

Abstract
Chemotherapeutics tumor resistance is a principal reason for treatment failure and clinical and experimental data indicate that multidrug transporters such as ATP-binding Cassette (ABC) B1 and ABCG2 play a leading role by preventing cytotoxic intracellular drug concentrations. Functional efflux inhibition of existing chemotherapeutics by these pumps continues to present a promising approach for treatment. A contributing factor to the failure of existing inhibitors in clinical applications is limited understanding of specific substrate/inhibitor/pump interactions. We have identified selective efflux inhibitors by profiling multiple ABC transporters against a library of small molecules to find molecular probes to further explore such interactions. In our primary screening protocol using JC-1 as a dual-pump fluorescent reporter substrate we identified a piperazine substituted pyrazolo[1,5-a]pyrimidine substructure with promise for selective efflux inhibition. As a result of a focused SAR-driven chemistry effort we describe compound 1 (CID44640177), an efflux inhibitor with selectivity toward ABCG2 over ABCB1. Compound 1 is also shown to potentiate the activity of mitoxantrone in vitro as well as preliminarily in vivo in an ABCG2 over-expressing tumor model. At least two analogs significantly reduce tumor size in combination with the chemotherapeutic topotecan. To our knowledge, low nanomolar chemoreversal activity coupled with direct evidence of efflux inhibition for ABCG2 is unprecedented.
Keywords: Multi-drug resistance, ABC Transporter, ABCG2, ABCB1, Efflux inhibition
Introduction
More than 50 members of the ATP-binding cassette (ABC) transporter super-family have been identified, and three major subfamilies (ABCB, ABCC, and ABCG) have been linked to human multidrug resistance (MDR). These efflux pumps are expressed in many human tumors where they likely contribute to chemotherapy resistance. The transporters ABCB1, ABCC1, and ABCG2 are highly expressed in the gut, liver, and kidneys and may restrict the oral bioavailability of administered drugs. ABCB1 and ABCG2 are also expressed in the epithelia of the brain and placenta and also in stem cells, where they perform a barrier function.1 They influence oral absorption and disposition of a wide variety of drugs, and as a result their expression levels have important consequences for susceptibility to drug-induced side effects, interactions, and treatment efficacy. The specific subclass members ABCB1 (Pgp, MDR1), ABCC1 (MRP1), and ABCG2 (BCRP, MXR) are known to significantly influence the efficacy of drugs and have unambiguously been shown to contribute to cancer multidrug resistance.2 Dual treatment with ABC transporter inhibitors in conjunction with chemotherapeutics is a common treatment strategy to circumvent MDR in cancers.3 Although a large number of compounds have been identified as possessing ABC transporter inhibitory properties, only a few of these agents are appropriate candidates for clinical use as MDR reversing agents.4 The clinical failures observed with the current class of drugs provide ample justification for identifying new classes of modulators and exploring the biology around them.
The development of ABC efflux transporter inhibitors is now in its third generation with the major focus still on ABCB1. Progress over the last decade has renewed interest in the efflux inhibition field and a variety of modulators have been identified. A large number of structurally and functionally diverse compounds act as substrates or modulators of these pumps.5 A representative subset of these compounds will be briefly discussed here. The first-generation of chemosensitizers were discovered from marketed drugs and included the calcium channel blockers verapamil and nicardipine, cyclosporin A, and progesterone; however, dose-related toxicity and solubility challenges prevented progress into the clinic. Second and third generation inhibitors were drawn predominantly from the derivatization of first-generation molecules, as well as from combinatorial chemistry, targeted primarily at ABCB1. Some of the higher profile examples include: i) the cyclosporin A derivative valspodar (PSC-833);6 ii) Vertex Pharmaceutical's biricodar (VX-710);7 iii) the anthranilamide-based modulators XR9051 (2),8 tariquidar (XR9576),9-10 XR9577,11 and WK-X-34;11 iv) acridone carboxamide derivative elacridar (GF120918);12 v) the heteroaryloxypropanolamines zosuquidar (LY335979),13 dofequidar (MS-209)14 and laniquidar;15 and vi) the diarylimidazole ontogen (OC144-093, ONT-093).16 The later generation inhibitors tended to be more potent and less toxic than the first generation compounds; however, multiple off-target issues remain.
As a specialty screening center in the National Institutes of Health Molecular Libraries Probe Production Centers Network (NIH MLPCN), the University of New Mexico Center for Molecular Discovery (UNMCMD) and its Chemistry Center partners are tasked with finding small molecule probe compounds for academic investigators seeking improved tools for interrogating biological systems. In collaboration with the University of Kansas Specialized Chemistry Center (KU SCC) we set out to develop new small molecule scaffolds with distinct efflux inhibition selectivity profiles based on multiplex transporter target assays. Early in the post-screen follow-up it was evident that ABCG2 was the desirable focus for a probe campaign based on promising preliminary selectivity. Although there has been significant progress with ABCB1 inhibitors, similar progress has not been achieved with ABCG2 inhibitors. An example was noted with the Aspergillus fumigates mycotoxin fumitremorgin C (FTC, 3) and its analogs Ko132, Ko134, and Ko143 (4) which have been demonstrated to be selective inhibitors for ABCG2.17-18 Other reported ABCG2 inhibitors engage non-selectively to include biricodar and nicardipine which are cross-pump inhibitors for ABCB1, ABCC1, and ABCG2.7,19 Further, specific relevance for ABCG2 as a clinical target has been well documented.20 This includes a mouse model using a human ovarian xenograft with Igrove1/T8 tumors,21 a system utilizing flavopiridol-resistant human breast cancer cells,22 FTC (3) and Ko143 (4) inhibition in vitro and mouse intestine model,17 and a phase I/II trial with lapatinib in glioblastoma multiforme.23
Given the absence of clinically relevant ABCB1 or ABCG2 specific inhibitors and as there remain gaps in understanding how inhibition of these efflux pumps can be best exploited for therapeutic gain, our team focused on vetting and optimizing novel hit scaffolds with promising preliminary ABCG2 or ABCB1 selectivity and potency. As part of that effort, several bench mark compounds were chosen for comparison during development of the pyrazolopyrimidinylpiperazine scaffold, 1. Bench mark compounds were chosen for differential selectivities on ABCB1, ABCC1 and ABCG2, so as to represent a broad panel against which analogs of 1 could be evaluated (Figure 1). For direct comparison of selective ABCG2 inhibition, both 3 and 4 were chosen.17-18 The submicromolar ABCB1 modulator 2 was chosen as it is known to reverse resistance to cytotoxic drugs such as doxorubicin and vincristine.8,24 Quinoline MK571 (5), a specific inhibitor of ABCC1, was necessary to gauge any ABCC1 activity.25 Also, reversan (6), identified as an active inhibitor of ABCB1 and ABCC1, was included as it contained a similar, pyrazolopyrimidine core.26
Figure 1.

Structures of small molecules chosen for direct experimental comparison. Probe compound CID44640177 (1), ABCB1 inhibitor XR9051 (2), ABCG2 inhibitors FTC (3) and Ko143 (4), ABCC1 inhibitor MK571 (5), and the pyrazolopyrimidine reversan (6).
