

Abstract
Background and Purpose
PD5 inhibitors have recently been reported to exert beneficial effects against ischaemia–reperfusion injury in several organs. However, there are few studies regarding their neuroprotective effects in brain ischaemia. The present study was designed to assess the effects of sildenafil against chemical hypoxia induced by malonate. Intrastriatal injection of malonate produces energy depletion and striatal lesions similar to that seen in cerebral ischaemia through mechanisms that involve generation of reactive oxygen species (ROS).
Experimental Approach
Volume lesion was analysed by cytochrome oxidase histochemistry. Generation of reactive species was determined by in situ visualization of superoxide production and nitrotyrosine measurement. Protein levels were determined by Western blot after subcellular fractionation.
Key Results
Sildenafil, given 30 min before malonate, significantly decreased the lesion volume in the rat. This protective effect cannot be attributed to any effect on ROS production but to the inhibition of downstream pathways. Thus, malonate induced the activation of apoptosis signal-regulating kinase-1 (ASK1) and two MAPK kinases, MKK3/6 and MKK7, which lead to an increased phosphorylation of JNK and p38 MAPK, effects that were blocked by sildenafil. Selective inhibitors of p38 and JNK (SB203580 or SP600125, respectively) were used in combination with malonate in order to evaluate the plausible implication of these pathways in the protection afforded by sildenafil. While inhibition of p38 provided a significant protection against malonate-induced neurotoxicity, inhibition of JNK did not.
Conclusions and Implications
Sildenafil protects against the chemical hypoxia induced by malonate through the regulation of the ASK1–MKK3/6–p38/MAPK signalling pathway.
Keywords: apoptosis signal-regulating kinase 1 (ASK1), malonate, MAPKs, PDE5, reactive oxygen species (ROS), sildenafil
Introduction
PDE5 is a selective enzyme that catalyses the breakdown of cGMP (Bender and Beavo, 2006) and has been found in several tissues and some brain regions (Bender and Beavo, 2004). PDE5 inhibitors, such as sildenafil, were initially approved for the treatment of erectile dysfunction and nowadays also for pulmonary arterial hypertension (Galie et al., 2005), due to their vasodilatory effects resulting from the activation of cGMP-dependent protein kinase, PKG (Archer et al., 1994). During the last few years, sildenafil has also been reported to exert beneficial effects against endothelial dysfunction induced by stroke in humans (Gori et al., 2005) and protects against ischaemia–reperfusion injury in the heart of rodents (Kukreja et al., 2004; Salloum et al., 2007) and other organs such as the intestine (Soydan et al., 2009), colon (Irkorucu et al., 2009), testicles (Beheshtian et al., 2008), kidney (Lledo-Garcia et al., 2009) or spinal cord (Kiymaz et al., 2008). Furthermore, in preclinical studies of stroke models with young and aged rats, delayed treatment with PDE5 inhibitors increased neurogenesis, angiogenesis and synaptogenesis, and improved functional outcomes compared with placebo (Zhang et al., 2005; 2006; Ding et al., 2008; Menniti et al., 2009). Based on these findings, safety studies in ischaemic stroke patients are being conducted prior to investigating the neurorestorative properties of sildenafil in humans (Silver et al., 2009).
Despite all these evidences, there are very few studies regarding the mechanism underlying the effects exerted by sildenafil in brain ischaemic stroke models. The present study was, therefore, undertaken to assess the plausible neuroprotective effects of sildenafil in a model of chemical hypoxia. For this, we used malonate, a reversible inhibitor of succinate dehydrogenase (SDH). Intrastriatal administration of this mitochondrial toxin produces both energy depletion and striatal lesions that share many features with those that accompany focal ischaemia (Schulz et al., 1998). The mechanisms of malonate-induced neuronal cell death comprise the generation of reactive oxygen species (ROS), secondary excitotoxicity and apoptosis (Greene and Greenamyre, 1996; Dedeoglu et al., 2002). Oxidative stress is known to activate specific cell signalling pathways, such as MAPKs, which contribute to the cellular damage seen after brain ischaemic insults (Saito et al., 2005; Yanagisawa et al., 2008). Among them, JNK and p38 MAPK, also known as stress-activated protein kinases (SAPKs), have recently been implicated in the toxic effects of malonate as well (Asanuma et al., 2004; Gomez-Lazaro et al., 2007).
Based on these premises, we focused our study on the effects of sildenafil on the intracellular signalling pathways activated by oxidative stress after a malonate-induced chemical hypoxia.
Methods
Drugs and chemicals
Sodium malonate dibasic monohydrate was from Sigma-Aldrich (Madrid, Spain); 1-[4-ethoxy-3-(6,7-dihydro-1-methyl-7-oxo-3-propyl-1H-pyrazolo [4,3-d]pyrimidin-5yl) phenylsulfonyl]-4-methylpiperazine citrate (sildenafil citrate, Viagra™) was from Pfizer (New York, NY, USA) and 4-[5-(4-Fluorophenyl)-2-[4-(methylsulphonyl)phenyl]-1H-imidazol-4-yl]pyridine hydrochloride (SB203580 hydrochloride) and Anthra[1–9-cd]pyrazol-6(2H)-one (SP600125) were purchased from Tocris (Biogen Científica SL, Madrid, Spain).
Animals, treatments and experimental design
All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). Experiments were carried out in male Wistar rats (220–270 g; Harlan Iberica, Barcelona, Spain). Rats (four per cage) were housed in constant conditions of humidity and temperature (22 ± 1°C) with a 12 h/12 h light–dark cycle (lights on at 7:00 h). Food and water were available ad libitum. The experiments were performed after approval of the protocol by the institutional Ethics Committee, in accordance with the law in force (European Directive 86/609/EEC and Real Decreto 1201/2005), following the Research Council's Guide for the Care and Use of Laboratory Animals. All efforts were made to minimize animal suffering and to reduce the number of animals used in the experiments.
Rats were anaesthetized with sodium pentobarbital (60 mg·kg−1 i.p.) and placed in a Kopf stereotaxic frame, with the incisor bar set at 3.3 mm below the interaural line. The skull was exposed, bregma pointed, and two holes were drilled at coordinates according to the atlas of Paxinos and Watson (1997): +0.6 mm AP, ± 3 mm ML. Animals were injected with 2 μL of malonate, SB203580 or SP600125 using a 10 μL Hamilton syringe with a 26-gauge needle lowered 5.5 mm from the skull. After injection, the needle was left in place for 2 min to allow complete diffusion of the injected volume. The body temperature of the animals was kept constant at 37°C using an electric blanket during surgery and until recovery. Animals were housed individually until they completely recovered from the anaesthesia.
In a first set of experiments, malonate (1.5 μmol in 2 μL, pH 7.4–7.6) or PBS was infused into the striatum, and animals were killed at different time points (10 min, 30 min, 1, 3, 6 or 9 h) for Western blot analysis. In a different set of experiments, rats were orally administered with saline or sildenafil (1.5 mg·kg−1) 30 min before malonate injection and killed 6 h or 72 h after treatment for Western blot and histochemistry analysis.
Solution of sildenafil was prepared as previously described (Puerta et al., 2010) by grinding Viagra™ tablets into powder and dissolved in distilled water. The drug solution was filtered (0.45 μm pore size) before oral administration. Dose was chosen to simulate the dose for a patient with 70 kg body weight after orally taking a 20 mg tablet of Viagra™ according to Reagan-Shaw et al. (2008) and is the same dose that has been shown to induce cardioprotection against ischaemia–reperfusion injury (Das et al. 2004) and to exert neuroprotective effects against other neurotoxins (Puerta et al., 2009; 2010).
In a different set of experiments, SP600125 (1 nmol in 2 μL, pH 7.4–7.6 in DMSO 1%) or SB203580 (1 nmol in 2 μL, pH 7.4–7.6 PBS), alone or in combination with malonate (1.5 μmol), was injected into the striatum and animals were killed 6 h or 72 h after treatment.
Cytochrome oxidase histochemistry and analysis of lesion size
Seventy-two hours after treatments, rats were killed, and brains were frozen immediately on dry ice and then sectioned (25 μm) on a cryostat. Every second section throughout the extent of the lesion was mounted on a polylysine-coated slide. Incubation medium consisted of 5 mg cytochrome c and 30 mg 3,3′-diaminobenzidine in 50 mL of 0.1 M phosphate buffer, pH 7.4. Slides were incubated for 90 min at 37°C and then removed to 4% neutral, buffered paraformaldehyde for 10 min. Sections were rinsed with distilled water, dehydrated, cleared in xylene and coverslipped (Goni-Allo et al., 2005). The lesioned area on each section was quantified using a video-based MCID image analysis system (Imaging Research, St. Catherines, Ontario, Canada). Area measurements were summed and multiplied by intersectional distance (50 μm) to determine lesion volume.
In situ detection of superoxide production
In situ visualization of superoxide production was assessed by hydroethidine histochemistry as previously described (Kim and Chan, 2002). Rats were administered with sildenafil 30 min prior malonate intrastriatal administration. Two hours later, 200 μL of PBS containing 1 mg·mL−1 hydroethidine (Molecular Probes, Invitrogen, Carlsbad, CA, USA) and 1% DMSO was administered through the carotid artery. Brains were collected 30 min later and frozen on dry ice. Midbrain sections (25 μm thick) were mounted onto gelatin-coated glass slides. Sections were incubated with DAPI (Merck, Darmstadt, Germany) in PBS for 15 min in a dark chamber and then were rinsed in distilled H2O and mounted with Aquamount (Shandon, Pittsburgh, PA, USA). Hydroethidine's oxidation product, ethidium accumulation, was examined by fluorescence microscopy (excitation 510 nm, emission 580 nm) and was quantified using the image analysis software AnalySISD 5.0 (Soft Imaging System, Olympus, Münster, Germany).
