

Abstract
Eukaryotic translation initiation factor 4F (eIF4F), comprising the cap-binding protein eIF4E, the helicase eIF4A, and the central scaffold eIF4G, is a convergence node for a complex signaling network that controls protein synthesis. Together with eIF3 and eIF4A/4B, eIF4G recruits ribosomal subunits to mRNAs and facilitates 5′ untranslated region unwinding. Mammalian eIF4G contains three HEAT domains and unstructured regions involved in protein-protein interactions. Despite detailed eIF4G structure data, the mechanisms controlling initiation scaffold formation remain obscure. We found a new, highly regulated eIF4B/-3 binding site within the HEAT-1/-2 interdomain linker, harboring two phosphorylation sites that we identified as substrates for Erk1/2 and casein kinase 2. Phorbol ester-induced sequential phosphorylation of both sites detached HEAT-2 from the complex with eIF4A/-4B/-3 and stimulated the association of HEAT-3 with the mitogen-activated protein kinase signal integrating kinase Mnk1. Our results provide a mechanistic link between intracellular signal transduction and dynamic initiation complex formation coordinated by flexible eIF4G structure.
INTRODUCTION
In eukaryotes, translation initiation is rate-limiting for protein synthesis and requires eukaryotic translation initiation factors (eIFs) for recruiting ribosomal subunits to mRNAs. The heterotrimeric complex eIF4F, comprised of the m7GTP cap-binding protein eIF4E, the ATP-dependent helicase eIF4A, and the scaffold eIF4G, recruits ribosomal subunits via eIF3 for mRNA scanning (1). The DEAD box helicase eIF4A is indispensable for unwinding/scanning during initiation at mammalian mRNAs with at least moderate 5′ untranslated region (5′UTR) structural complexity (reviewed in reference 2). It has two domains, both of which are involved in binding RNA and ATP cooperatively (3). A poor ATPase/helicase on its own, eIF4A is potently stimulated in a complex with its cofactor(s) eIF4B and eIF4G (4).
Mammalian eIF4G consists of three HEAT (Huntingtin/EF3/protein phosphatase 2A/Tor1) domains separated by flexible regions (5, 6) (Fig. 1A). eIF4A association with HEAT-1 and HEAT-2 (5, 7) and the N-terminal portion of the linker separating them (8) (Fig. 1A) sustains a conformational arrangement that favors eIF4A-RNA binding (9). In higher eukaryotes, eIF4G's C-terminal portion comprising HEAT-2 and HEAT-3 (Fig. 1A) interacts with eIF4A and the mitogen-activated protein kinase (MAPK)-signal integrating kinases Mnk1/2 (5) (Fig. 1A). Mutations in HEAT-2 decreased 48S preinitiation complex (PIC) formation ca. 3- to 4-fold, whereas mutations in HEAT-1 abolished it (10). Thus, HEAT-2 and HEAT-3 likely serve modulatory roles in translation and are dispensable for initiation (10); in fact, yeast eIF4G evolved with a loss of the C-terminal part of the interdomain linker, HEAT-2 and -3 (8).
Fig 1.

