

Abstract
Epidemiologic studies indicate that co-occurring substance use disorders and psychiatric disorders are frequently found in clinical practice. From a neurobiologic perspective, what do these two seemingly different groups of disorders have in common? Currently, several hypotheses are postulated to explain the high rates of comorbidity. Chronic alcohol and drug use may lead to neuroadaptation in the biologic systems mediating psychiatric disorders. Conversely, co-occurring psychiatric and substance use disorders (SUDs) may represent phenotypic expressions of common premorbid neurobiologic abnormalities. Similar alterations in the dopamine-mediated reward system and various neurotransmitter systems including glutamate, γ-aminobutyric acid, and serotonin are found in both SUDs and numerous psychiatric disorders. Stress and chronic distress with the resultant activation of the hypothalamic-pituitary-adrenal axis and stress system has also been implicated in the pathophysiology of both psychiatric disorders and SUDs. Better understanding the commonalities between the two groups of disorders should lead to more efficacious treatments and targeted prevention strategies.
Overview
Epidemiologic survey studies (the National Comorbidity Survey, the Epidemiologic Catchment Area study, and the National Epidemiological Survey on Alcohol and Related Conditions [NESARC]) have emphasized the prevalence of comorbid psychiatric and substance use disorders (SUDs) in community samples of adults. The most recent and largest (N>43,000) comorbidity study to date, NESARC, found strong associations between alcohol dependence and other SUDs and mood, anxiety, and personality disorders.1 As demonstrated in Figure 1,2 statistically significant positive associations were found between almost all drug use disorders and mood and anxiety disorders.3
Figure 1. OVERLAPPING CONDITIONS: SHARED VULNERABILITY2.
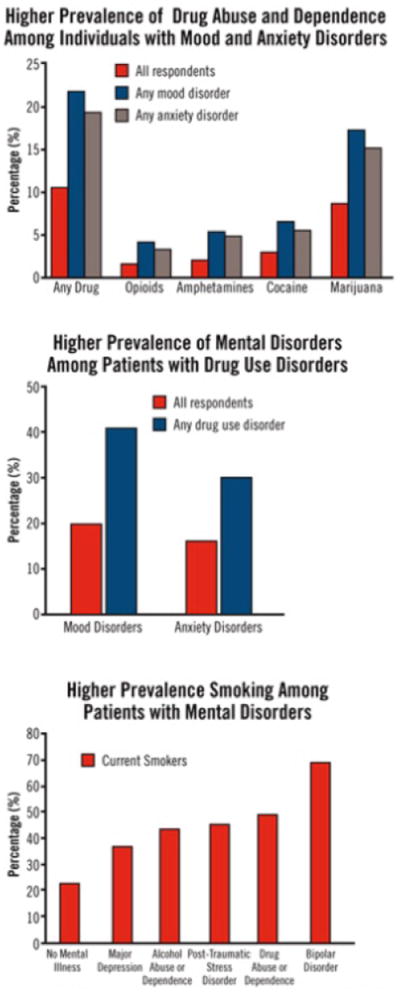
Because mood disorders increase vulnerability to drug abuse and addiction, the diagnosis and treatment of the mood disorder can reduce the risk of subsequent drug abuse. Because the inverse may also be true, the diagnosis and treatment of drug use disorders may reduce the risk of developing other mental illnesses, and if they do occur, lessen their severity or make them more amenable to effective treatment. Finally, >40% of the cigarettes smoked in the United States are smoked by individuals with a psychiatric disorder, such as major depressive disorder, alcoholism, posttraumatic stress disorder, schizophrenia, or bipolar disorder. Smoking by patients with mental illness contributes greatly to their increased morbidity and mortality.
Comorbidity: Addiction and Other Mental Illnesses. Research Report Series. US Department of Health and Human Services. National Institute of Health. National Institute on Drug Abuse. December 2008. NIH publication 08-5771. Reprinted with permission from the National Institute on Drug Abuse. Copyright 2008.
One of the overarching issues in the area of comorbidity is the basic question of etiologic relationships between substance use and other psychiatric disorders. Because acute intoxication and withdrawal from substances of abuse can mimic symptoms of psychiatric disorders, symptom overlap can be problematic for accurate diagnosis. Some of the high comorbidity reported in epidemiologic studies, which are not generally designed to differentiate substance-related and independent psychiatric symptoms, may be due to symptom overlap. In treatment settings, individuals presenting in acute withdrawal commonly complain of anxiety or depression symptoms which may be substance-induced states that will resolve with abstinence. Despite this caveat, even conservative estimates suggest that the comorbidity between psychiatric and SUDs is higher than would be expected by chance alone.
Etiologic Relationships: Neurobiologic Perspective
A growing body of evidence suggests that common neurobiologic pathways are involved in SUDs and many psychiatric disorders. Using a neurobiologic framework, some hypotheses can be postulated. First, SUDs and other psychiatric disorders are different symptomatic expressions of similar pre-existing neurobiologic abnormalities. Second, repeated drug administration leads to biologic changes (neuroadaptation) in systems mediating psychiatric disorders.4 The role of stress in development/relapse in SUDs and other psychiatric disorders is receiving increasing attention. Figure 2 provides a framework for the neurobiologic relationship between chronic stress, SUDs, and psychiatric comorbidity.5 Although this model conceptualizes chronic stress as the bridging construct, various genetic and environmental vulnerability factors contribute to the development of chronic stress and altered neurobiology.
Figure 2. SCHEMATIC MODEL OF CHRONIC DESTRESS AND PERPETUATION OF PSYCHIATRIC SYMPTOMS AND DRUG USE IN INDIVIDUALS WITH COMORBID DISORDERS5.