Materials and Methods
General information
The ABCB1 over-expressing drug-resistant cell line, CCRF-Adr 5000, and its parental CCRF-CEM cells were kindly provided by Dr. T. Efferth (Pharmaceutical Biology, German Cancer Research Center, Heidelberg, Germany). We have previously described the generation of the Jurkat-DNR ABCB1 over-expressing cell line.27 Ovarian ABCG2 over-expressing Ig-MXP3 and Igrov1/T8 cells as well as the parental Igrov1-sensitive cells were kindly provided by Dr. D. Ross (Department of Medicine, University of Maryland Greenebaum Cancer Center, Baltimore, MD). Cells were grown in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS, Hyclone, Logan, UT), 2 mM L-glutamine, 10 mM HEPES, 10 U mL−1 penicillin, 10 μg mL−1 streptomycin, and 4 μg mL−1 ciprofloxacin. Selective pressure for the ABCB1 over-expressing CCRF-ADR 5000 and Jurkat-DNR cells was maintained by growth in 20 nM daunorubicin hydrochloride (DNR). Selective pressure for the ABCG2 over-expressing Ig-MXP3 cells is maintained by treatment with 340 nM mitoxantrone dihydrochloride (MTX) for 1 hr. prior to harvest.
The fluorescent reporter dye JC-1 and cell type differentiation dye CellTrace™ Far Red DDAO-SE were obtained from Invitrogen™ (Carlsbad, CA). Nicardipine hydrochloride, DNR, MTX, topotecan hydrochloride hydrate (TPT) and FTC (3) were purchased from Sigma-Aldrich (St. Louis, MO). XR9051 (2), reversan (6), MK571 (5), and Ko143 (4) were purchased from Tocris Bioscience (Minneapolis, MN). Compounds ordered for SAR by commerce were purchased from ChemDiv (San Diego, CA) and Ryan Scientific (Mt. Pleasant, SC). Unless otherwise indicated, all compound solutions were maintained and diluted in DMSO prior to addition to assay wells. Final DMSO concentrations were no more than 1% (v/v). A Biomek® NX Multichannel (Beckman-Coulter, Brea, CA) was used for all cell and compound solution transfers for volumes greater than 1 μL. Low volume transfers (100 nL) were done via pintool (V&P Scientific, San Diego, CA). Compound dose-response plates were generated with the Biomek® NX Span-8 (Beckman-Coulter, Brea CA).
The HyperCyt® high throughput flow cytometry platform (IntelliCyt™, Albuquerque, NM) was used to sequentially sample cells from 384-well microplates (2 μL per sample) for flow cytometer presentation at a rate of ∼40 samples per minute.28-29 Flow cytometric analysis was performed on a CyAn™ flow cytometer (Beckman-Coulter, Brea, CA). The resulting time-gated data files were analyzed with HyperView® software to determine compound activity in each well. Inhibition response curves were fitted by Prism® software (GraphPad Software, Inc., La Jolla, CA) using nonlinear least-squares regression in a sigmoidal dose-response model with variable slope, also known as the four-parameter logistic equation. This type of time-gated flow cytometric data analysis was described in detail for a previous ABC transporter screen from our group.30
Primary assay conditions
To facilitate a shortened screening timeline the single point assay was performed as a duplex allowing for data from both cell lines to be collected in one screening campaign. The assay was conducted in 384-well format microplates in a total volume of 15.1 μL dispensed sequentially as follows: 1) JC-1 substrate (10 μL per well); 2) test compound (100 nL per well); 3) drug-resistant cells (5 μL per well). CCRF-Adr cells (ABCB1) were color-coded with 0.5 ng mL−1 CellTrace™ Far Red DDAO-SE for 15 minutes at room temperature, washed twice by centrifugation, and then combined with unlabeled Ig-MXP3 cells (ABCG2) in the assay buffer. Final in-well concentration of test compound was 6.6 μM, JC-1 concentration was ∼ 1 μM. JC-1 previously proved to be an ideal fluorescent reporter substrate for both ABCB1 and ABCG2.30 The cell concentration was 3 × 106 cells mL−1 (1:1 ratio of the two cell types). Nicardipine was used as an on-plate control for both pumps at 50 μM. The plate contents were mixed, rotated end-over-end at 4 RPM at 25 °C for 10 minutes, and then cell samples were immediately analyzed. This resulted in analysis of approximately 1,000 cells of each cell type from each well. Flow cytometric data of light scatter and fluorescence emission at 530 +/− 20 nm (488 nm excitation, FL1) and 665 +/− 10 nm (633 nm excitation, FL8) were collected.
The CCRF-Adr cell line proved optimal for the duplex-Far Red DDAO-SE staining protocol but in the single-plex follow-up we preferentially used the assay provider's ABCB1 over-expressing Jurkat-DNR cell line for confirmatory dose-response. Each cell line (Jurkat-DNR and Ig-MXP3) was run separately against all compounds (no differential cell staining) in dose-response. The protocol differed from the single point screen as describe here. Cells and reagents were added sequentially as follows: 1) PBS buffer (5 μL per well); 2) test compound (100 nL per well); 3) drug-resistant cells (10 μL per well) pre-exposed to the JC-1 substrate at 1 μM just prior to the well addition. Final in-well concentrations of test compound ranged from 50 μM to 69 nM over an 18 point dose-response and the cell concentration was 1 × 106 cells mL−1. Dose response IC50 values were average for multiple runs (average n of 2 to 4).
Chemoreversal secondary assay
Cells (ABCB1, Jurkat-DNR or ABCG2 Ig-MXP3) were incubated with the test compound in a 3 order of magnitude concentration range over three and seven day periods in the presence of the inhibitor and chemotherapeutic (ABCB1, 100 nM DNR or ABCG2 30 nM MTX), such that a cell concentration of at least 1 × 105 cells mL−1 was maintained. Cell viability was determined by trypan blue staining and enumeration under light microscopy. At day 3, wells with greater than 2 × 105 cells were refreshed with medium, to include readjustment of chemotherapeutic and inhibitor concentration. A chemoreversal index (Chemoreversal 50, CR50) was determined from the viability assessment. Using a similar approach, a direct cytotoxicity index (Toxic Dose 50, TD50) was determined by assessment of cell death of cells grown in media alone. Results were compared with the survival of parental cells in the presence of the selective agent (chemotherapeutic; 100% cell death), as well as survival of drug-resistant cells in the presence of the chemotherapeutic drug (control yields 100% viability). As previously described by our group for an ABCB1-reversal agent the difference between the CR50 and the TD50 affords an approximation of the in vitro therapeutic index for the test compound.31 The threshold for a “good” therapeutic window when comparing CR50 and TD50 somewhat depends on the endpoint use. For cancer treatment a low threshold, in the 10 fold (or greater) range, can still be considered acceptable due to the severity and life threatening nature of the disease.
Preliminary in vivo study
Igrov1/T8 cells were injected into the hind limbs of CB-17 SCID mice at a concentration of 1 × 107 cells in 200 μL, n of 3 per condition. The tumor was grown until the volume was in the range of 75 mm3 to 250 mm3. The volume of the tumor was verified with a Scienceware® Digi-Max™ slide caliper obtained from Sigma-Aldrich, and the tumor volumes were calculated by the equation: (W2/2)*L.21 Tumor-bearing mice were injected intratumorally with 150 nM topotecan alone, as well as with either 100 nM of 1 or 500 nM of 7. Injections were repeated daily and the size of the tumor was determined as a fraction of the starting size. After four days the mice were sacrificed and any recurring tumor was verified by light microscopy examination of histological sections. Mice were studied and maintained in accordance with guidelines of our Institutional Animal Research Committee at UNM HSC.