Nitrotyrosine (NT) measurement
For NT measurement, rats were orally administered with saline or sildenafil (1.5 mg·kg−1) 30 min before malonate injection and were killed 72 h later. Striatal homogenates were prepared in PBS containing a protease inhibitor cocktail set (Calbiochem, Darmstadt, Germany), 0.5% Nonidet P-40, 0.5% sodium deoxycholate and 0.1% SDS. The homogenates were centrifuged at 10 000× g for 10 min, and the supernatants were assayed for NT content using a Nitrotyrosine ELISA Kit (Hycult Biotechnology b.v., Uden, the Netherlands) according to the manufacturer's instructions.
Subcellular fractionation and Western blot analysis
For Western blot analysis, animals were killed by decapitation; brains were rapidly removed, placed on ice and 2 mm thick tissue section was taken (approximately 1 mm to either side of the injection zone). Striatal tissue was dissected out and fractionated into cytosolic and nuclear fractions according to established protocols (Garcia-Osta et al., 2004; Vijayvergiya et al., 2005; Pallotti and Lenaz, 2007), with some modifications. Briefly, tissues were gently homogenized by 20 strokes in a glass-Teflon Potter homogenizer on ice in 200 μL of buffer A (10 mM Tris, pH 7.4, 320 mM sucrose, 1 mM EDTA, 1 mM DTT, and 1 mg·mL−1 fatty-acid-free BSA) containing phosphatase inhibitors (Phosphatase Inhibitor Cocktail I, Sigma-Aldrich) and protease inhibitors (Protease Inhibitor Cocktail Set I, Animal-Free Aprotinin, Calbiochem). Homogenates were then centrifuged at 1500× g for 5 min at 4°C. Supernatants were centrifuged at 100 000× g for 1 h at 4°C and saved as cytosolic (S100) fractions. Pellets were resuspended in 80 μL of Buffer B (150 mM NaCl, 10 mM Tris, pH 8.5, 1.5 mM MgCl2, 0.5% Nonidet, 1 mM DTT, containing phosphatase and protease inhibitors) and were centrifuged twice at 1500× g for 5 min at 4°C to obtain the nuclear fraction. To verify the relative subcellular purification, each fraction was subjected to Western blotting for Thioredoxin 1 as a cytosolic marker using a rabbit monoclonal antibody anti-Thioredoxin I (2298; Cell Signaling Technology, Beverly, MA) and Lamin A/C as a nuclear marker using a rabbit monoclonal antibody anti Lamin A/C (2032 Cell Signaling Technology).
For DARPP-32 determinations, tissues were homogenized as previously described (Goni-Allo et al., 2008) to obtain whole cell extracts. Finally, protein was determined by Bradford protein assay (Bio-Rad, Hercules, CA, USA).
Proteins (20 μg) were separated by electrophoresis on a SDS-PAGE under reducing conditions. Membranes were probed using anti-DARPP-32 (AB1656, Chemicon, Millipore, Billerica, MA, USA); anti-p38 MAPK, anti-pASK1 (Ser83), anti-p-cjun (Ser63), anti-JNK (56G8), anti-pJNK, anti-Lamin A/C, anti-pMKK3/MKK6, anti-pMKK7 (9212, 3761, 9261, 9258, 9251, 2032, 9231, 4171, respectively, Cell Signaling Technology); anti-pp38 MAPK (1229-1; Epitomics, Burlingame, CA, USA), 1/1000 dilution or anti-β-actin monoclonal antibody (A1978; Sigma), 1/10 000 dilution. Blots were visualized using a chemiluminescense ECL Western blotting detection reagent (Amersham, Buckinghamshire, UK). Band intensity was estimated densitometrically on a GS-800 calibrated densitometer (Bio-Rd One). Note that blots were stripped and re-probed when necessary.
Data analysis
Results were expressed as mean ± SEM Comparisons among groups were made using Student's t-test or one-way anova followed by Tukey's test for multiple group comparisons. Treatment differences were considered statistically significant at P < 0.05. Data analyses were performed using the Statistical Program for the Social Sciences (SPSS for Windows, 15.0; SPSS, Chicago, IL, USA).
Results
Sildenafil prevents striatal lesions caused by malonate independent of ROS generation
To investigate the neuroprotective effects of sildenafil against neuronal death-induced by malonate, rats were administered with sildenafil (1.5 mg·kg−1 p.o.) 30 min before striatal stereotaxic injections of 1.5 μmol of malonate. Seventy-two hours later, rats were killed and tissue was prepared for histochemistry and Western blot analysis. As shown in Figure 1A, malonate produced a large striatal lesion that was reverted by sildenafil. Protective effect of this PDE5 inhibitor was further confirmed by Western blot analysis of striatal DARPP-32 protein levels. DARPP-32 is a marker of medium spiny GABAergic neurons, the dominant population of neurons in the striatum and the most vulnerable to excitotoxic lesions (Martinez-Serrano and Bjorklund, 1996). As depicted in Figure 1B, malonate produced a consistent loss of striatal DARPP-32 protein levels that was significantly prevented by sildenafil.
Figure 1.
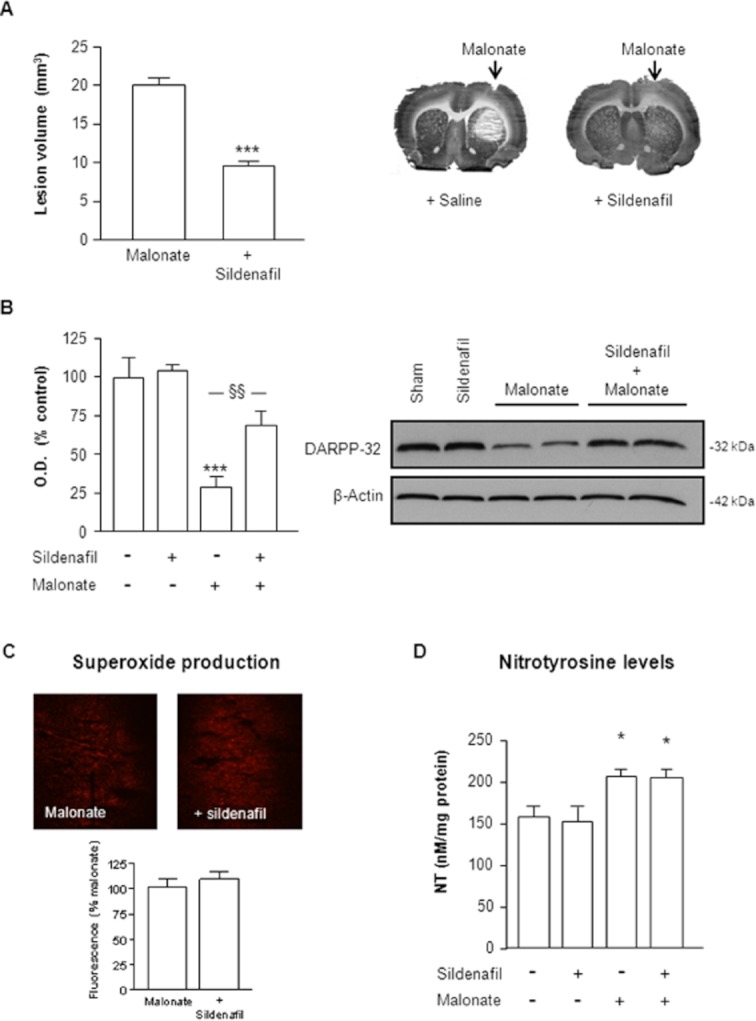
Neuroprotective effect of sildenafil against malonate injection. Sildenafil (1.5 mg·kg−1 p.o.) was administered 30 min before intrastriatal administration of malonate (1.5 μmol/2 μL). Rats were killed 72 h later. (A) Representative cytochrome oxidase-stained slices and quantification of malonate-induced striatal lesions (mm3) show that the neurotoxic effect of malonate is attenuated by sildenafil. Data are mean ± SEM (n = 9–13 animals per group). [t(20) = 6.233, P < 0.001]. (B) Quantitative measurement of DARPP-32 levels and representative Western blots showing DARPP-32 protein bands at 32 kDa. β-actin was used as equal loading control. Data are mean ± SEM (n = 5–8 animals per group). [F(3,20) = 7.516, P < 0.01]. ***P < 0.001 versus sham, §§P < 0.01 versus malonate. (C) Effect of sildenafil on malonate induced superoxide production. Quantitative measurement of fluorescence levels (bottom) and representative photomicrographs showing fluorescent ethidium signals (red) in the striatum 2.5 h after malonate injection. (D) Effect of sildenafil on malonate-induced nitrotyrosine (NT) formation in the striatum 72 h after treatments. Results are mean ± SEM, n = 5–8. Statistical analysis yielded the following result: [F(3,23) = 5.726;P < 0.01]. Different from the corresponding sham group: *P < 0.05.
It has been suggested that ROS generation by malonate plays a key role in the mechanisms underlying neurotoxicity (Fernandez-Gomez et al., 2005). We hypothesized, therefore, that protection afforded by sildenafil could be due to the inhibition of ROS production. Analysis of superoxide radical production using hydroethidine in situ detection revealed that in saline-injected rats, striatal superoxide and superoxide-derived oxidant production was minimal (data not shown). In contrast, ethidium fluorescence was increased 2.5 h after malonate injection, an effect not prevented by sildenafil (Figure 1C). This was further confirmed when striatal levels of NT, a highly reactive anion formed in the reaction of NO with superoxide radicals (Ischiropoulos and al-Mehdi, 1995), were measured 72 h after malonate injections (Figure 1D).
Malonate activates the ASK1/MAPKKs pathway: Effect of sildenafil
As sildenafil failed to block malonate-induced ROS production, we wondered whether sildenafil could be modulating ROS activation of the SAPKs pathway. Recent studies have shown that ASK1 is selectively required for sustained activation of the p38 and JNK SAPK induced by oxidative stress (Tobiume et al., 2001). Among the variety of ASK1 regulators, Akt is known to phosphorylate ASK1 on the serine 83, which is associated with a decrease in its kinase activity (Kim et al., 2001). Since sildenafil has been shown to activate Akt (Puerta et al., 2009), we investigated the phosphorylation status of ASK1 at Ser83. Western blot analysis showed that malonate significantly decreased the inhibitory phosphorylation of ASK1, effect that was prevented by sildenafil (Figure 2A).