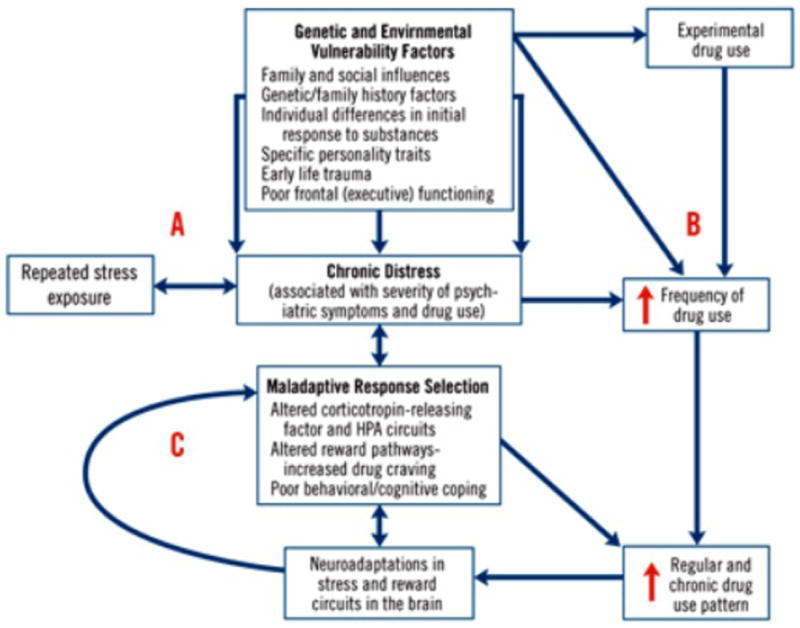
Various genetic and environmental vulnerability factors contribute to the development of psychiatric/emotional distress (A) and to drug abuse (B). Chronic distress is a common construct underlying both the disabling psychiatric symptoms associated with specific psychiatric disorders and the increasing distress associated with severity of substance use disorder. Chronic distress states are associated with selection of maladaptive responses, such as drug use, in order to attain desired goals or homeostasis. Maladaptive response selection mechanisms are associated with alterations in various neurotransmitter systems (including corticotropin-releasing factor and hypothalamic-pituitary-adrenal circuits), increased levels of stress-inducing drug craving, and poor adaptive coping, representing neuroadaptations in the stress and reward circuits (C). This mechanism contributes to the escalation of drug use to the chronic levels characteristic of dependence, supporting a feed-reward loop that leads to greater alterations in stress and reward systems. These alterations perpetuate chronic distress and susceptability to repeated stress exposures, thereby promoting a cycle of distress and drug use in individuals with comorbid disorders.
Brady KT, Sinha R. Co-occurring mental and substance use disorders: the neurobiological effects of chronic stress. Am J Psychiatry. 2005; 162(8): l483-1493. Reprinted with permission from the American Psychiatric Association. Copyright 2005.
HPA=hypothalamic-pituitary-adrenal
Corticotrophin releasing factor (CRF), one of the key hormones involved in the stress response, has been implicated in the pathophysiology of both psychiatric and SUDs.6,7 Animal studies suggest that CRF and noradrenergic pathways are involved in stress-induced reinstatement of drug-seeking behavior.8 Using animal models, potentiation of dopamine activity in mesolimbic reward circuits by activation of CRF circuitry has been demonstrated. Animal models of early life stress and chronic stress result in long-term changes in stress responses which can alter the sensitivity of the dopamine system to stress and increase susceptibility to self-administration of substances of abuse.9 This may provide the neurobiologic underpinnings of the well-established relationship between early life adversity and SUDs in adolescents and adults (Figure 2).5
While it is well established that dopamine neurotransmission is important to the rewarding properties of substances of abuse, data is accumulating to suggest that neuroadaptation in glutamatergic systems may underlie the long-term changes associated with chronic drug use. Glutamate is critical to synaptic plasticity and associative learning.10 Animal studies have demonstrated that glutamate release in neural projections from the prefrontal cortex underlies both stress and cocaine-primed reinstatement.11 It has been hypothesized that cellular adaptations in glutamatergic projections to the nucleus accumbens may promote drug seeking by decreasing the value of natural rewards, decreasing cognitive capacity to choose alternative behaviors and by enhancing the response to drug-related stimuli.12 Medications that alter glutamatergic transmission have been the focus of recent exploration in the treatment of addictions.
It is important to note that different substances of abuse have widely varying effects on neurobiologic systems. Cocaine and amphetamines have a stimulating effect on catecholaminergic systems. Opiate analgesic drugs act through multiple opiate receptors and nicotine acts through specific nicotinic receptors distributed throughout the central and peripheral nervous systems. g-aminobutyric acid (GABA)ergic and glutamatergic systems are particularly important in acute intoxication and withdrawal from alcohol and benzodiazepines. Clearly, the effects of acute intoxication and withdrawal differ for each of these drugs, and the relationship between substance use and psychiatric disorders also differs by both drug and disorder. However, there appear to be some common neurobiologic pathways operating across substances of abuse. Dopamine activity in the nucleus accumbens is implicated in the reinforcement of nearly all drugs of abuse. Furthermore, many drugs of abuse activate the hypothalamic-pituitary-adrenal (HPA) axis during use/abuse and alterations in the CRF/HPA and noradrenergic systems during acute withdrawal/abstinence are well documented.13
The following sections focus on major psychiatric disorders or classes of disorders (mood disorders, anxiety disorders, and schizophrenia). With the rapid development of technical advances in neurosciences, information concerning the molecular biology, neurotransmitter systems, and neural circuitry involved at the interface of mental illness and SUDs is rapidly growing. Research concerning mechanistic connections in the disorders to be discussed is particularly active.
