

Abstract
Background and Purpose
Hypertension increases cerebrovascular oxidative stress and inflammation and impairs vasomotor function. These pathological alterations lead to dysregulation of cerebral blood flow and exacerbate atherogenesis, increasing the morbidity of ischaemic cerebrovascular diseases and promoting vascular cognitive impairment. We aimed to test the hypothesis that increased production of the arachidonic acid metabolite 20-hydroxy-5,8,11,14-eicosatetraenoic acid (20-HETE) contributes to hypertension-induced cerebrovascular alterations.
Experimental Approach
We treated male spontaneously hypertensive rats (SHR) with HET0016 (N-hydroxy-N′-(4-butyl-2-methylphenyl)-formamidine), an inhibitor of 20-HETE synthesis. In middle cerebral arteries (MCAs) of SHRs, we focused on vasomotor responses and end points that are highly relevant for cellular reactive oxygen species (ROS) production, inflammatory cytokine expression and NF-κB activation.
Key Results
SHRs treated with HET0016 remained hypertensive (SHR + HET0016: 149 ± 8 mmHg, Wistar-Kyoto rat: 115 ± 4 mmHg; P < 0.05.), although their systolic blood pressure was decreased compared to untreated SHRs (191 ± 6 mmHg). In MCAs of SHRs, flow-induced constriction was increased, whereas ACh- and ATP-induced dilations were impaired. This functional impairment was reversed by treatment with HET0016. Treatment with HET0016 also significantly decreased oxidative stress in MCAs of SHRs (as shown by dihydroethidium staining and analysis of vascular 5-nitrotyrosine, 4-hydroxynonenal and carbonyl content) and inhibited cerebrovascular inflammation (shown by the reduced mRNA expression of TNFα, IL-1β and IL-6). Treatment of SHRs with HET0016 also attenuated vascular NF-κB activation. In vitro treatment with 20-HETE significantly increased vascular production of ROS and promoted NF-κB activation in cultured cerebromicrovascular endothelial cells.
Conclusions and Implications
Taken together, treatment with HET0016 confers anti-oxidative and anti-inflammatory effects in the cerebral arteries of SHRs by disrupting 20-HETE-mediated autocrine/paracrine signalling pathways in the vascular wall. It is likely that HET0016-induced decreases in blood pressure also potentiate the cerebrovascular protective effects of the drug.
Keywords: ROS, cytokines, cerebral blood flow, ischaemic stroke, vascular cognitive impairment, NF-κB, 20-HETE
Introduction
Hypertension increases cerebrovascular oxidative stress and inflammation and impairs vasomotor function (Barry, 1985; Mies et al., 1999; O'Sullivan et al., 2002; Amenta et al., 2003; Di Napoli and Papa, 2003; Beason-Held et al., 2007; Iadecola and Davisson, 2008). These pathological alterations lead to dysregulation of cerebral blood flow and eventually at a later phase may exacerbate atherogenesis in the cerebral arteries, which significantly increases the morbidity of ischaemic cerebrovascular diseases (Aiyagari and Gorelick, 2009) and promote the development of vascular cognitive impairment (Gorelick et al., 2011). Understanding the early mechanisms by which hypertension promotes cerebrovascular oxidative stress, inflammation and vasomotor dysfunction is critical, as it will enable the development of targeted therapeutic interventions for cerebrovascular protection in this high-risk patient population.
In recent years, evidence became available suggesting that hypertension is associated with an increased generation of 20-hydroxy-5,8,11,14-eicosatetraenoic acid (20-HETE) in the wall of cerebral vessels (Dunn et al., 2008). 20-HETE is a potent vasoconstrictor metabolite of arachidonic acid produced by cytochrome P450 Ω-hydroxylases and has a central role in regulation of cerebral blood flow by contributing to both pressure- and flow-dependent responses of cerebral arteries (Gebremedhin et al., 2000; Toth et al., 2011; Koller and Toth, 2012). Importantly, recent studies demonstrate that treatment of cultured endothelial cells in vitro with 20-HETE confers significant pro-inflammatory effects, including up-regulation of inflammatory cytokine production and activation of NF-κB (Cheng et al., 2008; 2010; Ishizuka et al., 2008). Yet, the in vivo pro-inflammatory effects of 20-HETE and the role of overproduction of 20-HETE in chronic cerebrovascular inflammation and in the development of vasomotor dysfunction in hypertension are not well understood.
The present study was undertaken to test the hypothesis that inhibition of 20-HETE production in hypertension confers cerebrovascular protection by attenuating inflammation and oxidative stress and restore vasomotor function. We chose to study spontaneously hypertensive rats (SHR), as this animal model exhibits well-described cerebrovascular dysfunction, vascular inflammation, 20-HETE overproduction and increased propensity of hypertension-induced brain damage, mimicking key aspects of essential hypertension in humans. To inhibit 20-HETE synthesis, the animals were chronically treated with N-hydroxy-N′-(4-butyl-2-methylphenyl)-formamidine (HET0016), a potent and specific inhibitor of the metabolism of arachidonic acid by cytochrome P450 (CYP) enzymes, which prevents formation of 20-HETE. We focused on end points that are highly relevant for vascular inflammation and the early mechanism contributing to the pathogenesis of atherosclerosis and vascular cognitive impairment, including cellular reactive oxygen species (ROS) production, inflammatory cytokine expression and NF-κB activation.
Materials and methods
Animals
In the present study, we used 12-week-old male spontaneously hypertensive rats (SHR, n = 16) and age-matched, normotensive male Wistar-Kyoto rats (WKY, n = 16) as controls. The animals were divided into two groups: (i) animals in vivo treated with HET0016 (10 mg·kg−1·per day, in 10% lecithin solution; intraperitoneally for 5 days (Renic et al., 2009; Wu et al., 2011) and (ii) control receiving vehicle. HET0016 (Figure 1) was reported to selectively inhibit the formation of 20-HETE by inhibiting CYP4A and CYP4F isoforms in SHR renal microsomes (IC50: 35.2 nM) and in human kidney (IC50: 8.9 nM; Miyata et al., 2001). Animals were housed individually on a 12 h light/dark cycle. All procedures were approved by the Institutional Animal Care and Use Committee of New York Medical College, and we consulted the ARRIVE and British Journal of Pharmacology guidelines for in vivo animal studies (Kilkenny et al., 2010; McGrath et al., 2010).
Figure 1.
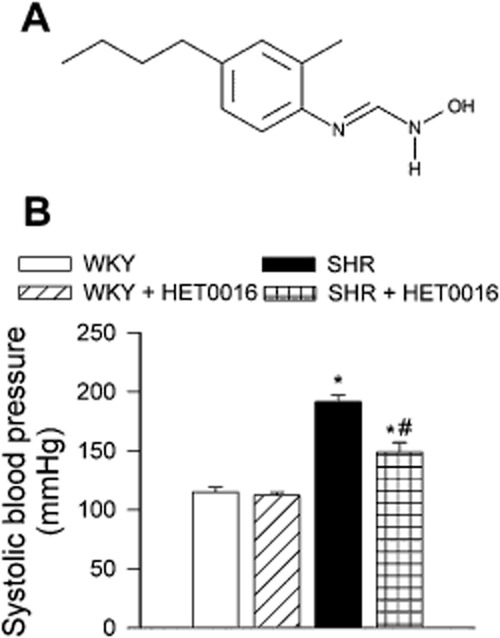
(A) Structure of HET0016. (B) The effect of HET0016 treatment (10 mg·kg−1, 5 days i.p.) on blood pressure in control Wistar-Kyoto (n = 6) and spontaneously hypertensive rats (n = 6). Data are mean ± SEM *P < 0.05 versus WKY; #P < 0.05 versus SHR.
Blood pressure measurements
Systolic blood pressure of rats in each experimental group was measured by the tail cuff method (CODA Non-Invasive Blood Pressure System, Kent Scientific Co., Torrington, CT, USA).
