

Abstract
Background and Purpose
L-gulonolactone oxidase-deficient (Gulo(-/-)) mice were used to study the effects of ascorbate deficiency on aortic relaxation by nitroglycerin (GTN) with focus on changes in the expression and activity of vascular aldehyde dehydrogenase-2 (ALDH2), which catalyses GTN bioactivation.
Experimental Approach
Ascorbate deficiency was induced in Gulo(-/-) mice by ascorbate deprivation for 4 weeks. Some of the animals were concomitantly treated with the proteasome inhibitor bortezomib and effects compared with ascorbate-supplemented Gulo(-/-), untreated or nitrate-tolerant wild-type mice. Aortic relaxation of the experimental groups to GTN, ACh and a NO donor was studied. Changes in mRNA and protein expression of vascular ALDH2 were quantified by qPCR and immunoblotting, respectively, and aortic GTN denitration rates determined.
Key Results
Like GTN treatment, ascorbate deprivation induced vascular tolerance to GTN that was associated with markedly decreased rates of GTN denitration. Ascorbate deficiency did not affect ALDH2 mRNA levels, but reduced ALDH2 protein expression and the total amount of ubiquitinated proteins to about 40% of wild-type controls. These effects were largely prevented by ascorbate supplementation or treating Gulo(-/-) mice with the 26S proteasome inhibitor bortezomib.
Conclusions and Implications
Our data indicate that ascorbate deficiency results in vascular tolerance to GTN via proteasomal degradation of ALDH2. The results support the view that impaired ALDH2-catalysed metabolism of GTN contributes significantly to the development of vascular nitrate tolerance and reveal a hitherto unrecognized protective effect of ascorbate in the vasculature.
Keywords: aldehyde dehydrogenase-2, ascorbate deficiency, bortezomib, genetic mouse model, nitrate tolerance, nitroglycerin, proteasomal degradation, vascular relaxation, vitamin C
Introduction
Organic nitrates, in particular nitroglycerin (GTN), have been used for decades to treat angina pectoris, but development of tolerance limits their usefulness and precludes continuous administration to patients. The proposed mechanisms underlying the development of vascular tolerance to GTN are manifold, including impaired bioactivation to NO or a related activator of soluble guanylate cyclase (Sage et al., 2000; Chen et al., 2002), desensitization of soluble guanylate cyclase (Schröder et al., 1988; Sayed et al., 2008) and oxidative stress associated with increased generation of superoxide in blood vessels leading to limited NO bioavailability due to peroxynitrite formation (Münzel et al., 1995). Although challenged by some studies (Hinz and Schröder, 1998; Csont et al., 2002; Hanspal et al., 2002), the oxidative stress concept is supported by several reports on beneficial effects of antioxidants like ascorbate in experimental and clinical studies of nitrate tolerance (Bassenge et al., 1998; Watanabe et al., 1998; McVeigh et al., 2002). This hypothesis is questioned, however, by a study showing that ascorbate did not prevent tolerance development (Milone et al., 1999).
There is a large body of evidence indicating that aldehyde dehydrogenase-2 (ALDH2) is a key enzyme of vascular GTN bioactivation and that exposure of blood vessels to GTN causes mechanism-based inactivation of ALDH2, resulting in impaired GTN bioactivation (Mayer and Beretta, 2008). Despite general agreement on the essential role of ALDH2 inactivation in nitrate tolerance, this concept has been questioned in a recent study showing that the haemodynamic response recovered more rapidly after GTN treatment than total vascular ALDH activity (D'Souza et al., 2011). Although GTN bioactivation is catalysed by cytosolic rather than mitochondrial ALDH2 (Beretta et al., 2012), exposure of blood vessels to the nitrate appears to result in mitochondrial oxidative stress. The oxidative stress concept has been combined with impaired GTN biotransformation in a unifying hypothesis explaining nitrate tolerance and endothelial dysfunction upon exposure of blood vessels to GTN (Münzel et al., 2011).
We have recently shown that ascorbate deprivation of guinea pigs causes vascular hyposensitivity to GTN (Wölkart et al., 2008; Wenzl et al., 2009). Despite the apparent involvement of impaired ALDH2-catalysed GTN biotransformation in both classical nitrate tolerance and GTN hyposensitivity induced by ascorbate deprivation, there was a remarkable difference between the two animal models. While residual GTN-triggered relaxation of nitrate-tolerant aortas was not further aggravated by ALDH2 inhibitors, the GTN response of ascorbate-deficient blood vessels was still sensitive to ALDH2 inhibition (Wölkart et al., 2008; Wenzl et al., 2009). Lack of antibodies and sequence information precluded further studies on the molecular mechanisms underlying this striking difference of the two nitrate tolerance models in guinea pigs.
Unlike guinea pigs and humans, which do not express L-gulonolactone oxidase (Gulo), the terminal enzyme of ascorbate biosynthesis (Linster and Van Schaftingen, 2007), mice and rats synthesize the vitamin endogenously, making them independent of dietary ascorbate. This is a major problem for researchers aiming to study the consequences of ascorbate deficiency in those rodents. To overcome this problem, we created mice that are unable to synthesize ascorbate by targeted deletion of the Gulo-encoding gene (Maeda et al., 2000) and found that impaired vasorelaxation to GTN in ascorbate deficiency is associated with down-regulation of vascular ALDH2 expression. In search for the underlying mechanism, we observed that ALDH2 mRNA levels were not significantly altered and, therefore, studied proteasomal degradation of ALDH2 as potential culprit. Taking into account that ascorbate deficiency may cause oxidative stress in the vasculature, we tested for reduced response of aortic rings to ACh and NO as a functional marker for superoxide generation, measured mRNA expression of xanthine oxidase (XO) and NADPH oxidases, and determined the redox status of the mice.