Anxiety Disorders
Anxiety disorders are the most common psychiatric disorders in the United States. In the NESARC, 17.7% of participants with an SUD in the past 12 months also met criteria for an anxiety disorder. Among people with any anxiety disorder in the past 12 months, ~15% had at least one SUD.14 The relationship between anxiety disorders and drug use disorders (OR=2.8) was stronger than the relationship between anxiety and alcohol use disorders (OR=1.7). The relationship between drug use and specific anxiety disorders were virtually all significantly positive (P<.05).
Numerous systems have been implicated in the underlying neurobiology of anxiety disorders. Both animal and human studies suggest that the amygdala and prefrontal cortex are involved in the mediation of anxiety and fear and increased amygdala activity in response to threat cues has been found across a range of anxiety disorders.15 The amygdala is also part of the mesolimbic reward system involved in drug reward.16 Animal studies suggest the anxiogenic effects of dopaminergic stimulants are the result of dopamine augmentation in the amygdala.17 During a functional magnetic resonance imaging (fMRI) perceptual processing task of angry and fearful faces, dextroamphetamine intensified the amygdala response.18
Examination of the neurochemical substrates of anxiety is complicated by high rates of symptom overlap and comorbidity with other psychiatric disorders. Positron emission tomography and single photon emission-computed tomography imaging has implicated GABA, glutamate, serotonin, adrenergic, dopamine, CRF, and a variety of neuropeptides (substance P, neuropeptide Y, galanin, orexin, oxytocin) in the underlying neurobiology of anxiety disorders.19,20 Many of these same neurotransmitter systems have been implicated in SUDs. Anxiety is a common symptom of substance withdrawal. Recent work demonstrated GABAergic, serotoninergic, and stress systems modulate the anxiety associated with alcohol withdrawal states.21 GABAergic receptors, the site of benzodiazepine action, are widely distributed throughout the brain with concentrations in areas implicated in anxiety including the medial prefrontal cortex (PFC), amygdala, and hippocampus.22 Global reduction of GABAA binding has been found in panic disorder with regional decreases in binding in (right orbitofrontal cortex and right insula) areas thought to be essential in the central mediation of anxiety.23 Reduced benzodiazepine receptor binding has also been found in the medial PFC patients with posttraumatic stress disorder (PTSD).24 Benzodiazepines, commonly used in the treatment of anxiety disorders, modulate GABAergic function in the amygdala and limbic structures. In a recent study,25 lorazepam significantly decreased blood-oxygen-level dependent fMRI activation in the amygdala and insula during an emotional face task. Chronic dysregulation within the GABAergic system likely contributes to the pathology of anxiety disorders and has been implicated in the pathophysiology of SUDs.
Noradrenergic systems are also involved in both anxiety disorders and SUDs. Stress selectively increases norepinephrine in the locus ceruleus, cerebral cortex, and limbic regions of hypothalamus, hippocampus, and amygdala.26 Some of the symptoms of anxiety disorders such as insomnia, startle, anxiety attacks, and autonomic hyperarousal are characteristic of increased noradrenergic activity. Opiates, alcohol, and benzodiazepines decrease noradrenergic and subsequent locus ceruleus firing likely accounting for reports of amelioration of hyperarousal and intrusive memories with use of these drugs.27
Abnormalities in HPA function may involve a subset of anxiety disorders, particularly PTSD. PTSD has been associated with increased CRF in the CSF, low 24h cortisol secretion, and hypersuppression of cortisol in low-dose dexamethasone suppression tests.20 Enhanced negative feedback inhibition of cortisol in the pituitary has been proposed as one model to explain these HPA alterations in PTSD.28 Evidence of HPA alternations and increased CRF in clinical anxiety has resulted in the exploration of CRF-1 receptor antagonists.20 This class of agents also decreases substance use in animal models of relapse.8
Primate studies29 of early life stress have demonstrated increased glutamate activity in brain regions associated with fear and anxiety implicating dysfunction of the glutamatergic system. Elevated glutamate levels (compared to creatine) were found in the anterior cingulate cortex of medication-naïve individuals with social anxiety compared to healthy controls, and the symptom severity was associated with the glutamate/creatine levels.30 In generalized anxiety disorder, the glutamate-release inhibitor riluzole had been shown to decrease anxiety symptoms.31 Dysfunction in glutamate-modulated neurotransmission from the PFC to the nucleus accumbens has been implicated in the principal features of addiction and compulsive drug seeking.12
Affective Disorders
Symptoms of mood instability and depression are among the most common psychiatric symptoms in individuals with SUDs. Using National Comorbidity Study data, the odds ratio for the relative risk of co-occurrence of SUDs with any affective disorder was 2.3, with major depressive disorder (MDD) was 2.7, and with bipolar disorder was 9.2.32 Bipolar disorder was the Axis I diagnosis most likely to co-occur with an SUD.
There are numerous clinical similarities and symptom overlap between affective disorders and SUDs. Depressive symptoms are commonly reported during acute and chronic withdrawal from drugs of abuse. Irritability, sleep difficulties, anxiety, and trouble with attention/concentration are associated with protracted withdrawal states as well as bipolar and depressive disorders. Acute intoxication with cocaine and other stimulants can mimic mania and hypomania with symptoms such as grandiosity, decreased need for sleep and paranoia being common to both disorders. As with the anxiety, neurobiologic similarities between affective disorders and SUDs are likely to contribute to both symptom overlap and high comorbidity.4 There is substantial data indicating that extrahypothalamic CRF and HPA axis abnormalities,7 alterations in catecholamines, serotonin GABA, and glutamate systems are associated with depressive disorders.33 Neuroadaptations associated with chronic drug abuse are associated with alterations in these neurotransmitter systems, especially during acute withdrawal states.4 There is a positive association between CRF/HPA response during acute drug withdrawal and withdrawal-related distress or depressive symptoms.34,35 There is also a growing body of evidence indicating that the HPA axis alterations associated with chronic drug can contribute to drug craving and relapse in SUDs.6 Studies5,36 in abstinent smokers, alcoholics, and polysubstance-dependent individuals have demonstrated that a blunted adrenocorticotropic hormone and/or cortisol response to a variety of stimuli is associated with drug or alcohol use in the follow-up period, suggesting an important relationship between changes in HPA axis function and relapse.