Regulation of Mnk1 and eIF3A/-4A/-4B binding with C-terminal eIF4G fragments by IDL length. (A) eIF4G domain organization. The interaction sites with other proteins, HEAT domains 1 to 3, and the IDL are indicated. (B) Structure of eIF4G truncation fragments used in the present study. All fragments are N-terminally (myc) and C-terminally (Flag) tagged. Major confirmed IDL phosphorylation sites are indicated. (C) myc-eIF4G expression and TPA-dependent activation of Pkc-α/Erk1/2 in HEK293 cells (top) and Mnk1-binding/phosphorylation of myc-eIF4G fragments (bottom). The eIF4G fragments are identified by their N-terminal amino acid number at the top. HEK293 cells were transfected (16 h), serum starved (24 h), and treated with either DMSO (0 min) or 200 nM TPA (30 min). Cell lysates were subjected to immunoblotting (top) or anti-Flag IP, followed by immunoblotting (bottom) with the indicated antibodies. The experiment was repeated four times; the results of a representative assay are shown. (D) Binding of eIF4G-IDL truncation variants with Mnk1 and initiation factors eIF4A/-4B in nonstimulated HEK293 cells. HEK293 cells were transfected with the indicated fragments (16 h) and serum starved (24 h), and the cell lysates were subjected to Flag-IP, followed by immunoblotting as indicated. Quantitative chemiluminescence measurements of the Mnk1 signal after Flag coimmunoprecipitation are shown in the lower panel. This experiment was repeated three times with consistent results; average binding values are shown. The relative binding efficiencies were normalized to Mnk1 binding to the 1085 IDL fragment. (E) Binding of eIF4A/-4B/-3A with diverse eIF4G IDL fragments indicates a role for the IDL in modulating protein-protein interactions with the C-terminal part of eIF4G. Flag-IP of the indicated IDL fragments containing HEAT-2/-3 domains and variable parts of the IDL. HEK293 cells were treated as described previously (see panel D). (F) Proposed model for intramolecular interactions governing eIF4A/-4B binding to the IDL/HEAT-2. (Left) The IDL (in the range of aa 1177 to 1192) establishes contacts with HEAT-3 (13) that prevent eIF4B association but favor eIF4A–HEAT-2 interactions. (Middle) Truncation of the IDL to 1222/1235 abolishes interactions with HEAT-3, enables eIF4B binding to eIF4G, and displaces eIF4A from HEAT-2. (Right) Without the IDL, eIF4B binding is abolished, and HEAT-2/eIF4A binding is unencumbered.
Topological and mechanistic studies of the eIF4A/-4G/-4B helicase complex and the regulation of ATP/ADP binding to eIF4A in vitro suggest a “dynamic network of multiple weak, but specific interactions” (8) that may direct mRNA unwinding. eIF4F assembly, recruitment of accessory factors, and their interactions with mRNAs in vivo is influenced by major mitogenic signaling pathways converging on translation machinery. Unraveling the role of mitogenic signals in eIF4F assembly, template unwinding, and/or scanning is impeded by a lack of information on eIF4G, the main signal integration module of the translation initiation apparatus. The interdomain linker (IDL) of eIF4G, which separates HEAT-1 and -2, harbors seven major confirmed phosphorylation sites (all of which are lost in yeast) (Fig. 1A). Due to its strategic placement between HEAT-1 and -2 eIF4A binding sites, the IDL may modulate dynamic protein-protein interactions in eIF4G's core.
We used eIF4G1 (referred to as eIF4G from here on) truncation variants, site-directed eIF4G mutagenesis, phosphoproteomic analysis, and other biochemical assays to investigate the regulation of dynamic eIF4G HEAT-2/-3 domain interactions with eIF4A/-4B and Mnk1a (referred to as Mnk1 from here on) by protein kinase C (Pkc)/Ras/Erk mitogenic signal transduction. Detailed mapping studies revealed a new, highly regulated binding site for eIF4B/eIF3 located at the IDL/HEAT-2 junction. Binding of eIF4A with eIF4G HEAT-2 is ATP/ADP dependent (8), whereas interactions of eIF4B/eIF3 with the newly identified binding site are not. The binding area contains two phosphorylation sites, S1232 and S1239, which were previously identified in vivo (11, 12). Our studies show that S1232 is an Erk1/2 substrate, whereas S1239 may be phosphorylated by casein kinase 2-α (Ck2-α). S1232 phosphorylation is partly controlled by the S1239 phospho-status and a conformational arrangement limiting Erk1/2-eIF4G binding. Pkc/Ras/Erk stimulation results in rapid dissociation of eIF4G HEAT-2 from the complex with eIF4A/-4B/-3, whereas HEAT-1 remains bound to the helicase complex under these conditions. The results presented here provide basic insight into molecular mechanisms of dynamic translation initiation control via signal transduction to eIF4G.
MATERIALS AND METHODS
Cell lines, DNA transfections, and eIF4G truncation variants.