Methods
Mice and experimental groups
Female and male Gulo(-/-) mice (Maeda et al., 2000) on C57BL/6 background were housed at the local animal facility in approved cages and kept on a regular 12 h dark/light cycle. They were fed standard mouse chow diet (Altromin 1324, containing <40 mg·kg−1 ascorbate; Königshofer Futtermittel Ebergassing, Austria) and provided drinking water ad libitum. Drinking water was supplemented with 330 mg ascorbate per litre and 0.01 mM EDTA, and renewed twice per week. This treatment was shown to provide enough ascorbate to prevent scurvy and maintain weight and health in Gulo(-/-) mice (Maeda et al., 2000; Parsons et al., 2006). At 3–4 months of age, Gulo(-/-) mice were randomly divided into three groups. While one group was left on ascorbate supplementation (ascorbate-supplemented), ascorbate deficiency was induced in the other groups by feeding ascorbate-free diet for 4 weeks (ascorbate-deficient). At the time point of ascorbate deprivation, one group of Gulo(-/-) mice was intraperitoneally injected the proteasome inhibitor bortezomib (0.5 mg·kg−1 body weight; Selleck Chemicals, Houston, TX, USA) once every 3 days over the 4 weeks deprivation period (bortezomib-treated ascorbate-deficient; Van Herck et al., 2010). For comparison, classical nitrate tolerance was induced in age-matched wild-type (WT; C57BL/6) mice by subcutaneous injection of GTN (20 mg·kg−1·per day; b.i.d.) for 4 days as described (Griesberger et al., 2011). WT mice kept on standard mouse chow and non-supplemented drinking water served as controls. At the end of the treatment period, animals were anaesthetized with 5% isoflurane and thereafter killed by cervical dislocation. Depth of anaesthesia was monitored by toe pinch reflex test. The descending aorta was removed and used for functional studies and determination of GTN denitration, latter measured as formation of 1,2- and 1,3- glycerol dinitrate (GDN). For determination of ascorbate and protein expression levels, tissues were frozen in liquid nitrogen and stored at −70°C until analysis. Animals were handled in accordance with the Austrian law on experimentation with laboratory animals (last amendment, 2004), which is based on the US National Institutes of Health guidelines. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Relaxation of aortic rings
For determination of isometric tension of aortic rings, a previously described protocol (Wölkart et al., 2008) was slightly modified. Rings of ∼3 mm width were mounted in 5 mL-organ baths filled with oxygenated Krebs–Henseleit solution (composition in mM: NaCl 118.4, NaHCO3 25, KCl 4.7, KH2PO4 1.2, CaCl2 2.5, MgCl2 1.2, d-glucose 10.1; pH 7.4). Care was taken to retain the endothelium for relaxation experiments. Tissues were equilibrated for 60 min by repeatedly adjusting tension to 0.5 g and changing the bath solution. Bathing solution was then replaced by a depolarizing solution containing 100 mM K+ for determination of maximal contraction. Rings that did not elicit adequate and stable contraction to high K+ were considered as damaged and discarded. After washout, tissues were precontracted with the thromboxane mimetic 9,11-dideoxy-11α,9α-epoxymethanoprostaglandin F2α (U-46619; Sigma; Vienna, Austria) to an equivalent level of ∼1.2 g (i.e. ∼90% of maximal contractile activity). Notably, vasoconstrictor response to 50 nM U-46619 did not significantly differ between experimental groups [mean total tone of all rings per group: 1.16 ± 0.02 g (WT), 1.14 ± 0.03 g (ascorbate-supplemented) and 1.17 ± 0.02 g (ascorbate-deficient, n = 14 mice per group)]. After reaching stable contraction, cumulative concentration-response curves were established using ACh (1 nM–10 μM; Sigma), GTN [1 nM–100 μM; Nitropol® ampoules (4.4 mM); G. Pohl-Boskamp GmbH, Hohenlockstedt, Germany] or 2,2-diethyl-1-nitroso-oxyhydrazine (DEA/NO; 1 nM–10 μM; Alexis Corp., Lausen, Switzerland). Some rings were incubated for 45 min with chloral hydrate (5 mM) before addition of agonists to test for the involvement of ALDH2 in GTN bioactivation. Contractile force corresponding to each concentration of the agonists was recorded and expressed as percent of precontraction (=baseline).
Determination of ascorbate levels
Plasma as well as homogenates of aortas and liver were acidified with meta-phosphoric acid and ascorbate levels measured by HPLC and UV detection at 264 nm using standard methods (Karlsen et al., 2007; Wenzl et al., 2009).
GTN denitration and ALDH2 protein expression in aorta
Vascular denitration of GTN to 1,2- and 1,3-GDN was determined by incubation of aortic slices with 14C-GTN and quantification of metabolites by radio thin-layer chromatography and liquid scintillation counting as described previously for guinea pig aorta (Wenzl et al., 2009). For determination of ALDH2 protein expression, sliced mouse aortas were homogenized with a glass potter in 50 mM Tris-HCl, 0.5 mM EDTA, 0.25 M sucrose buffer (pH 7.2), containing 0.1 mM phenylmethylsulfonyl fluoride and protease inhibitors (Roche Diagnostics GmbH, Graz, Austria). After centrifugation at 200× g at 4°C for 5 min, protein concentration was determined with a BCA protein assay kit (Thermo Scientific, purchased via THP, Vienna, Austria). For immunoblotting, 25 μg of total protein was separated on 12% SDS-polyacrylamide gels and transferred electrophoretically to nitrocellulose membranes. For detection of ALDH2 or β-actin, membranes were blocked with 5% non-fat dry milk in PBS containing 0.05% Tween-20 (vol/vol), while the membranes used for detection of ubiquitinated proteins were blocked with 5% non-fat dry milk in Tris-buffered saline containing 0.1% Tween-20 (vol/vol). After blocking, blots were incubated overnight with a primary polyclonal antibody to human ALDH2 (1:20 000 – kindly provided by Dr Henry Weiner), to β-actin (1:200 000 – Sigma-Aldrich, Vienna, Austria) or to ubiquitin (1:4000 – Dako Österreich GmbH, Vienna, Austria), washed and incubated for 1 h with a HRP-conjugated anti-rabbit (ALDH2 or ubiquitin) or anti-mouse (β-actin) IgG secondary antibody (both 1:5000 – Sigma-Aldrich). Bands were visualized with ECL Prime Western blot detection reagent (GE Healthcare, purchased via VWR, Vienna, Austria) using an E.A.S.Y. 1.3 HC camera (Herolab GmbH, Wiesloch, Germany) to detect ALDH2 and β-actin. For detection of ubiquitinated proteins, blots were exposed to X-ray films and all bands above 54 kDa used for quantification. Data are expressed as % of WT.