Recent findings from neuroimaging studies implicate similar alterations in frontal-limbic brain circuitry in SUDs and depressive disorders. Reduced frontal metabolism and decreased anterior cingulate activity has been reported in individuals with SUDs.37 Significant reduction in dopamine-2 receptors, particularly in frontal-striatal regions, has been noted in cocaine- and alcohol-dependent individuals.37 Reduced frontal-limbic metabolism and anterior cingulate (AC) hypoactivity have also been noted in depressive disorders.38 In addition, amygdala hyperactivity is associated with MDD38 and studies39,40 of individuals with SUDs have demonstrated amygdala activation associated with cue-induced drug craving. Under conditions of distress, cocaine-dependent individuals exhibit decreased activity in frontal regions such as the medial PFC and the AC, similar to that seen with negative mood-related changes in depressive disorders.41
The high rate of SUD comorbidity in people with bipolar disorder is striking. Nearly 50% of the population meeting criteria for bipolar disorder have alcohol use disorders making similarities in the neurobiology of alcohol dependence and bipolar disorder of particular interest.42 Accumulating evidence suggests that dysregulation of glutamatergic neurotransmission is important in both illnesses. Chronic exposure to ethanol is associated with upregulation of the glutamatergic N-methyl-D-aspartate (NMDA) receptor in animals as well as in postmortem brain tissue from human alcoholics.43 Alcohol-dependent patients in early abstinence report more rewarding effects after administration of the NMDA antagonist ketamine, as compared to healthy comparison subjects.44 Repeated cycles of intoxication and withdrawal from chronic ethanol results in increased levels of extracellular glutamate in multiple brain regions that become progressively larger with time.45 Chronic ethanol also upregulates glutamatergic alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors, an adaptation that may be especially relevant to relapse as glutamatergic projections from the amygdala and prefrontal cortex to the nucleus accumbens are critical for both stress- and drug-primed reinstatement.46 Accordingly, AMPA antagonists prevent reinstatement of ethanol-seeking behavior.47
The role for glutamate neurotransmission in bipolar disorder has begun to emerge from preclinical and neuroimaging data.48 Increases in brain glutamate levels have been demonstrated consistently by magnetic resonance spectroscopy in all phases of illness in bipolar disorder.49 Lithium treatment reverses the baseline elevation in glutamate in bipolar subjects,50 consistent with the preclinical finding that chronic, but not acute, treatment with lithium upregulates and stabilizes glutamate uptake.51 Though complicated by differences in mood status, medication history, and illness duration, neuroimaging studies have demonstrated consistent abnormalities of frontostriatal and frontolimbic networks in both bipolar disorder48 and alcohol use disorders.52 For example, decreases in gray matter volume in subregions of PFC, which project glutamatergic efferents to limbic and other subcortical structures, have been reported in both alcoholics53,54 and people with bipolar disorder.55,56 The loss of inhibitory tone by frontal cortical regions during planning and decision making has been suggested to mediate the increased impulsivity observed in addiction12 and bipolar disorder.57 These findings suggest that both bipolar disorder and alcohol dependence may share compromised inhibitory control alternating with periods of hyperexcitability in subcortical regions, associated with alterations in glutamate neurotransmission.
Schizophrenia
Alcohol and illicit drug use affects ~50% of schizophrenic patients.42 The most commonly abused substances are alcohol followed by cannabis and cocaine. Co-occurring SUDs are associated with a plethora of negative outcomes including higher rates of violence, incarceration, medication non-compliance, psychotic relapse, hospitalizations, and adverse health effects.58
The self-medication hypothesis suggests that individuals with schizophrenia may self-medicate with substances to reduce negative symptoms such as apathy and social withdrawal or to ameliorate uncomfortable side effects of antipsychotics.59 Although these factors likely play a role, alternatively, recent research60 theorizes that the underlying pathophysiology of schizophrenia affects the reward circuitry of the brain leading to vulnerability to addiction. Some studies61 indicate that the brain areas thought to be dysfunctional in schizophrenia are also part of the dopamine-mediated reward network and individuals with schizophrenia demonstrate dysfunction in the processing of rewards and punishments similar to individuals with SUDs. For example, schizophrenic patients overvalue immediate rewards and devalue delayed punishment compared to healthy controls. In this model of reward circuitry dysfunction, substance use may ameliorate the deficits in reward system allowing schizophrenic patients to experience more normal levels of reward by producing an increase in dopamine neuronal signal detection making vulnerability to SUDs an inherent aspect of neurobiology of schizophrenia.60 Additionally, in neurodevelopmental animal models of schizophrenia, increased addiction vulnerability may be related to dysfunction in glutamatergic input from the frontal cortex and hippocampus to the nucleus accumbens.62,63 The resulting dysregulated integration of dopamine and glutamate may also contribute to increased responsiveness to substances of abuse.
Recent neuroimaging studies support this model. Comparison of MRIs in schizophrenia with or without alcohol dependence, alcohol dependence alone, and matched controls revealed gray matter deficits in all patient groups with the greatest deficits in the comorbid group.64 Although the comorbid schizophrenic-alcoholic group had a five times lower lifetime alcohol consumption than the alcohol-dependent group alone without schizophrenia, the MRIs displayed the full detrimental effect of alcohol suggesting an interactive effect.