HEK293 cells (American Type Culture Collection) were maintained in Dulbecco modified Eagle medium supplemented with 10% fetal bovine serum (FBS) and nonessential amino acids, and were transfected with 16 μg of plasmid DNA using 40 μl of Lipofectamine 2000 (Invitrogen) per 15-cm petri dish. At 16 h posttransfection, the medium was changed to starvation medium (without FBS). After 24 h of serum starvation, the cells were treated with kinase inhibitors and/or activators, harvested, and lysed as described below. Construction of myc- and Flag-tagged eIF4G expression plasmids has been described previously (13). The eIF4G truncation variants used in the present study were generated by PCR amplification of corresponding fragments with the primers shown in Table S1 in the supplemental material. Single/double-site amino acid mutations were introduced by overlapping PCR as described earlier (13). The generation of stable HEK293 cell lines with tetracycline (Tet)-inducible expression of myc/Flag-tagged eIF4G has been described previously (14).
Kinase inhibitors and activators.
Inhibitors of Mek1 (UO126; Cell Signaling), Ck2-α (CX4945; Synkinase), and the protein kinase C (Pkc) activator 12-O-tetradecanoyl-phorbol-13-acetate (TPA; Sigma) were dissolved in dimethyl sulfoxide (DMSO) and used at the concentrations indicated. ADP, ATP, and adenosine 5′-[β,γ-imido]triphosphate (pNppA) (all Sigma) were dissolved in water to achieve the appropriate concentrations (see Fig. 7).
Fig 7.

Effect of ADP/ATP/pNppA on eIF4A/-4B binding to the eIF4G distal IDL-HEAT-2. HEK293 cells were transfected with eIF4G fragment 1245 (A) or 1192 (B) or with wt 1222 or 1222(SS1232/9AA) (C) (16 h). Lysates were subjected to Flag-IP, and the indicated concentrations of ADP/ATP/pNppA were added during the last 60 min of the IP procedure and to the following washes. The IPs were analyzed by immunoblotting with the indicated antibodies. The quantification of eIF4A relative binding is presented as eIF4A/eIF4G ratio. Each assay was repeated three times; a representative assay is shown.
Immunoprecipitation, immunoblotting, and antibodies.
Cell lysates were prepared using polysomal lysis buffer (PLB; 10 mM HEPES [pH 7.4], 100 mM KCI, 5 mM MgCI2, 0.5% Igepal CA-630 [Sigma], 3 mM dithiothreitol [DTT]). Prior to immunoprecipitation (IP), anti-Flag M2-agarose beads (Sigma) were washed twice by NT-2 solution (50 mM Tris-HCI [pH 7.5], 150 mM NaCl, 1 mM MgCI2, 0.05% Igepal CA-630) and blocked with 1% bovine serum albumin (BSA) in the same buffer for 1 h at 4°C. The blocking solution was removed, and 35 μl of agarose beads was incubated overnight at 4°C with cell lysates containing 6.5 mg of total protein in 1 ml of PLB. Then, the beads were washed five times with 0.9 ml of NT-2 solution, and 75 μl of NuPAGE 2× LDS loading buffer (Invitrogen) was added to each sample. Proteins were subjected to SDS-PAGE and immunoblot analyses as described previously (13). Membranes were blocked with 5% BSA in phosphate-buffered saline–Tween 20 (PBST) solution and probed with phospho-specific antibodies against p-Pkc-α(T638), p-Erk1/2(T202/Y204), p-eIF4G(S1148), p-eIF4B(S406), p-rpS6(SS235/6), and p-(S)-Pkc-substrate (all from Cell Signaling Technology) and p-eIF4G(S1232) (Novus Biologicals) or antibodies against c-myc (Sigma), mTorC1, Erk1/2, Mnk1, rpS6, eIF3A, eIF4A, eIF4B, and eIF4G1 (all from Cell Signaling Technology). Immunoblots were developed using SuperSignal West Pico- or Femto ECL kits (Thermo Scientific). For quantification, immunoblot signals were analyzed by chemiluminescence with a FluorChem FC2 imaging system (Cell Biosciences) and the AlphaEase FC program.
In vitro phosphorylation assays and phosphoproteomic analyses.