Figure 2.
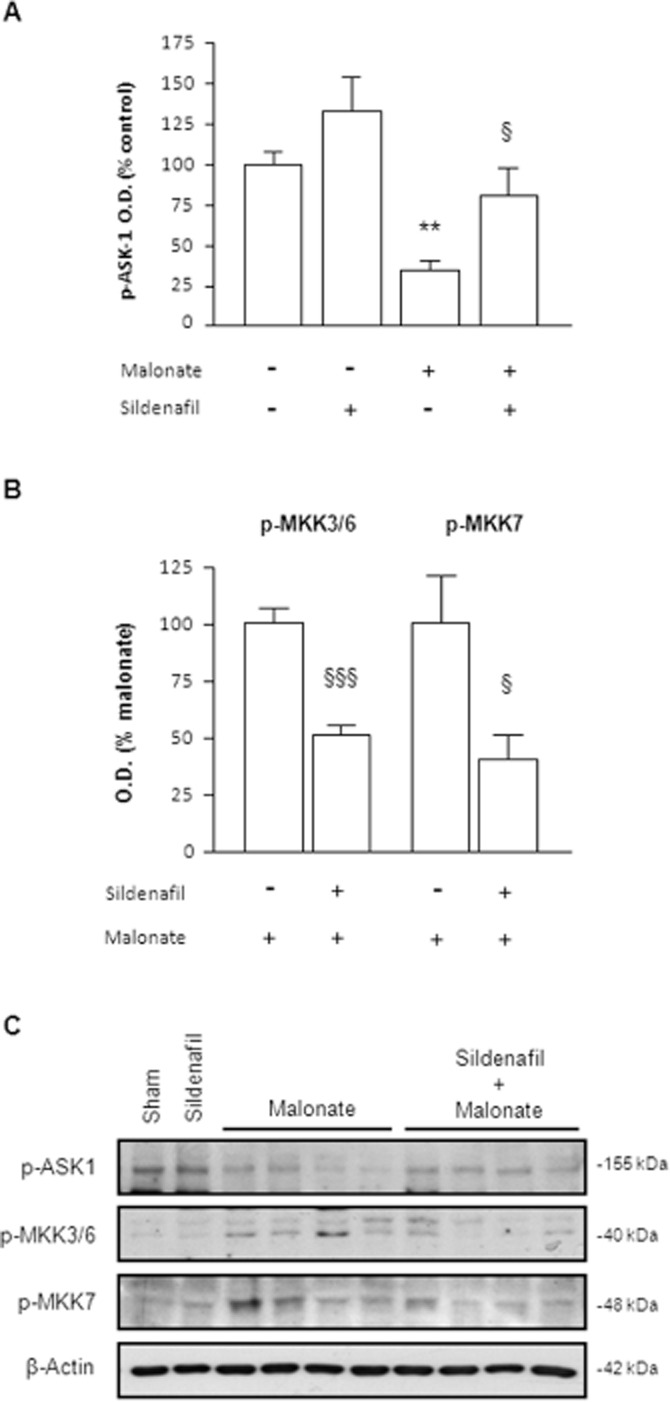
ASK1 activation is inhibited by sildenafil. (A) Quantitative measurements of optical density (O.D.) of p-ASK1 (Ser83). [F(3,21) = 9.505, P < 0.001]. (B) Quantitative measurements of Western blot analysis of the phosphorylation status of the p38-activating kinases MKK3 and MKK6 and the JNK-activating kinase MKK7. Note that values from sham and sildenafil groups were so weak that were not taken into account for quantitative measurement. Statistical analysis yielded the following results: for p-MKK3/6 [t(12) = 6.778, P < 0.001] and p-MKK7 [t(10) = 2.829, P < 0.05]. Results are mean ± SEM, n = 5–8. Different from the corresponding sham group: **P < 0.01. Different from malonate-only animals: §P < 0.05; §§§P < 0.001. (C) Representative blots showing that sildenafil reverted malonate-induced alterations on the phosphorylation status of ASK-1, MKK3/6 and MKK7.
Once activated, ASK1 phosphorylates the MAPK kinases MKK7 and MKK3/MKK6, which, in turn, promote JNK and p38 kinase activities respectively (Ichijo, 1997). As shown in Figure 2B, levels of p-MKK3/6 and p-MKK7 were significantly increased 6 h after malonate administration, an effect that was significantly decreased by sildenafil.
Time course activation of SAPKs pathway after malonate
Based on our results and those reported by others (Asanuma et al., 2004; Gomez-Lazaro et al., 2007), we examined the time course activation of JNK and p38 after malonate injections in nuclear and cytosolic fractions.
p38 MAPK is activated following phosphorylation at Thr180/Tyr182 by upstream MAPKKs. Upon activation, it translocates to the nucleus where it interacts with its targeted transcription factors (Raingeaud et al., 1996). As shown in Figure 3A, malonate activated p38 in a time-dependent manner. In particular, p-p38 and total p38 levels were significantly decreased in the cytosolic fraction by 30 min after malonate injections, whereas the opposite was found in the nuclear fraction, remaining above control levels up to 9 h later.
Figure 3.
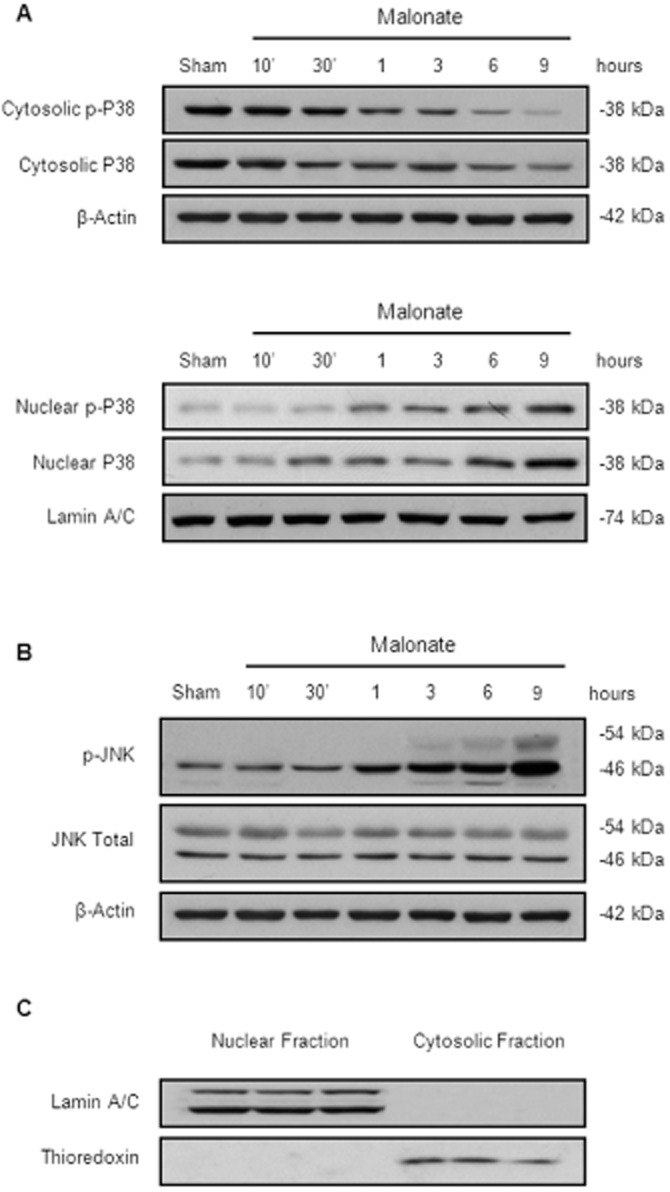
Time course activation of SAPK induced by malonate. Animals were killed at different time points after intrastriatal injection of malonate (1.5 μmol/2 μL). (A) Malonate induces p38 translocation to the nucleus in a time-dependent manner. Representative Western blots of cytosolic and nuclear fractions. β-actin and lamin A/C were used as equal loading control (n = 5 per group). (B) Representative Western blots showing the time course expression levels of p-JNK in the cytosol after malonate administration (n = 5 per group). (C) Representative blots showing the purity of our samples.
On the other hand, the activation of JNK (54 and 46 kDa), measured as a significant increase in the ratio between the phosphorylated and the unphosphorylated forms of JNK, was not evident until 3 h after malonate injection remaining significantly high for at least six more hours (Figure 3B). Figure 3C shows a representative blot used to assess the purity of our samples.
Effect of sildenafil on JNK activation induced by malonate
As MAPKs are activated in response to malonate, we next investigated whether sildenafil neuroprotection affected the activation of these pathways. In this case, we administered sildenafil 30 min prior to malonate injection and rats were killed 6 h later because we had previously observed a sustained increase of both, p38 and JNK, at this time point.
We first focused on JNK activation and found that sildenafil almost completely reversed the phosphorylation of JNK (Figure 4A). Within the nucleus, activated JNK controls the phosphorylation state of c-Jun as well as its transcriptional function (Bogoyevitch and Kobe, 2006). As expected, using a specific antibody for the phosphorylated form of c-Jun (p-c-Jun Ser63), we detected a significant activation of this transcription factor 6 h after malonate that was also reduced by sildenafil (Figure 4B).
Figure 4.