RNA extraction, reverse transcription and gene expression analysis
Total RNA was isolated from homogenized aortas using the GenElute™ Mammalian Total RNA Miniprep Kit (Sigma) including DNAse treatment of samples. RNA was transcribed to cDNA using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Vienna, Austria). qPCR analyses were performed with ∼25 ng cDNA using TaqMan® Universal PCR Master Mix and pre-designed TaqMan Gen Expression Assays. Reactions were carried out on a 7300 Real Time PCR System (Applied Biosystems, Vienna, Austria). Cycling conditions were as follows: 2 min at 50°C, 10 min at 95°C, 40 cycles of 15 s at 95°C and for 1 min at 60°C. Data were analysed according to the 2-ΔΔCt method using cyclophilin D as reference gene. Lack of amplification was verified in no-template controls.
Determination of antioxidant status
Plasma total antioxidative status was determined using a commercially available kit (TAS NX2331, Randox, Crumlin, UK) following the manufacturer's instructions. The reaction is based on inhibition of metmyoglobin- and H2O2-induced oxidation of 2,2'-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) by plasma antioxidants (Miller et al., 1993). In addition, the antioxidative capacity of acetonitrile-deproteinized plasma samples was measured using the 2,2-diphenyl-1-picryl-hydrazyl (DPPH) method described recently (Chrzczanowicz et al., 2008).
Statistical analysis
Data are presented as mean values ± SEM of n experiments. Concentration-response curves established with different ring segments from a single animal were averaged and counted as individual experiment. Individual concentration-response curves were fitted to a Hill-type model giving estimates of agonist potency (EC50) and efficacy (Emax). To account for the biphasic characteristic of GTN-induced vasorelaxation, EC50 values were calculated for the high-affinity component (1 nM–1 μM GTN). Analysis of variance (anova) with post hoc Bonferroni–Dunn test was used for comparison between groups using StatView® (Version 5.0; SAS Institute Inc., Cary, NC, USA). Significance was assumed at P < 0.05.
Results
Body weights and ascorbate levels
In line with previous reports on the Gulo(-/-) mouse strain (Maeda et al., 2000; Parsons et al., 2006), ascorbate deprivation for 4 weeks caused slight weight loss in Gulo(-/-) mice as compared to ascorbate-supplemented or WT animals (Table 1). As further shown in the table, ascorbate levels in plasma, aorta and liver were markedly decreased in ascorbate-deficient Gulo(-/-) mice as compared to WT animals. In plasma and aortas of ascorbate-supplemented Gulo(-/-) mice, ascorbate levels were similar to WT controls, but in liver, the levels were significantly lower than in WT.
Table 1.
Body weight and ascorbate levels in plasma, aorta and liver of ascorbate-deficient and -supplemented Gulo(-/-) mice in comparison to WT
| Genotype | WT | Gulo(-/-) | |
|---|---|---|---|
| asc-deficient | asc-supplemented | ||
| Body weight (g) | 26 ± 0.4 | 23 ± 0.71 | 26 ± 0.72 |
| Plasma ascorbate (μM) | 132 ± 8.1 | 11 ± 4.81 | 95 ± 222 |
| Aortic ascorbate (pmol·mg−1) | 93 ± 27 | 5 ± 21 | 74 ± 142 |
| Liver ascorbate (pmol·mg−1) | 710 ± 50 | 127 ± 71 | 350 ± 702 |
Data are mean values ± SEM of 12–15 (body weight), 8 (plasma), 5 (aorta) or 9 (liver) duplicate determinations.
P < 0.05 versus WT.
P < 0.05 versus ascorbate-deficient (anova).
asc, ascorbate.
Effect of ascorbate deficiency on vascular relaxation to GTN
As shown in Figure 1A, GTN caused concentration-dependent relaxation of U-46619-precontracted aortas isolated from WT mice. In line with a previous observation in mice (Chen et al., 2005), the GTN response was biphasic with high- (<1 μM; Figure 1B) and low- (>1 μM) affinity components. The EC50 value for the high-affinity component of relaxation of WT aorta was 40 ± 6 nM (unfilled squares in Figure 1B). As expected, the high-affinity component of GTN relaxation was considerably diminished in nitrate-tolerant animals (filled squares in Figure 1B). Similarly, ascorbate deficiency led to fivefold lower relaxing potency of GTN with a corresponding EC50 value of 200 ± 50 nM (unfilled diamonds in Figure 1B; P < 0.05) and a significant decrease in maximal relaxation induced by 1 μM GTN (Emax = −24 ± 3% as compared to −48 ± 3% in untreated WT). The low-affinity response to GTN was also reduced in nitrate-tolerant WT and ascorbate-deficient Gulo(-/-) mice, though the effect was less pronounced. The apparent GTN tolerance of ascorbate-deficient aortas was fully prevented by ascorbate supplementation of Gulo(-/-) mice (unfilled circles in Figure 1A and B) or proteasome inhibition by bortezomib (filled diamonds in Figure 1A and B). The corresponding EC50 values for the high-affinity response (57 ± 15 nM and 64 ± 16 nM, respectively) and maximal relaxation to 1 μM GTN (Emax = −49 ± 4% and −44 ± 8%, respectively) were not significantly different from WT controls. Preincubation with the ALDH inhibitor chloral hydrate caused a pronounced right-ward shift of the GTN concentration-response curve of untreated WT, ascorbate-supplemented or bortezomib-treated ascorbate-deficient Gulo(-/-) aortas, while the effect of chloral hydrate on relaxation of nitrate-tolerant or ascorbate-deficient blood vessels was either abolished (nitrate-tolerant) or very small in magnitude (ascorbate-deficient; Supporting Information Figure S1).
Figure 1.

GTN (A, B), ACh (C) and DEA/NO (D) concentration-response curves of aortic rings from untreated WT (open squares), nitrate-tolerant WT (filled squares), ascorbate-supplemented (open circles), ascorbate-deficient (open diamonds) and bortezomib-treated ascorbate-deficient (filled diamonds) Gulo(-/-) mice. For clarity, the high-affinity pathway of the GTN curve is illustrated separately in panel B. Rings were precontracted with the thromboxane mimetic U-46619. In panels A, B and D, data are mean values ± SEM of nine (untreated WT, ascorbate-supplemented, ascorbate-deficient) or five (WT, nitrate-tolerant, bortezomib-treated ascorbate-deficient) animals. In panel C, data are mean values ± SEM of five animals. Concentration-response curves of different ring segments from a single animal were averaged and counted as individual experiment. *P < 0.05 versus ascorbate-deficient animals. The effect of nitrate tolerance is not indicated for reasons of clarity. NS, non-significant.