To determine the specific activity of recombinant human glutathione S-transferase (GST)–Erk2 (SignalChem) in the phosphorylation of eIF4G, 40 μl of lysate from HEK293 cells that were serum starved (24 h) and treated with 20 μM UO126 (2 h) was mixed with 10 μl of kinase assay buffer (25 mM morpholinepropanesulfonic acid [pH 7.2], 12.5 mM β-glycerol phosphate, 25 mM MgCl2, 5 mM EGTA, 2 mM EDTA, 0.25 mM DTT) and incubated with 1 μl of recombinant GST-Erk2 (0.1 μg/μl) in the presence of 2 mM ATP for 0, 15, 30, or 60 min at 20°C. Reactions (10-μl aliquots) were stopped by adding 10 μl of 4× loading buffer, followed by heating for 1 min at 95°C. In vitro phosphorylation of recombinant GST-eIF4G(Ct) (13) by recombinant GST-Erk2 was performed in 50 μl of a mixture containing 150 nM recombinant GST-Ct in kinase assay buffer, 2 mM ATP, and 2× Halt phosphatase inhibitor cocktail (Thermo Scientific). The reaction mixture was incubated at 20°C with 1 μl of 0.1 μg of GST-Erk2/μl for various intervals. Aliquots (10 μl) taken at 0, 30, 60, or 120 min were mixed with 10 μl of 4× loading buffer and heated for 1 min at 95°C to stop the enzymatic reaction. Samples were subjected to SDS-PAGE, followed by immunoblotting with p-eIF4G(Ser1232) antibodies. Large-scale in vitro phosphorylation for phosphoproteomic analysis was performed with 50 μg of recombinant GST-Ct per sample. Four reaction mixtures in kinase assay buffer were phosphorylated by 1 μg of GST–Ck1-α, GST–CK1-δ, or GST–Ck2-α (SignalChem) or incubated with 2 mM ATP without kinase for 4 h at room temperature. Phosphorylated GST-Ct was purified by GST pulldown by incubation with 150 μl of glutathione-Sepharose 4B (GE Healthcare) at 4°C (2 h). Sepharose beads were washed twice with 1 ml of NT-2 solution containing 1.5× Halt phosphatase inhibitor cocktail and twice with 1 ml of 50 mM NH4HCO3 and subjected to phosphoproteomics. Identification of the eIF4G residues phosphorylated in vitro was performed by liquid chromatography-tandem mass spectrometry at the Duke University Proteomics Core Facility.
RESULTS
The eIF4G IDL assumes autoinhibitory conformations that block Mnk1 binding.
Earlier, we showed that the interdomain linker (IDL) in eIF4G controls Mnk1-eIF4G binding (13). Based on these findings, we systematically evaluated the IDL's role in coordinating protein-protein interactions in the C-terminal part of eIF4G. To this end, we constructed various IDL truncation variants (Fig. 1B; named by the N-terminal amino acid number according to GenBank accession no. EAW78266). The longest variant, 1085, features the seven major confirmed IDL phosphorylation sites; the shortest fragment, 1245, lacks the IDL entirely (and 10 N-terminal amino acids of HEAT-2) (Fig. 1B). To enable biochemical assays, all eIF4G-IDL truncations were N-terminally (myc) and C-terminally (Flag) tagged.
To investigate Mnk1-binding and phosphorylation of eIF4G-IDL fragments after Pkc/Ras/Erk stimulation with the phorbol ester TPA, total lysates and Flag-IPs from cells transfected with specific IDL truncations were probed by immunoblot (Fig. 1C). The tagged fragments were expressed at similar levels and TPA stimulation for 30 min induced Erk1/2 and Pkc-α phosphorylation (Fig. 1C, top panel). Flag-IP revealed eIF4G phosphorylation on S1148, S1186, and S1232 according to their inclusion in the IDL truncations (Fig. 1C, bottom panel). Phosphorylation of S1148 was not detected in the 1146 fragment, presumably due to truncation of the substrate context. In accordance with previous data (13), the phosphorylation of S1148 did not respond to TPA (Fig. 1C, lanes 2 and 3). In contrast, phosphorylation of S1186 (by Pkc-α [13]) and S1232 were TPA responsive in all fragments containing these sites (Fig. 1C, bottom panel).