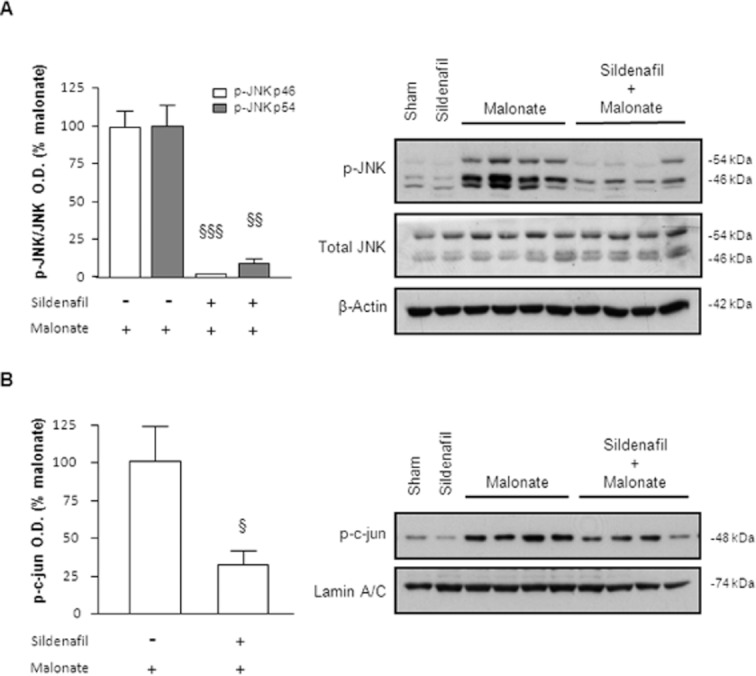
Sildenafil reverses JNK activation caused by malonate. Sildenafil (1.5 mg·kg−1 p.o.) was administered 30 min before malonate injection (1.5 μmol/2 μL), and rats were killed 6 h later. (A) Quantitative measurement of p-JNK/JNK levels and representative Western blots showing that sildenafil inhibits the phosphorylation of JNK (46 and 54 kDa) induced by malonate. Note that the levels of p-JNK from sham and sildenafil groups were so weak that were not taken into account for statistical analysis. Data are means ± SEM (n = 5–9 animals for each group). Data analysed by Student's t-test revealed significant differences: for p-JNK 46 kDa [t(12) = 12.77, P < 0.001] and for p-JNK 54 kDa [t(12) = 8.330, P < 0.01]. (B) Sildenafil inhibits the increase in c-jun phosphorylation at Ser63 shown in nuclear fractions. Data are mean ± SEM (n = 6 animals per group) [t(11) = 2.950, P < 0.05]. §P < 0.05, §§P < 0.01, §§§P < 0.001 versus malonate.
JNK inhibition does not protect against malonate-induced cell death
In parallel experiments, we investigated whether the inhibition of JNK would result in a corresponding decrease of malonate neurotoxicity. For this, rats were treated with the reversible ATP competitive JNK inhibitor, SP600125 (1 nmol/2 μL), in combination with malonate. As seen in figure 5A, the administration of SP600125 resulted in a significant decrease of both p-JNK and p-cjun, indicating an effective blockage of JNK activity. This effect, however, was not accompanied by any reduction on the infarct size induced by malonate (Figure 5B). Furthermore, no effect on p38 translocation to the nucleus was observed (Figure 5C and 5D).
Figure 5.
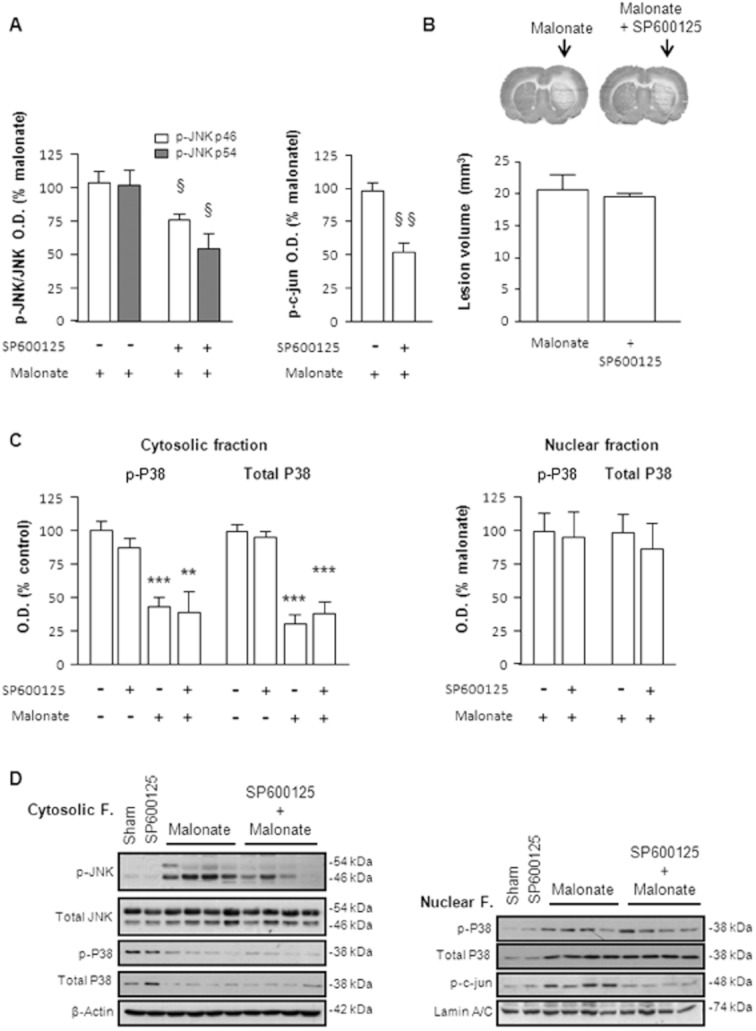
Effect of the JNK inhibitor, SP600125, on malonate induced neurotoxicity. SP600125 (1 nmol/2 μL) was administered intrastriatally in combination with malonate (1.5 μmol/2 μL). (A) Quantitative measurement of optical density showing that SP600125 produced a half/significant decrease in JNK and c-jun phosphorylation caused by malonate. Data are mean ± SEM (n = 5–8). Data analysed by Student's t-test revealed significant differences: for p-JNK 46 kDa [t(8) = 2.565, P < 0.05], for p-JNK 54 kDa [t(9) = 2.721, P < 0.05] and for p-c-jun [t(10) = 4.544, P < 0.05]. (B) Representative cytochrome oxidase-stained slices and quantification of malonate-induced striatal lesions (mm3) show that SP600125 failed to inhibit the neurotoxic effect of malonate observed 72 h after treatment. (C) SP600125 had no effect on p38 translocation to the nucleus. (D) Representative blots from nuclear and cytosolic fractions showing the effects of SP600125 on malonate-induced activation of the SAPKs pathway 6 h after treatment.
Effect of sildenafil on p38 activation induced by malonate
We next investigated the effect of sildenafil on the activation of p38 observed after malonate injection. As described before, sildenafil was administered 30 min prior to malonate, and rats were killed 6 h later. As shown in Figure 6, sildenafil prevented p38 and p-p38 reductions in the cytosol by inhibiting their translocation to the nucleus.
Figure 6.
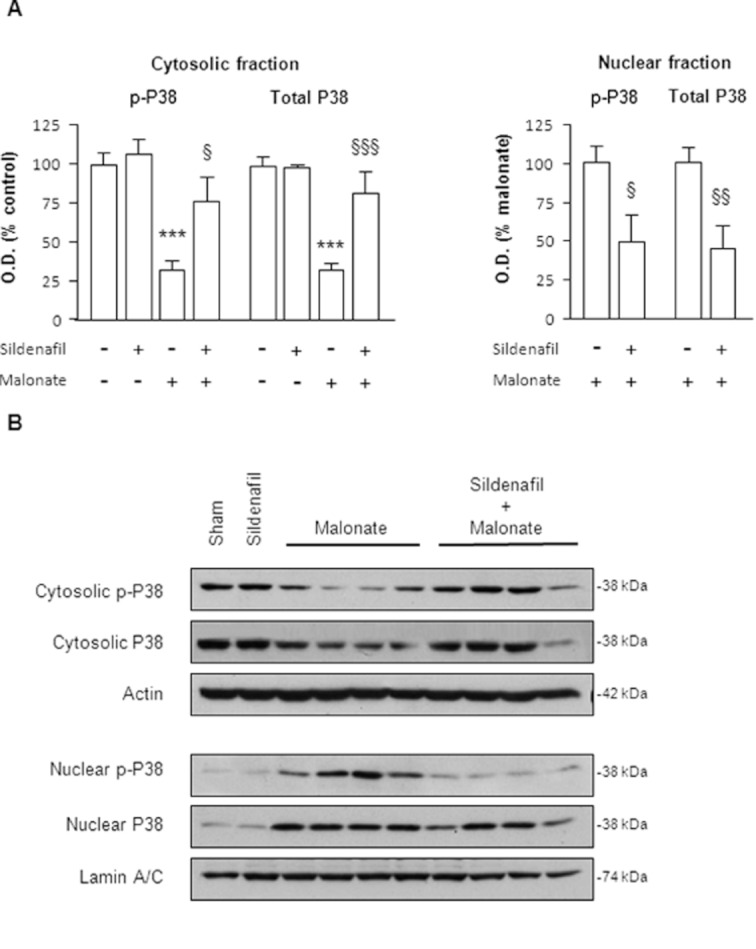
Sildenafil reverses p38 activation caused by malonate. Sildenafil (1.5 mg·kg−1 p.o.) was administered 30 min before malonate injection (1.5 μmol/2 μL), and rats were killed 6 h later. (A) Quantitative measurement of p-p38 and p38 levels in both cytosolic and nuclear fractions. Note that nuclear values from sham and sildenafil groups were so weak that were not taken into account for statistical analysis. Data are mean ± SEM (n = 6–9). Statistical analysis yielded the following results: for cytosolic p-p38 [F(3,31) = 12.73, P < 0.001] and total p38 [F(3,31) = 17.81, P < 0.001], for nuclear p-p38 [t(13) = 2.224, P < 0.05] and total p38 [t(11) = 3.038, P < 0.01]. ***P < 0.001 versus sham; §P < 0.05, §§P < 0.01, §§§P < 0.001 versus malonate. (B) Representative blots from nuclear and cytosolic fractions showing that sildenafil blocks p38 translocation induced by malonate.