Isolation of middle cerebral arteries (MCAs) and assessment of vasomotor function
At the end of the experimental period, the animals were killed under anaesthesia (pentobarbital sodium, 50 mg·kg−1 i.p.) by decapitation and segments of the MCAs were isolated using microsurgery instruments, as reported (Ungvari et al., 1999; Toth et al., 2011). Branches of the MCAs were mounted onto two glass micropipettes in a vessel chamber and pressurized to 80 mmHg. The hydrodynamic resistance of the micropipettes was matched. Inflow and outflow pressures were controlled and measured by a pressure servo-control system (Living Systems Instrumentation, Burlington, VE, USA). Inner vascular diameter was measured with a video micrometer system and continuously recorded using a computerized data acquisition system. All vessels were allowed to stabilize for 60 min in oxygenated (21% O2-5% CO2-75% N2) Krebs' buffer (at 37°C) as reported previously. After the equilibration period, during which spontaneous myogenic tone developed, flow-induced vascular responses were assessed. Intraluminal flow was controlled by creating a pressure gradient through the vessel (from ΔP = 0 to 40 mmHg, corresponding to Q = 0 to 320 μL·min−1 intraluminal flow) keeping intraluminal pressure constant. Intraluminal flow was measured with a micro-flow meter as described (Toth et al., 2011). To assess the role of thromboxane/prostaglandin endoperoxide (TP) receptors, the vessels were incubated with SQ29,548 ([1S-[1α,2α(Z),3α,4α]]-7-[3-[[2-[(phenylamino)carbonyl]hydrazino]methyl]-7-oxabicyclo[2.2.1]hept-2-yl]-5-heptenoic acid) (10−6 mol·L−1), a specific antagonist of TP receptors and flow-induced responses were re-assessed. In addition, constrictions of MCAs to the thromboxane A2 analogue U46619 ((Z)-7-[(1S,4R,5R,6S)-5-[(E,3S)-3-hydroxyoct-1-enyl]-3-oxabicyclo[2.2.1]heptan-6-yl]hept-5-enoic acid; 10−7 mol·L−1) were assessed. To assess endothelial function, vascular responses to cumulative doses of ACh (10−8 to 10−5 mol·L−1) and ATP (10−8 to 10−5 mol·L−1) were obtained. To analyse the role of NO, endothelium-mediated responses were reassessed in the presence of NOS inhibitor L-NAME (Nω-nitro-L-arginine methyl ester; 10−4 mol·L−1). At the end of each experiment, the passive diameters of the vessels were measured at 80 mmHg intraluminal pressure in the presence of Ca2+-free PSS containing the L-type Ca2+ channel inhibitor nifedipine (10−5 mol·L−1) to achieve maximal vasodilatation.
Measurement of vascular ROS production
O2.- production in the MCA from rats in each experimental group was determined using dihydroethidium (DHE), an oxidative fluorescent dye, as previously reported (Bailey-Downs et al., 2012). Freshly harvested vessels were incubated with DHE (3 × 10−6 mol·L−1; at 37°C for 30 min). After three washes with PBS, the vessels were embedded in OCT medium and cryosectioned. Microscopic images of O2.- specific red fluorescence were captured at 20× magnification and analysed using Image J software as previously reported (a public domain, Java-based image processing program developed at the National Institutes of Health; http://rsbweb.nih.gov/ij/download.html) (Bailey-Downs et al., 2012). The mean fluorescence intensities of DHE-stained nuclei in the endothelium and medial layer were calculated for each vessel. Thereafter, the intensity values for each animal in the group were averaged.
In separate experiments, isolated vascular segments were incubated with 20-HETE (10−7 mol·L−1) in the absence or presence of apocynin (4′-hydroxy-3′-methoxyacetophenone; 3 × 10−4 mol·L−1; for 4 h), an inhibitor of ROS production by NADPH oxidases. Vascular ROS production was assessed fluorometrically by a Tecan Infinite M200 (Tecan Group Ag, Mannedorf, Switzerland) plate reader using the Amplex Red/horseradish peroxidase assay (Life Technologies, Grand Island, NY, USA), as described (Csiszar et al., 2007).
Determination of protein carbonylation and vascular 5-nitrotyrosine (5-NT) and 4-hydroxynonenal (4-HNE) content
As additional markers of oxidative/nitrosative stress in hypertension, carbonyl derivatives of protein oxidation, 5-NT (a marker for peroxynitrite action) and 4-HNE (a marker of lipid peroxidation) were assessed in pooled vascular tissues using the OxiSelect Protein Carbonyl, Nitrotyrosine and HNE-His Adduct ELISA Kits (Cell Biolabs, San Diego, CA, USA), according to the manufacturer's guidelines.
Quantitative real-time reverse-transcription (RT)-PCR
A quantitative real-time RT-PCR technique was used to analyse mRNA expression of the following NF-κB target genes and inflammatory markers in MCAs of rats from each experimental group: Nos2 (inducible NOS, iNOS), Il6 (IL-6), Il1b (IL-1β) and Tnfa (TNF-α), using a Strategen MX3000 (Strategene, La Jolla, CA, USA) as previously reported (Bailey-Downs et al., 2012) ( Table 1). In separate experiments, mRNA expression of inflammatory markers was assessed in vascular segments treated in vitro in organoid culture with exogenous 20-HETE (10−6 mol·L−1, for 18 h). Total RNA was isolated with a Mini RNA Isolation Kit (Zymo Research, Orange, CA, USA) and was reverse transcribed using Superscript III RT (Invitrogen, Life Technologies) as described previously. Amplification efficiencies were determined using a dilution series of a standard vascular sample. Quantification was performed using the efficiency-corrected ΔΔCq method. The relative quantities of the reference genes Hprt, Ywhaz, B2m and Actb were determined and a normalization factor was calculated based on the geometric mean for internal normalization. Fidelity of the PCR reaction was determined by melting temperature analysis and visualization of the product on a 2% agarose gel.
Table 1.
Oligonucleotides for real-time RT-PCR
| mRNA targets | Description | Sense | Antisense |
|---|---|---|---|
| Tnfa | Tumour necrosis factor alpha (TNF-α) | AACCACCAAGCAGAGGAG | CTTGATGGCGGAGAGGAG |
| Il6 | Interleukin 6 (IL-6) | TACCCCAACTTCCAATGC | GATACCCATCGACAGGAT |
| Il1b | Interleukin 1 beta (IL-1β) | CAGCAATGGTCGGGAC | ATAGGTAAGTGGTTGCCT |
| Nos2 | Inducible nitric oxide synthase (iNOS) | TCCCGAAACGCTACACT | CAATCCACAACTCGCT |
| Hprt | Hypoxanthine phosphoribosyltransferase 1 | AAGACAGCGGCAAGTTGAATC | AAGGGACGCAGCAACAGAC |
| Actb | Beta actin | GAAGTGTGACGTTGACAT | ACATCTGCTGGAAGGTG |
| B2m | Beta-2-microglobin | ATTCACACCCACCGAGAC | GGATCTGGAGTTAAACTGGTC |
| Ywhaz | Tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein, zeta polypeptide 1 | AACTGCCTACATATTGGT | CACACAGACTACACTCAT |
Nuclear extraction and NF-κB binding activity assay
Nuclei were extracted from freshly isolated carotid arteries using the Nuclear Extraction kit from Active Motif (Carlsbad, CA, USA) as reported (Csiszar et al., 2005). In brief, carotid arteries isolated from each experimental group were homogenized with a dounce tissue homogenizer in 500 μL ice-cold hypotonic lysis buffer followed by two centrifugation steps (500 g, for 30 s, 4°C) to exclude tissue debris. Then, nuclear proteins were extracted and the protein concentrations in samples were equalized, as reported (Csiszar et al., 2005). Using the nuclear extract obtained, NF-κB binding activity was assayed using the TransAM NF-κB ELISA kit (Active Motif) as reported (Csiszar et al., 2005).