Representative synthesis
Probe compound 1 and many analogues were synthesized by the method shown (Figure 2, sequence A-C). Commercial substituted aminopyrazoles 90 (R1 = phenyl) were treated with the appropriate dialkylmalonate or β-ketoester to give intermediate 91, followed by chlorination to afford the pyrazolo[1,5-a]pyrimidine core intermediate 92. Installation of the piperazine moiety afforded 1 directly. In some cases, a Suzuki or Molander type coupling was preferred to install aryl functionality at a late stage in the synthesis (Figure 2, sequence D-F). More detailed synthetic methods and spectral data can be found in supplementary material.
Figure 2.

Representative synthetic route for compound 1. (A) methyl 3-(furan-2-yl)-3-oxopropanoate, AcOH, 100°C, 2 hr (65% yield). (B) POCl3, BnEt3NCl, PhNMe2, CH3CN, 80°C, 16 hr (84% yield). (C) furan-3-yl(piperazin-1-yl)methanone, DIPEA, CH3CN, 100 °C, 16 hr (99% yield). (D) diethylmalonate (21% yield) NaOEt, EtOH, 80°C, 3 hr (75% yield). (E) POCl3, N,N-dimethylaniline, 115°C, 16 hr (42% yield). (F) potassium aryltrifluoroborate salt, Pd(OAc)2, RuPhos, Na2CO3, EtOH, MWI, 90 °C, 6 hr.
Results and Discussion
Primary screening
A high throughput, no wash, duplex assay was constructed in which both the ABCB1 and ABCG2 transporters were evaluated in parallel using fluorescent JC-1 as the efflux reporting substrate. ABCB1 over-expressing CCRF-Adr cells were color-coded to allow their distinction from Ig-MXP3 ABCG2 over-expressing cells as previously described.30 Figure 3 briefly summarizes the primary, duplex screening protocol. The primary screening results were uploaded as PubChem BioAssay Database Identifiers (AID) 1325 and 1326 for ABCG2 and ABCB1 respectively (Summary AID 1818).32 A total of 194,393 Molecular Libraries Small Molecule Repository (MLSMR, http://mlsmr.glpg.com) compounds were tested with Z′ values of 0.74 ± 0.10 and 0.64 ± 0.13 for ABCB1 and ABCG2 respectively. A total of 200 and 130 actives were noted in ABCG2 and ABCB1 respectively. Compounds were deemed active if the percent inhibition was greater than 80%. A subsequent cherry pick resulted in single point confirmatory testing of 273 compounds (AIDs 1453 and 1451) resulting in 16 and 18 actives in ABCG2 and ABCB1, respectively. As a fluorescence counter-screen, a set of related 488/530 nm fluorescence compound profiling data was also associated with the SMR cherry pick set in which compound fluorescence was assessed in the absence of JC-1 to rule out false positives versus actual efflux inhibitors. These data were uploaded as two AIDs (1480 and 1483) where the 273 compounds were tested with 89 and 83 compounds noted as active (i.e. fluorescent) in ABCG2 and ABCB1, respectively. Based on this single point screening data confirmatory dose-response analysis was subsequently performed on 40 compounds (AIDs 1690 and 1689) resulting in 16 actives for ABCB1 and 9 actives for ABCG2.
Figure 3.

General scheme for the duplex HTS flow cytometric screening campaign. (A) In 384 well format, 1 μM JC-1 in PBS is added to the assay wells. (B) A volume of 100 nL of test compound is added to each well via pintool transfer (final concentration of 6.6 μM). (C) A 3 × 106 cells mL−1 mixture of both cell lines is added to each well. The ABCB1 cell line was previously color-coded with CellTrace™ Far Red DDAO-SE prior to mixture with the unlabeled ABCG2 line. (D) Flow cytometric data of light scatter and fluorescence emission at 530 +/− 20 nm (488 nm excitation, FL1) and 665 +/− 10 nm (633 nm excitation, FL8) are then collected via HyperCyt®. Each population is gated in FL8 allowing for analysis of FL1 in individual time bins for each cell line. The JC-1 retention can then be quantified as an indication of efflux inhibition by test compound(s). The FL1 versus time excerpt shown represents 24 binned wells of a 384 well plate.
Efflux inhibition driven SAR
No discernable structure activity relationship (SAR) was revealed through the first round of cherry pick analysis or the powder resupply, and many of the compounds were observed to be fluorescent artifacts. However, preliminary chemoreversal secondary screening efforts confirmed activity of several compounds, including 7 (CID1434724), where micromolar potentiation and low toxicity were observed but with little pump specificity. Secondary potentiation data for compound 7 and fourteen other confirmed compounds were reported in AIDs 2830 and 2833. Resynthesis, purification and retesting of 7 confirmed the efflux inhibition, and a series of compounds similar in structure was ordered around this original hit. These 31 compounds were tested in dose-response in the two efflux pump over-expressing cell lines: Jurkat-DNR (ABCB1) and Ig-MXP3 (ABCG2). A few compounds showed modest ABCG2 selectivity, but gaps in the collection did not resolve the structural functionality responsible for any significant efflux potency or selectivity towards ABCG2 or ABCB1. Due to the diversity of structural changes present in the commercial set, additional compounds were needed to construct meaningful SAR. The commercial powder set contained several members with conserved functionality that provided the basis for establishing a methodical SAR assessment.
The pyrazolo[1,5-a]pyrimidine core was preserved, and exchange of the peripheral substituents were surveyed, depicted as shaded regions (Figure 4, panel A). Exploratory commercial SAR expansion resulted in compounds with selectivity profiles significantly biased toward ABCG2. The initial set of hit-related compounds screened from the MLSMR and purchased from vendors predominately possessed structural differences in R1 - R3 and the furan ring of R4. Based on these structural variations around the core, we chose functional groups that would fill in the SAR gaps and further reveal pharmacological preferences based on steric interactions, lipophilicity, hydrogen bond donating or accepting character, and modulating the electronic nature of aryl substituents. Of the hits that were identified through this endeavor, 8 (CID1441553) had attractive efflux potency towards ABCG2 and marginal selectivity over ABCB1 (Figure 5, panel B). The KU SCC launched an SAR campaign aimed at further understanding the origin of potency and selectivity and set out to optimize the compound profile to meet the MLPCN probe criteria for potency and selectivity in the efflux assay. A suitable probe was defined as a compound that effected micromolar potentiation with a toxic-dose50/chemoreversal50 (TD50/CR50) ratio of greater than 10 and with overall toxicity greater than 15 μM.
Figure 4.

Scaffold modification summary from primary hit to probe compound. (A) Screening hit compound 7 (CID1434724) and regions of targeted SAR optimization (shaded areas). (B) Compound 8 (CID1441553) obtained from first-generation SAR optimization. (C) SAR refinement of ABCG2 selectivity leading to compound 1 (CID44640177).
Figure 5.