In accordance with previous findings (13), Mnk1 binding to eIF4G was strongly enhanced upon IDL truncation, suggesting that the IDL in the range of aa 1085 to 1245 inherently restricts the association of eIF4G with Mnk1 (Fig. 1C). Mapping of this interaction with serial IDL truncations indicated two inhibitory regions spanning aa 1146 to 1177 (Fig. 1C, lanes 4 and 5) and aa 1222 to 1245 (Fig. 1C, lanes 10 and 11). The differences in Mnk1 binding may be explained by specific sequences in these regions, by the phosphorylation state of corresponding fragments, or by the superposition of both. To discriminate between these possibilities, we first investigated Mnk1-eIF4G binding with fragments in transfected, nonstimulated cells (Fig. 1D). For a better understanding of the inhibitory properties of the IDL, we added two IDL truncations: fragment 1162 (to narrow the upstream inhibitory region in aa 1146 to 1177 to 15 aa) and fragment 1431, containing only the HEAT-3 domain (including the Mnk1 binding site), to reveal possible inhibitory properties of HEAT-2 for Mnk1 binding. Quantitative assessment of Mnk1 binding with all indicated IDL fragments confirmed and extended earlier findings (Fig. 1D, bottom panel).
Mnk1 binding to fragments 1431 (HEAT-3 domain alone) and 1245 (lacking the IDL) was similar, suggesting that restrictive Mnk1-eIF4G interaction is not due to HEAT-2 (Fig. 1D). Relative to 1431, Mnk1 binding to the 1177 fragment dropped ∼8-fold, whereas binding to the 1085 fragment was reduced ∼180-fold (Fig. 1D). Since, apart from the Mnks, the C-terminal part of eIF4G establishes critical interactions with eIF4A, we tested whether the IDL truncations affected eIF4G's scaffolding functions more broadly. We found that eIF4A interactions with its known binding site in HEAT-2 (Fig. 1A) covary with Mnk1 binding, whereas eIF4B and -3A showed an obverse interaction pattern (Fig. 1D and E). Consistent with our results, it was recently shown that HEAT-2 directly interacts with eIF4A and that an IDL fragment (aa 994 to 1138) binds eIF3 and eIF4H (a truncated eIF4B homolog) (8).
Binding of eIF4A/-4B with eIF4G HEAT-2 is coordinated by the IDL.
To explain the inhibitory properties of the IDL for Mnk1 binding, we previously proposed that the linker region surrounding the Pkc-α phosphorylation site (S1186) interacts with the AA-box 1 in HEAT-3 (Fig. 1F, left panel) (13). To evaluate whether such IDL interactions may exert control over binding of eIF4A/-4B with eIF4G, we compared eIF4A coimmunoprecipitation with eIF4G fragments containing variable portions of the IDL and HEAT-2/-3 domains (Fig. 1E). Fragments 1177 and 1192 bound to eIF4A almost as strongly as the 1245 construct (Fig. 1E, lanes 3, 4, and 7). Such binding, however, was abolished with the 1085, 1222, and 1235 fragments (Fig. 1E, lanes 2, 5, and 6). Truncation of the IDL to fragments 1222 and 1235 not only abolished eIF4A binding to HEAT-2/-3 but also enabled binding of eIF4B, which did not interact with any of the other fragments tested (Fig. 1E, lanes 2, 5, and 6). Coimmunoprecipitation with the 1222 fragment, but not the 1235 fragment, also yielded eIF3A, suggesting that eIF3 associates with the distal IDL in eIF4G indirectly through eIF4B (Fig. 1E).
