

Abstract
Background and Purpose
The dual role of the constitutive androstane receptor (CAR) as both a xenosensor and a regulator of endogenous energy metabolism (lipogenesis and gluconeogenesis) has recently gained acceptance. Here, we investigated the effects of 4-[(4R,6R)-4,6-diphenyl-1,3-dioxan-2-yl]-N,N-dimethylaniline (transpDMA), an effective CAR activator, on the gluconeogenic genes phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6Pase) in rat livers.
Experimental Approach
The effects of transpDMA were investigated in normal and high-fat diet-fed Wistar rats using real-time PCR, Western blotting, chromatin immunoprecipitation assays (ChIP), glucose tolerance test and insulin tolerance test.
Key Results
The expression of the gluconeogenic enzymes PEPCK and G6Pase was repressed by transpDMA treatment under fasting conditions. Long-term CAR activation by transpDMA significantly reduced fasting blood glucose levels and improved glucose homeostasis and insulin sensitivity in high-fat diet-fed rats. The metabolic benefits of CAR activation by transpDMA may have resulted from the inhibition of hepatic gluconeogenic genes. ChIP assays demonstrated that transpDMA prevented the binding of forkhead box O1 (FOXO1) to insulin response sequences in the PEPCK and G6Pase gene promoters in rat livers. Moreover, transpDMA-activated CAR inhibited hepatocyte nuclear factor-4α (HNF4α) transactivation by competing with HNF4α for binding to the specific binding element (DR1-site) in the gluconeogenic gene promoters.
Conclusions and Implications
Our results provide evidence to support the conclusion that transpDMA inhibits the gluconeogenic genes PEPCK and G6Pase through suppression of HNF4α and FOXO1 transcriptional activity.
Keywords: CAR, gluconeogenesis, FOXO1, HNF4α, PEPCK, G6Pase
Introduction
The liver is the major site of endogenous glucose production through either gluconeogenesis or glycogenolysis. Gluconeogenesis is largely responsible for the overproduction of glucose in patients with type 2 diabetes mellitus (Wajngot et al., 2001). Thus, inhibition of the gluconeogenesis pathway may be part of a strategy against the hyperglycaemia associated with type 2 diabetes mellitus. Phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6Pase) are key gluconeogenic enzymes. These enzymes are regulated in the liver at the transcriptional level and are highly activated during fasting and suppressed in the fed state and by insulin (Ropelle et al., 2009). The PEPCK and G6Pase promoters have been extensively studied and are known to be regulated by cross-talk between two transcriptional factors, specifically forkhead box O1 (FOXO1, also known as FKHR) and hepatocyte nuclear factor-4α (HNF4α, NR2A1). HNF4α is central to glucose metabolism because it is a key activator of basal expression of the PEPCK and G6Pase genes (Odom et al., 2004). Both of these genes contain functional HNF4α-binding sites (DR1 motif) in their promoters. FOXO1 also plays an important role in glucose homeostasis (Barthel et al., 2005). The unphosphorylated form of FOXO1 localizes to the nucleus and interacts with insulin response sequences (IRS) in the promoters of gluconeogenic genes, thereby activating their transcription. Insulin suppresses FOXO1 transcriptional activity via Akt phosphorylation-mediated protein degradation (Matsuzaki et al., 2003).
The constitutive androstane receptor (CAR, NR1I3; receptor nomenclature follows Alexander et al., 2011), expressed mainly in the liver, plays a central role in detoxification processes by regulating the expression of a set of genes involved in metabolism of xenobiotics and drugs (Kachaylo et al., 2011). CAR also regulates other physiologically important enzymes, and the crosstalk between hepatic gluconeogenesis and CAR has gained increased attention. PEPCK and G6Pase genes are repressed in response to CAR activators, and this repression is CAR dependent (Ueda et al., 2002). The CAR-mediated repression of these genes seems to involve several molecular mechanisms. CAR inhibits HNF4α activity by competing with HNF4α for binding to the DR1 motif and the common coactivators PPARγ coactivator 1α and glucocorticoid receptor-interacting protein 1 (Miao et al., 2006). CAR can also bind to FOXO1 and suppress its transcriptional activity by preventing FOXO1 binding to the IRS in the PEPCK and G6Pase promoters (Kodama et al., 2004). The in vivo significance of CAR-mediated suppression of gluconeogenesis was recently shown. CAR activation suppresses glucose production, stimulates glucose uptake, improves glucose tolerance and insulin sensitivity, and prevents obesity in ob/ob mice and high-fat diet-fed wild-type mice (Dong et al., 2009; Gao et al., 2009). These findings may establish CAR as a potential therapeutic target for metabolic diseases, including obesity and type 2 diabetes mellitus. Because CAR is a potential therapeutic target for metabolic diseases, a search for selective CAR modulators may provide novel therapeutic tools for the management of metabolic disorders.
In our previous studies, the effects of the cis isoform of 2,4,6-triphenyldioxane-1,3 (cisTPD) were investigated. cisTPD is a highly effective activator of CAR and its target genes in rat liver, but it is not able to activate CAR in mice (Pustylnyak et al., 2011a; 2011b). We have demonstrated that a single 10 mg kg−1 body weight i.p. injection of cisTPD evoked a significant decrease in the expression of the key hepatic gluconeogenic genes PEPCK and G6Pase through a diminished association of HNF4α and FOXO1 with their specific binding sites in the gene promoters (Kachaylo et al., 2012a). Moreover, long-term cisTPD treatment reduced fasting blood glucose levels in both normal diet- and high-fat diet-fed rats and improved glucose tolerance in high-fat diet-fed rats (Kachaylo et al., 2012b). The aim of the present study was to examine the effects and mechanism of action of the cisTPD analogue 4-[(4R,6R)-4,6-diphenyl-1,3-dioxan-2-yl]-N,N-dimethylaniline (transpDMA) on the hepatic gluconeogenic CAR-regulated genes PEPCK and G6Pase in rats. Our previous results have shown that cisTPD and transpDMA have similar potency on the CAR target, the 2B subfamily of cytochrome P450 (CYP2B) in rat liver (Pustylnyak et al., 2011a).