Establishment and characterization of a primary cerebromicrovascular endothelial cell culture
To assess the direct effect of 20-HETE on endothelial NF-κB activation, we established a primary cerebromicrovascular endothelial cell (CMVEC) culture. In brief, the brain of a control rat was removed aseptically, rinsed in ice cold PBS and minced into ≍1 mm squares. The tissue was washed twice in ice-cold 1X PBS by low-speed centrifugation (50× g, 2–3 min). The diced tissue was digested in a solution of collagenase (800 U·g−1 tissue), hyaluronidase (2.5 U·g−1 tissue) and elastase (3 U·g−1 tissue) in 1 mL PBS/100 mg tissue for 45 min at 37°C in rotating humid incubator. The digested tissue was passed through a 100 um cell strainer to remove undigested blocks. The single cell lysate was centrifuged for 2 min at 70× g. After removing the supernatant, carefully, the pellet was washed twice in cold PBS supplemented with 2.5% fetal calf serum, and the suspension centrifuged at 300× g, for 5 min at 4C. To create an endothelial cell-enriched fraction, the cell suspension was gradient centrifuged by using OptiPrep solution (Axis-Shield Pic, Dundee, Scotland, UK). Briefly, the cell pellet was resuspended in HBSS and mixed with 40% iodixanol thoroughly (final concentration: 17% (w/v) iodixanol solution; ρ = 1.096 g·mL−1). 2 mL of HBSS was layered on top and centrifuged at 400× g for 15 min at 20°C. Endothelial cells, which banded at the interface between HBSS and the 17% iodixanol layer, were collected. The endothelial cell-enriched fraction was incubated for 30 min at 4C in dark with anti-CD31/PE (BD Biosciences, San Jose, CA, USA), anti-MCAM/FITC (BD Biosciences). After washing the cells twice with MACS Buffer (Milltenyi Biotech, Cambridge, MA, USA), anti-FITC magnetic bead labelled and anti-PE magnetic bead labelled secondary antibodies were used for 15 min at room temperature. Endothelial cells were collected by magnetic separation using the MACS LD magnetic separation columns according to the manufacturer's guidelines (Milltenyi Biotech, Cambridge, MA, USA). The endothelial fraction was cultured on fibronectin-coated plates in endothelial growth medium (Cell Application, San Diego, CA, USA) for 10 days. Endothelial cells were phenotypically characterized by flow cytometry (GUAVA 8HT, Merck Millipore, Billerica, MA, USA). Briefly, antibodies against five different endothelial specific markers were used (anti-CD31-PE, anti-erythropoietin receptor-APC, anti-VEGF R2-PerCP, anti-ICAM-fluorescein, anti-CD146-PE) and isotype-specific antibody labelled fractions served as negative controls. Flow cytometric analysis showed that after the third cycle of immunomagnetic selection, there were virtually no CD31-, CD146-, EpoR- and VEGFR2- cells in the resultant cell populations. All antibodies were purchased from R&D Systems (R&D Systems, Minneapolis, MN, USA).
Transient transfection and NF-κB reporter gene assay
The direct effect of treatment with 20-HETE (10−6 mol·L−1, for 4 h) on transcriptional activity of NF-κB in CMVECs was tested by a reporter gene assay as described (Csiszar et al., 2006b). To inhibit TP receptors, we pretreated CMVECs with SQ19,548 (10−6 mol·L−1). We used a NF-κB reporter comprised of a NF-κB response element upstream of firefly luciferase (NF-κB-Luc, Stratagene) and a renilla luciferase plasmid under the control of the cytomegalovirus promoter (as an internal control). Transfections in CMVECs were performed using the Amaxa Nucleofector technology (Amaxa, Gaithersburg, MD, USA), as we have previously reported (Csiszar et al., 2004; 2005; 2006a). Firefly and renilla luciferase activities were assessed after 24 h using the Dual Luciferase Reporter Assay Kit (Promega, Madison, WI, USA) and a Tecan Infinite M200 plate reader.
Statistical analysis
Statistical analysis was performed by one-way anova. P < 0.05 was considered significant. Data are expressed as mean ± SEM.
Results
Blood pressure
Systolic blood pressure, measured by the tail cuff method, was significantly higher in SHRs than in control WKY rats (Figure 1B). In WKY rats, treatment with HET0016 did not result in any significant change in systolic blood pressure. SHRs treated with HET0016 remained hypertensive (P < 0.05 vs. WKY), although their systolic blood pressure was moderately decreased compared to untreated SHRs (Figure 1B).
In vivo treatment with HET0016 inhibits flow-induced constriction and improves endothelial function in cerebral arteries of SHRs
Increases in intraluminal flow elicit constriction in both rodent MCAs and human intracerebral arteries, which is mediated predominantly by 20-HETE (Toth et al., 2011). We assessed flow-mediated vasoconstriction in isolated MCAs to bioassay 20-HETE production in the vascular tissue. Here we confirm that in response to stepwise increases in intraluminal flow (controlled by creating a pressure gradient through the vessel from ΔP = 0 to 40 mmHg), segments of MCAs isolated from control WKY rats exhibit significant vasoconstriction (Figure 2A). Flow-induced constriction in MCAs isolated from SHRs was significantly increased compared to responses of vessels from control rats (Figure 2A). Treatment of SHRs with HET0016 abolished flow-induced constriction of MCAs (Figure 2A), showing that the treatment regimen used effectively disrupted 20-HETE signalling in the cerebral vasculature. 20-HETE is known to bind to the TP receptor, which mediates its vasoconstrictor effect. Accordingly, we found that treatment with the TP receptor antagonist SQ 29 548 abolished flow-induced constriction in MCAs isolated from SHRs (Figure 2, upper panel). Constriction of cerebral vessels to the TP receptor agonist U46619 did not differ between WKY and SHR (Figure 2B, lower panel), and it was not affected by HET 0016 (data not shown).
Figure 2.
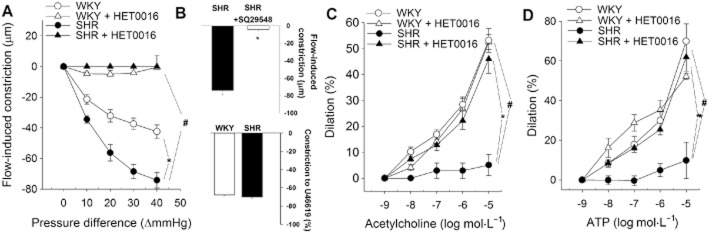
Inhibition of 20-HETE production improves vascular function in MCA of SHR. (A) Flow-induced constriction in MCAs isolated from control WKY rats, SHRs, WKYs and SHRs treated with HET0016 (SHR + HET0016; 10 mg·kg−1·per day, i.p., n = 6 for each data point). Intraluminal flow was controlled by creating a pressure gradient through the vessel (see Methods). (B, above): Treatment with the TP receptor antagonist SQ29,548 (1 μmol·L−1) inhibits flow-induced constriction in MCAs of SHRs (n = 7). Data are mean ± SEM *P < 0.05 versus control; #P < 0.05 versus untreated. (B, below): U46619 (10−7 mol·L−1), a thromboxane A2 analogue elicits comparable constriction in MCAs of WKY and SHR (n = 7). ACh- (C) and ATP (D)-induced dilations of MCAs isolated from control WKY rats and SHRs and WKYs and SHRs treated with HET0016 (n = 6 for each data point). Data are mean ± SEM *P < 0.05 versus control; #P < 0.05 versus untreated.
The endothelium-dependent vasodilators ACh (Figure 2C) and ATP (Figure 2D) elicited concentration-dependent dilation in MCAs of control rats. In agreement with the results of previous studies, we found that ACh- and ATP-dependent dilations were inhibited by the NOS inhibitor L-NAME (data not shown). ACh- and ATP-induced dilation in MCAs isolated from SHRs was significantly reduced compared to responses of vessels from control rats (Figure 2C, D respectively). Treatment of SHRs with HET0016 significantly improved endothelial function in MCAs, normalizing vascular dilations to ACh (Figure 2C) and ATP (Figure 2D). Treatment of WKY by HET 0016 did not affect endothelium-dependent vasodilation in MCAs (Figure 2C, D).
