary Material
Acknowledgments
This work has been supported by National Institutes of Health Medical Scientist Training Program Award (T32 GM007309), US Public Health Service Grants (MH018501), and from the National Institutes of Health (NS38377). T.M.D. is the Leonard and Madlyn Abramson Professor in Neurodegenerative Diseases. The authors would like to acknowledge Bob Cole and Lauren Hitt (Johns Hopkins University Mass Spectrometry Core) in identifying parkin sulfhydration sites. The authors acknowledge the joint participation by the Adrienne Helis Malvin Medical Research Foundation through its direct engagement in the continuous active conduct of medical research in conjunction with The Johns Hopkins Hospital and the Johns Hopkins University School of Medicine and the Foundation’s Parkinson’s Disease Program No. M-1.
Footnotes
Competing Financial Interests:
The authors declare no competing financial interests.
Author Contributions:
M.S.V., B.D.P., and N.S. designed, performed, and analyzed experiments. R.X., F.R., H.S.K., S.K., and Y.I.L. assisted with experimental design and data analysis. M.S.V., B.D.P, A.M.S., generated plasmid constructs. V.L.D. and T.M.D. provided experimental support, data analysis, and edited the manuscript. M.S.V. and S.H.S. wrote the manuscript.
References
- 1.Martin I, Dawson VL, Dawson TM. Recent advances in the genetics of Parkinson’s disease. Annu Rev Genomics Hum Genet. 2011;12:301–325. doi: 10.1146/annurev-genom-082410-101440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hatano T, Kubo S, Sato S, Hattori N. Pathogenesis of familial Parkinson’s disease: new insights based on monogenic forms of Parkinson’s disease. Journal of Neurochemistry. 2009;111:1075–1093. doi: 10.1111/j.1471-4159.2009.06403.x. [DOI] [PubMed] [Google Scholar]
- 3.Chung KKK, et al. S-nitrosylation of parkin regulates ubiquitination and compromises parkin’s protective function. Science (New York, NY) 2004;304:1328–1331. doi: 10.1126/science.1093891. [DOI] [PubMed] [Google Scholar]
- 4.Yao D, et al. Nitrosative stress linked to sporadic Parkinson’s disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:10810–10814. doi: 10.1073/pnas.0404161101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Szabó C. Hydrogen sulphide and its therapeutic potential. Nature Reviews Drug Discovery. 2007;6:917–935. doi: 10.1038/nrd2425. [DOI] [PubMed] [Google Scholar]
- 6.Vandiver MS, Snyder SH. Hydrogen sulfide: a gasotransmitter of clinical relevance. J Mol Med (Berl) 2012;90:255–263. doi: 10.1007/s00109-012-0873-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hu LF, Lu M, Hon Wong PT, Bian JS. Hydrogen sulfide: neurophysiology and neuropathology. Antioxidants & redox signaling. 2011;15:405–419. doi: 10.1089/ars.2010.3517. [DOI] [PubMed] [Google Scholar]
- 8.Li L, Rose P, Moore PK. Hydrogen sulfide and cell signaling. Annual review of pharmacology and toxicology. 2011;51:169–187. doi: 10.1146/annurev-pharmtox-010510-100505. [DOI] [PubMed] [Google Scholar]
- 9.Mustafa AK, et al. H2S signals through protein S-sulfhydration. Science Signaling. 2009;2:ra72. doi: 10.1126/scisignal.2000464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jaffrey SR, Snyder SH. The biotin switch method for the detection of S-nitrosylated proteins. Science’s STKE: signal transduction knowledge environment. 2001;2001 doi: 10.1126/stke.2001.86.pl1. pl1. [DOI] [PubMed] [Google Scholar]
- 11.Sen N, et al. Hydrogen sulfide-linked sulfhydration of NF-kB mediates its anti-apoptotic actions. Molecular Cell. 2011;45:13–24. doi: 10.1016/j.molcel.2011.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li L, et al. Characterization of a novel, water-soluble hydrogen sulfide-releasing molecule (GYY4137): new insights into the biology of hydrogen sulfide. Circulation. 2008;117:2351–2360. doi: 10.1161/CIRCULATIONAHA.107.753467. [DOI] [PubMed] [Google Scholar]