Methods
Experimental animals
All animal care and experimental procedures were approved by the Animal Care and Use Committee of the Institute of Molecular Biology and Biophysics SB RAMS. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 50 animals were used in the experiments described here. Male Wistar rats (150–200 g) were supplied by the Institute of Clinical Immunology SB RAMS (Novosibirsk, Russia). Animals were acclimated for 1 week and allowed free access to food and water. Animals were treated i.p. with cisTPD or transpDMA (a single injection of 5 or 10 mg kg−1 body weight in corn oil), fasted 20 h later and decapitated 18 h after fasting began. Control animals received only an equal volume of corn oil. Three rats were used in each treatment group.
Rats were randomly divided into six groups of five rats each to examine the long-term effects of cisTPD and transpDMA on glucose metabolism and homeostasis. Control group rats were fed a normal diet (5% fat of kJ intake) for 8 weeks. The high-fat diet (HFD) group rats received a HFD (45% fat of kJ intake) for 8 weeks. The cisTPD group rats were fed a normal diet and injected weekly i.p. with a single dose of 10 mg kg−1 body weight of cisTPD in corn oil for 8 weeks. The transpDMA group rats were fed a normal diet and injected i.p. with 10 mg kg−1 body weight of transpDMA in corn oil for 8 weeks as a single weekly dose. The HFD + cisTPD rats received HFD for 8 weeks and were i.p. injected with 10 mg kg−1 body weight of cisTPD in corn oil as a single weekly dose. This dosage regimen significantly decreased gluconeogenic gene expression and improved glucose tolerance (Kachaylo et al., 2012b). The HFD + transpDMA rats received HFD for 8 weeks and were i.p. injected with 10 mg kg−1 body weight of transpDMA in corn oil as a single weekly dose (similar to cisTPD). After 8 weeks, all groups of rats were fasted for 16 h, and a fasting blood glucose analysis was performed using OneTouch Select glucometer (LifeScan, Inc., Milpitas, CA, USA).
Glucose tolerance test (GTT) and insulin tolerance test (ITT)
Rats were fasted for 16 h before receiving an i.p. injection of d-glucose at 2 g kg−1 body weight or an i.p. injection of human regular insulin at 0.75 units kg−1 body weight. Blood glucose was measured by tail bleed at 0, 20, 40, 60 and 120 min after the glucose load with OneTouch Select glucometer (LifeScan, Inc.).
cDNA synthesis and real-time PCR
Total RNA was isolated from rat livers frozen in liquid nitrogen using the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's protocol. The concentration and purity of the RNA were determined by measuring the absorbance at 260 and 280 nm, with a correction for background at 320 nm, and RNA integrity was examined by visualizing the 18S and 28S rRNA bands on a denaturing agarose (1%) gel. Total RNA (1μg samples) was used for the synthesis of single-stranded cDNA. First-strand cDNA synthesis was carried out using of the QuantiTect Reverse Transcription Kit (Qiagen) according to the manufacturer's protocol. Expression levels of the genes were measured by real-time PCR using the Maxima SYBR Green/ROX qPCR Master Mix (Fermentas, Vilnius, Lithuania). Real-time PCR was carried out on an IQ5 real-time PCR system (Bio-Rad Laboratories, Hercules, CA, USA). Signals were normalized to the housekeeping gene 18S rRNA as an endogenous internal control. Gene-specific oligonucleotide primers were used for CYP2B1, CYP2B2, PEPCK, G6Pase and 18S rRNA (Kachaylo et al., 2012a). The fold change in the target gene normalized to the level of 18S rRNA and the control was calculated based on PCR efficiency and Ct.
Preparation of crude liver extracts and microsomal proteins
For the preparation of crude liver extracts, livers were rinsed in cold phosphate-buffered saline and suspended in lysis buffer (50 mM Tris-HCl, pH 7.4; 150 mM NaCl; 1% Triton X-100; 0.5% NP-40; 1 mM DTT; and 1 mM EDTA) supplemented with Complete Mini Protease Inhibitor Cocktail (Roche Diagnostics, Basle, Switzerland). The livers were homogenized. Homogenates were incubated on ice for 30 min and centrifuged at 5000 g for 10 min to remove insoluble precipitates. Supernatants were used as crude liver protein extracts. Crude liver extracts were collected and stored at −80°C. The protein concentrations in crude liver extracts were determined using the Bradford method with bovine serum albumin as a standard. The microsomal protein fractions were isolated from freshly excised liver tissues by standard differential centrifugation (Burke et al., 1985). The protein concentrations in the microsomes were measured using the Lowry method with a bovine albumin solution as a standard. The microsomal fractions were collected and stored at −80°C. The activity of 7-pentoxyresorufin O-dealkylase (PROD) was measured at 37°C by fluorimetry (Burke et al., 1985).