Our results suggest a model, wherein interactions of the IDL (in the aa 1177 to 1192 range) with HEAT-3 favor eIF4A binding to HEAT-2 (Fig. 1F, left panel). Upon IDL truncation to 1222, an eIF4B-binding site within the IDL becomes accessible and eIF4B-IDL binding displaces eIF4A from HEAT-2 (Fig. 1F, middle panel). Elimination of the IDL prevents eIF4B binding by deleting the eIF4B binding site in the distal IDL and enables unencumbered HEAT-2-eIF4A interactions (Fig. 1F, right panel). This suggests an influence of intra/intermolecular interactions (i) between the IDL and HEAT-3 and (ii) between eIF4B/-3 and a new eIF4B-binding site within aa 1222 to 1245 of eIF4G, on assembly of eIF4G/-4A/-4B helicase complexes with HEAT-2 and on Mnk1 binding to HEAT-3.
eIF4G(S1232) is phosphorylated by Erk1/2 upon TPA stimulation.
The very C-terminal part of the IDL (aa 1222 to 1235) and the adjacent HEAT-2 domain (aa 1236 to 1245) mediate autoinhibitory functions for Mnk1 binding to HEAT-3 (Fig. 1C and D) and eIF4A binding to HEAT-2 (Fig. 1D and E) and contain an eIF4B binding site (Fig. 1E). Since these interactions were characterized based on artificial IDL truncations, we sought to determine what features of natural eIF4G may coordinate dynamic protein-protein interactions in this region of eIF4G. Within the aa 1222 to 1245 span, there are two major, confirmed phosphorylation sites at S1232 and S1239 (Fig. 1B and 2A). HEAT-2 contains a putative Erk1/2 binding motif (Fig. 2A) and S1232 is in optimal context for Erk1/2 MAPKs (Fig. 2B). Therefore, we sought to confirm earlier indications that eIF4G(S1232) phosphorylation, which is sensitive to the Mek1 inhibitor UO126, is phosphorylated by Erk1/2 (13). We tested whether recombinant Erk2 induces eIF4G(S1232) phosphorylation by incubating lysates from serum-starved, UO126-treated HEK293 cells with GST-Erk2 (Fig. 2C). Recombinant Erk2 was used in these assays because Erk2 coimmunoprecipitated with eIF4G to a greater extent than Erk1 (Fig. 2F) and because Erk1 and -2 possess identical substrate specificity in vitro. Endogenous eIF4G(S1232) phosphorylation was abolished in UO126-treated HEK293 cell lysates (Fig. 2C). None of the endogenous kinases present in UO126-treated cells were able to phosphorylate S1232 (evident from incubation of cell lysates with 2 mM ATP alone) (Fig. 2C). Only the addition of recombinant GST-Erk2 restored efficient eIF4G(S1232) phosphorylation (Fig. 2C). This was confirmed by in vitro phosphorylation of a recombinant C-terminal fragment of eIF4G (GST-Ct; aa 683 to 1600) upon incubation with GST-Erk2 in the presence of ATP (Fig. 2D).
Fig 2.
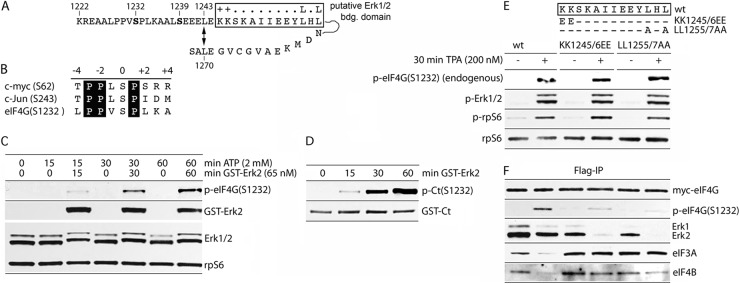
Erk2 phosphorylation of eIF4G(S1232) in vitro and identification of an Erk1/2 binding site in eIF4G. (A) eIF4G sequence surrounding SS1232/9 and a putative Erk1/2 binding site (boxed). A proposed α1/-2 helix stacking interaction (L1243↔L1270), based on HEAT-2 structure (8), is shown. (B) eIF4G(S1232) context compared to Erk1/2 substrates in c-myc/c-Jun. Letters in black boxes represent sequences strongly preferred by Erk1/2. (C) Phosphorylation of endogenous eIF4G(S1232) by recombinant GST-Erk2. Lysates of serum-starved (24 h) and UO126 (20 μM)-treated (2 h) HEK293 cells were incubated with recombinant GST-Erk2 (65 nM) in the presence of 2 mM ATP for the indicated times. The samples were tested by immunoblot as indicated. (D) In vitro phosphorylation of recombinant GST-Ct (150 nM; in kinase assay buffer) with GST-Erk2. (E) Mutational analysis of a putative Erk1/2 binding site in eIF4G. The proposed Erk1/2 docking site and two double mutants used in our studies are shown atop. HEK293 cells were transfected with wt fragment 1222 or the 1222(KK1245/6EE) or 1222(LL1255/7AA) mutant (16 h), serum starved (24 h), and treated with DMSO (−) or TPA (200 nM; +) (30 min). Cell lysates were subjected to immunoblot with the indicated antibodies (E) or IP/immunoblot (F). The experiment was repeated three times; a representative assay is shown.