Effects of ascorbate deficiency on vascular relaxation to ACh and DEA/NO
Since long-term ascorbate deficiency may cause oxidative stress in the vasculature and compromise NO-mediated vascular relaxation through superoxide generation, we compared relaxation to ACh and exogenously applied NO of untreated or nitrate-tolerant WT aortas with that of blood vessels isolated from ascorbate-deficient, bortezomib-treated ascorbate-deficient and ascorbate-supplemented Gulo(-/-) mice. As shown in Figure 1C, ACh relaxed untreated WT (open squares), nitrate-tolerant WT (filled squares), ascorbate-deficient (open diamonds), ascorbate-deficient bortezomib-treated (filled diamonds) and ascorbate-supplemented (open circles) aortas with EC50 values of 200 ± 68, 200 ± 53, 260 ± 68, 140 ± 30 and 150 ± 40 nM, respectively. The corresponding Emax values were −66 ± 4, −59 ± 4, −60 ± 3, −70 ± 4 and −68 ± 5%. The differences between experimental groups were not statistically significant, indicating that even 4 weeks of ascorbate deprivation does not cause significant endothelial dysfunction in mice. Figure 1D shows that the response to the direct NO donor DEA/NO was neither affected by ascorbate deficiency or classical nitrate tolerance. The calculated EC50 values were 128 ± 30, 186 ± 31, 199 ± 88, 176 ± 16 and 87 ± 25 nM for untreated WT, nitrate-tolerant, ascorbate-deficient, bortezomib-treated ascorbate-deficient and -supplemented aortas, respectively.
Vascular GTN reductase activity
To test for the effect of ascorbate deprivation on vascular GTN biotransformation, we measured the rates of 1,2- and 1,3-GDN formation. Aortic rings of WT and ascorbate-supplemented Gulo(-/-) mice exhibited similar rates of GTN denitration (∼0.7 pmol 1,2-GDN × min−1 × g·wet weight−1; see Table 2). Formation of the 1,3-isomer was very low, with 1,2-/1,3-GDN ratios of 17.1 and 23.5 for WT and ascorbate-supplemented Gulo(-/-) mice, respectively, confirming previous reports (Chen et al., 2002; Wölkart et al., 2008; Wenzl et al., 2009). Ascorbate deprivation of the mice led to reduction of denitration rates to about 25% of WT controls that was accompanied by a decrease in the 1,2-/1,3-GDN ratio from 22 to 3.4.
Table 2.
Rates of GTN denitration of aortic rings from WT, ascorbate-supplemented (asc-suppl.) and ascorbate-deficient (asc-def.) Gulo(-/-) mice
| Genotype | 1,2-GDN | 1,3-GDN |
|---|---|---|
| (pmol·min−1·g−1) | ||
| WT | 0.77 ± 0.177 | 0.05 ± 0.025 |
| Gulo(-/-), asc-suppl. | 0.66 ± 0.162 | 0.03 ± 0.044 |
| Gulo(-/-), asc-def. | 0.17 ± 0.0571,2 | 0.05 ± 0.026 |
Data are mean values ± SEM of four animals.
P < 0.05 compared with WT animals.
P < 0.05 compared with ascorbate-supplemented mice (anova).
asc, ascorbate.
Vascular ALDH2 expression
The role of ALDH2 in the development of vascular tolerance to GTN was further studied by measuring ALDH2 mRNA and protein levels in aortas of the different experimental groups. As shown in Figure 2A, ALDH2 mRNA expression was not altered by any experimental intervention, suggesting that neither classical nitrate tolerance nor ascorbate deficiency significantly affected vascular ALDH2 gene transcription. However, ascorbate deprivation of Gulo(-/-) mice for 4 weeks reduced aortic ALDH2 protein levels to 38% of WT controls (Figure 2B). This effect was largely reversed by ascorbate supplementation or treatment with bortezomib. In contrast, classical nitrate tolerance led to a much less-pronounced decrease in aortic ALDH2 protein expression (83.4% of WT). This value agrees well with most published data including results obtained with human arterial and venous blood vessels (Hink et al., 2007; Szöcs et al., 2007; Wenzel et al., 2007). However, one study found that ALDH2 expression was markedly reduced to 20% of controls in vena cava of nitrate-tolerant rats (D'Souza et al., 2011).
Figure 2.
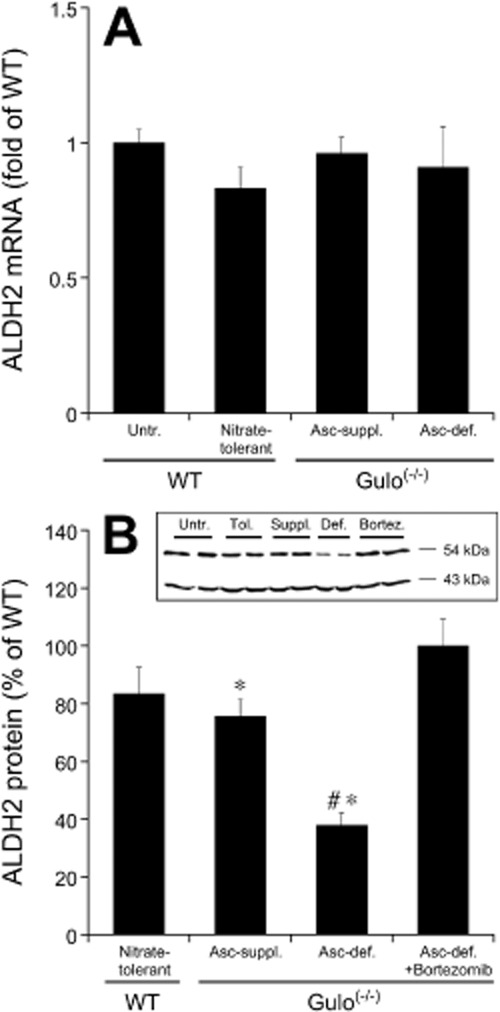
Aortic ALDH2 mRNA (A) and protein (B) levels of nitrate-tolerant WT, ascorbate-supplemented (Asc-suppl.), ascorbate-deficient (Asc-def.) and bortezomib-treated ascorbate-deficient Gulo(-/-) aortas relative to untreated WT controls. mRNA levels were analysed according to the 2-ΔΔCt method using cyclophilin D as reference gene. ALDH2 protein expression was analysed by immunoblotting and quantified densitometrically with band intensities of samples from untreated WT mice (applied on the same gels) set to 100%. A representative blot is shown as inset. Data are mean values ± SEM of four (A) or 6–9 (B) animals. *P < 0.05 compared with untreated (non-tolerant) WT animals. #P < 0.05 compared with ascorbate-supplemented mice.