Approximately 60% to 90% of individuals with chronic schizophrenia have co-occurring nicotine dependence.65 Nicotine interacts with the mesolimbic, dopaminergic, and glutamatergic neural networks involved in schizophrenia. A prospective study66 of >14,000 adolescents found that subjects who smoked more than half a pack per day at the initial evaluations were significantly more likely to be hospitalized for schizophrenia during the follow-up period. Smoking in vulnerable adolescents may represent self-medication of premorbid symptoms and be a sign of disordered nicotinic transmission in schizophrenia. Alternatively, chronic stimulation of the mesolimbic dopamine pathways may be involved in the development of schizophrenia. Nicotine administration improves several abnormalities associated with schizophrenia, including dysfunction in smooth pursuit eye movement67 and deficits in inhibitory gating P-50-evoked response to repeated auditory stimuli.68 Deficits in visuospatial working memory have been found among smokers with and without schizophrenia.69 After prolonged smoking cessation, schizophrenic smokers showed further decline in visuospatial memory while non-schizophrenic smokers improved.
Conclusion
The nature of the relationship between psychiatric disorders and SUDs is complex and multifaceted. There is a great deal of overlap in the neurobiologic systems involved in the pathophysiology of psychiatric disorders and SUDs. Neuroadaptations in brain stress and reward pathways associated with chronic stress may predispose or unmask a vulnerability to psychiatric disorders, SUDs, or both. Emerging evidence concerning abnormalities in glutamatergic function in bipolar disorder, schizophrenia, and other psychiatric disorders may mediate vulnerability to the development of SUDs. Evidence concerning negative emotional states and stress-related drug seeking/craving provides additional insight into emotional distress states and drug use in individuals with SUDs.
While the focus of this article has been on neurobiologic connections between psychiatric disorders and SUDs, it is important to note that this is just one facet of a complex issue. Further exploration of overlapping neural circuitry and mechanistic relationships will be essential in guiding treatment and prevention efforts. However, improvement in our understanding of co-occurring disorders will only be useful if there is a treatment system in place able to implement these findings. Clearly, change at public policy levels will be necessary to maximize the benefits derived from neurobiologic explorations so that these findings can be utilized to improve the lives of individuals with comorbidity.
Acknowledgments
This work was supported in part by the Ralph H. Johnson VA Medical Center.
Disclosure: Dr. Hartwell receives grant support from the Global Research Awards for Nicotine Dependence and grant and salary support from the National Institutes of Health’s Office of Research on Women’s Health. Dr. Tolliver receives grant and salary support from Forest and the National Institutes of Health’s National Institute on Alcohol Abuse and Alcoholism. Dr. Brady is consultant to Orexigen Therapeutics.
References
- 1.Hasin DS, Stinson FS, Ogburn E, Grant BF. Prevalence, correlates, disability, and comorbidity of DSM-IV alcohol abuse and dependence in the United States: results from the National Epidemiologic Survey on Alcohol and Related Conditions. Arch Gen Psychiatry. 2007;64(7):830–842. doi: 10.1001/archpsyc.64.7.830. [DOI] [PubMed] [Google Scholar]
- 2.Comorbidity: Addiction and Other Mental Illnesses. Research Report Series. US Department of Health and Human Services. National Institute of Health. National Institute on Drug Abuse; Dec, 2008. NIH publication 08-5771. [Google Scholar]
- 3.Conway KP, Compton W, Stinson FS, Grant BF. Lifetime comorbidity of DSM-IV mood and anxiety disorders and specific drug use disorders: results from the National Epidemiologic Survey on Alcohol and Related Conditions. J Clin Psychiatry. 2006;67(2):247–257. doi: 10.4088/jcp.v67n0211. [DOI] [PubMed] [Google Scholar]
- 4.Markou A, Kosten TR, Koob GF. Neurobiological similarities in depression and drug dependence: a self-medication hypothesis. Neuropsychopharmacology. 1998;18(3):135–174. doi: 10.1016/S0893-133X(97)00113-9. [DOI] [PubMed] [Google Scholar]
- 5.Brady KT, Sinha R. Co-occurring mental and substance use disorders: the neurobiological effects of chronic stress. Am J Psychiatry. 2005;162(8):1483–1493. doi: 10.1176/appi.ajp.162.8.1483. [DOI] [PubMed] [Google Scholar]
- 6.Sinha R. How does stress increase risk of drug abuse and relapse? Psychopharmacology (Berl) 2001;158(4):343–359. doi: 10.1007/s002130100917. [DOI] [PubMed] [Google Scholar]
- 7.Nemeroff CB. The corticotropin-releasing factor (CRF) hypothesis of depression: new findings and new directions. Mol Psychiatry. 1996;1(4):336–342. [PubMed] [Google Scholar]