In vivo treatment with HET0016 attenuates oxidative stress in cerebral arteries of SHRs
Representative fluorescent images of cross sections of DHE-stained MCAs isolated from control WKY, SHR and HET0016-treated WKY and SHRs are shown in Figure 3A. Analysis of nuclear DHE fluorescent intensities indicated that, compared to vessels from control WKY rats, O2.- production was significantly increased in MCAs of SHRs (Figure 3B). Increased vascular O2.- generation in SHRs was significantly reduced by HET0016 treatment (Figure 3B). In contrast, in WKYs, HET0016 did not significantly alter vascular ROS production (Figure 3B). To further substantiate the role of increased levels of 20-HETE in induction of ROS production, we treated detector vessels with exogenous 20-HETE. We found that administration of 20-HETE significantly increased vascular ROS production (Figure 3C). The pro-oxidative effect of 20-HETE was abolished by pretreatment of the vessels with the NAD(P)H-oxidase inhibitor apocynin (Figure 3C).
Figure 3.
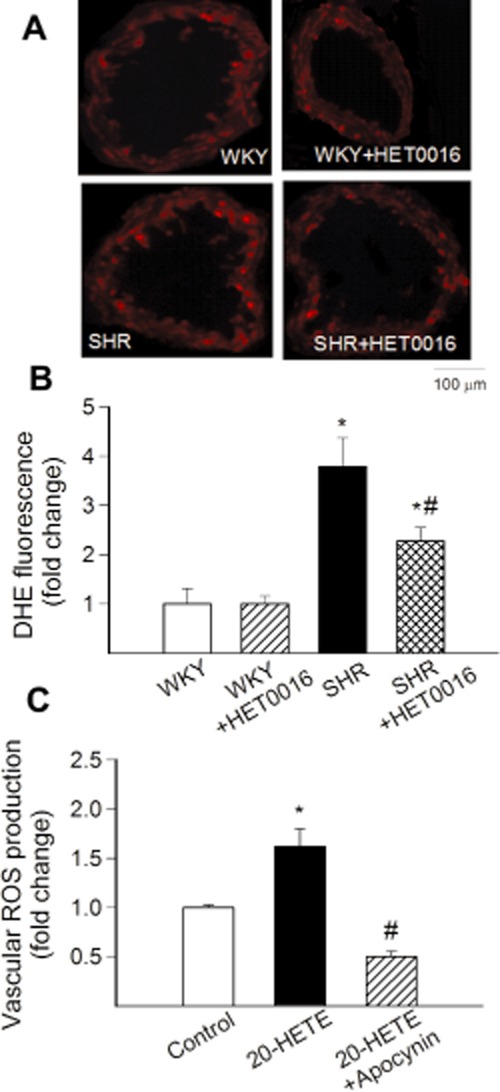
In vivo treatment with HET0016 attenuates oxidative stress in MCA of SHR. (A) Representative images showing red nuclear DHE fluorescence, representing cellular O2.- production, in sections of MCAs of control WKY rats, SHRs and treated WKY and SHRs with HET0016 (10 mg·kg−1·per day, i.p.). (B) Summary data for nuclear DHE fluorescence intensities (n = 6 for each group). (C) Summary data showing the effect of 20-HETE (10−7 mol·L−1) on production of ROS of vascular ring preparations (n = 7) in the absence and presence of NADPH oxidase inhibitor apocynin (3 × 10−4 mol·L−1). Data are mean ± SEM *P < 0.05 versus control; #P < 0.05 versus untreated.
Consistent with the presence of hypertension-related oxidative stress, vascular 5-NT, 4-HNE and protein carbonyl content was increased in SHRs (Figure 4A, B and C respectively). Treatment with HET0016 significantly decreased vascular 5-NT and 4-HNE content in SHRs (Figure 4A and B respectively). Treatment with HET0016 also abolished the difference between protein carbonyl content in the vascular wall in SHRs and WKY controls (Figure 4C).
Figure 4.
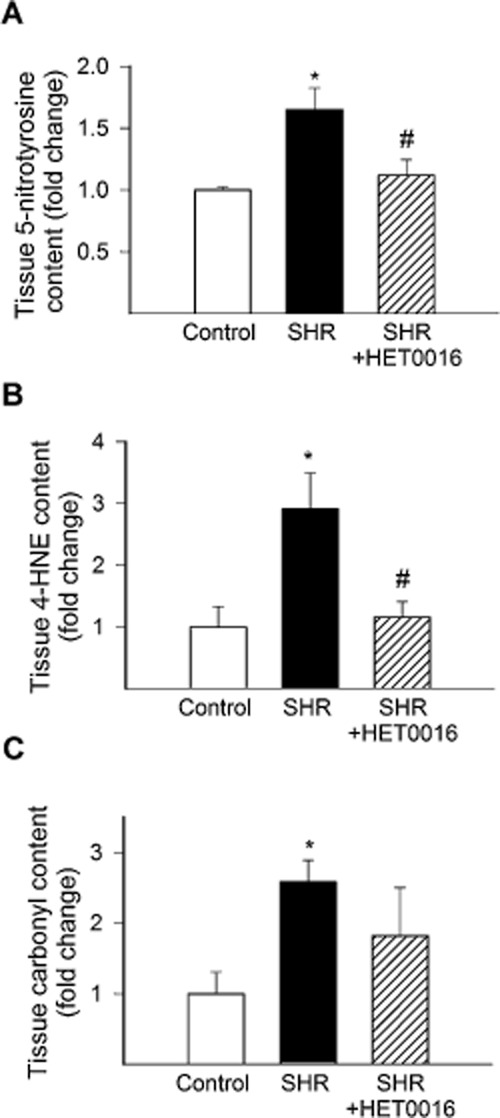
In vivo treatment with HET0016 attenuates vascular oxidative/nitrosative damage in SHRs. (A)–(C) show the effects of treatment with HET0016 on markers of vascular oxidative/nitrosative stress in SHRs, including 5-NT (a marker for peroxynitrite action, C), 4-HNE (a marker of lipid peroxidation, D) and carbonyl derivatives of protein oxidation (E). Data are mean ± SEM *P < 0.05 versus Control; #P < 0.05 versus untreated (n = 6 for each data point).
Role of 20-HETE in vascular NF-κB activation
We found that hypertension-induced oxidative stress is associated with an increased nuclear NF-κB binding activity in SHR arteries (Figure 5A). Treatment of SHRs with HET0016 attenuated vascular NF-κB binding activity (Figure 5A), suggesting that increased 20-HETE production has an important role in regulation of vascular inflammatory processes.
Figure 5.
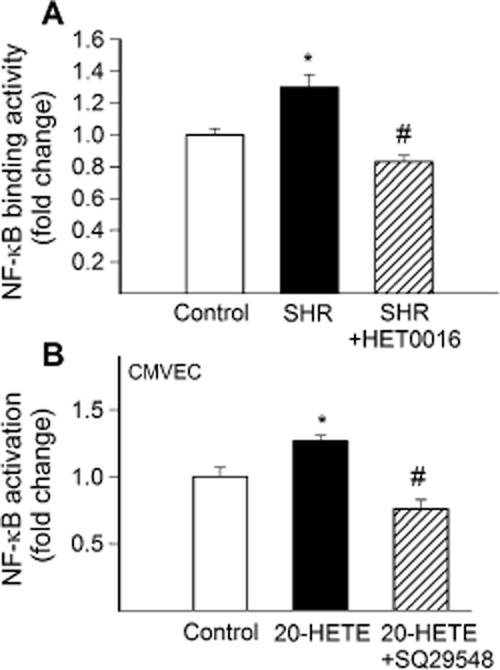
In vivo treatment with HET0016 attenuates vascular NF-κB activation in SHR. (A) shows NF-κB binding activity in nuclear extracts from carotid arteries isolated from control WKY rats, SHRs and SHRs treated with HET0016 (SHR + HET0016; 10 mg·kg−1·per day, i.p., n = 6 for each data point). (B) effect of 20-HETE (10−7 mol·L−1) on NF-κB activation measured by dual-luciferase reporter assay in cultured rat cerebromicrovascular endothelial cells (CMVEC) in the absence and presence of TP receptor blocker SQ 29548 (10−6 mol·L−1). Data are mean ± SEM *P < 0.05 versus control; #P < 0.05 versus untreated.