Effect of ascorbate deficiency on vascular levels of ubiquitinated proteins
Since the protective effect of bortezomib suggested that the loss of ALDH2 is due to activation of the proteasome in ascorbate-deficient blood vessels, we measured the levels of ubiquitinated proteins in aortic lysates. As shown in Figure 3, the total amount of ubiquitinated proteins was reduced in ascorbate-deficient aortas to 37 ± 8.0% of untreated WT controls and this effect was fully restored by ascorbate supplementation or treating Gulo(-/-) mice with bortezomib. Note that the reduction in ubiquitinated protein levels in ascorbate-deprived aortas was virtually identical to the degree of ALDH2 down-regulation (cf. Figures 2B and 3). However, it should be noted that signal intensity of the inevitably overexposed lanes may have been underestimated due to lack of linear response.
Figure 3.
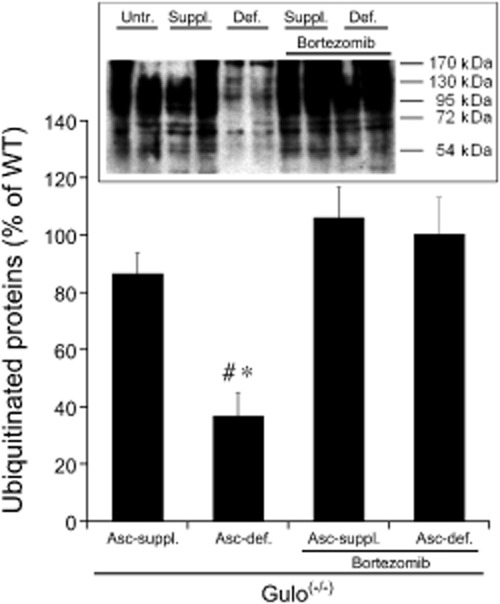
Levels of ubiquitinated proteins in lysates of aortas from ascorbate-supplemented (Asc-suppl.) and ascorbate-deficient (Asc-def.) Gulo(-/-) mice with and without bortezomib treatment relative to untreated WT controls. A representative blot is shown as inset. All bands >54 kDa (MW of ALDH2) were included in the densitometric quantification. Average band intensities of samples from untreated WT mice (applied on the same gels) were set to 100%. Data are mean values ± SEM of five animals. *P < 0.05 compared with untreated WT animals. #P < 0.05 compared with ascorbate-supplemented mice.
Vascular mRNA levels of NADPH oxidases (NOX2 and NOX4) and XO
Considering the function of ascorbate as major endogenous antioxidant, we measured mRNA expression of potential sources of vascular superoxide production, that is, NADPH oxidases (NOX2 and NOX4) and XO in aortas of ascorbate-deficient and -supplemented Gulo(-/-) mice in comparison to WT. There was no change in the expression levels of XO, NOX2 or NOX4 mRNAs in any experimental group (see Supporting Information Table S1).
Redox state
As shown in Figure 4, two established methods provided no evidence for a significant change in the systemic redox status of ascorbate-deficient Gulo(-/-) mice as compared to WT or ascorbate-supplemented controls.
Figure 4.
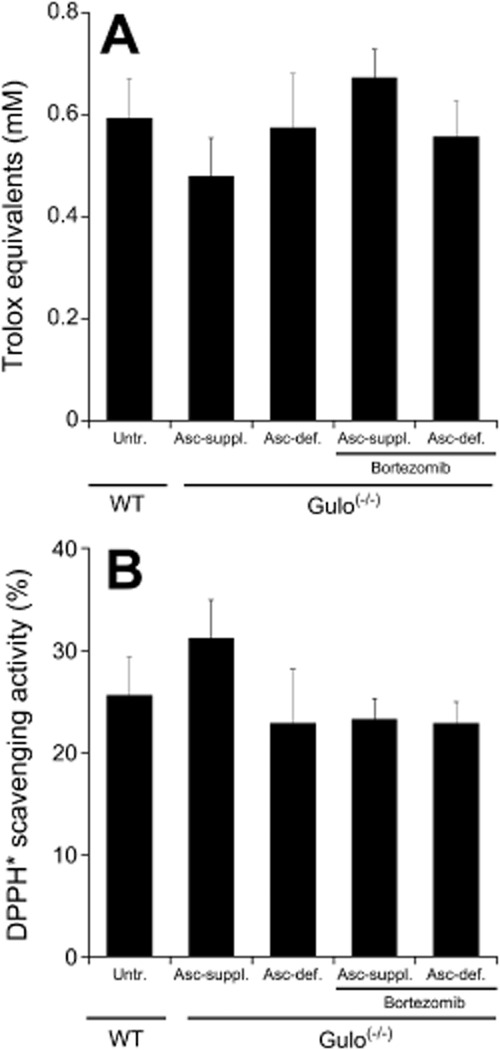
Redox status of ascorbate-supplemented (Asc-suppl.) and ascorbate-deficient (Asc-def.) Gulo(-/-) mice with and without bortezomib treatment relative to untreated WT controls. (A) Total antioxidant status of plasma was determined using a commercially available kit as described in Methods. Results are expressed as Trolox equivalents. (B) Antioxidant capacity of plasma samples was measured as 2,2-diphenyl-1-picryl-hydrazyl (DPPH) scavenging activity. 100% refers to complete loss of DPPH absorbance at 550 nm, that is, complete reduction of the radical. Data are mean values ± SEM of plasma samples obtained from five mice.