Efflux inhibition and chemotherapeutic potentiation of 1 (CID 44640177). (A) A representative curve showing efflux inhibition of ABCB1 in Jurkat-DNR cells (closed circles). The average IC50 (n = 3) is 4.65 ± 0.74 μM. (B) A representative curve showing efflux inhibition of ABCG2 in Ig-MXP3 cells (open circles). The average IC50 (n = 2) is 0.13 ± 0.03 μM (C) Potentiation of daunorubicin (DNR) mediated killing in Jurkat-DNR cells with 1 (n = 2 per data point). The CR50 (closed triangles) is 0.55 μM while the TD50 (closed squares) is 5.52 μM. Minimum allowable toxicity is set at 15 μM, thus the toxicity here is below the cut-off. (D) Potentiation of mitoxantrone (MTX) mediated killing in Ig-MXP3 cells (n = 2 per data point). The CR50 (open triangles) is 0.31 μM while the TD50 (open squares) is 18.30 μM. The minimum toxicity and the CR50/TD50 ratio (equal to 59) meet the cut-off criteria for a desirable compound in the chemoreversal secondary ABCG2 assay.
Of the approximately 160 compounds assessed in the primary efflux dose-response assay (AIDs 489002, 489003, 504566, and 504569), 126 were synthesized by the KU SCC. Several compounds in the purchased collection contained a 3-chlorophenyl substituent at R1, analogous to the R1 moiety present in hit 8. As such, an initial series of compounds was prepared with this feature maintained while adjusting R2-R4 (Table 1). One compound cluster was constructed with R1-R3 groups (11-25) identical to that of the parent hit 8 while modulating R4. Notably, the substitution of the acyl-2 furan for acyl-3-furan (11) produced a 7-fold enhancement in selectivity for ABCG2, predominately due to erosion of ABCB1 potency, while only modestly attenuating ABCG2 potency as compared to the parent hit. Improved ABCG2 potency was achieved with installation of an acyl-3-pyridine; however, the selectivity deteriorated essentially to pan inhibition (20). Following incorporation of the acyl-3-furan as the more optimal R4 substituent, a survey was then done on the R2 group while holding constant R1 and R3(26-32). Substantial potency for ABCG2 was gained when R4 was 3-pyridine (30); however, once again, selectivity was negatively impacted.
Table 1.
SAR expansion on initial hit compound 8 (CID1441553).
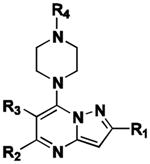
|
IC50 (μM)a | |||||||
|---|---|---|---|---|---|---|---|---|
|
|
|
|||||||
| Cpd | CID | R1 | R2 | R3 | R4 | ABCB1 | ABCG2 | ∼Fold G2 Selectiveb |
| 8 | CID1441553 | 3-Cl-Ph | Ph | H | CO-2-furyl | 6.18 ± 4.92 | 0.96 ± 0.37 | 6.4 |
| 9 | CID644556 | 3-Cl-Ph | Me | Me | CO-2-furyl | 8.40 ± 0.70 | 2.65 ± 1.07 | 3.2 |
| 10 | CID652994 | 3-Cl-Ph | Me | H | CO-2-furyl | 5.88 ± 1.17 | 2.78 ± 0.82 | 2.1 |
| 11 | CID44640182 | 3-Cl-Ph | Ph | H | CO-3-furyl | 8.58 ± 0.63 | 1.19 ± 0.26 | 7.2 |
| 12 | CID44607976 | 3-Cl-Ph | Ph | H | CO-3-thiophene | 3.97 ± 0.80 | 1.40 ± 0.32 | 2.8 |
| 13 | CID44602407 | 3-Cl-Ph | Ph | H | CO-Ph | 3.42 ± 1.64 | 1.45 ± 0.59 | 2.4 |
| 14 | CID45105078 | 3-Cl-Ph | Ph | H | CO-2-MeO-Ph | 4.73 ± 1.78 | 4.92 ± 4.28 | 1.0 |
| 15 | CID45105074 | 3-Cl-Ph | Ph | H | CO-3-MeO-Ph | 2.64 ± 0.64 | 4.45 ± 4.68 | 0.6 |
| 16 | CID45105073 | 3-Cl-Ph | Ph | H | CO-4-MeO-Ph | 2.89 ± 0.55 | 2.82 ± 1.91 | 1.0 |
| 17 | CID45105075 | 3-Cl-Ph | Ph | H | CO-2-Cl-Ph | 1.85 ± 0.75 | 1.31 ± 0.23 | 1.4 |
| 18 | CID45105080 | 3-Cl-Ph | Ph | H | CO-3-Cl-Ph | 4.19 ± 1.88 | 2.95 ± 0.57 | 1.4 |
| 19 | CID45105081 | 3-Cl-Ph | Ph | H | CO-4-Cl-Ph | 2.36 ± 0.70 | 2.67 ± 0.85 | 0.9 |
| 20 | CID44623842 | 3-Cl-Ph | Ph | H | CO-3-pyridyl | 0.57 ± 0.08 | 0.23 ± 0.15 | 2.5 |
| 21 | CID44630540 | 3-Cl-Ph | Ph | H | CO-4-pyridyl | 1.16 ± 0.30 | 1.03 ± 0.23 | 1.1 |
| 22 | CID45281172 | 3-Cl-Ph | Ph | H | CO-cyclohexyl | 3.31 ± 1.86 | 2.67 ± 1.26 | 1.2 |
| 23 | CID44602405 | 3-Cl-Ph | Ph | H | COCH3 | 5.67 ± 2.21 | 6.07 ± 0.73 | 0.9 |
| 24 | CID45105082 | 3-Cl-Ph | Ph | H | CO-Bn | 4.68 ± 2.69 | 5.76 ± 3.40 | 0.8 |
| 25 | CID44607585 | 3-Cl-Ph | Ph | H | SO2-Ph | >50 | >50 | NA |
| 26 | CID44631077 | 3-Cl-Ph | 2-F-Ph | H | CO-3-furyl | 4.69 ± 1.50 | 1.93 ± 0.48 | 2.4 |
| 27 | CID44629741 | 3-Cl-Ph | 4-F-Ph | H | CO-3-furyl | 5.65 ± 0.11 | 2.41 ± 0.52 | 2.3 |
| 28 | CID44630538 | 3-Cl-Ph | 3-MeO-Ph | H | CO-3-furyl | 2.17 ± 1.02 | 1.14 ± 0.54 | 1.9 |
| 29 | CID44629740 | 3-Cl-Ph | 4-MeO-Ph | H | CO-3-furyl | 4.12 ± 1.05 | 1.71 ± 0.58 | 2.4 |
| 30 | CID44629742 | 3-Cl-Ph | 3-pyridyl | H | CO-3-furyl | 0.77 ± 0.28 | 0.27 ± 0.12 | 2.9 |
| 31 | CID44630541 | 3-Cl-Ph | 4-pyridyl | H | CO-3-furyl | 3.68 ± 0.35 | 1.46 ± 0.35 | 2.5 |
| 32 | CID44631078 | 3-Cl-Ph | ethynyl | H | CO-3-furyl | 10.16±3.99 | 2.36 ± 0.53 | 4.3 |
| 33 | CID44623844 | 4-Br-Ph | Ph | H | CO-3-furyl | 1.31 ± 0.35 | 0.76 ± 0.43 | 1.7 |
| 34 | CID44640183 | 2-MeO-Ph | Ph | H | CO-3-furyl | 2.17 ± 0.76 | 0.45 ± 0.17 | 4.8 |
| 35 | CID44623840 | 3-MeO-Ph | Ph | H | CO-3-furyl | 2.26 ± 0.83 | 1.14 ± 0.59 | 2.0 |
| 36 | CID44607592 | 4-MeO-Ph | Ph | H | CO-3-furyl | 1.44 ± 0.70 | 1.16 ± 0.66 | 1.2 |
| 37 | CID44640176 | 2-MeO-Ph | Ph | H | CO-2-furyl | 3.00 ± 0.57 | 1.03 ± 0.17 | 2.9 |
| 38 | CID44968166 | 3-MeO-Ph | Ph | H | CO-2-furyl | 4.77 ± 0.63 | 1.40 ± 0.32 | 3.4 |
| 39 | CID492424 | 4-Cl-Ph | Ph | H | CO-2-furyl | 3.64 ± 1.47 | 1.37 ± 0.53 | 2.7 |
| 40 | CID45105079 | 3-Me-Ph | Ph | H | CO-2-furyl | 3.88 ± 1.67 | 2.46 ± 1.09 | 1.6 |
| 41 | CID44640179 | 2-MeO-Ph | Ph | H | CO-Ph | 2.31 ± 0.79 | 0.76 ± 0.06 | 3.0 |
| 42 | CID44968164 | 3-F-Ph | Ph | H | CO-Ph | 4.34 ± 2.09 | 2.27 ± 0.06 | 1.9 |
| 43 | CID44968158 | 4-F-Ph | Ph | H | CO-Ph | 3.99 ± 2.31 | 2.03 ± 0.11 | 2.0 |
| 1 | CID44640177 | Ph | 2-furyl | H | CO-3-furyl | 4.65 ± 0.74 | 0.13 ± 0.03 | 35.8 |
| 44 | CID46905002 | Ph | 2-MeO-Ph | H | CO-3-furyl | 4.52 | 3.66 ± 2.54 | 1.2 |
| 45 | CID46905009 | Ph | 4-MeO-Ph | H | CO-3-furyl | 4.55 | 2.37 ± 1.80 | 1.9 |
| 46 | CID46904993 | Ph | 2-F-Ph | H | CO-3-furyl | 5.61 | 0.56 | 10.0 |
| 47 | CID46905008 | Ph | 3-F-Ph | H | CO-3-furyl | 7.07 | 1.68 ± 0.61 | 4.2 |
| 48 | CID46904994 | Ph | 4-F-Ph | H | CO-3-furyl | 11.82 | 6.45 | 1.8 |