To further substantiate the role of increased levels of 20-HETE in induction of vascular NF-κB activation, we treated CMVECs with exogenous 20-HETE (10−7 mol·L−1, for 24 h). We found that administration of 20-HETE significantly increased transcriptional activity of NF-κB in CMVECs, and this effect could be inhibited by the TP receptor blocker SQ29,548 (Figure 5B). Treatment of arterial segments maintained in organoid culture with exogenous 20-HETE (10−6 mol·L−1, for 18 h) also increased the mRNA expression of the inflammatory markers Icam1 (by ∼84%), and Mcp1 (by ∼195%) in the vascular wall.
In vivo treatment with HET0016 attenuates vascular inflammation in SHRs
We found that hypertension-induced oxidative stress and NF-κB activation are associated with significantly increased mRNA expression of the inflammatory markers TNF-α (Figure 6A), IL-1β (Figure 6B) and IL-6 (Figure 6C) in MCAs of SHRs. In contrast, treatment with HET0016 in SHRs attenuated cerebrovascular inflammation induced by hypertension, down-regulating the expression of TNF-α (Figure 6A), IL-1β (Figure 6B) and IL-6 (Figure 6C). Expression of iNOS also tended to decrease in HET0016-treated SHRs, yet, the difference did not reach statistical significance (Figure 6D). In WKYs, treatment with HET0016 did not significantly alter vascular expression of inflammatory markers (not shown).
Figure 6.
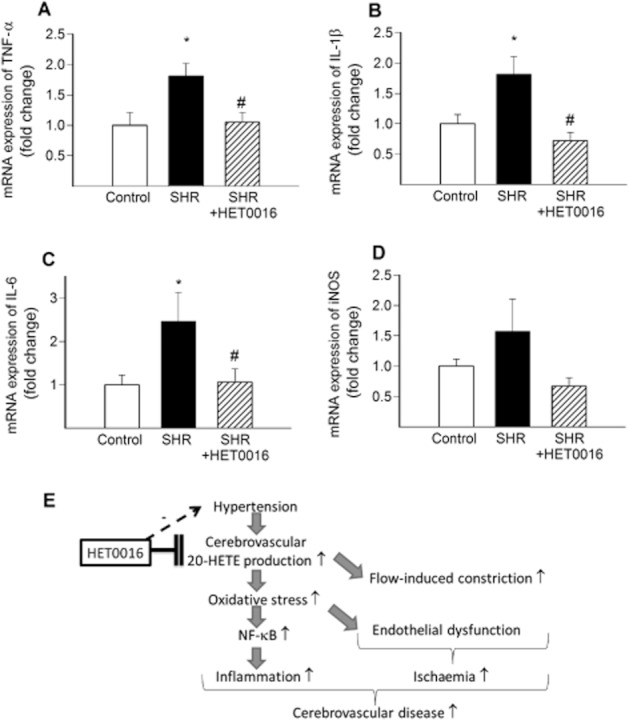
In vivo treatment with HET0016 attenuates inflammation in MCA of SHR. Quantitative real-time RT-PCR data showing mRNA expression of TNF-α (A), IL-1β (B), IL-6 (C) and iNOS (D) in MCAs isolated from control WKY rats, SHRs and SHRs treated with HET0016 (SHR + HET0016; 10 mg·kg−1·per 5 days, i.p., n = 6 for each data point). Data are mean ± SEM *P < 0.05 versus control; #P < 0.05 versus untreated. (E) Scheme depicting the proposed mechanisms underlying the cerebrovascular protective effects of HET0016 in hypertension. Accordingly, in the wall of cerebral arteries, hypertension increases 20-HETE production, which results in increased flow-induced constriction and promotes endothelial dysfunction via enhancing vascular oxidative stress. Increased oxidative stress also promotes NF-κB activation and vascular inflammation, exacerbating the development of cerebrovascular diseases. HET0016 inhibits 20-HETE synthesis in the vascular wall and thereby confers anti-oxidative and anti-inflammatory cerebrovascular protective effects in hypertension. HET0016 also decreases blood pressure likely via its renal action, which is also likely to contribute indirectly to its cerebrovascular protective effects.
Discussion and conclusions
20-HETE, generated locally in the wall of arteries branching off the circle of Willis, is a key mediator involved in the regulation of cerebrovascular resistance (Gebremedhin et al., 2000). Recently, we demonstrated that 20-HETE mediates flow-induced constriction of cerebral arteries via TP receptors, a newly described mechanism involved in the autoregulation of cerebral blood flow (Toth et al., 2011; Koller and Toth, 2012). Here we demonstrate for the first time that flow-induced, 20-HETE-mediated constriction of MCAs is substantially augmented in hypertension (Figure 2A), consistent with an up-regulation of 20-HETE synthesizing cytochrome P450 Ω-hydroxylases system in various rodent models of hypertension, including the stroke-prone SHR (Dunn et al., 2008), SHR (a trend was reported by Dunn et al. (2008), the difference fell short of significance due to the large variation in the data), Sprague-Dawley rats with 5alpha-dihydrotestosterone-induced hypertension (Singh et al., 2007) and angiotensin II-infused mice (P. Toth and Z. Ungvari, unpubl. data, 2012). The findings that administration of SQ29548 (Figure 2B) abolished flow-induced vasomotor responses is consistent with the view that TP receptors are involved in 20-HETE-induced signalling in the vascular smooth muscle cells. Indeed, SQ29548 also abolishes vasoconstriction induced by exogenous 20-HETE (P. Toth and Z. Ungvari, unpubl. data, 2012). Because we found that the specific thromboxane A2 analogue U46619 elicits comparable vasoconstriction in vessels isolated from WKY rats and SHRs (Figure 2B), it is unlikely that hypertension alters TP receptor number, sensitivity or TP receptor-mediated signalling in vascular smooth muscle cells. Recently, flow-induced constriction was found in carotid arteries, as well (Craig and Martin, 2012). Because vasomotor function of carotid arteries also affects cerebral blood flow their response in hypertension should be clarified in the future.
In addition to promoting flow-induced vasoconstriction, increased 20-HETE production in hypertension also exacerbates endothelial dysfunction. Accordingly, we found that HET0016 treatment significantly improves ACh- and ATP-induced dilations in SHRs suggesting that elevated production of 20-HETE contributes to endothelial dysfunction in the cerebral vasculature in hypertension (Singh et al., 2007; Cheng et al., 2008; 2010; Dunn et al., 2008). Increased flow-induced constriction and endothelial vasodilator dysfunction are likely to limit cerebral flow reserve, rendering the organism vulnerable to regional cerebral ischemia by increasing vascular resistance. The concept that increased 20-HETE production may contribute to cerebral ischaemic damage is supported by the findings that treatment with HET0016 decreases infarct volume and improves cerebral blood flow after MCA occlusion (Poloyac et al., 2006; Dunn et al., 2008; Renic et al., 2009). The mechanisms by which increased production of 20-HETE in the vascular wall impair endothelial function are likely multifaceted and likely involve increased oxidative stress (see below) and perhaps uncoupling of eNOS (Cheng et al., 2010) and/or alterations in AKT and AMPK signalling pathways (Inoue et al., 2009).