SDS-PAGE electrophoresis and Western blotting
The crude liver protein extract (60 μg) or 30 μg of microsomal proteins was loaded in each lane, separated by 10% SDS-PAGE and transferred to a nitrocellulose membrane. Membranes were stained with Ponceau S to verify loading and transfer efficiency. Immunodetection was performed with polyclonal anti-CYP2B1, anti-PEPCK (sc-32879, Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), anti-G6Pase (sc-25840, Santa Cruz Biotechnology), anti-CAR (sc-13065, Santa Cruz Biotechnology), anti-HNF-4α (sc-8987, Santa Cruz Biotechnology), anti-FKHR (FOXO1) (sc-11350, Santa Cruz Biotechnology) or anti-human β-actin (Sigma-Aldrich, St. Louis, MO, USA) antibodies. Polyclonal antibodies against rat CYP2B1 were prepared in rabbits by immunization with the purified CYP2B1 protein. β-Actin was used as a loading control. The bands were visualized using the Visualizer Spray and Glow ECL Western Blotting Detection System (Millipore, Billerica, MA, USA) or colorimetry with 4-chloro-1-naphthol (MP Biomedicals, Santa Ana, CA, USA) as a substrate.
Chromatin immunoprecipitation (ChIP) assay
ChIP assays were performed in accordance with a previously described protocol (Kachaylo et al., 2012a). In brief, livers were rinsed in cold phosphate-buffered saline with 0.4% NP-40 and homogenized in the same solution. Homogenates were treated with 1% formaldehyde to cross-link the chromatin. Fixed cells were collected by centrifugation, washed, homogenized in cell lysis buffer, and incubated on ice for 30 min. Lysates were sonicated on ice with three pulses of 20 s and 20% amplitude in a Microson™ Ultrasonic Liquid Processor XL-2000 cell disrupter (Qsonica LLC, Newtown, CT, USA), yielding chromatin fragments of 500–1000 BP. Samples were centrifuged to remove debris and the supernatants were collected. As a positive control (Input), the supernatants were retained. The supernatants were diluted 10-fold with dilution buffer. The diluted lysates were precleared by incubating them with 20 μL mL−1 protein G agarose/salmon sperm DNA (Millipore) for 1 h at 4°C on a rotating plate. Cleared lysates were subjected to an overnight incubation with either the appropriate antibody or a normal rabbit IgG at 4°C under rotation. Immunocomplexes were precipitated by adding protein G agarose/salmon sperm DNA (Millipore) for 1 h at 4°C with shaking. Samples were then washed with low-salt buffer, high-salt buffer, LiCl buffer and twice with 10 mM Tris-HCL, pH 7.5; and 1 mM EDTA buffer. The immunoselected chromatin was eluted in elution buffer. The samples were further incubated for 6 h at 65°C to reverse the cross-linking. After protease digestion, the DNA was purified by phenol/chloroform extraction. PCR amplification was performed using primers specific for the rat CYP2B2 gene promoter [phenobarbital responsive element module (PBREM) site: F: 5′-CGTGGACACAACCTTCAAG-3′ and R: 5′-GAGCAAGGTCCTGGTGTC-5′]; the PEPCK gene promoter (DR1 site: F: 5′-CAGAGCTGAATTCCCTTC-3′ and R: 5′-AGCTGTGAGGTGTCAC-3′; IRS site: F: 5′-GGGAGTGACACCTCA-3′, R: 5′-GTGTGCCAGTGGCTGC-3′); and the G6Pase gene promoter (DR1 and IRS sites: F: 5′-TTATCAGTTGCCAGGTGGG-3′, R: 5′-CCAAAGTCGTGGAGCACGTTC-3′).
Data analysis
Data are presented as the mean ± SD. Statistical analyses were performed using GraphPad Prism 5.0 (GraphPad Software, Inc., San Diego, CA, USA). A one-way ANOVA followed by Dunnett's post hoc test was used to compare each treated group to the control (for more than two groups). A two-way ANOVA followed by Bonferroni's post hoc test was used for multiple comparisons between diets with and without compound treatments. A P-value < 0.05 was considered statistically significant.
Materials
cisTPD and transpDMA (Figure 1) were synthesized as described by Griengl and Geppert (1976). The identity of the compound was verified by NMR spectroscopy, and LC/MS analysis showed the preparation to be 97–98% pure. All chemicals and solvents were of analytical grade and obtained from commercial sources.
Figure 1.
Structures of the compounds tested. cisTPD, (4S,6R)-2,4,6-triphenyl-1,3-dioxane; transpDMA, 4-[(4R,6R)-4,6-diphenyl-1,3-dioxan-2-yl]-N,N-dimethylaniline.
Results
Effects of compounds on hepatic CYP2B in rats
Total RNA was isolated from rat livers 48 h after treatment with cisTPD, transpDMA or the vehicle, and mRNA levels of CYP2B1 and CYP2B2 were measured by real-time PCR. The animals were subjected to fasting for 18 h before decapitation. As expected from our previous results (Pustylnyak et al., 2011a), hepatic CYP2B1/2 gene expression, which are typical CAR target genes, was induced in a dose-dependent manner by cisTPD and transpDMA (Figure 2A). Next, we examined the effect of the compounds on the protein levels in rat livers. The amounts of CYP2B1/2 proteins were investigated by Western blotting analysis (Figure 2B). These proteins were expressed at basal levels in the livers of vehicle-treated rats but hepatic CYP2B1/2 was expressed at markedly higher levels in rats exposed to cisTPD and transpDMA than in control rats. In parallel, the inducibility of CYP2B in rat livers by cisTPD and transpDMA was estimated by determining PROD, a sensitive marker of enzymic activity. As shown in Figure 2C, treatment with cisTPD and transpDMA increased PROD activity in a dose-dependent manner.