Malonate neurotoxicity is mediated via p38 MAPK
The significance of p38 MAPK activation in malonate neurotoxicity was confirmed by administrating SB203580, a highly specific inhibitor of p38 MAPK (Cuenda et al., 1995). Treatment with SB203580 (1 nmol/2 μL) significantly decreased the histological lesion caused by malonate (Figure 7A). Of notice, SB203580 not only inhibited p38 activity but also blocked the phosphorylation of JNK and c-jun caused by malonate (Figure 7B). These results not only support the key role of p38 translocation in malonate neurotoxicity but also point to its inhibition by sildenafil as a plausible mechanism underlying its cytoprotective effects.
Figure 7.
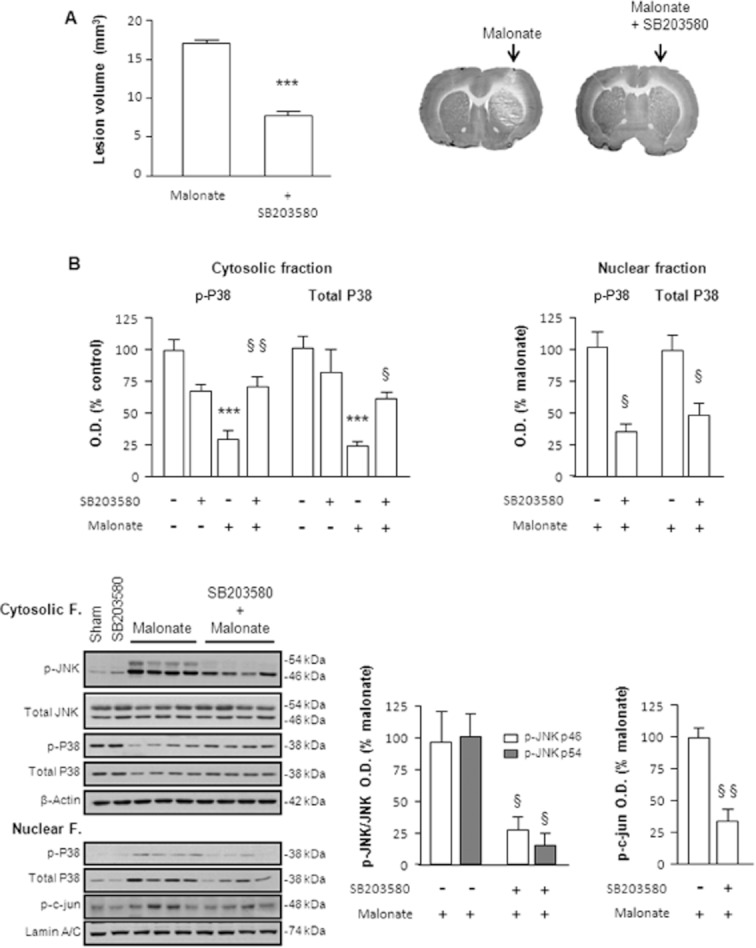
Activation of p38 is involved in malonate-induced cell death. Rats were administered intrastriatally with malonate (1.5 μmol/2 μL) alone or in combination with the p38 inhibitor, SB203580 (1 nmol/2 μL). (A) Representative cytochrome oxidase-stained slices and quantification of malonate-induced striatal lesions (mm3) showing that SB203580 significantly inhibited the neurotoxic effect of malonate 72 h after treatment. Data are mean ± SEM (n = 6) [t(11) = 12.89, P < 0.001]. (B) Quantitative measurements and representative blots showing that SB203580 inhibited not only p38 activation but also the activation of the JNK pathway induced by malonate. For this, rats were killed 6 h after treatment. Statistical analysis yielded the following results: for cytosolic p-p38 [F(3,30) = 13.29, P < 0.001] and total p38 [F(3,20) = 16.60, P < 0.001], for nuclear p-p38 [t(12) = 5.319, P < 0.05] and total p38 [t(12) = 3.409, P < 0.05], for p-JNK/JNK (46 kDa) [t(11) = 2.531, P < 0.05] and p-JNK/JNK (54 kDa) [t(11) = 4.591, P < 0.05] and for p-c-jun [t(8) = 5.292, P < 0.01]. Different from the corresponding sham group: ***P < 0.001. Different from malonate-only animals: §P < 0.05; §§P < 0.01.
Discussion
Neuroprotective effects of PDE5 inhibitors have been described in several models of neurological disorders such as Parkinson's disease (Picconi et al., 2011; Tozzi et al., 2012); Alzheimer's disease (Cuadrado-Tejedor et al., 2011) and mitochondrial neurotoxicity (Puerta et al., 2010). In addition, they improve striatal synaptic plasticity under physiological conditions (Calabresi et al., 1999). Lately, and encouraged by the results obtained in other organs, new studies are focusing on the potential role of sildenafil, and other PDE5 inhibitors, in the treatment of cerebral ischaemia. Indeed, recent reports have shown that sildenafil improves the outcome of rats after stroke by enhancing angiogenesis, neurogenesis and improving neurologic function (Zhang et al., 2005; 2006; Ding et al., 2008).
Intrastriatal administration of the reversible succinate dehydrogenase inhibitor, malonate, in rats closely mimics some of the main pathological features of focal ischaemia (Schulz et al., 1998). The mechanisms leading to neuronal death in this model involve secondary excitotoxicity, generation of ROS and apoptosis (Greene and Greenamyre, 1996; Dedeoglu et al., 2002). In the present study, we demonstrated that sildenafil, given at a dose comparable with that used in humans and administered 30 min before malonate injection, attenuated striatal toxicity. Under our experimental conditions, malonate produced histological lesions, assessed by cytochrome oxidase histochemistry, and a consistent loss of striatal DARPP-32 levels, effects that were significantly prevented by sildenafil.
Oxidative stress has long been considered as the major cause of tissue injury after cerebral ischaemia (Saito et al., 2005) and is considered to play a key role in malonate-induced neurotoxicity (Fernandez-Gomez et al., 2005). In an attempt to understand the mechanisms underlying the protective effects afforded by sildenafil, we analysed the effect of this PDE5 inhibitor on ROS production. As expected, malonate increased ROS production; but, in contrast to previous reports (Koupparis et al., 2005; Muzaffar et al., 2005; Bivalacqua et al., 2009; Puerta et al., 2012), sildenafil failed to prevent malonate-induced rise in ROS production. Furthermore, sildenafil also failed to reduce the increased NT concentrations found 72 h after malonate. Fernandez-Gomez et al. (2005) have suggested that malonate requires very few minutes to disrupt mitochondrial redox status. This fast and excessive production of ROS could overwhelm sildenafil antioxidant capacity when the PDE5i is given shortly before malonate administration.
Upon ROS formation, many intracellular pathways are activated. Among them, ASK1 is selectively required for sustained activation of the SAPKs pathway induced by oxidative stress (Tobiume et al., 2001; Sekine et al., 2006). The activity of this kinase can be regulated by a number of different ASK1-interacting proteins (Hsieh and Papaconstantinou, 2006; Hattori et al., 2009). For instance, phosphorylation of ASK1 on Ser83, negatively regulates its proapoptotic function and, in turn, leads to the enhancement of cell survival (Kim et al., 2001). Our results show that malonate decreases the inhibitory phosphorylation of ASK1, which can indirectly indicate an increase on its activity. By contrast, sildenafil treated animals kept the levels of inactive ASK1 similar to those found in the sham group. Our group and others have previously shown that sildenafil increases Akt phosphorylation in the brain (Wang et al., 2005; Puerta et al., 2009), an effect that depends on the PI3K signal transduction pathway. Akt is a serine/threonine kinase known to phosphorylate ASK1 on Ser83; and therefore, although not directly addressed in this work, the PI3K/Akt/Ask1 pathway could account for the protection afforded by sildenafil against malonate.
Once activated, ASK1 can selectively activate JNK and p38 MAPKs, leading to neuronal death through the activation of different MAPKKs (Hattori et al., 2009). MKK3 and MKK6 are the main MAPKKs that phosphorylate and activate p38 MAPK (Enslen et al., 1998); whereas MKK7 is responsible for JNK activation (Bogoyevitch and Kobe, 2006). We therefore analysed the phosphorylation levels of MKK3/6 and MKK7 after malonate and found they were significantly increased. Of notice, sildenafil inhibited the activation of MKK3/6 and MKK7 induced by malonate possibly as a consequence of ASK1 inhibition.
MAPKs play important roles in cellular response to different stimuli (Cowan, 2003). Among the MAPKs, JNK and p38 are often implicated in cell death (Xia et al., 1995). A growing body of evidence has demonstrated that JNK and p38 are activated following cerebral ischaemia and contribute to ischaemic neuronal death (Irving and Bamford, 2002 Borsello et al., 2003; Toledo-Pereyra et al., 2008). Recently, JNK and p38 have also been implicated in the toxic effects of malonate (Asanuma et al., 2004; Gomez-Lazaro et al., 2007). Taking this into account and based on our previous results, we focused on the plausible role of SAPKs inhibition in the neuroprotective effects of sildenafil against chemical hypoxia caused by malonate. We addressed this issue, by analysing the activation pattern of JNK and p38 following malonate administration. We observed an important activation of p38, which translocates to the nucleus within the first few minutes. Although starting later, we also confirmed that malonate induces the phosphorylation of JNK that, as well as p38, remains activated up to 9 h after malonate. Noteworthy, sildenafil inhibited the activation of both SAPKs. Furthermore, sildenafil prevented the activation of c-jun, a downstream effector of JNK implicated in stress responses and apoptosis (Behrens et al., 1999). Although some authors have suggested that p38 phosphorylation could be mediating the angiogenic effects of sildenafil (Pyriochou et al., 2007), our findings are in line with the observations of Caretti et al. (2008), who reported that sildenafil prevents p38 phosphorylation in a model of chronic hypoxia and also with the results reported by Zhao et al. (2011), who showed that sildenafil inhibits the activation of both JNK and p38 caused during LPS-induced pro-inflammatory response in vitro.