There is overwhelming evidence that in hypertension, increased oxidative stress contributes to the development of endothelial dysfunction in virtually all vascular beds studied, including the cerebral circulation (Heistad et al., 1990; Huang and Koller, 1996; Rajagopalan et al., 1996; Dunn et al., 2008). Important in that regard is that inhibition of 20-HETE significantly reduces hypertension-induced cerebrovascular oxidative stress (Dunn et al., 2008; Figures 3 and 4), an effect, which is likely contributing to the improved cerebrovascular endothelial function in HET0016-treated SHRs. The findings that administration of exogenous 20-HETE in vitro results in NADPH oxidase activation and increased ROS production (Figure 6; Medhora et al., 2008; Zeng et al., 2010; Jacobs et al., 2012) support the concept that increased levels of 20-HETE in the vascular wall are causally related to cerebrovascular oxidative stress in hypertension and that HET0016 exerts anti-oxidative effects directly in the cerebrovasculature. Furthermore, increased vascular 20-HETE production induced by adenoviral overexpression of CYP4A2 in rats also results in up-regulation of NADPH oxidase, increased ROS production and endothelial dysfunction (Wang et al., 2006). 20-HETE binds to the TP receptor. In that regard, it is interesting that treatment with the TP antagonist terutroban also prevents hypertension-induced vascular oxidative stress and endothelial dysfunction in rats, an effect that is independent of changes in blood pressure (Gelosa et al., 2010; 2011).
In addition to its direct vascular effect, HET0016 is also likely to confer cerebrovascular protection in hypertension indirectly, by lowering blood pressure. Accordingly, inhibition of 20-HETE production was shown to decrease blood pressure in various animal models of hypertension (Sacerdoti et al., 1989; Wang et al., 2001). Treatment of SHRs with HET0016 also results in a significant decrease in blood pressure (Figure 1B). The underlying mechanisms of this antihypertensive effect are likely multifaceted. 20-HETE is known to participate in the tubuloglomerular feedback (Zou et al., 1994) and in the pressure natriuresis mechanisms in the kidneys by regulating Na+ transport (Williams et al., 2007). It is likely that inhibition of renal 20-HETE synthesis by HET0016 contributes significantly to its antihypertensive effect. It is logical to assume that HET0016-induced decreases in blood pressure also contribute to the attenuation of cerebrovascular oxidative stress in SHRs. In support of this concept, previous studies demonstrated that acute exposure of isolated peripheral arteries to high intraluminal pressure increases vascular ROS production by activating NADPH oxidases (Huang et al., 1998; Ungvari et al., 2003). We have recently confirmed that acute exposure of MCAs to high pressure also increases ROS production in the vascular wall (Toth and Ungvari, manuscript in preparation, 2012). It is possible that the effects of increased 20-HETE levels and high pressure-related increases in wall tension converge on a common signalling pathway for NADPH oxidase activation. There is also a large body of evidence linking hypertension and activation of the renin-angiotensin system to vascular oxidative stress (Rajagopalan et al., 1996; Ungvari et al., 2004; Bagi et al., 2011). In that regard, it is significant that 20-HETE signalling and the renin-angiotensin II system crosstalk (Sodhi et al., 2010; Cheng et al., 2012) and that angiotensin II enhances the production of 20-HETE (Alonso-Galicia et al., 2002). Taken together, HET0016 likely exerts direct anti-oxidative effects in the cerebrovasculature by inhibiting 20-HETE synthesis in the vascular wall and this effect is potentiated by its indirect, anti-hypertensive effect. To further characterize the direct vascular effects of HET0016, future studies should investigate its cerebrovascular effects in an animal model of hypertension, in which blood pressure can be kept constant (e.g. mice with controlled angiotensin II administration).
Previous studies indicate that hypertension-induced oxidative stress promotes chronic low-grade inflammation in the vasculature, at least in part, by activating the redox sensitive transcription factor NF-κB (Ando et al., 2004; Csiszar et al., 2005; Zhou et al., 2005; Riou et al., 2007). In line with these findings, we found that hypertension-induced vascular ROS production is associated with an up-regulation of NF-κB target genes and other inflammatory mediators and increased NF-κB activation in the vascular wall (Figures 5 and 6). These pro-inflammatory alterations are believed to have a pathogenic role in development of atherosclerosis, ischaemic cerebrovascular diseases and vascular cognitive impairment associated with hypertension (Barry, 1985; Mies et al., 1999; O'Sullivan et al., 2002; Amenta et al., 2003; Di Napoli and Papa, 2003; Beason-Held et al., 2007; Iadecola and Davisson, 2008). Here we show for the first time that hypertension-induced vascular inflammatory alterations are attenuated by HET0016 (Figures 5 and 6). Because administration of exogenous 20-HETE results in significant NF-κB activation in cultured cerebromicrovascular endothelial cells in a TP receptor-dependent manner (Figure 5B), it is likely that disruption of the 20-HETE/TP receptor/NF-κB signalling pathway in the vascular wall is responsible for the observed anti-inflammatory effects of HET0016 treatment. Previous findings also support this concept, showing that 20-HETE treatment stimulates NF-κB activation and the production of inflammatory cytokines in cultured human umbilical vein endothelial cells (Ishizuka et al., 2008). Furthermore, treatment of hypertensive rats with a TP receptor antagonist also attenuates expression of pro-inflammatory cytokines, including IL-1β and improves endothelial function in hypertensive rats (Gelosa et al., 2010; 2011).
In conclusion, our studies provide evidence that in hypertension treatment with HET0016 confers cerebrovascular protective effects, by attenuating cerebrovascular oxidative stress and inflammation and improving vasomotor function (Figure 6E). Further studies are warranted to determine whether pharmacological interventions that disrupt 20-HETE signalling prevent development of cerebrovascular diseases and vascular cognitive impairment known to be associated with hypertension.
Acknowledgments
We gratefully thank Dr Michal L Schwartzman (Department of Pharmacology, New York Medical College) for generously providing us with HET0016. This work was supported by grants from the American Heart Association (to PT, AC, ZU and AK), the Hungarian National Science Research Fund OTKA (K71591, K67984 and MHT-2011 to AK), the Hungarian National Development Agency (TÁMOP/SROP-4.2.1/b-10/2/KONV-2010-0012 and SROP-4.2.2.A-11/1/KONV-2012-0024 to ZU and AK), the Oklahoma Center for the Advancement of Science and Technology (to AC, ZU, WES), the NIH (P01 HL-43023 to AK, AG031085 to AC; AT006526 to ZU; AG038747, NS056218 and P01 AG11370 to WES) and the Ellison Medical Foundation (to WES). The authors would like to express their gratitude for the support of the Donald W. Reynolds Foundation, which funds aging research at the University of Oklahoma Health Sciences Center under its Aging and Quality of Life Program.
Glossary
- 20-HETE
20-hydroxy-5,8,11,14-eicosatetraenoic acid
- CYP 450
cytochrome P450
- DHE
dihydroethidium
- HET0016
N-hydroxy-N′-(4-butyl-2-methylphenyl)-formamidine
- MCA
middle cerebral artery
- ROS
reactive oxygen species
- SHR
spontaneously hypertensive rat
- WKY
Wistar-Kyoto rat
Conflict of interest
No conflict of interest to disclose.