Figure 2.
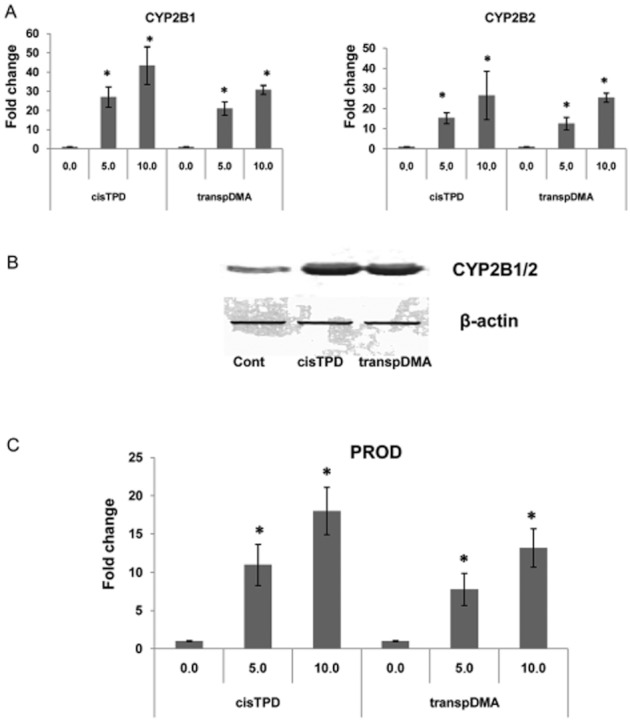
Dose-dependent effects of cisTPD and transpDMA on CAR target gene CYP2B1/2 expression (A), protein levels (B) and activity (C) in rat livers. Total RNAs were prepared and subjected to RT-PCR for measurement of mRNA. Microsomal proteins were prepared from rat livers and subjected to Western blotting analysis and CYP2B-specific PROD activity. Fold change was expressed by normalizing the corresponding value of the untreated rats to unity. Bars represent the mean ± SD. Three animals were used in each treatment group. *P < 0.05, significantly different from vehicle-treated animals; one-way ANOVA followed by Dunnett's post hoc test.
Effects of the compounds on hepatic gluconeogenic genes in rats
We analysed the gene expression levels of PEPCK and G6Pase after 48 h of treatment under fasting conditions to examine the effects of cisTPD and transpDMA on gluconeogenic genes. Treatment of rats with CAR activators resulted in a dose-dependent decrease in the mRNA levels of both genes (Figure 3A). The decrease in PEPCK and G6Pase gene expression levels paralleled changes in the protein levels (Figure 3B, C). Western blot analysis demonstrated a basal level of hepatic PEPCK and G6Pase protein in the livers of vehicle-treated rats. Hepatic PEPCK and G6Pase were markedly decreased in rats exposed to both compounds relative to the levels found in control rats.
Figure 3.

Dose-dependent effects of cisTPD and transpDMA on PEPCK and G6Pase gene expression (A) and protein levels (B, C) in rat livers. Total RNAs were prepared and subjected to RT-PCR for measurement of mRNA. Crude liver extracts were prepared from rat livers and subjected to Western blot analysis. The protein bands were analysed by the computerized densitometric program Total Lab. The intensities of the signals were determined from the areas under the curves for each peak, and the data were plotted. PEPCK and G6Pase were normalized to β-actin as an internal control. Fold change was expressed by normalizing the corresponding value of the vehicle-treated rats to unity. Bars represent the mean ± SD. Three animals were used in each treatment group. *P < 0.05, significantly different from control animals; one-way ANOVA followed by Dunnett's post hoc test.
Recruitment of CAR, HNF4α and FOXO1 to their respective binding sites in vivo
We examined the effect of treatment with cisTPD and transpDMA on the association of CAR, HNF4α and FOXO1 with the promoters of the CYP2B2, PEPCK and G6Pase genes. Association of these transcriptional factors with the promoters was determined by a ChIP assay using antibodies against CAR, HNF4α and FOXO1. An increase in CAR recruitment to the CYP2B2 gene promoter (PBREM) was detected after cisTPD and transpDMA treatment (Figure 4A). Binding of HNF4α (DR1 site) and FOXO1 (IRS site) to the PEPCK and G6Pase promoters was detected in control rat livers (Figure 4B, C). cisTPD and transpDMA treatment inhibited the binding of HNF4α and FOXO1 to the promoters. Nevertheless, treatment with both compounds induced the recruitment of CAR to the DR1 motif, an HNF4α-specific binding site (Figure 4B, C). Moreover, the results of the Western blotting analysis showed that the protein bands corresponding to CAR, HNF4α and FOXO1 were unchanged in the crude liver extracts relative to the control and compound-treated rats (Figure 5).
Figure 4.