Discussion and conclusions
In the present study, we used the Gulo(-/-) genetic mouse model to clarify the mechanism underlying the development of nitrate tolerance in ascorbate deficiency described previously in guinea pigs (Wölkart et al., 2008; Wenzl et al., 2009). Upon 4 weeks of feeding with ascorbate-free diet, ascorbate plasma levels of Gulo(-/-) mice were reduced to approximately 20% of ascorbate-supplemented controls or WT (see Table 1). Decreased ascorbate plasma levels were associated with impaired relaxation of isolated aortas in response to GTN (see Figure 1).
The organ bath experiments showed that the high-affinity response to GTN was abolished in both classical nitrate tolerance and upon ascorbate deprivation of Gulo(-/-) mice, whereas relaxation of blood vessels from ascorbate-supplemented mice was identical to WT (see Figure 1B). The responsiveness of aortic rings to ACh and DEA/NO was not affected by ascorbate deprivation (see Figure 1C and D). Together with the about fourfold reduced rates of 1,2-GDN formation measured in ascorbate-deficient aortas (see Table 2), these results indicate that ascorbate deprivation interferes specifically with GTN bioactivation. Thus, the hyposensitivity of ascorbate-deficient blood vessels to GTN resembles classical nitrate tolerance, which is also associated with reduced vascular ALDH2 activity (Sydow et al., 2004; Wölkart et al., 2008). However, while exposure of blood vessels to GTN results in mechanism-based inactivation of ALDH2 (Mayer and Beretta, 2008) with only 20–30% reduction of protein levels [see Figure 2B and (Hink et al., 2007; Szöcs et al., 2007; Wenzel et al., 2007)], ascorbate deficiency led to markedly reduced vascular ALDH2 protein levels. Surprisingly, this effect was not accompanied by reduced ALDH2 mRNA expression (see Figure 2A), pointing to a post-transcriptional mechanism. Significant reduction of ubiquitinated protein levels (see Figure 3) suggested that activation of the proteasome might cause degradation of ALDH2 in ascorbate-deprived aortas. This hypothesis was tested with bortezomib, a selective inhibitor of the 26S proteasome (Papandreou, 2005). Indeed, treatment with bortezomib prevented all effects of ascorbate deprivation, that is, the decrease in ubiquitinated protein levels, ALDH2 down-regulation and the development of nitrate tolerance, strongly suggesting that impaired GTN bioactivation in ascorbate-deficient aortas was due to proteasomal degradation of vascular ALDH2. This is apparently not a consequence of ALDH2 inactivation because the effect of classical nitrate tolerance, which leads to inactivation of the enzyme, was much less pronounced. Moreover, inhibition of the proteasome with bortezomib completely prevented the effects of ascorbate deficiency and led to normal relaxation response to GTN, indicating that ALDH2 was active in the ascorbate-deficient vessels.
It is well established that the ubiquitin-proteasome system is critically involved in the regulation of cardiovascular function and various pathologies (Willis et al., 2010). An important function of the proteasome appears to be degradation of oxidized proteins formed in conditions of oxidative stress (Shang and Taylor, 2011). Effects of ascorbate on proteasomal activity have not been reported so far, but ascorbate supplementation was shown to prevent protein ubiquitination caused by oxidative stress in atherogenesis (Hermann et al., 2003). Since ubiquitination targets proteins towards proteasomal degradation, this may reflect a related mechanism by which vascular ascorbate protects blood vessels from oxidative injury. Considering the function of ascorbate as endogenous antioxidant and the role of oxidative stress in activation of the proteasome, we carefully studied whether ascorbate deprivation of Gulo(-/-) mice led to systemic or vascular oxidative stress. A previous study found that the plasma antioxidant capacity of ascorbate-deficient Gulo(-/-) mice was significantly reduced as compared to ascorbate-supplemented controls (Maeda et al., 2000). However, using spectrophotometric methods instead of the chemiluminescence technique used by Maeda et al., we observed no changes of the antioxidant status or the antioxidative capacity of the plasma (see Figure 4). It has been reported that the assays are sensitive to different factors and yield divergent results upon ascorbate infusion (Waring et al., 2003), rendering it difficult to judge which measurements correctly reflect the redox status of the animals. The results of the present study suggest that an antioxidative defence system efficiently compensates the systemic lack of ascorbate in vivo. These data do not exclude local oxidative stress in the vasculature, but normal relaxation to DEA/NO and ACh as well as unchanged NADPH oxidase and XO expression levels appear to exclude significant formation of superoxide in ascorbate-deficient blood vessels. Of note, these measurements were made in the absence of GTN, which is known to trigger oxidative stress in the vasculature and the oxidative stress response could be enhanced under conditions of ascorbate deficiency. Thus, despite the lack of experimental evidence, we cannot exclude a role of oxidative stress with certainty. In addition, it is conceivable that activation of the proteasome involves selective oxidation of highly redox-sensitive proteins that are normally protected by ascorbate. It should be noted that we cannot exclude that bortezomib acted through increased expression of antioxidant enzymes as has been reported for a rat model of alcoholic liver disease (Bardag-Gorce et al., 2011).
Taken together, our results reveal a novel mechanism of vascular tolerance to GTN that is caused by ascorbate deprivation. In contrast to classical nitrate tolerance, which essentially involves oxidative inactivation of ALDH2 by GTN, ascorbate deficiency leads to impaired GTN bioactivation through proteasomal degradation of ALDH2. Since nitrate tolerance was only observed upon severe ascorbate deprivation, it appears unlikely that the therapeutic effects of GTN are compromised in ascorbate-deficient subjects. However, regulation of ALDH2 expression could contribute to the well-documented beneficial effects of ascorbate in nitrate tolerance (Mayer and Beretta, 2008) and disorders associated with overproduction of reactive aldehydes that are detoxified by ALDH2 (Chen et al., 2008). Future work is necessary to address these issues and to clarify the molecular mechanism underlying activation of the proteasome in ascorbate-deprived blood vessels.
Acknowledgments
This work was supported by the Austrian Science Fund (FWF): P20669, P21693 and W901 DK Molecular Enzymology (to BM) and by the National Institute of Health: grant number HL42630 (to NM).