In order to elucidate the implication of SAPKs inhibition on the protection afforded by sildenafil, we next studied the effect of SP600125 and SB203580, selective inhibitors of JNK and p38 respectively. Interestingly, the administration of the reversible ATP competitive inhibitor of JNK, SP600125 (Bennett et al., 2001), failed to protect the cellular loss caused by malonate. These data suggest that, at least in this context, JNK activation and subsequent c-jun phosphorylation is not relevant in malonate induced cell death. Otherwise, the importance of p38 activation in malonate neurotoxicity was confirmed using the selective inhibitor SB203580, as it significantly reduced the infarct size. Of note, SB203580 also reduced the activation of JNK and c-jun induced by malonate. This reduction could be attributed to a side effect of SB203580, as it was previously described that high doses of this inhibitor can affect JNK activation (Whitmarsh et al., 1997). As reviewed by Boutros et al. (2008), MAPK pathways are not independent from each other but contain a series of overlapping signalling mechanism. Therefore, although not described before, we cannot preclude a possible crosstalk between p38 and JNK in this model.
The physiological actions of NO are primarily mediated through stimulation of soluble guanylate cyclase, which results in accumulation of cGMP and subsequent activation of PKG (for review, see Schlossmann et al., 2003). Sildenafil by inhibiting the enzymatic hydrolysis of cGMP by PDE5, maintains the tissue accumulation of cGMP, which also leads to downstream activation of PKG. Noteworthy, inhibition of nitric oxide formation has been proven to prevent cell death after a brain ischaemic insult or malonate treatment (Zhang et al., 1996; Connop et al., 1997; Matthews et al., 1997; Schulz et al., 1997; Zhao et al., 2000; Willmot et al., 2005) and so does PDE5 inhibition (Kiymaz et al., 2008). These apparent disparate findings may be explained as follows. NO reacts avidly with superoxide radicals to form the cytotoxic molecule peroxynitrite playing a deleterious role in malonate and ischaemia-induced neurotoxicity (Schulz et al., 1996; Matthews et al., 1997; Dohi et al., 2003). PDE5 inhibition, by acting downstream nitric oxide formation, does not promote peroxynitrite formation but rather activates/inhibits signalling pathways, like those reported in this study, that protect against malonate-induced cell death.
In conclusion, our findings show that sildenafil exerts neuroprotection against a chemical hypoxia induced by the mitochondrial toxin, malonate, in rats. This protective effect is independent of any change in ROS production but appears to be related to an inhibition of the ASK1–MKK3/6–p38 pathway (Figure 8). Future studies using other animal models of stroke are warranted to exploit the possible therapeutic potential of sildenafil in preventing neuronal cell death following ischaemia/reperfusion.
Figure 8.
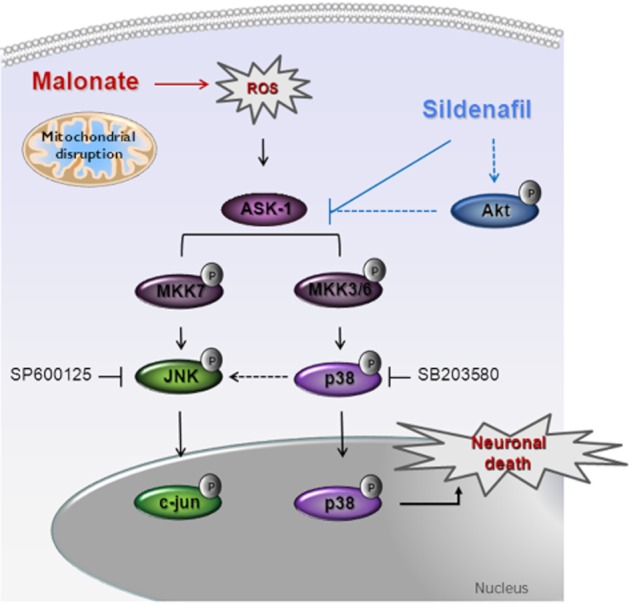
Proposed mechanisms underlying sildenafil neuroprotection against malonate-induced neurotoxicity. Inhibition of succinate dehydrogenase by malonate administration induces mitochondrial dysfunction, which triggers the generation of ROS. The increase in ROS production leads to the activation of ASK1, which, in turn, activates JNK and p38. Nuclear translocation of p38 contributes to neuronal death. Sildenafil afforded protection is independent of any change in ROS production but appears to be related to the inhibition of the ASK1–MKK3/6–p38 pathway.
Acknowledgments
The authors are grateful to Sandra Lizaso and Mari Luz Muro for technical assistance. They would also like to thank ‘Amigos de la Universidad de Navarra’ for a fellowship to LO and LB-M and Ramon Areces Foundation for a fellowship to EP. This work was supported by grants from the Ministerio de Ciencia e Innovación (SAF2008-05143-C03-03).
Glossary
- ASK1
apoptosis signal-regulating kinase 1
- MAPKKs
MAPK kinases
- PKG
cGMP-dependent protein kinase
- ROS
reactive oxygen species
- SAPKs
stress-activated protein kinases
- SDH
succinate dehydrogenase
Conflict of interest
None of the authors have any actual or potential conflict of interest including any financial, personal or other relationships with other people or organizations that could inappropriately influence their work.
References
- Archer SL, Huang JM, Hampl V, Nelson DP, Shultz PJ, Weir EK. Nitric oxide and cGMP cause vasorelaxation by activation of a charybdotoxin-sensitive K channel by cGMP-dependent protein kinase. Proc Natl Acad Sci U S A. 1994;91:7583–7587. doi: 10.1073/pnas.91.16.7583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asanuma T, Inanami O, Tabu K, Waki K, Kon Y, Kuwabara M. Protection against malonate-induced ischemic brain injury in rat by a cell-permeable peptidic c-Jun N-terminal kinase inhibitor, (L)-HIV-TAT48-57-PP-JBD20, observed by the apparent diffusion coefficient mapping magnetic resonance imaging method. Neurosci Lett. 2004;359:57–60. doi: 10.1016/j.neulet.2004.02.009. [DOI] [PubMed] [Google Scholar]
- Beheshtian A, Salmasi AH, Payabvash S, Kiumehr S, Ghazinezami B, Rahimpour S, et al. Protective effects of sildenafil administration on testicular torsion/detorsion damage in rats. World J Urol. 2008;26:197–202. doi: 10.1007/s00345-008-0243-6. [DOI] [PubMed] [Google Scholar]
- Behrens A, Sibilia M, Wagner EF. Amino-terminal phosphorylation of c-Jun regulates stress-induced apoptosis and cellular proliferation. Nat Genet. 1999;21:326–329. doi: 10.1038/6854. [DOI] [PubMed] [Google Scholar]
- Bender AT, Beavo JA. Specific localized expression of cGMP PDEs in Purkinje neurons and macrophages. Neurochem Int. 2004;45:853–857. doi: 10.1016/j.neuint.2004.03.015. [DOI] [PubMed] [Google Scholar]
- Bender AT, Beavo JA. Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev. 2006;58:488–520. doi: 10.1124/pr.58.3.5. [DOI] [PubMed] [Google Scholar]
- Bennett BL, Sasaki DT, Murray BW, O'Leary EC, Sakata ST, Xu W, et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci U S A. 2001;98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bivalacqua TJ, Sussan TE, Gebska MA, Strong TD, Berkowitz DE, Biswal S, et al. Sildenafil inhibits superoxide formation and prevents endothelial dysfunction in a mouse model of secondhand smoke induced erectile dysfunction. J Urol. 2009;181:899–906. doi: 10.1016/j.juro.2008.10.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogoyevitch MA, Kobe B. Uses for JNK: the many and varied substrates of the c-Jun N-terminal kinases. Microbiol Mol Biol Rev. 2006;70:1061–1095. doi: 10.1128/MMBR.00025-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borsello T, Clarke PG, Hirt L, Vercelli A, Repici M, Schorderet DF, et al. A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat Med. 2003;9:1180–1186. doi: 10.1038/nm911. [DOI] [PubMed] [Google Scholar]
- Boutros T, Chevet E, Metrakos P. Mitogen-activated protein (MAP) kinase/MAP kinase phosphatase regulation: roles in cell growth, death, and cancer. Pharmacol Rev. 2008;60:261–310. doi: 10.1124/pr.107.00106. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Gubellini P, Centonze D, Sancesario G, Morello M, Giorgi M, et al. A critical role of the nitric oxide/cGMP pathway in corticostriatal long-term depression. J Neurosci. 1999;19:2489–2499. doi: 10.1523/JNEUROSCI.19-07-02489.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caretti A, Bianciardi P, Ronchi R, Fantacci M, Guazzi M, Samaja M. Phosphodiesterase-5 inhibition abolishes neuron apoptosis induced by chronic hypoxia independently of hypoxia-inducible factor-1alpha signaling. Exp Biol Med (Maywood) 2008;233:1222–1230. doi: 10.3181/0802-RM-73. [DOI] [PubMed] [Google Scholar]
- Connop BP, Boegman RJ, Beninger RJ, Jhamandas K. Malonate-induced degeneration of basal forebrain cholinergic neurons: attenuation by lamotrigine, MK-801, and 7-nitroindazole. J Neurochem. 1997;68:1191–1199. doi: 10.1046/j.1471-4159.1997.68031191.x. [DOI] [PubMed] [Google Scholar]
- Cowan KJ. Mitogen-activated protein kinases: new signaling pathways functioning in cellular responses to environmental stress. J Exp Biol. 2003;206:1107–1115. doi: 10.1242/jeb.00220. [DOI] [PubMed] [Google Scholar]