- 8.Shalev U, Grimm JW, Shaham Y. Neurobiology of relapse to heroin and cocaine: a review. Pharmacol Rev. 2002;54(1):1–42. doi: 10.1124/pr.54.1.1. [DOI] [PubMed] [Google Scholar]
- 9.Piazza PV, Le Moal M. The role of stress in drug self-administration. Trends Pharmacol Sci. 1998;19(2):67–74. doi: 10.1016/s0165-6147(97)01115-2. [DOI] [PubMed] [Google Scholar]
- 10.Antzoulatos E, Byrne J. Learning insights transmitted by glutamate. Trends Neurosci. 2004;27(9):550–560. doi: 10.1016/j.tins.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 11.Kalivas P, McFarland K, Bowers S, Szumlinski K, Zheng-Xiong X, Baker D. Glutamate transmission and addiction to cocaine. Ann N Y Acad Sci. 2003;1003:169–175. doi: 10.1196/annals.1300.009. [DOI] [PubMed] [Google Scholar]
- 12.Kalivas PW, Volkow ND. The neural basis of addiction: a pathology of motivation and choice. Am J Psychiatry. 2005;162(8):1403–1413. doi: 10.1176/appi.ajp.162.8.1403. [DOI] [PubMed] [Google Scholar]
- 13.Koob GF, Ahmed SH, Boutrel B, et al. Neurobiological mechanisms in the transition from drug use to drug dependence. Neurosci Biobehav Rev. 2004;27(8):739–749. doi: 10.1016/j.neubiorev.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 14.Grant BF, Stinson FS, Dawson DA, et al. Prevalence and co-occurrence of substance use disorders and independent mood and anxiety disorders: Results from the National Epidemiologic Survey on Alcohol and Related Conditions. Arch Gen Psych. 2004;61(8):807–816. doi: 10.1001/archpsyc.61.8.807. [DOI] [PubMed] [Google Scholar]
- 15.Anand A, Shekhar A. Brain imaging studies in mood and anxiety disorders: special emphasis on the amygdala. Ann N Y Acad Sci. 2003;985:370–388. doi: 10.1111/j.1749-6632.2003.tb07095.x. [DOI] [PubMed] [Google Scholar]
- 16.Koob GF. The neurobiology of addiction: a neuroadaptational view relevant for diagnosis. Addiction. 2006;101(suppl 1):23–30. doi: 10.1111/j.1360-0443.2006.01586.x. [DOI] [PubMed] [Google Scholar]
- 17.Rosenkranz JA, Grace AA. Modulation of basolateral amygdala neuronal firing and afferent drive by dopamine receptor activation in vivo. J Neurosci. 1999;19(24):11027–11039. doi: 10.1523/JNEUROSCI.19-24-11027.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hariri AR, Mattay VS, Tessitore A, Fera F, Smith WG, Weinberger DR. Dextroamphetamine modulates the response of the human amygdala. Neuropsychopharmacology. 2002;27(6):1036–1040. doi: 10.1016/S0893-133X(02)00373-1. [DOI] [PubMed] [Google Scholar]
- 19.Nutt DJ, Stein DJ. Understanding the neurobiology of comorbidity in anxiety disorders. CNS Spectr. 2006;11(10 suppl 12):13–20. doi: 10.1017/s1092852900025803. [DOI] [PubMed] [Google Scholar]
- 20.Mathew SJ, Price RB, Charney DS. Recent advances in the neurobiology of anxiety disorders: implications for novel therapeutics. Am J Med Gen C Semin Med Genet. 2008;148(2):89–98. doi: 10.1002/ajmg.c.30172. [DOI] [PubMed] [Google Scholar]
- 21.Faingold CL, Knapp DJ, Chester JA, Gonzalez LP. Integrative neurobiology of the alcohol withdrawal syndrome--from anxiety to seizures. Alcohol Clin Exp Res. 2004;28(2):268–278. doi: 10.1097/01.alc.0000113421.41962.8d. [DOI] [PubMed] [Google Scholar]
- 22.Roy-Byrne PP. The GABA-benzodiazepine receptor complex: structure, function, and role in anxiety. J Clin Psychiatry. 2005;2(66 suppl):14–20. [PubMed] [Google Scholar]
- 23.Malizia AL, Cunningham VJ, Bell CJ, Liddle PF, Jones T, Nutt DJ. Decreased brain GABAA-Benzodiazepine receptor binding in panic disorder: preliminary results from a quantitative PET study. Arch Gen Psychiatry. 1998;55(8):715–720. doi: 10.1001/archpsyc.55.8.715. [DOI] [PubMed] [Google Scholar]
- 24.Bremner JD, Innis RB, Southwick SM, Staib L, Zoghbi S, Charney DS. Decreased benzodiazepine receptor binding in prefrontal cortex in combat-related posttraumatic stress disorder. Am J Psychiatry. 2000;157(7):1120–1126. doi: 10.1176/appi.ajp.157.7.1120. [DOI] [PubMed] [Google Scholar]
- 25.Paulus MP, Feinstein JS, Castillo G, Simmons AN, Stein MB. Dose-dependent decrease of activation in bilateral amygdala and insula by lorazepam during emotion processing. Arch Gen Psychiatry. 2005;62(3):282–288. doi: 10.1001/archpsyc.62.3.282. [DOI] [PubMed] [Google Scholar]
- 26.Bremner JD, Krystal JH, Southwick SM, Charney DS. Noradrenergic mechanisms in stress and anxiety: I. preclinical studies. Synapse. 1996;23(1):28–38. doi: 10.1002/(SICI)1098-2396(199605)23:1<28::AID-SYN4>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 27.Neumeister A, Daher RJ, Charney DS. Anxiety disorders: noradrenergic neurotransmission. Handb Exp Pharmacol. 2005;(169):205–223. doi: 10.1007/3-540-28082-0_8. [DOI] [PubMed] [Google Scholar]
- 28.Yehuda R. Advances in understanding neuroendocrine alterations in PTSD and their therapeutic implications. Ann N Y Acad Sci. 2006;1071:137–166. doi: 10.1196/annals.1364.012. [DOI] [PubMed] [Google Scholar]
- 29.Mathew SJ, Shungu DC, Mao X, et al. A magnetic resonance spectroscopic imaging study of adult nonhuman primates exposed to early-life stressors. Biol Psychiatry. 2003;54(7):727–735. doi: 10.1016/s0006-3223(03)00004-0. [DOI] [PubMed] [Google Scholar]