Glossary
- ALDH2
aldehyde dehydrogenase-2
- DEA/NO
2,2-diethyl-1-nitroso-oxyhydrazine
- GDN
glycerol dinitrate
- GTN
glycerol trinitrate (nitroglycerin)
- Gulo
L-gulonolactone oxidase
- WT
wild type
- XO
xanthine oxidase
Conflict of interest
None declared.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 Effect of chloral hydrate (CH, 5 mM, filled symbols) on the GTN concentration-response of aortic rings from untreated WT (squares), nitrate-tolerant WT (triangles), ascorbate-supplemented (circles), ascorbate-deficient (diamonds) and bortezomib-treated ascorbate-deficient (right-angled triangles) Gulo(-/-) mice (A). For clarity the high affinity pathway of the GTN response in the absence or presence of CH for each experimental group is illustrated separately in panels B-F (up to 1 μM GTN). Aortic rings were incubated for 45 min with CH before the addition of GTN. Data are mean values ± SEM of 9 (untreated WT, ascorbate-supplemented, ascorbate-deficient) or 5 (WT, nitrate-tolerant, bortezomib-treated ascorbatedeficient) animals. Concentration-response curves of different ring segments from a single animal were averaged and counted as individual experiment. *P < 0.05. NS, non-significant
Table S1 Aortic mRNA expression of NOX2, NOX4, and XO in wildtype (WT), ascorbate-supplemented (asc-suppl.) and ascorbate-deficient (asc-def.) Gulo(-/-) mice.
References
- Bardag-Gorce F, Oliva J, Lin A, Li J, French BA, French SW. Proteasome inhibitor up regulates liver antioxidative enzymes in rat model of alcoholic liver disease. Exp Mol Pathol. 2011;90:123–130. doi: 10.1016/j.yexmp.2010.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassenge E, Fink N, Skatchkov M, Fink B. Dietary supplement with vitamin C prevents nitrate tolerance. J Clin Invest. 1998;102:67–71. doi: 10.1172/JCI977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beretta M, Wölkart G, Schernthaner M, Griesberger M, Neubauer R, Schmidt K, et al. Vascular bioactivation of nitroglycerin is catalyzed by cytosolic aldehyde dehydrogenase-2. Circ Res. 2012;110:385–393. doi: 10.1161/CIRCRESAHA.111.245837. [DOI] [PubMed] [Google Scholar]
- Chen CH, Budas GR, Churchill EN, Disatnik MH, Hurley TD, Mochly-Rosen D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science. 2008;321:1493–1495. doi: 10.1126/science.1158554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Zhang J, Stamler JS. Identification of the enzymatic mechanism of nitroglycerin bioactivation. Proc Natl Acad Sci U S A. 2002;99:8306–8311. doi: 10.1073/pnas.122225199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Foster MW, Zhang J, Mao L, Rockman HA, Kawamoto T, et al. An essential role for mitochondrial aldehyde dehydrogenase in nitroglycerin bioactivation. Proc Natl Acad Sci U S A. 2005;102:12159–12164. doi: 10.1073/pnas.0503723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrzczanowicz J, Gawron A, Zwolinska A, De Graft-Johnson J, Krajewski W, Krol M, et al. Simple method for determining human serum 2,2-diphenyl-1-picryl-hydrazyl (DPPH) radical scavenging activity – possible application in clinical studies on dietary antioxidants. Clin Chem Lab Med. 2008;46:342–349. doi: 10.1515/CCLM.2008.062. [DOI] [PubMed] [Google Scholar]
- Csont T, Csonka C, Ónody A, Görbe A, Dux L, Schulz R, et al. Nitrate tolerance does not increase production of peroxynitrite in the heart. Am J Physiol. 2002;283:H69–H76. doi: 10.1152/ajpheart.00817.2001. [DOI] [PubMed] [Google Scholar]
- D'Souza Y, Dowlatshahi S, Bennett BM. Changes in aldehyde dehydrogenase 2 expression in rat blood vessels during glyceryl trinitrate tolerance development and reversal. Br J Pharmacol. 2011;164:632–643. doi: 10.1111/j.1476-5381.2011.01448.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griesberger M, Kollau A, Wölkart G, Wenzl MV, Beretta M, Russwurm M, et al. Bioactivation of pentaerythrityl tetranitrate by mitochondrial aldehyde dehydrogenase. Mol Pharmacol. 2011;79:541–548. doi: 10.1124/mol.110.069138. [DOI] [PubMed] [Google Scholar]
- Hanspal IS, Magid KS, Webb DJ, Megson IL. The effect of oxidative stress on endothelium-dependent and nitric oxide donor-induced relaxation: implications for nitrate tolerance. Nitric Oxide Biol Chem. 2002;6:263–270. doi: 10.1006/niox.2001.0412. [DOI] [PubMed] [Google Scholar]
- Hermann J, Gulati R, Napoli C, Woodrum JE, Lerman LO, Rodriguez-Porcel M, et al. Oxidative stress-related increase in ubiquitination in early coronary atherogenesis. FASEB J. 2003;17:1730–1732. doi: 10.1096/fj.02-0841fje. [DOI] [PubMed] [Google Scholar]
- Hink U, Daiber A, Kayhan N, Trischler J, Kraatz C, Oelze M, et al. Oxidative inhibition of the mitochondrial aldehyde dehydrogenase promotes nitroglycerin tolerance in human blood vessels. J Am Coll Cardiol. 2007;50:2226–2232. doi: 10.1016/j.jacc.2007.08.031. [DOI] [PubMed] [Google Scholar]
- Hinz B, Schröder H. Vitamin C attenuates nitrate tolerance independently of its antioxidant effect. FEBS Lett. 1998;428:97–99. doi: 10.1016/s0014-5793(98)00506-7. [DOI] [PubMed] [Google Scholar]
- Karlsen A, Blomhoff R, Gundersen TE. Stability of whole blood and plasma ascorbic acid. Eur J Clin Nutr. 2007;61:1233–1236. doi: 10.1038/sj.ejcn.1602655. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linster CL, Van Schaftingen E. Vitamin C: biosynthesis, recycling and degradation in mammals. FEBS J. 2007;274:1–22. doi: 10.1111/j.1742-4658.2006.05607.x. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVeigh GE, Hamilton P, Wilson M, Hanratty CG, Leahey WJ, Devine AB, et al. Platelet nitric oxide and superoxide release during the development of nitrate tolerance – effect of supplemental ascorbate. Circulation. 2002;106:208–213. doi: 10.1161/01.cir.0000021600.84149.78. [DOI] [PubMed] [Google Scholar]