References
- Aiyagari V, Gorelick PB. Management of blood pressure for acute and recurrent stroke. Stroke. 2009;40:2251–2256. doi: 10.1161/STROKEAHA.108.531574. [DOI] [PubMed] [Google Scholar]
- Alonso-Galicia M, Maier KG, Greene AS, Cowley AW, Jr, Roman RJ. Role of 20-hydroxyeicosatetraenoic acid in the renal and vasoconstrictor actions of angiotensin II. Am J Physiol Regul Integr Comp Physiol. 2002;283:R60–R68. doi: 10.1152/ajpregu.00664.2001. [DOI] [PubMed] [Google Scholar]
- Amenta F, Di Tullio MA, Tomassoni D. Arterial hypertension and brain damage – evidence from animal models (review) Clin Exp Hypertens. 2003;25:359–380. doi: 10.1081/ceh-120023545. [DOI] [PubMed] [Google Scholar]
- Ando H, Zhou J, Macova M, Imboden H, Saavedra JM. Angiotensin II AT1 receptor blockade reverses pathological hypertrophy and inflammation in brain microvessels of spontaneously hypertensive rats. Stroke. 2004;35:1726–1731. doi: 10.1161/01.STR.0000129788.26346.18. [DOI] [PubMed] [Google Scholar]
- Bagi Z, Feher A, Cassuto J, Akula K, Labinskyy N, Kaley G, et al. Increased availability of angiotensin AT 1 receptors leads to sustained arterial constriction to angiotensin II in diabetes – role for Rho-kinase activation. Br J Pharmacol. 2011;163:1059–1068. doi: 10.1111/j.1476-5381.2011.01307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey-Downs LC, Mitschelen M, Sosnowska D, Toth P, Pinto JT, Ballabh P, et al. Liver-specific knockdown of IGF-1 decreases vascular oxidative stress resistance by impairing the Nrf2-dependent antioxidant response: a novel model of vascular aging. J Gerontol A Biol Sci Med Sci. 2012;67:313–329. doi: 10.1093/gerona/glr164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry DI. Cerebral blood flow in hypertension. J Cardiovasc Pharmacol. 1985;7(Suppl. 2):S94–S98. doi: 10.1097/00005344-198507002-00018. [DOI] [PubMed] [Google Scholar]
- Beason-Held LL, Moghekar A, Zonderman AB, Kraut MA, Resnick SM. Longitudinal changes in cerebral blood flow in the older hypertensive brain. Stroke. 2007;38:1766–1773. doi: 10.1161/STROKEAHA.106.477109. [DOI] [PubMed] [Google Scholar]
- Cheng J, Ou JS, Singh H, Falck JR, Narsimhaswamy D, Pritchard KA, Jr, et al. 20-hydroxyeicosatetraenoic acid causes endothelial dysfunction via eNOS uncoupling. Am J Physiol Heart Circ Physiol. 2008;294:H1018–H1026. doi: 10.1152/ajpheart.01172.2007. [DOI] [PubMed] [Google Scholar]
- Cheng J, Wu CC, Gotlinger KH, Zhang F, Falck JR, Narsimhaswamy D, et al. 20-hydroxy-5,8,11,14-eicosatetraenoic acid mediates endothelial dysfunction via IkappaB kinase-dependent endothelial nitric-oxide synthase uncoupling. J Pharmacol Exp Ther. 2010;332:57–65. doi: 10.1124/jpet.109.159863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J, Garcia V, Ding Y, Wu CC, Thakar K, Falck JR, et al. Induction of angiotensin-converting enzyme and activation of the renin-angiotensin system contribute to 20-hydroxyeicosatetraenoic acid-mediated endothelial dysfunction. Arterioscler Thromb Vasc Biol. 2012;32:1917–1924. doi: 10.1161/ATVBAHA.112.248344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig J, Martin W. Dominance of flow-mediated constriction over flow-mediated dilatation in the rat carotid artery. Br J Pharmacol. 2012;167:527–536. doi: 10.1111/j.1476-5381.2012.02006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csiszar A, Ungvari Z, Koller A, Edwards JG, Kaley G. Proinflammatory phenotype of coronary arteries promotes endothelial apoptosis in aging. Physiol Genomics. 2004;17:21–30. doi: 10.1152/physiolgenomics.00136.2003. [DOI] [PubMed] [Google Scholar]
- Csiszar A, Smith KE, Koller A, Kaley G, Edwards JG, Ungvari Z. Regulation of bone morphogenetic protein-2 expression in endothelial cells: role of nuclear factor-kappaB activation by tumor necrosis factor-alpha, H2O2, and high intravascular pressure. Circulation. 2005;111:2364–2372. doi: 10.1161/01.CIR.0000164201.40634.1D. [DOI] [PubMed] [Google Scholar]
- Csiszar A, Ahmad M, Smith KE, Labinskyy N, Gao Q, Kaley G, et al. Bone morphogenetic protein-2 induces proinflammatory endothelial phenotype. Am J Pathol. 2006a;168:629–638. doi: 10.2353/ajpath.2006.050284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csiszar A, Smith K, Labinskyy N, Orosz Z, Rivera A, Ungvari Z. Resveratrol attenuates TNF-{alpha}-induced activation of coronary arterial endothelial cells: role of NF-{kappa}B inhibition. Am J Physiol. 2006b;291:H1694–H1699. doi: 10.1152/ajpheart.00340.2006. [DOI] [PubMed] [Google Scholar]
- Csiszar A, Labinskyy N, Zhao X, Hu F, Serpillon S, Huang Z, et al. Vascular superoxide and hydrogen peroxide production and oxidative stress resistance in two closely related rodent species with disparate longevity. Aging Cell. 2007;6:783–797. doi: 10.1111/j.1474-9726.2007.00339.x. [DOI] [PubMed] [Google Scholar]
- Di Napoli M, Papa F. Association between blood pressure and C-reactive protein levels in acute ischemic stroke. Hypertension. 2003;42:1117–1123. doi: 10.1161/01.HYP.0000100669.00771.6E. [DOI] [PubMed] [Google Scholar]
- Dunn KM, Renic M, Flasch AK, Harder DR, Falck J, Roman RJ. Elevated production of 20-HETE in the cerebral vasculature contributes to severity of ischemic stroke and oxidative stress in spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol. 2008;295:H2455–H2465. doi: 10.1152/ajpheart.00512.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebremedhin D, Lange AR, Lowry TF, Taheri MR, Birks EK, Hudetz AG, et al. Production of 20-HETE and its role in autoregulation of cerebral blood flow. Circ Res. 2000;87:60–65. doi: 10.1161/01.res.87.1.60. [DOI] [PubMed] [Google Scholar]
- Gelosa P, Ballerio R, Banfi C, Nobili E, Gianella A, Pignieri A, et al. Terutroban, a thromboxane/prostaglandin endoperoxide receptor antagonist, increases survival in stroke-prone rats by preventing systemic inflammation and endothelial dysfunction: comparison with aspirin and rosuvastatin. J Pharmacol Exp Ther. 2010;334:199–205. doi: 10.1124/jpet.110.165787. [DOI] [PubMed] [Google Scholar]
- Gelosa P, Sevin G, Pignieri A, Budelli S, Castiglioni L, Blanc-Guillemaud V, et al. Terutroban, a thromboxane/prostaglandin endoperoxide receptor antagonist, prevents hypertensive vascular hypertrophy and fibrosis. Am J Physiol Heart Circ Physiol. 2011;300:H762–H768. doi: 10.1152/ajpheart.00880.2010. [DOI] [PubMed] [Google Scholar]
- Gorelick PB, Scuteri A, Black SE, Decarli C, Greenberg SM, Iadecola C, et al. Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2011;42:2672–2713. doi: 10.1161/STR.0b013e3182299496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heistad DD, Mayhan WG, Coyle P, Baumbach GL. Impaired dilatation of cerebral arterioles in chronic hypertension. Blood Vessels. 1990;27:258–262. doi: 10.1159/000158817. [DOI] [PubMed] [Google Scholar]
- Huang A, Koller A. Both nitric oxide and prostaglandin-mediated responses are impaired in skeletal muscle arterioles of hypertensive rats. J Hypertens. 1996;14:887–895. doi: 10.1097/00004872-199607000-00012. [DOI] [PubMed] [Google Scholar]