| 49 | CID46905006 | Ph | tBu | H | CO-3-furyl | 5.62 | 11.50 | 0.5 |
Efflux inhibition activity (IC50) of the JC-1 substrate is reported using Jurkat-DNR cells over-expressing ABCB1 and Ig-MXP3 cells over-expressing ABCG2. Replicates of two to four are reported as averages.
Selectivity is indicated by the quotient of the average ABCB1 IC50 and the average of ABCG2 IC50.
Alterations in the 3-chlorophenyl R1 substituent were then made while assessing three R4 head groups, specifically alternating between acyl-2-furan, acyl-3-furan, or benzoyl functionalities (33-43). No substantial improvements were noted with these changes; however, when R1 was changed from 3-chlorophenyl to phenyl, and R2 was varied (1, 44-49), it was discovered that a 2-furan at R2 in concert with the optimized acyl-3-furan afforded a significant boost in both ABCG2 potency as well as overall ABCG2 selectivity (1, ABCB1 EC50 = 4.65 μM; ABCG2 EC50 = 0.13 μM, selectivity = 36 fold). Representative dose-response curves for 1 are compared (Figure 5).
With this information in hand, the team followed up with an SAR effort aimed at demonstrating supportive SAR for compounds bearing an R2 = 2-furyl group while also attempting to improve upon the profile of the most promising analog, 1 (Figure 4, panel C). The modified scaffold, represented by 1, was further studied by adjusting physiochemical and spatial elements in R1 (Supplemental Table S1, compounds 1, 50-60). Replacing the phenyl ring of the lead with a t-butyl group erased much of the gains towards ABCG2 selectivity (50). Traditional phenyl replacements such as thiophene or furan were tolerated, but only led to modest selectivity and potencies. The installation of a 4-chlorophenyl substituent led to a reduced inhibition of ABCB1, resulting in selectivity in the efflux assay of 22-fold (54); however, the change also marginalized the potency on ABCG2. Renovating the phenyl substituent with electron donating groups was not beneficial.
An examination of 2-furan replacements at R2 was also undertaken (Supplemental Table S1, compounds 1, 44-49, 61-66). Simple alkyl units such as methyl or t-butyl degraded potency and fold-selectivity for both transporters. Notably, use of t-butyl actually reversed selectivity for ABCB1, albeit at the expense of potency (49). Some R2 revisions resulted in impressive ABCG2 selectivities and potencies. The choice of 2-F-phenyl (46) slightly degraded potency for ABCB1 as compared to the parent (1), leading to a 10-fold selectivity in favor of ABCG2. For the fluorinated series (46-48), the potency for both transporters decreased as the fluorine atom was migrated from the 2- to 3- to 4-position of the aromatic R2 ring. Interestingly, the use of a 3-MeO-phenyl group impeded potency for ABCB1 activity while retaining submicromolar ABCG2 potency on par with the parent, leading to an improved 83-fold selectivity between the transporters (65). In this series (44, 45, 65), however, a trend was not observed as the substituent was shifted from each position. Additional compounds prepared with the 3-MeO-phenyl group at R2 did not show a consistent SAR (data not shown).
The commercial set of compounds contained a few scaffolds bearing a methyl group at R3. SAR data generated in the early experimental phases demonstrated some benefit to the presence of small alkyl groups at R3; however, this was highly dependent on the identity of groups at R1, R2 and R4. To understand the functionality changes around 1, one analogue was prepared to quickly evaluate the effect of this substitution pattern in concert with our chosen functionalities at R1, R2 and R4). ABCB1 potency was encouragingly impaired, but not without also effecting G2, resulting in a marginal selectivity profile (data not shown).
Attention was then turned to investigating the effect of different R4 functionality appended to the piperazine (Supplemental Table S1, compounds 1, 67-76). In earlier SAR sets, activity was found to be sensitive to the identity of R4 and the pairing of groups at R2 and R3. In the context of our new lead, 1, we wanted to better understand the effect of R4 with the chosen substituents. It was confirmed that an acyl-3-furan was preferred to an acyl-2-furan (67), and simple alkyl substitution of the 3-furan (70-72) or a larger benzofuran (73), while tolerated, did not reveal any benefits. However, the most influential effects on ABCB1 were observed when the acyl furan was exchanged for a benzyl ester (76). While ABCG2 potency was compromised compared to the lead, ABCB1 potency was completely lost, yielding a selectivity of ∼ 19 fold.
In a more aggressive effort, the entire “top piece” of the scaffold, consisting of the piperazine and the R4 group, was modified (Supplemental Table S2, compounds 77-86). Ring-opened piperazine equivalents, truncated amino groups, piperidine amides, ring-expanded amines (not shown) and various structural variations on a theme did not produce a profile superior to that which had already been observed.
In the process of evaluating these structural modifications, several compounds were prepared singly to target possible oversights in SAR, as every possible R1-R4 combination cannot be prepared and assessed in a timely way. Others were targeted as a means of inserting the best combinations as gleaned from the preceding generations of SAR. These compounds were more recently pursued to probe specific structural combinations and are summarized. (Supplemental Table S1, compounds 87-89). Data obtained early in the project had indicated that the acetyl group at R4 was more advantageous than other changes that had been surveyed (including the benzyl ester modification), though later refinement of these data does not now stand out as particular SAR of interest. Based on the information available at the time, substituted phenyl derivatives with varying electronic features at R1 were incorporated with the acetyl R4 group in place (87, 88). No advantages were found.