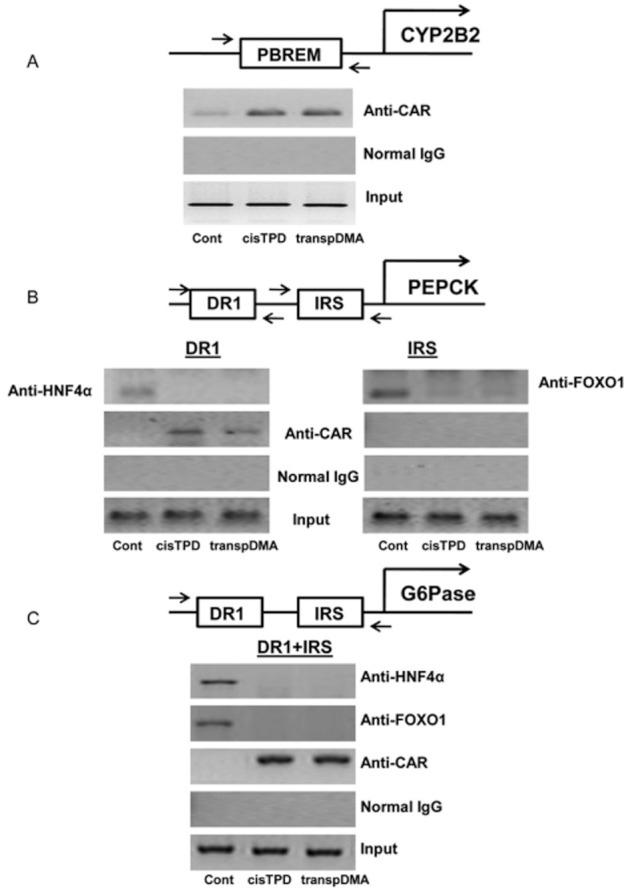
Recruitment of CAR, HNF4α and FOXO1 to CYP2B2 (A), PEPCK (B) and G6Pase (C) gene promoters in rat livers in response to cisTPD and transpDMA treatment. Chromatin was isolated and pre-cleared as described. Pre-cleared chromatin was immunoprecipitated using antibodies against CAR, HNF4α, FOXO1 or normal rabbit IgG. The DNA regions were amplified by PCR and resolved on agarose gels.
Figure 5.
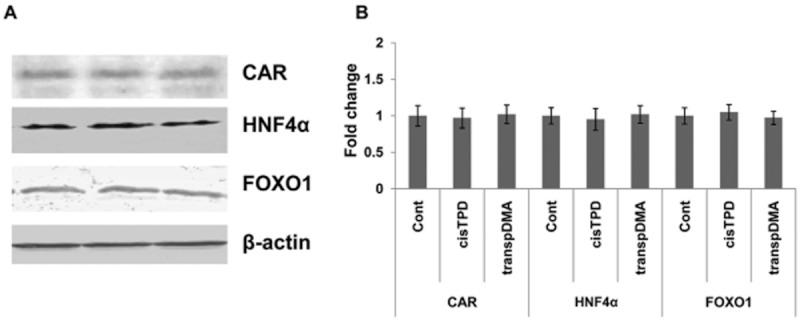
Effect of cisTPD and transpDMA on CAR, HNF4α and FOXO1 proteins in rat livers. Crude liver extracts were prepared from rat livers and subjected to Western blotting analysis (A). The protein bands were analysed by the computerized densitometric program Total Lab. The intensities of the signals were determined from the areas under the curves for each peak, and the data were plotted (B). β-actin was used as an internal control to which PEPCK and G6Pase were normalized. Fold change was expressed by normalizing the corresponding value of the vehicle-treated rats to unity. Bars represent the mean ± SD Three animals were used in each treatment group. No significant differences between groups were observed; one-way ANOVA.
Effect of long-term compound treatment on glucose metabolism in HFD rats
We employed six rat groups, including normal diet-fed or HFD-fed rats with or without cisTPD or transpDMA treatment, to investigate how long-term CAR activation by cisTPD and transpDMA influenced glucose metabolism. Fasting blood glucose levels in rats were monitored in response to 8 weeks of treatment. Long-term cisTPD treatment significantly decreased fasting blood glucose levels compared with vehicle-treated animals (Figure 6A). However, transpDMA treatment did not produce a significant change in fasting blood glucose levels compared with vehicle-treated animals. We also examined the effect of long-term CAR activation in HFD rats. Rats were fed the HFD for 8 weeks and concurrently treated with cisTPD or transpDMA (10 mg kg-−1, once a week) or vehicle. Fasting blood glucose levels were significantly increased by the HFD compared with the control group rats (Figure 6A). Treatment with cisTPD and transpDMA in HFD rats decreased the level of fasting blood glucose to control group levels (Figure 6A). Moreover, as shown in Figure 6B, cisTPD or transpDMA significantly inhibited a gain in body weight in the HFD rats after 8 weeks of treatment. By the end of week 8, the cisTPD- and transpDMA-treated HFD groups had lower body weights compared with the untreated HFD group. We further examined the effects of the compounds on insulin sensitivity. The treatment of HFD rats with cisTPD or transpDMA for 8 weeks significantly improved insulin sensitivity, as confirmed by both GTT (Figure 7A) and ITT (Figure 7B). Taken together, these results indicate that the CAR activators cisTPD and transpDMA may improve insulin sensitivity through the suppression of hepatic gluconeogenic gene expression.
Figure 6.