- Huang A, Sun D, Kaley G, Koller A. Superoxide released to high intra-arteriolar pressure reduces nitric oxide-mediated shear stress- and agonist-induced dilations. Circ Res. 1998;83:960–965. doi: 10.1161/01.res.83.9.960. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Davisson RL. Hypertension and cerebrovascular dysfunction. Cell Metab. 2008;7:476–484. doi: 10.1016/j.cmet.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Sodhi K, Puri N, Gotlinger KH, Cao J, Rezzani R, et al. Endothelial-specific CYP4A2 overexpression leads to renal injury and hypertension via increased production of 20-HETE. Am J Physiol Renal Physiol. 2009;297:F875–F884. doi: 10.1152/ajprenal.00364.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizuka T, Cheng J, Singh H, Vitto MD, Manthati VL, Falck JR, et al. 20-Hydroxyeicosatetraenoic acid stimulates nuclear factor-kappaB activation and the production of inflammatory cytokines in human endothelial cells. J Pharmacol Exp Ther. 2008;324:103–110. doi: 10.1124/jpet.107.130336. [DOI] [PubMed] [Google Scholar]
- Jacobs ER, Bodiga S, Ali I, Falck AM, Falck JR, Medhora M, et al. Tissue protection and endothelial cell signaling by 20-HETE analogs in intact ex vivo lung slices. Exp Cell Res. 2012;318:2143–2152. doi: 10.1016/j.yexcr.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koller A, Toth P. Contribution of flow-dependent vasomotor mechanisms to the autoregulation of cerebral blood flow. J Vasc Res. 2012;49:375–389. doi: 10.1159/000338747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medhora M, Chen Y, Gruenloh S, Harland D, Bodiga S, Zielonka J, et al. 20-HETE increases superoxide production and activates NAPDH oxidase in pulmonary artery endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2008;294:L902–L911. doi: 10.1152/ajplung.00278.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mies G, Hermann D, Ganten U, Hossmann KA. Hemodynamics and metabolism in stroke-prone spontaneously hypertensive rats before manifestation of brain infarcts. J Cereb Blood Flow Metab. 1999;19:1238–1246. doi: 10.1097/00004647-199911000-00008. [DOI] [PubMed] [Google Scholar]
- Miyata N, Taniguchi K, Seki T, Ishimoto T, Sato-Watanabe M, Yasuda Y, et al. HET0016, a potent and selective inhibitor of 20-HETE synthesizing enzyme. Br J Pharmacol. 2001;133:325–329. doi: 10.1038/sj.bjp.0704101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Sullivan M, Lythgoe DJ, Pereira AC, Summers PE, Jarosz JM, Williams SC, et al. Patterns of cerebral blood flow reduction in patients with ischemic leukoaraiosis. Neurology. 2002;59:321–326. doi: 10.1212/wnl.59.3.321. [DOI] [PubMed] [Google Scholar]
- Poloyac SM, Zhang Y, Bies RR, Kochanek PM, Graham SH. Protective effect of the 20-HETE inhibitor HET0016 on brain damage after temporary focal ischemia. J Cereb Blood Flow Metab. 2006;26:1551–1561. doi: 10.1038/sj.jcbfm.9600309. [DOI] [PubMed] [Google Scholar]
- Rajagopalan S, Kurz S, Munzel T, Tarpey M, Freeman BA, Griendling KK, et al. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest. 1996;97:1916–1923. doi: 10.1172/JCI118623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renic M, Klaus JA, Omura T, Kawashima N, Onishi M, Miyata N, et al. Effect of 20-HETE inhibition on infarct volume and cerebral blood flow after transient middle cerebral artery occlusion. J Cereb Blood Flow Metab. 2009;29:629–639. doi: 10.1038/jcbfm.2008.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riou S, Mees B, Esposito B, Merval R, Vilar J, Stengel D, et al. High pressure promotes monocyte adhesion to the vascular wall. Circ Res. 2007;100:1226–1233. doi: 10.1161/01.RES.0000265231.59354.2c. [DOI] [PubMed] [Google Scholar]
- Sacerdoti D, Escalante B, Abraham NG, McGiff JC, Levere RD, Schwartzman ML. Treatment with tin prevents the development of hypertension in spontaneously hypertensive rats. Science. 1989;243:388–390. doi: 10.1126/science.2492116. [DOI] [PubMed] [Google Scholar]
- Singh H, Cheng J, Deng H, Kemp R, Ishizuka T, Nasjletti A, et al. Vascular cytochrome P450 4A expression and 20-hydroxyeicosatetraenoic acid synthesis contribute to endothelial dysfunction in androgen-induced hypertension. Hypertension. 2007;50:123–129. doi: 10.1161/HYPERTENSIONAHA.107.089599. [DOI] [PubMed] [Google Scholar]
- Sodhi K, Wu CC, Cheng J, Gotlinger K, Inoue K, Goli M, et al. CYP4A2-induced hypertension is 20-hydroxyeicosatetraenoic acid- and angiotensin II-dependent. Hypertension. 2010;56:871–878. doi: 10.1161/HYPERTENSIONAHA.110.154559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth P, Rozsa B, Springo Z, Doczi T, Koller A. Isolated human and rat cerebral arteries constrict to increases in flow: role of 20-HETE and TP receptors. J Cereb Blood Flow Metab. 2011;31:2096–2105. doi: 10.1038/jcbfm.2011.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungvari Z, Pacher P, Kecskemeti V, Koller A. Fluoxetine dilates isolated small cerebral arteries of rats and attenuates constrictions to serotonin, norepinephrine, and a voltage- dependent Ca(2+) channel opener. Stroke. 1999;30:1949–1954. doi: 10.1161/01.str.30.9.1949. [DOI] [PubMed] [Google Scholar]
- Ungvari Z, Csiszar A, Huang A, Kaminski PM, Wolin MS, Koller A. High pressure induces superoxide production in isolated arteries via protein kinase C-dependent activation of NAD(P)H oxidase. Circulation. 2003;108:1253–1258. doi: 10.1161/01.CIR.0000079165.84309.4D. [DOI] [PubMed] [Google Scholar]
- Ungvari Z, Csiszar A, Kaminski PM, Wolin MS, Koller A. Chronic high pressure-induced arterial oxidative stress: involvement of protein kinase C-dependent NAD(P)H oxidase and local renin-angiotensin system. Am J Pathol. 2004;165:219–226. doi: 10.1016/S0002-9440(10)63290-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JS, Singh H, Zhang F, Ishizuka T, Deng H, Kemp R, et al. Endothelial dysfunction and hypertension in rats transduced with CYP4A2 adenovirus. Circ Res. 2006;98:962–969. doi: 10.1161/01.RES.0000217283.98806.a6. [DOI] [PubMed] [Google Scholar]
- Wang MH, Zhang F, Marji J, Zand BA, Nasjletti A, Laniado-Schwartzman M. CYP4A1 antisense oligonucleotide reduces mesenteric vascular reactivity and blood pressure in SHR. Am J Physiol Regul Integr Comp Physiol. 2001;280:R255–R261. doi: 10.1152/ajpregu.2001.280.1.R255. [DOI] [PubMed] [Google Scholar]
- Williams JM, Sarkis A, Lopez B, Ryan RP, Flasch AK, Roman RJ. Elevations in renal interstitial hydrostatic pressure and 20-hydroxyeicosatetraenoic acid contribute to pressure natriuresis. Hypertension. 2007;49:687–694. doi: 10.1161/01.HYP.0000255753.89363.47. [DOI] [PubMed] [Google Scholar]
- Wu CC, Cheng J, Zhang FF, Gotlinger KH, Kelkar M, Zhang Y, et al. Androgen-dependent hypertension is mediated by 20-hydroxy-5,8,11,14-eicosatetraenoic acid-induced vascular dysfunction: role of inhibitor of kappaB kinase. Hypertension. 2011;57:788–794. doi: 10.1161/HYPERTENSIONAHA.110.161570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Q, Han Y, Bao Y, Li W, Li X, Shen X, et al. 20-HETE increases NADPH oxidase-derived ROS production and stimulates the L-type Ca2+ channel via a PKC-dependent mechanism in cardiomyocytes. Am J Physiol Heart Circ Physiol. 2010;299:H1109–H1117. doi: 10.1152/ajpheart.00067.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Ando H, Macova M, Dou J, Saavedra JM. Angiotensin II AT1 receptor blockade abolishes brain microvascular inflammation and heat shock protein responses in hypertensive rats. J Cereb Blood Flow Metab. 2005;25:878–886. doi: 10.1038/sj.jcbfm.9600082. [DOI] [PubMed] [Google Scholar]
- Zou AP, Imig JD, Ortiz de Montellano PR, Sui Z, Falck JR, Roman RJ. Effect of P-450 omega-hydroxylase metabolites of arachidonic acid on tubuloglomerular feedback. Am J Physiol. 1994;266(6 Pt 2):F934–F941. doi: 10.1152/ajprenal.1994.266.6.F934. [DOI] [PubMed] [Google Scholar]