- Cuadrado-Tejedor M, Hervias I, Ricobaraza A, Puerta E, Perez-Roldan JM, Garcia-Barroso C, et al. Sildenafil restores cognitive function without affecting beta-amyloid burden in a mouse model of Alzheimer's disease. Br J Pharmacol. 2011;164:2029–2041. doi: 10.1111/j.1476-5381.2011.01517.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuenda A, Rouse J, Doza YN, Meier R, Cohen P, Gallagher TF, et al. SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett. 1995;364:229–233. doi: 10.1016/0014-5793(95)00357-f. [DOI] [PubMed] [Google Scholar]
- Das A, Ockaili R, Salloum F, Kukreja RC. Protein kinase C plays an essential role in sildenafil-induced cardioprotection in rabbits. Am J Physiol Heart Circ Physiol. 2004;286:1455–1460. doi: 10.1152/ajpheart.01040.2003. [DOI] [PubMed] [Google Scholar]
- Dedeoglu A, Ferrante RJ, Andreassen OA, Dillmann WH, Beal MF. Mice Overexpressing 70-kDa Heat Shock Protein Show Increased Resistance to Malonate and 3-Nitropropionic Acid. Exp Neurol. 2002;176:262–265. doi: 10.1006/exnr.2002.7933. [DOI] [PubMed] [Google Scholar]
- Ding G, Jiang Q, Li L, Zhang L, Zhang ZG, Ledbetter KA, et al. Magnetic resonance imaging investigation of axonal remodeling and angiogenesis after embolic stroke in sildenafil-treated rats. J Cereb Blood Flow Metab. 2008;28:1440–1448. doi: 10.1038/jcbfm.2008.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohi K, Ohtaki H, Inn R, Ikeda Y, Shioda HS, Aruga T. Peroxynitrite and caspase-3 expression after ischemia/reperfusion in mouse cardiac arrest model. Acta Neurochir Suppl. 2003;86:87–91. doi: 10.1007/978-3-7091-0651-8_20. [DOI] [PubMed] [Google Scholar]
- Enslen H, Raingeaud J, Davis RJ. Selective activation of p38 mitogen-activated protein (MAP) kinase isoforms by the MAP kinase kinases MKK3 and MKK6. J Biol Chem. 1998;273:1741–1748. doi: 10.1074/jbc.273.3.1741. [DOI] [PubMed] [Google Scholar]
- Fernandez-Gomez FJ, Galindo MF, Gomez-Lazaro M, Yuste VJ, Comella JX, Aguirre N, et al. Malonate induces cell death via mitochondrial potential collapse and delayed swelling through an ROS-dependent pathway. Br J Pharmacol. 2005;144:528–537. doi: 10.1038/sj.bjp.0706069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galie N, Ghofrani HA, Torbicki A, Barst RJ, Rubin LJ, Badesch D, et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med. 2005;353:2148–2157. doi: 10.1056/NEJMoa050010. [DOI] [PubMed] [Google Scholar]
- Garcia-Osta A, Del Rio J, Frechilla D. Increased CRE-binding activity and tryptophan hydroxylase mRNA expression induced by 3,4-methylenedioxymethamphetamine (MDMA, ‘ecstasy’) in the rat frontal cortex but not in the hippocampus. Brain Res Mol Brain Res. 2004;126:181–187. doi: 10.1016/j.molbrainres.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Gomez-Lazaro M, Galindo MF, Melero-Fernandez de Mera RM, Fernandez-Gomez FJ, Concannon CG, Segura MF, et al. Reactive oxygen species and p38 mitogen-activated protein kinase activate Bax to induce mitochondrial cytochrome c release and apoptosis in response to malonate. Mol Pharmacol. 2007;71:736–743. doi: 10.1124/mol.106.030718. [DOI] [PubMed] [Google Scholar]
- Goni-Allo B, Ramos M, Jordan J, Aguirre N. In vivo studies on the protective role of minocycline against excitotoxicity caused by malonate or N-methyl-d-aspartate. Exp Neurol. 2005;191:326–330. doi: 10.1016/j.expneurol.2004.10.010. [DOI] [PubMed] [Google Scholar]
- Goni-Allo B, Puerta E, Mathuna BO, Hervias I, Lasheras B, de la Torre R, et al. On the role of tyrosine and peripheral metabolism in 3,4-methylenedioxymethamphetamine-induced serotonin neurotoxicity in rats. Neuropharmacology. 2008;54:885–900. doi: 10.1016/j.neuropharm.2008.01.007. [DOI] [PubMed] [Google Scholar]
- Gori T, Sicuro S, Dragoni S, Donati G, Forconi S, Parker JD. Sildenafil prevents endothelial dysfunction induced by ischemia and reperfusion via opening of adenosine triphosphate-sensitive potassium channels: a human in vivo study. Circulation. 2005;111:742–746. doi: 10.1161/01.CIR.0000155252.23933.2D. [DOI] [PubMed] [Google Scholar]
- Greene JG, Greenamyre JT. Manipulation of membrane potential modulates malonate-induced striatal excitotoxicity in vivo. J Neurochem. 1996;66:637–643. doi: 10.1046/j.1471-4159.1996.66020637.x. [DOI] [PubMed] [Google Scholar]
- Hattori K, Naguro I, Runchel C, Ichijo H. The roles of ASK family proteins in stress responses and diseases. Cell Commun Signal. 2009;7:9. doi: 10.1186/1478-811X-7-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh CC, Papaconstantinou J. Thioredoxin-ASK1 complex levels regulate ROS-mediated p38 MAPK pathway activity in livers of aged and long-lived Snell dwarf mice. FASEB J. 2006;20:259–268. doi: 10.1096/fj.05-4376com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichijo H. Induction of Apoptosis by ASK1, a Mammalian MAPKKK That Activates SAPK/JNK and p38 Signaling Pathways. Science. 1997;275:90–94. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- Irkorucu O, Ucan BH, Cakmak GK, Emre AU, Tascilar O, Ofluoglu E, et al. Does sildenafil reverse the adverse effects of ischemia on ischemic colon anastomosis: yes, ‘no’. Int J Surg. 2009;7:39–43. doi: 10.1016/j.ijsu.2008.10.003. [DOI] [PubMed] [Google Scholar]
- Irving EA, Bamford M. Role of mitogen- and stress-activated kinases in ischemic injury. J Cereb Blood Flow Metab. 2002;22:631–647. doi: 10.1097/00004647-200206000-00001. [DOI] [PubMed] [Google Scholar]
- Ischiropoulos H, al-Mehdi AB. Peroxynitrite-mediated oxidative protein modifications. FEBS Lett. 1995;364:279–282. doi: 10.1016/0014-5793(95)00307-u. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim AH, Khursigara G, Sun X, Franke TF, Chao MV. Akt phosphorylates and negatively regulates apoptosis signal-regulating kinase 1. Mol Cell Biol. 2001;21:893–901. doi: 10.1128/MCB.21.3.893-901.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim GW, Chan PH. Involvement of superoxide in excitotoxicity and DNA fragmentation in striatal vulnerability in mice after treatment with the mitochondrial toxin, 3-nitropropionic acid. J Cereb Blood Flow Metab. 2002;22:798–809. doi: 10.1097/00004647-200207000-00005. [DOI] [PubMed] [Google Scholar]
- Kiymaz N, Yilmaz N, Mumcu C, Anlar O, Ozen S, Kayaoglu CR. Protective effect of sildenafil (Viagra) in transient spinal cord ischemia. Pediatr Neurosurg. 2008;44:22–28. doi: 10.1159/000110658. [DOI] [PubMed] [Google Scholar]
- Koupparis AJ, Jeremy JY, Muzaffar S, Persad R, Shukla N. Sildenafil inhibits the formation of superoxide and the expression of gp47 NAD[P]H oxidase induced by the thromboxane A2 mimetic, U46619, in corpus cavernosal smooth muscle cells. BJU Int. 2005;96:423–427. doi: 10.1111/j.1464-410X.2005.05643.x. [DOI] [PubMed] [Google Scholar]
- Kukreja RC, Ockaili R, Salloum F, Yin C, Hawkins J, Das A, et al. Cardioprotection with phosphodiesterase-5 inhibition–a novel preconditioning strategy. J Mol Cell Cardiol. 2004;36:165–173. doi: 10.1016/j.yjmcc.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Lledo-Garcia E, Subira-Rios D, Rodriguez-Martinez D, Dulin E, Alvarez-Fernandez E, Hernandez-Fernandez C, et al. Sildenafil as a protecting drug for warm ischemic kidney transplants: experimental results. J Urol. 2009;182:1222–1225. doi: 10.1016/j.juro.2009.05.006. [DOI] [PubMed] [Google Scholar]
- Martinez-Serrano A, Bjorklund A. Protection of the neostriatum against excitotoxic damage by neurotrophin-producing, genetically modified neural stem cells. J Neurosci. 1996;16:4604–4616. doi: 10.1523/JNEUROSCI.16-15-04604.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews RT, Yang L, Beal MF. S-Methylthiocitrulline, a neuronal nitric oxide synthase inhibitor, protects against malonate and MPTP neurotoxicity. Exp Neurol. 1997;143:282–286. doi: 10.1006/exnr.1996.6406. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menniti FS, Ren J, Coskran TM, Liu J, Morton D, Sietsma DK, et al. Phosphodiesterase 5A inhibitors improve functional recovery after stroke in rats: optimized dosing regimen with implications for mechanism. J Pharmacol Exp Ther. 2009;331:842–850. doi: 10.1124/jpet.109.156919. [DOI] [PubMed] [Google Scholar]