Prior art comparison and potentiation
In parallel to the above efforts, select compounds were also assessed in secondary assays; however, this chemoreversal assay based on potentiation of a given chemotherapeutic is a very low throughput assay and compound data from this assay could not be used to drive the SAR program. Key data have been collected (AIDs 504476 and 504477) for some of the most promising compounds (Table 2). Evaluation of probe compound 7 showed submicromolar effective potentiated killing of both over-expressing cell lines with preference for ABCG2 from a toxicity perspective, though the degree of selectivity observed in the cell killing assay is removed (1.8 fold in chemoreversal vs. 36-fold in efflux assay). Though clear SAR trends could not be gleaned from the chemoreversal and toxicity data, these experiments provided the basis for evaluating how select compounds would perform in a cellular context. Most of the compounds evaluated in the chemoreversal assay for ABCB1 registered in the 100 to 600 nM range, with only a few outliers. For ABCG2, the CR50 was more broad, ranging from ∼ 20 to 1400 nM. These data, coupled with several compounds that showed TD50 values > 100, provided support for the selection of compounds for preliminary in vivo studies.
Table 2.
Potentiation data and prior art comparison.
| ABCB1 | ABCG2 | ||||||
|---|---|---|---|---|---|---|---|
|
|
|
||||||
| Cpd | CID | IC50a (μM) | CR50b (μM) | TD50c (μM) | IC50a (μM) | CR50b (μM) | TD50c (μM) |
| 1 | CID44640177 | 4.65 | 0.55 | 5.52 | 0.13 | 0.31 | 18.30 |
| 8 | CID1441553 | 6.18 | 0.25 | 6.77 | 0.96 | 0.14 | 6.00 |
| 9 | CID644556 | 8.40 | 1.58 | >100 | 2.65 | 0.17 | 47.40 |
| 15 | CID45105074 | 2.64 | 0.35 | >100 | 4.45 | 0.21 | >100 |
| 20 | CID44623842 | 0.57 | 0.17 | 4.72 | 0.23 | 0.13 | 10.30 |
| 21 | CID44630540 | 1.16 | 0.25 | 10.90 | 1.03 | 0.15 | 28.30 |
| 24 | CID45105082 | 4.68 | 0.51 | 17.80 | 5.76 | 0.60 | >100 |
| 30 | CID44629742 | 0.77 | 0.20 | 5.33 | 0.27 | 0.02 | 5.77 |
| 31 | CID44630541 | 3.68 | 0.60 | 13.50 | 1.46 | 0.89 | 57.80 |
| 34 | CID44640183 | 2.17 | 0.45 | 4.37 | 0.45 | 0.49 | 6.50 |
| 36 | CID44607592 | 1.44 | 0.23 | 5.54 | 1.16 | 0.38 | >100 |
| 44 | CID46905002 | 4.52 | 0.17 | 3.73 | 3.66 | 0.68 | 7.62 |
| 52 | CID46905000 | 1.70 | 0.19 | 1.93 | 2.35 | 1.21 | >100 |
| 53 | CID45105077 | 6.42 | 0.53 | 8.53 | 2.55 | 1.38 | 10.80 |
| 56 | CID45281176 | 7.40 | 0.47 | 5.52 | 2.26 | 1.00 | >100 |
| 57 | CID45489721 | 3.45 | 5.77 | >100 | 0.89 | 0.76 | >100 |
| 61 | CID46905005 | 1.60 | 0.42 | 7.33 | 1.40 | 0.76 | 18.30 |
| 65 | CID46904996 | 16.42 | 0.18 | 5.77 | 0.20 | 0.68 | 67.80 |
| 74 | CID46173053 | 3.64 | 0.19 | 10.10 | 0.52 | 0.93 | >100 |
| 87 | CID46912089 | 0.80 | 0.08 | 17.60 | 0.56 | 0.58 | >100 |
| 88 | CID46912090 | 1.64 | 0.32 | 49.30 | 1.90 | 1.18 | >100 |
| 2 | XR9051 | 0.61 | 0.01 | 1.97 | 2.27 | 0.65 | 21.40 |
| 4 | Ko143 | >50 | 0.99 | 2.84 | 13.55 | 5.94 | 20.00 |
| 5 | MK571 | >50 | 20.40 | 20.40 | >50 | 57.40 | 57.40 |
| 6 | reversan | 4.41 | 0.22 | 7.04 | 0.84 | 3.61 | >100 |
Efflux inhibition activity (IC50) of the JC-1 substrate is reported using Jurkat-DNR cells over-expressing ABCB1 and Ig-MXP3 cells over-expressing ABCG2. Replicates of two to four are reported as averages (standard deviation has been removed in this table for clarity).
Chemoreversal50 values are calculated as compared to day zero viability across varied dose of inhibitor and constant dose of chemotherapeutic (100 nM DNR for ABCB1 and 30 nM MTX for ABCG2). Each data point was the average of two replicates.
Toxic-dose50 values are calculated as compared to day zero viability across varied dose of inhibitor without chemotherapeutic.
Prior art for 1 includes 2 (XR9051) and MK571 which were chosen to verify both ABCB1 activity and counterscreen ABCC1 activity, respectively in our system (Table 2). Compound 2 inhibits the efflux of JC-1 in both ABCB1 and ABCG2 over-expressing cell lines (0.61 ± 0.43 and 2.27 ± 2.05 μM respectively). Potentiation data indicate submicromolar chemoreversal in both Jurkat-DNR and Ig-MXP3 cells with a bias toward ABCB1 at 10 nM as compared to 650 nM for ABCG2. Not surprisingly, we didn't observe any inhibition of ABCB1 or ABCG2 with MK571 (5) up to 50 μM and activity noted in the secondary assays mirrored toxicity indicating no potentiation. Direct comparison of reversan (6) in our efflux inhibition system shows low micromolar inhibition of both ABCB1 and ABCG2 (4.41 ± 2.90 and 0.84 ± 0.03 μM respectively) with moderate selectivity for ABCG2. In our chemoreversal potentiation assay, 6 showed micromolar activity and no ABCG2 selectivity (0.22 and 3.61 μM in ABCB1 and ABCG2, respectively). This was coupled with significant toxicity in the Jurkat-DNR cell line. FTC (3) showed no activity in either cell line in the efflux inhibition assay and was not tested in the potentiation assay. Ko143 (4) has been shown to potentiate mitoxantrone (MTX) at nanomolar levels in ABCG2 over-expressing cells 17. In our potentiation assay there appeared to be a selectivity for ABCB1 over ABCG2 (CR50 = 0.99 and 5.94 μM respectively) with considerable toxicity in both cell lines. The efflux inhibition activity did not emulate this, showing no activity for ABCB1 and only 13.55 μM inhibition for ABCG2, potentially indicating a binding site difference versus JC-1. Probe compound 1 demonstrates a 36-fold better inhibition of JC-1 efflux for ABCG2 over ABCB1, thus establishing its usefulness for biochemical exploration. This result, coupled with the noted cellular activity in the potentiation assay justifies the overall utility of 1 as a probe for ABCG2. Compound 1 showed greater potency and ABCG2 selectivity than any of the aforementioned literature precedent compounds in the efflux inhibition screening conditions. Only 2 appears to have better activity in the potentiation assay although with reversed selectivity toward ABCB1.