Next, we analyzed the role of a putative Erk1/2 binding domain in eIF4G (aa 1245 to 1257) (Fig. 2A) in S1232 phosphorylation. Consensus Erk1/2 docking motifs contain two adjacent positively charged aa (Fig. 2A, ++) and two hydrophobic amino acids separated by one amino acid (Fig. 2A, L.L). To test a role of this sequence in Erk1/2 binding to eIF4G and in S1232 phosphorylation, we compared 1222 IDL fragments to the wild-type (wt) sequence or with KK1245/6EE or LL1255/7AA substitutions during TPA stimulation (Fig. 2E). Immunoblot of lysates revealed intact phosphorylation of endogenous eIF4G(S1232), Erk1/2, and rpS6 after TPA treatment (Fig. 2E). Flag-IP of the ectopic, Flag-tagged eIF4G fragments, however, indicated substantially diminished Erk1/2-eIF4G binding and S1232 phosphorylation with both mutant isoforms (Fig. 2F). Interestingly, phosphorylation of S1232 in the wt fragment inhibited the interaction of eIF4G with eIF3A and -4B; both double mutations in the Erk1/2 docking motif abolished this effect (Fig. 2F). Our findings suggest that a properly configured binding motif controls Erk1/2 positioning relative to its substrate site at eIF4G(S1232).
eIF4G(S1239) is phosphorylated by casein kinase 2 in vitro.
Prior analyses confirmed that eIF4G(S1239) is phosphorylated in vivo (11). Since S1239 is in direct proximity to S1232 and the Erk1/2 binding site in HEAT-2 (Fig. 2A), may influence S1232 phosphorylation by Erk1/2, and/or may participate in coordination of protein-protein interactions with the IDL/HEAT-2, we sought to identify the kinase(s) responsible for eIF4G(S1239) phosphorylation. Downstream sequences of S1239 contain several acidic and no basic residues, suggesting that it may be a substrate for acidophylic casein kinases. Indeed, alignment with the optimal substrate consensus for the casein kinases Ck1-α (15), Ck1-δ (16), and Ck2-α (17) revealed similarities with the eIF4G(S1239) context (Fig. 3A).
Fig 3.