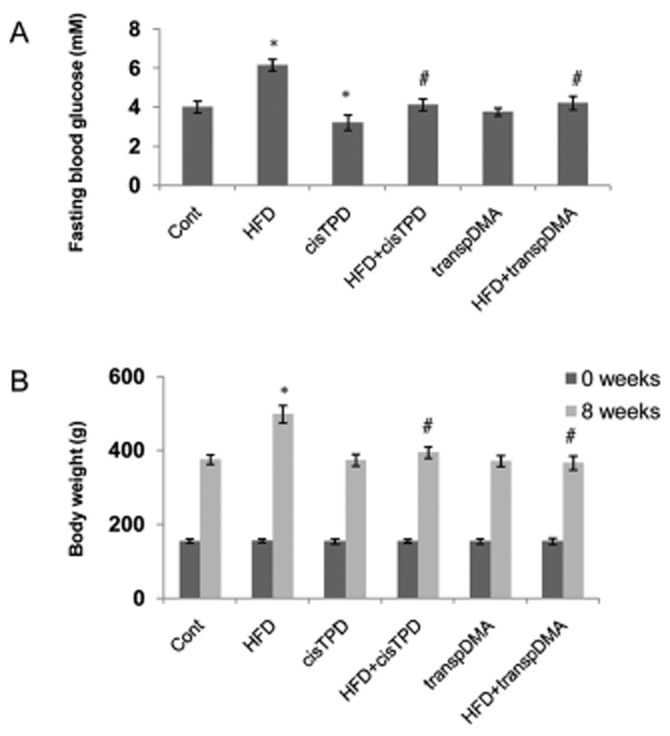
Effect of long-term cisTPD and transpDMA treatment on fasting blood glucose (A) and body weight (B) at the end of 8 weeks of a normal diet (Cont) or HFD. Bars represent the mean ± SD Five animals were used in each treatment group. *P < 0.05, significantly different from control animals; #P < 0.05, significantly different from HFD animals; two-way ANOVA followed by Bonferroni's post hoc test.
Figure 7.
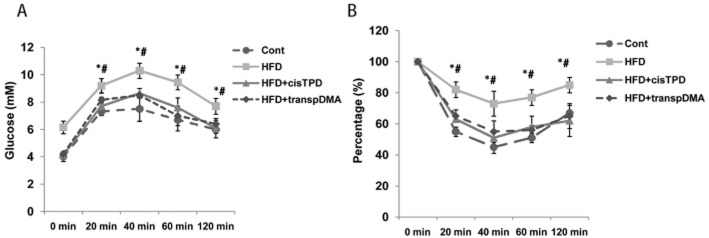
Effect of long-term cisTPD and transpDMA treatment on insulin sensitivity. Rats were fed a normal diet (Cont) or HFD for 8 weeks and treated with cisTPD or transpDMA as a single weekly dose. After 16 h of fasting, GTT (A) and ITT (B) were performed. Bars represent the mean ± SD Five animals were used in each treatment group. *P < 0.05, significantly differentfrom the HFD + cisTPD group; #P < 0.05, significantly different from the HFD + transpDMA group; two-way ANOVA followed by Bonferroni's post hoc test.
Discussion and conclusions
The constitutive androstane receptor was initially characterized as a xenosensor regulating the response to xenochemicals. Recent studies have suggested that the CAR is also involved in energy metabolism (lipogenesis and gluconeogenesis), showing anti-diabetic and anti-obesity effects (Dong et al., 2009; Gao et al., 2009). It is tempting to speculate that the pharmacological modulation of CAR may be beneficial in managing metabolic diseases. Thus, the development of CAR activators for use as potential therapeutic tools in the treatment and prevention of these metabolic disorders holds promise. In the present study, the effects of transpDMA on the hepatic gluconeogenic genes PEPCK and G6Pase in rat livers were studied in reference to cisTPD.
CYP2B genes are commonly used biomarkers to measure CAR activation. We have demonstrated that cisTPD and transpDMA treatment markedly increased the mRNA and protein levels and specific activity of CYP2B. For CYP2B genes, transcriptional activation occurs upon CAR binding to a 51-base pair phenobarbital-responsive enhancer module (PBREM), which contains two nuclear receptor DR4 sites (NR1 and NR2) in the 5'-flanking sequence of the genes (Pustylnyak et al., 2007). ChIP was used to examine the recruitment of CAR to the CAR target gene CYP2B2 promoter region and demonstrated that the recruitment of CAR to its binding site (PBREM) on the target gene was increased in response to both compounds. In contrast to CYP2B, mRNA and protein levels of rat PEPCK and G6Pase were decreased by treatment with these compounds. Moreover, our data suggest that long-term CAR activation by transpDMA and cisTPD inhibited glucose production and improved glucose homeostasis and insulin sensitivity in HFD rats. Taken together, these results indicate that the CAR activators cisTPD and transpDMA may suppress hepatic gluconeogenesis via inhibition of gluconeogenic genes. However, this hypothesis needs to be investigated in more detail.
The two transcriptional factors HNF4α and FOXO1 play key roles in the regulation of gluconeogenesis (Puigserver et al., 2003; Rhee et al., 2003; Hirota et al., 2008). HNF-4α is central to glucose metabolism because it is a key positive regulator of the basal hepatic expression of PEPCK and G6Pase genes (Yoon et al., 2001). FOXO1 is a member of the forkhead family of transcription factors and regulates glucose metabolism in the adult liver, especially during fasting (Puigserver et al., 2003; Accili and Arden, 2004). FOXO1 regulates the expression of the PEPCK and G6Pase genes by directly binding to its target DNA sequence and through interaction with nuclear receptors. FOXO1 activity is regulated by phosphorylation and acetylation. Insulin induces the phosphorylation of FOXO1 through the PI3K-Akt signalling pathway. Phosphorylated FOXO1 is excluded from the nucleus, thereby attenuating its transcriptional activity (Matsuzaki et al., 2003). Several reports have suggested that reducing FOXO1 activity may represent a potential strategy for treatment of type 2 diabetes mellitus (Nakae et al., 2001; Altomonte et al., 2003; Qu et al., 2006; Nagashima et al., 2010). In our previous study using ChIP assays, we demonstrated that cisTPD-activated CAR inhibited HNF-4α transactivation by competing with HNF-4α for binding to the HNF-4α-binding element (DR1-site) in gluconeogenic gene promoters (Kachaylo et al., 2012a). Moreover, we showed that cisTPD treatment prevented the binding of FoxO1 to IRS in the PEPCK and G6Pase gene promoters in rat livers. Similar to our previous studies (Kachaylo et al., 2012a), we demonstrated that transpDMA-activated CAR produced a significant decrease in the expression of the key hepatic gluconeogenic genes PEPCK and G6Pase through a diminished association of HNF4α and FOXO1 with their specific binding sites in the gene promoters. However, the protein levels of both transcription factors remained unchanged. These results agree with previous studies showing that activated CAR can regulate gluconeogenic genes through an HNF4α-specific binding site in their promoters (Miao et al., 2006) and bind to FOXO1 and suppress its transcriptional activity by preventing it from binding to IRS in the promoters of target genes (Kodama et al., 2004).