Preliminary in vivo mouse data
To specifically demonstrate the direct effect of a chemotherapeutic agent relevant to these studies, we administered an intratumoral dose of topotecan (TPT, 150 nM). This concentration of TPT was slow to kill non-resistant (ABCG2 non-expressing) parental tumor cells, but not the drug resistant (ABCG2 expressing) tumor cells (EC50 for parental is 7 nM vs. 311 nM for resistant cells). To demonstrate efficacy of the inhibitors, we grew ABCG2 resistant tumor cells to a volume between 75 and 250 mm3 in CB-17 SCID mice. Tumor size was determined immediately prior to the first injection. Tumor-bearing mice were injected with 150 nM TPT and either 100 nM of 1 or 500 nM of 7. Such treatment dramatically reduced tumor size the over a 96 hour observatory period (Figure 6), indicating that tumor sensitivity of TPT returned when either of the ABCG2-blocking compounds were present (p < 0.001). A concentration of 150 nM TPT alone did not affect the growth of tumors (data not shown). Compounds 1 and 7 were chosen as representative molecules from this scaffold, with 7 being the initial hit from the screening campaign and 1 being the efflux inhibition optimized molecule. As previously stated, the low throughput nature of the in vitro chemoreversal secondary assay did not allow for exhaustive testing of all members in the scaffold and thus specific correlation between efflux inhibition and potentiated activity of known chemotherapeutics was not readily accessible. However, similar in vivo activity of 1 and 7 (note there is a five-fold concentration difference for the two, see Figure 6) despite dissimilar efflux inhibition profiles indicates that the efflux reporting via JC-1 may not represent activity for both MTX and DNR in Ig-MXP3 and Jurkat-DNR cell lines respectively. This in conjunction with the reported in vitro chemoreversal data in Supplemental Table S1 indicates a need for further analysis of the in vitro to in vivo correlation. A subsequent manuscript detailing activity of molecules from this scaffold in this animal model is forthcoming.
Figure 6.

Response of ABCG2 resistant Igrov1/T8 derived tumors in mice to combination therapy of 150 nM topotecan (TPT). The tumor size at 0, 24, 48, 72, and 96 hr is indicated (n = 3) along with the standard error of the mean (SEM). The significant difference between the mean values from 0 to 96 hr is indicated by an asterisk (p < 0.001). Inhibitor concentration was selected based on potentiation efficacy balanced with apparent cellular toxicity. Significant tumor reduction was noted in both cases. (A) Compound 7 (original MLSMR hit) at 500 nM in conjunction with TPT. (B) Probe compound 1 at 100 nM with TPT.
Summary
As a result of a duplex, high-throughput flow cytometric screening campaign and subsequent medicinal chemistry optimization we report herein the discovery of an ABCG2 efflux inhibitor 1 which demonstrates a 36-fold selectivity over ABCB1 toward blocking efflux of the fluorescent substrate JC-1. Furthermore, in the same JC-1 efflux reporter system, 1 maintains approximately 100-fold higher potency in ABCG2 than the prior art Ko143. Subsequent in vitro assays using the known chemotherapeutic mitoxantrone demonstrate the ability of 1 to potentiate cell death of the ABCG2 expressing Ig-MXP3 cells at submicromolar concentration levels. We believe that selective transport inhibitors may allow for more targeted and effective therapy. Preliminary in vivo mouse model data indicate dramatic tumor reduction with co-treatment of TPT with 1. Future studies will focus on several related scaffold members as well as on selectivity profiles in both in vitro and in vivo potentiation.
Supplementary Material
Acknowledgments
Financial Support: This work was supported by the Nation Institutes of Health grants U54MH084690 (LAS, UNMCMD), R03 MH081228-01A1 (RSL), and U54HG0050311 (JA, KU SCC).
Footnotes
Supporting Information Available: This material is available free of charge via the Internet.
References
- 1.Sarkadi B, Homolya L, Szakacs G, Varadi A. Human multidrug resistance ABCB and ABCG transporters: Participation in a chemoimmunity defense system. Physiol Rev. 2006;86:1179–1236. doi: 10.1152/physrev.00037.2005. [DOI] [PubMed] [Google Scholar]
- 2.Krishna R, Mayer LD. Multidrug resistance (MDR) in cancer. Mechanisms, reversal using modulators of MDR and the role of MDR modulators in influencing the pharmacokinetics of anticancer drugs. Eur J Pharm Sci. 2000;11:265–283. doi: 10.1016/s0928-0987(00)00114-7. [DOI] [PubMed] [Google Scholar]
- 3.Gillet JP, Efferth T, Remacle J. Chemotherapy-induced resistance by ATP-binding cassette transporter genes. Biochim Biophys Acta. 2007;1775:237–262. doi: 10.1016/j.bbcan.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 4.Szakacs G, Varadi A, Ozvegy-Laczka C, Sarkadi B. The role of ABC transporters in drug absorption, distribution, metabolism, excretion and toxicity (ADME-Tox) Drug Discov Today. 2008;13:379–393. doi: 10.1016/j.drudis.2007.12.010. [DOI] [PubMed] [Google Scholar]
- 5.Eckford PDW, Sharom FJ. ABC efflux pump-based resistance to chemotherapy drugs. Chem Rev. 2009;109:2989–3011. doi: 10.1021/cr9000226. [DOI] [PubMed] [Google Scholar]
- 6.Atadja P, Watanabe T, Xu H, Cohen D. PSC-833, a frontier in modulation of P-glycoprotein mediated multidrug resistance. Cancer Metastasis Rev. 1998;17:163–168. doi: 10.1023/a:1006046201497. [DOI] [PubMed] [Google Scholar]
- 7.Germann UA, Shlyakhter D, Mason VS, Zelle RE, Duffy JP, Galullo V, et al. Cellular and biochemical characterization of VX-710 as a chemosensitizer: Reversal of P-glycoprotein-mediated multidrug resistance in vitro. Anti-Cancer Drugs. 1997;8:125–140. doi: 10.1097/00001813-199702000-00004. [DOI] [PubMed] [Google Scholar]
- 8.Dale IL, Tuffley W, Callaghan R, Holmes JA, Martin K, Luscombe M, et al. Reversal of P-glycoprotein-mediated multidrug resistance by XR9051, a novel diketopiperazine derivative. Br J Cancer. 1998;78:885–892. doi: 10.1038/bjc.1998.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mistry P, Stewart AJ, Dangerfield W, Okiji S, Liddle C, Bootle D, et al. In vitro and in vivo reversal of P-glycoprotein-mediated multidrug resistance by a novel potent modulator, XR9576. Cancer Res. 2001;61:749–758. [PubMed] [Google Scholar]