- Muzaffar S, Shukla N, Srivastava A, Angelini GD, Jeremy JY. Sildenafil citrate and sildenafil nitrate (NCX 911) are potent inhibitors of superoxide formation and gp91phox expression in porcine pulmonary artery endothelial cells. Br J Pharmacol. 2005;146:109–117. doi: 10.1038/sj.bjp.0706305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallotti F, Lenaz G. Isolation and subfractionation of mitochondria from animal cells and tissue culture lines. Methods Cell Biol. 2007;80:3–44. doi: 10.1016/S0091-679X(06)80001-4. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. New York: Academic Press; 1997. [Google Scholar]
- Picconi B, Bagetta V, Ghiglieri V, Paille V, Di Filippo M, Pendolino V, et al. Inhibition of phosphodiesterases rescues striatal long-term depression and reduces levodopa-induced dyskinesia. Brain. 2011;134(Pt 2):375–387. doi: 10.1093/brain/awq342. [DOI] [PubMed] [Google Scholar]
- Puerta E, Hervias I, Goni-Allo B, Lasheras B, Jordan J, Aguirre N. Phosphodiesterase 5 inhibitors prevent 3,4-methylenedioxymethamphetamine-induced 5-HT deficits in the rat. J Neurochem. 2009;108:755–766. doi: 10.1111/j.1471-4159.2008.05825.x. [DOI] [PubMed] [Google Scholar]
- Puerta E, Hervias I, Barros-Minones L, Jordan J, Ricobaraza A, Cuadrado-Tejedor M, et al. Sildenafil protects against 3-nitropropionic acid neurotoxicity through the modulation of calpain, CREB, and BDNF. Neurobiol Dis. 2010;38:237–245. doi: 10.1016/j.nbd.2010.01.013. [DOI] [PubMed] [Google Scholar]
- Puerta E, Barros-Minones L, Hervias I, Gomez-Rodriguez V, Orejana L, Pizarro N, et al. Long-lasting neuroprotective effect of sildenafil against 3,4-methylenedioxymethamphetamine- induced 5-hydroxytryptamine deficits in the rat brain. J Neurosci Res. 2012;90:518–528. doi: 10.1002/jnr.22759. [DOI] [PubMed] [Google Scholar]
- Pyriochou A, Zhou Z, Koika V, Petrou C, Cordopatis P, Sessa WC, et al. The phosphodiesterase 5 inhibitor sildenafil stimulates angiogenesis through a protein kinase G/MAPK pathway. J Cell Physiol. 2007;211:197–204. doi: 10.1002/jcp.20929. [DOI] [PubMed] [Google Scholar]
- Raingeaud J, Whitmarsh AJ, Barrett T, Derijard B, Davis RJ. MKK3- and MKK6-regulated gene expression is mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Mol Cell Biol. 1996;16:1247–1255. doi: 10.1128/mcb.16.3.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J. 2008;22:659–661. doi: 10.1096/fj.07-9574LSF. [DOI] [PubMed] [Google Scholar]
- Saito A, Maier CM, Narasimhan P, Nishi T, Song YS, Yu F, et al. Oxidative stress and neuronal death/survival signaling in cerebral ischemia. Mol Neurobiol. 2005;31:105–116. doi: 10.1385/MN:31:1-3:105. [DOI] [PubMed] [Google Scholar]
- Salloum FN, Takenoshita Y, Ockaili RA, Daoud VP, Chou E, Yoshida K, et al. Sildenafil and vardenafil but not nitroglycerin limit myocardial infarction through opening of mitochondrial K(ATP) channels when administered at reperfusion following ischemia in rabbits. J Mol Cell Cardiol. 2007;42:453–458. doi: 10.1016/j.yjmcc.2006.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlossmann J, Feil R, Hofmann F. Signaling through NO and cGMP-dependent protein kinases. Ann Med. 2003;35:21–27. doi: 10.1080/07853890310004093. [DOI] [PubMed] [Google Scholar]
- Schulz JB, Huang PL, Matthews RT, Passov D, Fishman MC, Beal MF. Striatal malonate lesions are attenuated in neuronal nitric oxide synthase knockout mice. J Neurochem. 1996;67:430–433. doi: 10.1046/j.1471-4159.1996.67010430.x. [DOI] [PubMed] [Google Scholar]
- Schulz JB, Matthews RT, Klockgether T, Dichgans J, Beal MF. The role of mitochondrial dysfunction and neuronal nitric oxide in animal models of neurodegenerative diseases. Mol Cell Biochem. 1997;174:193–197. [PubMed] [Google Scholar]
- Schulz JB, Weller M, Matthews RT, Heneka MT, Groscurth P, Martinou JC, et al. Extended therapeutic window for caspase inhibition and synergy with MK-801 in the treatment of cerebral histotoxic hypoxia. Cell Death Differ. 1998;5:847–857. doi: 10.1038/sj.cdd.4400420. [DOI] [PubMed] [Google Scholar]
- Sekine Y, Takeda K, Ichijo H. The ASK1-MAP kinase signaling in ER stress and neurodegenerative diseases. Curr Mol Med. 2006;6:87–97. doi: 10.2174/156652406775574541. [DOI] [PubMed] [Google Scholar]
- Silver B, McCarthy S, Lu M, Mitsias P, Russman AN, Katramados A, et al. Sildenafil treatment of subacute ischemic stroke: a safety study at 25-mg daily for 2 weeks. J Stroke Cerebrovasc Dis. 2009;18:381–383. doi: 10.1016/j.jstrokecerebrovasdis.2009.01.007. [DOI] [PubMed] [Google Scholar]
- Soydan G, Sokmensuer C, Kilinc K, Tuncer M. The effects of sildenafil on the functional and structural changes of ileum induced by intestinal ischemia-reperfusion in rats. Eur J Pharmacol. 2009;610:87–92. doi: 10.1016/j.ejphar.2009.03.038. [DOI] [PubMed] [Google Scholar]
- Tobiume K, Matsuzawa A, Takahashi T, Nishitoh H, Morita K, Takeda K, et al. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep. 2001;2:222–228. doi: 10.1093/embo-reports/kve046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo-Pereyra LH, Lopez-Neblina F, Toledo AH. Protein kinases in organ ischemia and reperfusion. J Invest Surg. 2008;21:215–226. doi: 10.1080/08941930802130149. [DOI] [PubMed] [Google Scholar]
- Tozzi A, Costa C, Siliquini S, Tantucci M, Picconi B, Kurz A, et al. Mechanisms underlying altered striatal synaptic plasticity in old A53T-alpha synuclein overexpressing mice. Neurobiol Aging. 2012;33:1792–1799. doi: 10.1016/j.neurobiolaging.2011.05.002. [DOI] [PubMed] [Google Scholar]
- Vijayvergiya C, Beal MF, Buck J, Manfredi G. Mutant superoxide dismutase 1 forms aggregates in the brain mitochondrial matrix of amyotrophic lateral sclerosis mice. J Neurosci. 2005;25:2463–2470. doi: 10.1523/JNEUROSCI.4385-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Gang Zhang Z, Lan Zhang R, Chopp M. Activation of the PI3-K/Akt pathway mediates cGMP enhanced-neurogenesis in the adult progenitor cells derived from the subventricular zone. J Cereb Blood Flow Metab. 2005;25:1150–1158. doi: 10.1038/sj.jcbfm.9600112. [DOI] [PubMed] [Google Scholar]
- Whitmarsh AJ, Yang SH, Su MS, Sharrocks AD, Davis RJ. Role of p38 and JNK mitogen-activated protein kinases in the activation of ternary complex factors. Mol Cell Biol. 1997;17:2360–2371. doi: 10.1128/mcb.17.5.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willmot M, Gibson C, Gray L, Murphy S, Bath P. Nitric oxide synthase inhibitors in experimental ischemic stroke and their effects on infarct size and cerebral blood flow: a systematic review. Free Radic Biol Med. 2005;39:412–425. doi: 10.1016/j.freeradbiomed.2005.03.028. [DOI] [PubMed] [Google Scholar]
- Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- Yanagisawa D, Kitamura Y, Inden M, Takata K, Taniguchi T, Morikawa S, et al. DJ-1 protects against neurodegeneration caused by focal cerebral ischemia and reperfusion in rats. J Cereb Blood Flow Metab. 2008;28:563–578. doi: 10.1038/sj.jcbfm.9600553. [DOI] [PubMed] [Google Scholar]
- Zhang L, Zhang RL, Wang Y, Zhang C, Zhang ZG, Meng H, et al. Functional recovery in aged and young rats after embolic stroke: treatment with a phosphodiesterase type 5 inhibitor. Stroke. 2005;36:847–852. doi: 10.1161/01.STR.0000158923.19956.73. [DOI] [PubMed] [Google Scholar]
- Zhang RL, Zhang Z, Zhang L, Wang Y, Zhang C, Chopp M. Delayed treatment with sildenafil enhances neurogenesis and improves functional recovery in aged rats after focal cerebral ischemia. J Neurosci Res. 2006;83:1213–1219. doi: 10.1002/jnr.20813. [DOI] [PubMed] [Google Scholar]
- Zhang ZG, Reif D, Macdonald J, Tang WX, Kamp DK, Gentile RJ, et al. ARL 17477, a potent and selective neuronal NOS inhibitor decreases infarct volume after transient middle cerebral artery occlusion in rats. J Cereb Blood Flow Metab. 1996;16:599–604. doi: 10.1097/00004647-199607000-00009. [DOI] [PubMed] [Google Scholar]
- Zhao S, Zhang L, Lian G, Wang X, Zhang H, Yao X, et al. Sildenafil attenuates LPS-induced pro-inflammatory responses through down-regulation of intracellular ROS-related MAPK/NF-kappaB signaling pathways in N9 microglia. Int Immunopharmacol. 2011;11:468–474. doi: 10.1016/j.intimp.2010.12.017. [DOI] [PubMed] [Google Scholar]
- Zhao X, Haensel C, Araki E, Ross ME, Iadecola C. Gene-dosing effect and persistence of reduction in ischemic brain injury in mice lacking inducible nitric oxide synthase. Brain Res. 2000;872:215–218. doi: 10.1016/s0006-8993(00)02459-8. [DOI] [PubMed] [Google Scholar]