- 30.Phan KL, Fitzgerald DA, Cortese BM, Seraji-Bozorgzad N, Tancer ME, Moore GJ. Anterior cingulate neurochemistry in social anxiety disorder: 1H-MRS at 4 Tesla. Neuroreport. 2005;16(2):183–186. doi: 10.1097/00001756-200502080-00024. [DOI] [PubMed] [Google Scholar]
- 31.Mathew SJ, Amiel JM, Coplan JD, Fitterling HA, Sackeim HA, Gorman JM. Open-label trial of riluzole in generalized anxiety disorder. Am J Psychiatry. 2005;162(12):2379–2381. doi: 10.1176/appi.ajp.162.12.2379. [DOI] [PubMed] [Google Scholar]
- 32.Kessler RC, McGonagle KA, Zhao S, et al. Lifetime and 12-month prevalence of DSM-III-R psychiatric disorders in the United States. Results from the National Comorbidity Survey. Arch Gen Psychiatry. 1994;51(1):8–19. doi: 10.1001/archpsyc.1994.03950010008002. [DOI] [PubMed] [Google Scholar]
- 33.Sanacora G, Gueorguieva R, Epperson CN, et al. Subtype-specific alterations of gamma-aminobutyric acid and glutamate in patients with major depression. Arch Gen Psychiatry. 2004;61(7):705–713. doi: 10.1001/archpsyc.61.7.705. [DOI] [PubMed] [Google Scholar]
- 34.Frederick SL, Reus VI, Ginsberg D, Hall SM, Munoz RF, Ellman G. Cortisol and response to dexamethasone as predictors of withdrawal distress and abstinence success in smokers. Biol Psychiatry. 1998;43(7):525–530. doi: 10.1016/S0006-3223(97)00423-X. [DOI] [PubMed] [Google Scholar]
- 35.Elman I, Breiter HC, Gollub RL, et al. Depressive symptomatology and cocaine-induced pituitary-adrenal axis activation in individuals with cocaine dependence. Drug Alcohol Depend. 1999;56(1):39–45. doi: 10.1016/s0376-8716(99)00009-5. [DOI] [PubMed] [Google Scholar]
- 36.Sinha R, Garcia M, Paliwal P, Kreek MJ, Rounsaville BJ. Stress-induced cocaine craving and hypothalamic-pituitary-adrenal responses are predictive of cocaine relapse outcomes. Arch Gen Psychiatry. 2006;63(3):324–331. doi: 10.1001/archpsyc.63.3.324. [DOI] [PubMed] [Google Scholar]
- 37.Volkow ND, Fowler JS. Addiction, a disease of compulsion and drive: involvement of the orbitofrontal cortex. Cereb Cortex. 2000;10(3):318–325. doi: 10.1093/cercor/10.3.318. [DOI] [PubMed] [Google Scholar]
- 38.Davidson RJ, Lewis DA, Alloy LB, et al. Neural and behavioral substrates of mood and mood regulation. Biol Psychiatry. 2002;52(6):478–502. doi: 10.1016/s0006-3223(02)01458-0. [DOI] [PubMed] [Google Scholar]
- 39.Childress AR, Mozley PD, McElgin W, Fitzgerald J, Reivich M, O’Brien CP. Limbic activation during cue-induced cocaine craving. Am J Psychiatry. 1999;156(1):11–18. doi: 10.1176/ajp.156.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kilts CD, Gross RE, Ely TD, Drexler KP. The neural correlates of cue-induced craving in cocaine-dependent women. Am J Psychiatry. 2004;161(2):233–241. doi: 10.1176/appi.ajp.161.2.233. see comment. [DOI] [PubMed] [Google Scholar]
- 41.George MS, Ketter TA, Parekh PI, Horwitz B, Herscovitch P, Post RM. Brain activity during transient sadness and happiness in healthy women. Am J Psychiatry. 1995;152(3):341–351. doi: 10.1176/ajp.152.3.341. [DOI] [PubMed] [Google Scholar]
- 42.Regier DA, Farmer ME, Rae DS, et al. Comorbidity of mental disorders with alcohol and other drug abuse. Results from the Epidemiologic Catchment Area (ECA) Study. JAMA. 1990;264(19):2511–2518. [PubMed] [Google Scholar]
- 43.Tsai G, Coyle JT. The role of glutamatergic neurotransmission in the pathophysiology of alcoholism. Annu Rev Med. 1998;49:173–184. doi: 10.1146/annurev.med.49.1.173. [DOI] [PubMed] [Google Scholar]
- 44.Krystal JH, Petrakis IL, Limoncelli D, et al. Altered NMDA glutamate receptor antagonist response in recovering ethanol-dependent patients. Neuropsychopharmacology. 2003;28(11):2020–2028. doi: 10.1038/sj.npp.1300252. [DOI] [PubMed] [Google Scholar]
- 45.Dalchour A, DeWitte P. Effects of acamprosate on excitatory amino acids during multiple ethanol withdrawal periods. Alcohol Clin Exp Res. 2003;27(3):465–470. doi: 10.1097/01.ALC.0000056617.68874.18. [DOI] [PubMed] [Google Scholar]
- 46.McFarland K, Lapish CC, Kalivas PW. Prefrontal glutamate release into the core of the nucleus accumbens mediates cocaine-induced reinstatement of drug-seeking behavior. J Neurosci. 2003;23(8):3531–3537. doi: 10.1523/JNEUROSCI.23-08-03531.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sanchis-Segura C, Borchardt T, Vengeliene V, et al. Involvement of the AMPA receptor GluR-C subunit in alcohol-seeking behavior and relapse. J Neurosci. 2006;26(4):1231–1238. doi: 10.1523/JNEUROSCI.4237-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Strakowski SM, Delbello MP, Adler CM. The functional neuroanatomy of bipolar disorder: a review of neuroimaging findings. Mol Psychiatry. 2005;10(1):105–116. doi: 10.1038/sj.mp.4001585. [DOI] [PubMed] [Google Scholar]