- 10.Stewart A, Steiner J, Mellows G, Laguda B, Norris D, Bevan P. Phase I trial of XR9576 in healthy volunteers demonstrates modulation of P-glycoprotein in CD56+ lymphocytes after oral and intravenous administration. Clin Cancer Res. 2000;6:4186–4191. [PubMed] [Google Scholar]
- 11.Jekerle V, Klinkhammer W, Scollard DA, Breitbach K, Reilly RM, Piquette-Miller M, et al. In vitro and in vivo evaluation of WK-X-34, a novel inhibitor of P-glycoprotein and BCRP, using radio imaging techniques. Int J Cancer. 2006;119:414–422. doi: 10.1002/ijc.21827. [DOI] [PubMed] [Google Scholar]
- 12.Hyafil F, Vergely C, Du Vignaud P, Grand-Perret T. In vitro and in vivo reversal of multidrug resistance by GF120918, an acridonecarboxamide derivative. Cancer Res. 1993;53:4595–4602. [PubMed] [Google Scholar]
- 13.Gerrard G, Payne E, Baker RJ, Jones DT, Potter M, Prentice HG, et al. Clinical effects and P-glycoprotein inhibition in patients with acute myeloid leukemia treated with zosuquidar trihydrochloride, daunorubicin and cytarabine. Haematologica. 2004;89:782–790. [PubMed] [Google Scholar]
- 14.Saeki T, Nomizu T, Toi M, Ito Y, Noguchi S, Kobayashi T, et al. Dofequidar fumarate (MS-209) in combination with cyclophosphamide, doxorubicin, and fluorouracil for patients with advanced or recurrent breast cancer. J Clin Oncol. 2007;25:411–417. doi: 10.1200/JCO.2006.08.1646. [DOI] [PubMed] [Google Scholar]
- 15.van Zuylen L, Sparreboom A, van der Gaast A, Nooter K, Eskens FALM, Brouwer E, et al. Disposition of docetaxel in the presence of P-glycoprotein inhibition by intravenous administration of R101933. Eur J Cancer. 2002;38:1090–1099. doi: 10.1016/s0959-8049(02)00035-7. [DOI] [PubMed] [Google Scholar]
- 16.Guns ES, Denyssevych T, Dixon R, Bally MB, Mayer L. Drug interaction studies between paclitaxel (Taxol) and OC144-093 - A new modulator of MDR in cancer chemotherapy. Eur J Drug Metab Pharmacokinet. 2002;27:119–126. doi: 10.1007/BF03190426. [DOI] [PubMed] [Google Scholar]
- 17.Allen JD, Van Loevezijn A, Lakhai JM, Van der Valk M, Van Tellingen O, Reid G, et al. Potent and specific inhibition of the breast cancer resistance protein multidrug transporter in vitro and in mouse intestine by a novel analogue of fumitremorgin C. Mol Cancer Ther. 2002(1):417–425. [PubMed] [Google Scholar]
- 18.Rabindran SK, Ross DD, Doyle LA, Yang W, Greenberger LM. Fumitremorgin C reverses multidrug resistance in cells transfected with the breast cancer resistance protein. Cancer Res. 2000;60:47–50. [PubMed] [Google Scholar]
- 19.Abe T, Koike K, Ohga T, Kubo T, Wada M, Kohno K, et al. Chemosensitisation of spontaneous multidrug resistance by a 1,4-dihydropyridine analogue and verapamil in human glioma cell lines overexpressing MRP or MDR1. Br J Cancer. 1995;72:418–423. doi: 10.1038/bjc.1995.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robey RW, Polgar O, Deeken J, To KW, Bates SE. ABCG2: Determining its relevance in clinical drug resistance. Cancer Metastasis Rev. 2007;26:39–57. doi: 10.1007/s10555-007-9042-6. [DOI] [PubMed] [Google Scholar]
- 21.Garimella TS, Ross DD, Eiseman JL, Mondick JT, Joseph E, Nakanishi T, et al. Plasma pharmacokinetics and tissue distribution of the breast cancer resistance protein (BCRP/ABCG2) inhibitor fumitremorgin C in SCID mice bearing T8 tumors. Cancer Chemother Pharmacol. 2005;55:101–109. doi: 10.1007/s00280-004-0866-2. [DOI] [PubMed] [Google Scholar]
- 22.Robey RW, Medina-Perez WY, Nishiyama K, Lahusen T, Miyake K, Litman T, et al. Overexpression of the ATP-binding cassette half-transporter, ABCG2 (MXR/BCRP/ABCP1), in flavopiridol-resistant human breast cancer cells. Clin Cancer Res. 2001;7:145–152. [PubMed] [Google Scholar]
- 23.Thiessen B, Stewart C, Tsao M, Kamel-Reid S, Schaiquevich P, Mason W, et al. A phase I/II trial of GW572016 (lapatinib) in recurrent glioblastoma multiforme: Clinical outcomes, pharmacokinetics and molecular correlation. Cancer Chemother Pharmacol. 2010;65:353–361. doi: 10.1007/s00280-009-1041-6. [DOI] [PubMed] [Google Scholar]
- 24.Mistry P, Plumb J, Eccles S, Watson S, Dale I, Ryder H, et al. In vivo efficacy of XR9051, a potent modulator of P-glycoprotein mediated multidrug resistance. Br J Cancer. 1999;79:1672–1678. doi: 10.1038/sj.bjc.6690267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vellenga E, Tuyt L, Wierenga BJ, Muller M, Dokter W. Interleukin-6 production by activated human monocytic cells is enhanced by MK-571, a specific inhibitor of the multi-drug resistance protein-1. Br J Pharmacol. 1999;127:441–448. doi: 10.1038/sj.bjp.0702577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burkhart CA, Watt F, Murray J, Pajic M, Prokvolit A, Xue C, et al. Small-molecule multidrug resistance-associated protein 1 inhibitor reversan increases the therapeutic index of chemotherapy in mouse models of neuroblastoma. Cancer Res. 2009;69:6573–6580. doi: 10.1158/0008-5472.CAN-09-1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Estes DA, Lovato DM, Khawaja HM, Winter SS, Larson RS. Genetic alterations determine chemotherapy resistance in childhood T-ALL: modelling in stage-specific cell lines and correlation with diagnostic patient samples. Br J Haematol. 2007;139:20–30. doi: 10.1111/j.1365-2141.2007.06763.x. [DOI] [PubMed] [Google Scholar]
- 28.Kuckuck FW, Edwards BS, Sklar LA. High throughput flow cytometry. Cytometry. 2001;44:83–90. [PubMed] [Google Scholar]
- 29.Ramirez S, Aiken Charity T, Andrzejewski B, Sklar Larry A, Edwards Bruce S. High-throughput flow cytometry: validation in microvolume bioassays. Cytometry. 2003;53:55–65. doi: 10.1002/cyto.a.10035. [DOI] [PubMed] [Google Scholar]
- 30.Ivnitski-Steele I, Larson RS, Lovato DM, Khawaja HM, Winter SS, Oprea TI, et al. High-throughput flow cytometry to detect selective inhibitors of ABCB1, ABCC1, and ABCG2 transporters. Assay Drug Dev Technol. 2008;6:263–276. doi: 10.1089/adt.2007.107. [DOI] [PubMed] [Google Scholar]
- 31.Winter SS, Lovato DM, Khawaja HM, Edwards BS, Steele ID, Young SM, et al. High-throughput screening for daunorubicin-mediated drug resistance identifies mometasone furoate as a novel ABCB1-reversal agent. J Biomol Screen. 2008;13:185–193. doi: 10.1177/1087057108314610. [DOI] [PubMed] [Google Scholar]
- 32.National Center for Biotechnology Information. PubChem BioAssay Database. AID=1818 Source=University of New Mexico Center for Molecular Discovery. http://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=1818&loc=Dea_ras.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.