- Maeda N, Hagihara H, Nakata Y, Hiller S, Wilder J, Reddick R. Aortic wall damage in mice unable to synthesize ascorbic acid. Proc Natl Acad Sci U S A. 2000;97:841–846. doi: 10.1073/pnas.97.2.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer B, Beretta M. The enigma of nitroglycerin bioactivation and nitrate tolerance: news, views, and troubles. Br J Pharmacol. 2008;155:170–184. doi: 10.1038/bjp.2008.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller NJ, Rice-Evans C, Davies MJ, Gopinathan V, Milner A. A novel method for measuring antioxidant capacity and its application to monitoring the antioxidant status in premature neonates. Clin Sci. 1993;84:407–412. doi: 10.1042/cs0840407. [DOI] [PubMed] [Google Scholar]
- Milone SD, Pace-Asciak CR, Reynaud D, Azevedo ER, Newton GE, Parker JD. Biochemical, hemodynamic, and vascular evidence concerning the free radical hypothesis of nitrate tolerance. J Cardiovasc Pharmacol. 1999;33:685–690. doi: 10.1097/00005344-199905000-00002. [DOI] [PubMed] [Google Scholar]
- Münzel T, Sayegh H, Freeman BA, Tarpey MM, Harrison DG. Evidence for enhanced vascular superoxide anion production in nitrate tolerance – a novel mechanism underlying tolerance and cross-tolerance. J Clin Invest. 1995;95:187–194. doi: 10.1172/JCI117637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Münzel T, Daiber A, Gori T. Nitrate therapy: new aspects concerning molecular action and tolerance. Circulation. 2011;123:2132–2144. doi: 10.1161/CIRCULATIONAHA.110.981407. [DOI] [PubMed] [Google Scholar]
- Papandreou CN. The proteasome as a target for cancer treatment: focus on bortezomib. Am J Cancer. 2005;4:359–372. [Google Scholar]
- Parsons KK, Maeda N, Yamauchi M, Banes AJ, Koller BH. Ascorbic acid-independent synthesis of collagen in mice. Am J Physiol. 2006;290:E1131–E1139. doi: 10.1152/ajpendo.00339.2005. [DOI] [PubMed] [Google Scholar]
- Sage PR, De la Lande IS, Stafford I, Bennett CL, Phillipov G, Stubberfield J, et al. Nitroglycerin tolerance in human vessels: evidence for impaired nitroglycerin bioconversion. Circulation. 2000;102:2810–2815. doi: 10.1161/01.cir.102.23.2810. [DOI] [PubMed] [Google Scholar]
- Sayed N, Kim DD, Fioramonti X, Iwahashi T, Durán WN, Beuve A. Nitroglycerin-induced S-nitrosylation and desensitization of soluble guanylyl cyclase contribute to nitrate tolerance. Circ Res. 2008;103:557–559. doi: 10.1161/CIRCRESAHA.108.175133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder H, Leitman DC, Bennett BM, Waldman SA, Murad F. Glyceryl trinitrate-induced desensitization of guanylate cyclase in cultured rat lung fibroblasts. J Pharmacol Exp Ther. 1988;245:413–418. [PubMed] [Google Scholar]
- Shang F, Taylor A. Ubiquitin-proteasome pathway and cellular responses to oxidative stress. Free Radic Biol Med. 2011;51:5–16. doi: 10.1016/j.freeradbiomed.2011.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sydow K, Daiber A, Oelze M, Chen ZP, August M, Wendt M, et al. Central role of mitochondrial aldehyde dehydrogenase and reactive oxygen species in nitroglycerin tolerance. J Clin Invest. 2004;113:482–489. doi: 10.1172/JCI19267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szöcs K, Lassegue B, Wenzel P, Wendt M, Daiber A, Oelze M, et al. Increased superoxide production in nitrate tolerance is associated with NAD(P)H oxidase and aldehyde dehydrogenase 2 downregulation. J Mol Cell Cardiol. 2007;42:1111–1118. doi: 10.1016/j.yjmcc.2007.03.904. [DOI] [PubMed] [Google Scholar]
- Van Herck JL, De Meyer GRY, Martinet W, Bult H, Vrints CJ, Herman AG. Proteasome inhibitor bortezomib promotes a rupture-prone plaque phenotype in ApoE-deficient mice. Basic Res Cardiol. 2010;105:39–50. doi: 10.1007/s00395-009-0054-y. [DOI] [PubMed] [Google Scholar]
- Waring WS, Mishra V, Maxwell SRJ. Comparison of spectrophotometric and enhanced chemiluminescent assays of serum antioxidant capacity. Clin Chim Acta. 2003;338:67–71. doi: 10.1016/j.cccn.2003.07.013. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Kakihana M, Ohtsuka S, Sugishita Y. Randomized, double-blind, placebo-controlled study of the preventive effect of supplemental oral vitamin C on attenuation of development of nitrate tolerance. J Am Coll Cardiol. 1998;31:1323–1329. doi: 10.1016/s0735-1097(98)00085-0. [DOI] [PubMed] [Google Scholar]
- Wenzel P, Oelze M, Coldewey M, Hortmann M, Seeling A, Hink U, et al. Heme oxygenase-1: a novel key player in the development of tolerance in response to organic nitrates. Arterioscler Thromb Vasc Biol. 2007;27:1729–1735. doi: 10.1161/ATVBAHA.107.143909. [DOI] [PubMed] [Google Scholar]
- Wenzl MV, Wölkart G, Stessel H, Beretta M, Schmidt K, Mayer B. Different effects of ascorbate deprivation and classical vascular nitrate tolerance on aldehyde dehydrogenase-catalysed bioactivation of nitroglycerin. Br J Pharmacol. 2009;156:1248–1255. doi: 10.1111/j.1476-5381.2009.00126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis MS, Townley-Tilson WHD, Kang EY, Homeister JW, Patterson C. Sent to destroy: the ubiquitin proteasome system regulates cell signaling and protein quality control in cardiovascular development and disease. Circ Res. 2010;106:463–478. doi: 10.1161/CIRCRESAHA.109.208801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wölkart G, Wenzl MV, Beretta M, Stessel H, Schmidt K, Mayer B. Vascular tolerance to nitroglycerin in ascorbate deficiency. Cardiovasc Res. 2008;79:304–312. doi: 10.1093/cvr/cvn107. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.