- 13.Corti O, et al. The p38 subunit of the aminoacyl-tRNA synthetase complex is a Parkin substrate: linking protein biosynthesis and neurodegeneration. Hum Mol Genet. 2003;12:1427–1437. doi: 10.1093/hmg/ddg159. [DOI] [PubMed] [Google Scholar]
- 14.Choi JW, et al. Splicing variant of AIMP2 as an effective target against chemoresistant ovarian cancer. J Mol Cell Biol. 2012;4:164–173. doi: 10.1093/jmcb/mjs018. [DOI] [PubMed] [Google Scholar]
- 15.Ko HS, et al. Phosphorylation by the c-Abl protein tyrosine kinase inhibits parkin’s ubiquitination and protective function. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:16691–16696. doi: 10.1073/pnas.1006083107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hyun DH, et al. Effect of wild-type or mutant Parkin on oxidative damage, nitric oxide, antioxidant defenses, and the proteasome. The Journal of biological chemistry. 2002;277:28572–28577. doi: 10.1074/jbc.M200666200. [DOI] [PubMed] [Google Scholar]
- 17.Nakamura T, Lipton Sa. S-nitrosylation of critical protein thiols mediates protein misfolding and mitochondrial dysfunction in neurodegenerative diseases. Antioxidants & redox signaling. 2011;14:1479–1492. doi: 10.1089/ars.2010.3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsang AH, et al. S-nitrosylation of XIAP compromises neuronal survival in Parkinson’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:4900–4905. doi: 10.1073/pnas.0810595106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fang J, Nakamura T, Cho DH, Gu Z, Lipton SA. S-nitrosylation of peroxiredoxin 2 promotes oxidative stress-induced neuronal cell death in Parkinson’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:18742–18747. doi: 10.1073/pnas.0705904104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kida K, et al. Inhaled hydrogen sulfide prevents neurodegeneration and movement disorder in a mouse model of Parkinson’s disease. Antioxidants & Redox Signaling. 2011;15:343–352. doi: 10.1089/ars.2010.3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu LF, et al. Neuroprotective effects of hydrogen sulfide on Parkinson’s disease rat models. Aging Cell. 2010;9:135–146. doi: 10.1111/j.1474-9726.2009.00543.x. [DOI] [PubMed] [Google Scholar]
- 22.Lu M, et al. The Neuroprotection of Hydrogen Sulfide Against MPTP-Induced Dopaminergic Neuron Degeneration Involves Uncoupling Protein 2 Rather Than ATP-Sensitive Potassium Channels. Antioxidants & redox signaling. 2012 doi: 10.1089/ars.2011.4507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee M, et al. Effects of hydrogen sulfide-releasing L-DOPA derivatives on glial activation: potential for treating Parkinson disease. The Journal of Biological Chemistry. 2010;285:17318–17328. doi: 10.1074/jbc.M110.115261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Storch A, et al. Randomized, double-blind, placebo-controlled trial on symptomatic effects of coenzyme Q(10) in Parkinson disease. Arch Neurol. 2007;64:938–944. doi: 10.1001/archneur.64.7.nct60005. [DOI] [PubMed] [Google Scholar]
- 25.Snow BJ, et al. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson’s disease. Mov Disord. 2010;25:1670–1674. doi: 10.1002/mds.23148. [DOI] [PubMed] [Google Scholar]
- 26.Imam SZ, et al. Novel regulation of parkin function through c-Abl-mediated tyrosine phosphorylation: implications for Parkinson’s disease. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2011;31:157–163. doi: 10.1523/JNEUROSCI.1833-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shin JH, et al. PARIS (ZNF746) repression of PGC-1α contributes to neurodegeneration in Parkinson’s disease. Cell. 2011;144:689–702. doi: 10.1016/j.cell.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 29.Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem. 2003;75:4646–4658. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.