Thus, our findings provide evidence to support the conclusion that transpDMA, similar to cisTPD, inhibits the gluconeogenic genes PEPCK and G6Pase through the suppression of HNF4α and FOXO1 transcriptional activity. These results suggest that transpDMA may be a novel potential therapeutic tool for the regulation of gluconeogenesis. However, this study focused on only a few key molecules of the gluconeogenic pathway. Therefore, additional biochemical and dynamic analyses are necessary. Moreover, there is no evidence that transpDMA can activate human CAR. Further investigation will be required to determine the species-specific effects of this compound in humans and investigate the use of this compound as a therapeutic tool for the treatment and prevention of metabolic disorders.
Acknowledgments
This work was supported by the Russian Federal Program of the Ministry of Education and Science of the Russian Federation ‘Scientific and pedagogical manpower of innovational Russia’ GK 14.740.11.1054 and GK 16.740.11.0631 to V.O.P.
Glossary
- CAR
constitutive androstane receptor
- ChIP
chromatin immunoprecipitation
- cisTPD
cis isoform of 2,4,6-triphenyldioxane-1,3
- CYP2B
cytochromes P450 2B subfamily
- FOXO1
forkhead box O1
- G6Pase
glucose-6-phosphatase
- GTT
Glucose tolerance test
- HFD
high-fat diet
- HNF4α
hepatocyte nuclear factor-4α
- IRS
insulin response sequences
- ITT
insulin tolerance test
- PBREM
phenobarbital responsive element module
- PEPCK
phosphoenolpyruvate carboxykinase
- PROD
7-pentoxyresorufin O-dealkylase
- transpDMA
4-[(4R,6R)-4,6-diphenyl-1,3-dioxan-2-yl]-N,N-dimethylaniline
Conflicts of interest
There are no conflicts of interest.
References
- Accili D, Arden KC. FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell. 2004;117:421–426. doi: 10.1016/s0092-8674(04)00452-0. [DOI] [PubMed] [Google Scholar]
- Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altomonte J, Richter A, Harbaran S, Suriawinata J, Nakae J, Thung SN, et al. Inhibition of Foxo1 function is associated with improved fasting glycemia in diabetic mice. Am J Physiol Endocrinol Metab. 2003;285:E718–E728. doi: 10.1152/ajpendo.00156.2003. [DOI] [PubMed] [Google Scholar]
- Barthel A, Schmoll D, Unterman TG. FoxO proteins in insulin action and metabolism. Trends Endocrinol Metab. 2005;16:183–189. doi: 10.1016/j.tem.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Burke MD, Thompson S, Elcombe C, Halpert J, Haaparanta T, Mayer RT. Ethoxy-, pentoxy- and benzyloxyphenoxazones and homologues: a series of substrates to distinguish between different induced cytochromes P-450. Biochem Pharmacol. 1985;34:3337–3345. doi: 10.1016/0006-2952(85)90355-7. [DOI] [PubMed] [Google Scholar]
- Dong B, Saha PK, Huang W, Chen W, Abu-Elheiga LA, Wakil SJ, et al. Activation of nuclear receptor CAR ameliorates diabetes and fatty liver disease. Proc Natl Acad Sci U S A. 2009;106:18831–18836. doi: 10.1073/pnas.0909731106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, He J, Zhai Y, Wada T, Xie W. The constitutive androstane receptor is an anti-obesity nuclear receptor that improves insulin sensitivity. J Biol Chem. 2009;284:25984–25992. doi: 10.1074/jbc.M109.016808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griengl H, Geppert KP. Prins-reaktionen mit arylaldehyden und Arylolefine. Monatshafte fur Chemie. 1976;107:421–431. [Google Scholar]
- Hirota K, Sakamaki J, Ishida J, Shimamoto Y, Nishihara S, Kodama N, et al. A combination of HNF-4 and Foxo1 is required for reciprocal transcriptional regulation of glucokinase and glucose-6-phosphatase genes in response to fasting and feeding. J Biol Chem. 2008;283:32432–32441. doi: 10.1074/jbc.M806179200. [DOI] [PubMed] [Google Scholar]
- Kachaylo E, Yarushkin A, Pustylnyak V. Constitutive androstane receptor activation by 2,4,6-triphenyldioxane-1,3 suppresses the expression of the gluconeogenic genes. Eur J Pharmacol. 2012a;679:139–143. doi: 10.1016/j.ejphar.2012.01.007. [DOI] [PubMed] [Google Scholar]
- Kachaylo EM, Pustylnyak VO, Lyakhovich VV, Gulyaeva LF. Constitutive androstane receptor (CAR) is a xenosensor and target for therapy. Biochemistry (Mosc) 2011;76:1087–1097. doi: 10.1134/S0006297911100026. [DOI] [PubMed] [Google Scholar]