- 49.Yildiz-Yesiloglu A, Ankerst DP. Neurochemical alterations of the brain in bipolar disorder and their implications for pathophysiology: a systematic review of the in vivo proton magnetic resonance spectroscopy findings. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30(6):969–995. doi: 10.1016/j.pnpbp.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 50.Friedman SD, Dager SR, Parow A, et al. Lithium and valproic acid treatment effects on brain chemistry in bipolar disorder. Biol Psychiatry. 2004;56(5):340–348. doi: 10.1016/j.biopsych.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 51.Dixon JF, Hokin LE. Lithium acutely inhibits and chronically up-regulates and stabilizes glutamate uptake by presynaptic nerve endings in mouse cerebral cortex. Proc Natl Acad Sci U S A. 1998;95(14):8363–8368. doi: 10.1073/pnas.95.14.8363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goldstein RZ, Volkow ND. Drug addiction and its underlying neurobiological basis: neuroimaging evidence for the involvement of the frontal cortex. Am J Psychiatry. 2002;159(10):1642–1652. doi: 10.1176/appi.ajp.159.10.1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jernigan TL, Butters N, DiTraglia G, et al. Reduced cerebral grey matter observed in alcoholics using magnetic resonance imaging. Alcohol Clin Exp Res. 1991;15(3):418–427. doi: 10.1111/j.1530-0277.1991.tb00540.x. [DOI] [PubMed] [Google Scholar]
- 54.Pfefferbaum A, Sullivan EV, Mathalon DH, Lim KO. Frontal lobe volume loss observed with magnetic resonance imaging in older chronic alcoholics. Alcohol Clin Exp Res. 1997;21(3):521–529. doi: 10.1111/j.1530-0277.1997.tb03798.x. [DOI] [PubMed] [Google Scholar]
- 55.Drevets WC, Price JL, Simpson JR, Jr, et al. Subgenual prefrontal cortex abnormalities in mood disorders. Nature. 1997;386(6627):824–827. doi: 10.1038/386824a0. [DOI] [PubMed] [Google Scholar]
- 56.Lopez-Larson MP, DelBello MP, Zimmerman ME, Schwiers ML, Strakowski SM. Regional prefrontal gray and white matter abnormalities in bipolar disorder. Biol Psychiatry. 2002;52(2):93–100. doi: 10.1016/s0006-3223(02)01350-1. [DOI] [PubMed] [Google Scholar]
- 57.Najt P, Perez J, Sanches M, Peluso MA, Glahn D, Soares JC. Impulsivity and bipolar disorder. Eur Neuropsychopharmacol. 2007;17(5):313–320. doi: 10.1016/j.euroneuro.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 58.Drake RE, McHugo GJ, Xie H, Fox M, Packard J, Helmstetter B. Ten-year recovery outcomes for clients with co-occurring schizophrenia and substance use disorders. Schizophr Bull. 2006;32(3):464–473. doi: 10.1093/schbul/sbj064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Goswami S, Mattoo SK, Basu D, Singh G. Substance-abusing schizophrenics: do they self-medicate? Am J Addict. 2004;13(2):139–150. doi: 10.1080/10550490490435795. [DOI] [PubMed] [Google Scholar]
- 60.Green AI. Pharmacotherapy for schizophrenia and co-occurring substance use disorders. Neurotox Res. 2007;11(1):33–40. doi: 10.1007/BF03033480. [DOI] [PubMed] [Google Scholar]
- 61.Krystal JH, D’Souza DC, Gallinat J, et al. The vulnerability to alcohol and substance abuse in individuals diagnosed with schizophrenia. Neurotox Res. 2006;10(3-4):235–252. doi: 10.1007/BF03033360. [DOI] [PubMed] [Google Scholar]
- 62.Conroy SK, Rodd Z, Chambers RA. Ethanol sensitization in a neurodevelopmental lesion model of schizophrenia in rats. Pharmacol Biochem Behav. 2007;86(2):386–394. doi: 10.1016/j.pbb.2006.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chambers RA, Krystal JH, Self DW. A neurobiological basis for substance abuse comorbidity in schizophrenia. Biol Psychiatry. 2001;50(2):71–83. doi: 10.1016/s0006-3223(01)01134-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mathalon DH, Pfefferbaum A, Lim KO, Rosenbloom MJ, Sullivan EV. Compounded brain volume deficits in schizophrenia-alcoholism comorbidity. Arch Gen Psychiatry. 2003;60(3):245–252. doi: 10.1001/archpsyc.60.3.245. [DOI] [PubMed] [Google Scholar]
- 65.George TP, Vessicchio JC, Sacco KA, et al. A placebo-controlled trial of bupropion combined with nicotine patch for smoking cessation in schizophrenia. Biol Psychiatry. 2008;63(11):1092–1096. doi: 10.1016/j.biopsych.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Weiser M, Reichenberg A, Grotto I, et al. Higher rates of cigarette smoking in male adolescents before the onset of schizophrenia: a historical-prospective cohort study. Am J Psychiatry. 2004;161(7):1219–1223. doi: 10.1176/appi.ajp.161.7.1219. [DOI] [PubMed] [Google Scholar]
- 67.Olincy A, Ross RG, Young DA, Roath M, Freedman R. Improvement in smooth pursuit eye movements after cigarette smoking in schizophrenic patients. Neuropsychopharmacology. 1998;18(3):175–185. doi: 10.1016/S0893-133X(97)00095-X. [DOI] [PubMed] [Google Scholar]
- 68.Adler LE, Hoffer LD, Wiser A, Freedman R. Normalization of auditory physiology by cigarette smoking in schizophrenic patients. Am J Psychiatry. 1993;150(12):1856–1861. doi: 10.1176/ajp.150.12.1856. [DOI] [PubMed] [Google Scholar]
- 69.George TP, Vessicchio JC, Termine A, et al. Effects of smoking abstinence on visuospatial working memory function in schizophrenia. Neuropsychopharmacology. 2002;26(1):75–85. doi: 10.1016/S0893-133X(01)00296-2. [DOI] [PubMed] [Google Scholar]