- Kachaylo EM, Yarushkin AA, Pustylnyak VO. Long-term constitutive androstane receptor activation by 2,4,6-triphenyldioxane-1,3 improves glucose metabolism in high-fat diet rats. Biochem Anal Biochem. 2012b:S4. doi: 10.4172/2161-1009.S4-001. [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodama S, Koike C, Negishi M, Yamamoto Y. Nuclear receptors CAR and PXR cross talk with FOXO1 to regulate genes that encode drug-metabolizing and gluconeogenic enzymes. Mol Cell Biol. 2004;24:7931–7940. doi: 10.1128/MCB.24.18.7931-7940.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki H, Daitoku H, Hatta M, Tanaka K, Fukamizu A. Insulin-induced phosphorylation of FKHR (Foxo1) targets to proteasomal degradation. Proc Natl Acad Sci U S A. 2003;100:11285–11290. doi: 10.1073/pnas.1934283100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao J, Fang S, Bae Y, Kemper JK. Functional inhibitory cross-talk between constitutive androstane receptor and hepatic nuclear factor-4 in hepatic lipid/glucose metabolism is mediated by competition for binding to the DR1 motif and to the common coactivators, GRIP-1 and PGC-1alpha. J Biol Chem. 2006;281:14537–14546. doi: 10.1074/jbc.M510713200. [DOI] [PubMed] [Google Scholar]
- Nagashima T, Shigematsu N, Maruki R, Urano Y, Tanaka H, Shimaya A, et al. Discovery of novel forkhead box O1 inhibitors for treating type 2 diabetes: improvement of fasting glycemia in diabetic db/db mice. Mol Pharmacol. 2010;78:961–970. doi: 10.1124/mol.110.065714. [DOI] [PubMed] [Google Scholar]
- Nakae J, Kitamura T, Silver DL, Accili D. The forkhead transcription factor Foxo1 (Fkhr) confers insulin sensitivity onto glucose-6-phosphatase expression. J Clin Invest. 2001;108:1359–1367. doi: 10.1172/JCI12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odom DT, Zizlsperger N, Gordon DB, Bell GW, Rinaldi NJ, Murray HL, et al. Control of pancreas and liver gene expression by HNF transcription factors. Science. 2004;303:1378–1381. doi: 10.1126/science.1089769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puigserver P, Rhee J, Donovan J, Walkey CJ, Yoon JC, Oriente F, et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature. 2003;423:550–555. doi: 10.1038/nature01667. [DOI] [PubMed] [Google Scholar]
- Pustylnyak V, Yarushkin A, Kachaylo E, Slynko N, Lyakhovich V, Gulyaeva L. Effect of several analogs of 2,4,6-triphenyldioxane-1,3 on constitutive androstane receptor activation. Chem Biol Interact. 2011a;192:177–183. doi: 10.1016/j.cbi.2011.03.005. [DOI] [PubMed] [Google Scholar]
- Pustylnyak V, Kazakova Y, Yarushkin A, Slynko N, Gulyaeva L. Effect of several analogs of 2,4,6-triphenyldioxane-1,3 on CYP2B induction in mouse liver. Chem Biol Interact. 2011b;194:134–138. doi: 10.1016/j.cbi.2011.09.003. [DOI] [PubMed] [Google Scholar]
- Pustylnyak VO, Gulyaeva LF, Lyakhovich VV. Induction of cytochrome P4502B: role of regulatory elements and nuclear receptors. Biochemistry (Mosc) 2007;72:608–617. doi: 10.1134/s000629790706003x. [DOI] [PubMed] [Google Scholar]
- Qu S, Altomonte J, Perdomo G, He J, Fan Y, Kamagate A, et al. Aberrant Forkhead box O1 function is associated with impaired hepatic metabolism. Endocrinology. 2006;147:5641–5652. doi: 10.1210/en.2006-0541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee J, Inoue Y, Yoon JC, Puigserver P, Fan M, Gonzalez FJ, et al. Regulation of hepatic fasting response by PPARgamma coactivator-1alpha (PGC-1): requirement for hepatocyte nuclear factor 4alpha in gluconeogenesis. Proc Natl Acad Sci U S A. 2003;100:4012–4017. doi: 10.1073/pnas.0730870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ropelle ER, Pauli JR, Cintra DE, Frederico MJ, de Pinho RA, Velloso LA, et al. Acute exercise modulates the Foxo1/PGC-1alpha pathway in the liver of diet-induced obesity rats. J Physiol. 2009;587:2069–2076. doi: 10.1113/jphysiol.2008.164202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda A, Hamadeh HK, Webb HK, Yamamoto Y, Sueyoshi T, Afshari CA, et al. Diverse roles of the nuclear orphan receptor CAR in regulating hepatic genes in response to phenobarbital. Mol Pharmacol. 2002;61:1–6. doi: 10.1124/mol.61.1.1. [DOI] [PubMed] [Google Scholar]
- Wajngot A, Chandramouli V, Schumann WC, Ekberg K, Jones PK, Efendic S, et al. Quantitative contributions of gluconeogenesis to glucose production during fasting in type 2 diabetes mellitus. Metabolism. 2001;50:47–52. doi: 10.1053/meta.2001.19422. [DOI] [PubMed] [Google Scholar]
- Yoon JC, Puigserver P, Chen G, Donovan J, Wu Z, Rhee J, et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature. 2001;413:131–138. doi: 10.1038/35093050. [DOI] [PubMed] [Google Scholar]