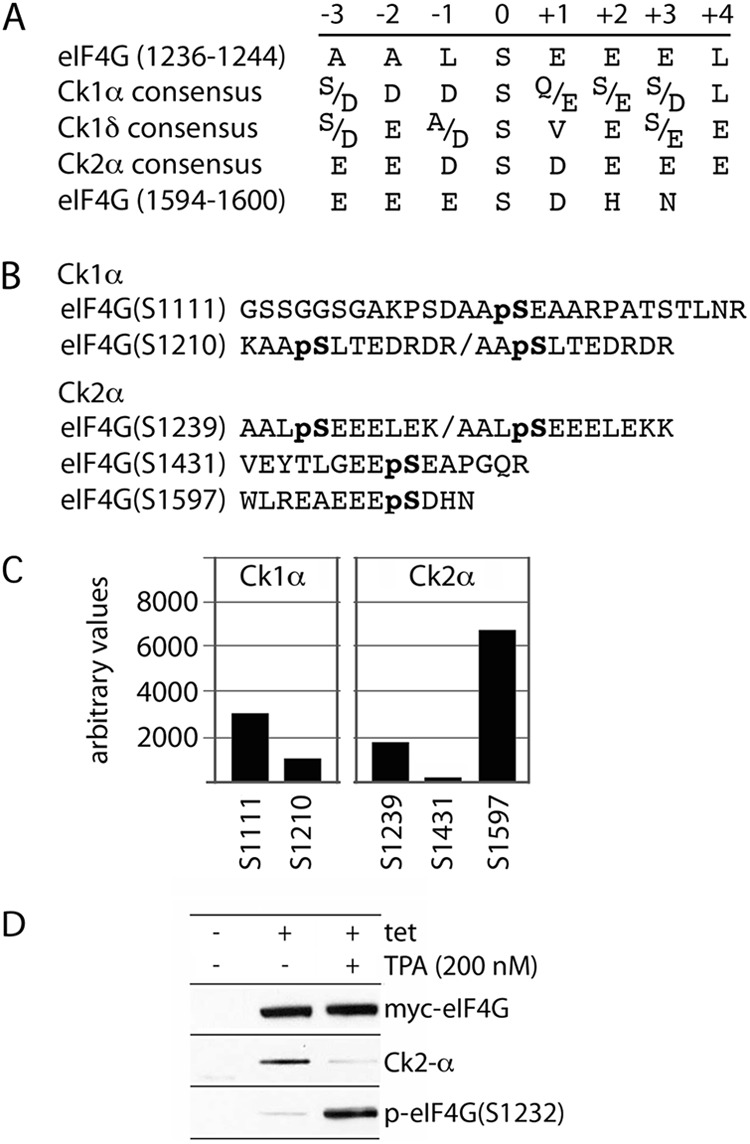
In vitro phosphorylation of eIF4G by Ck2-α. (A) Substrate consensus of Ck1-α, Ck1-δ, and Ck2-α compared to eIF4G(S1239) and (S1597). (B) Sequences of major in vitro phosphorylated eIF4G peptides determined from tandem mass spectrometry; “pS” denotes p-Ser residues. Only phosphopeptides with >95% probability of identification and a Mascot ion score of >25 are presented. (C) Quantitative phosphoproteomic analysis of relative phosphorylation intensities of major sites in Ck1-α and Ck2-α in vitro-phosphorylated recombinant eIF4G(Ct). The relative intensity is the intensity ratio of the phosphopeptide phosphorylated in the presence of recombinant kinase/ATP to the intensity of the same peptide incubated with ATP alone. (D) Direct binding of Ck2-α with eIF4G in Tet-induced HEK293 cells expressing eIF4G after mock or TPA stimulation.
To test an involvement of Cks in eIF4G(S1239) phosphorylation, we performed in vitro phosphorylation of GST-Ct by recombinant Ck1-α, Ck1-δ, or Ck2-α. Since a specific p-S1239 antibody is not available, the in vitro-phosphorylated products were subjected to quantitative phosphoproteomic analysis. Major eIF4G phospho-peptides resulted from the incubation with Ck1-α and Ck2-α only (Fig. 3B), suggesting that Ck1-δ is not an eIF4G kinase. Ck1-α and Ck2-α yielded two and three nonoverlapping phospho-peptides, respectively (Fig. 3B). eIF4G(S1239) was phosphorylated specifically by Ck2-α, suggesting that it may be a substrate for Ck2-α in vivo (Fig. 3C). Interestingly, a major Ck2-α site may reside in eIF4G(S1597) (Fig. 3C), within the binding motif for the Mnks, in accordance with the sequence context preference for Ck2-α (Fig. 3A). This site, and the biological significance of Ck2-α-mediated phosphorylation of S1597, is the subject of a separate study. To investigate the direct interactions between eIF4G and Ck2-α in vivo, we tested the Flag immunoprecipitates from Tet-inducible HEK293 cells expressing myc-eIF4G(197-1600)-Flag (14) (Fig. 3D). This assay confirmed eIF4G-Ck2-α binding, which was diminished upon TPA stimulation, similar to Erk1/2 binding to eIF4G (Fig. 3D).
Phosphorylation of eIF4G(S1232) is influenced by the phospho status of S1239.
We investigated whether the rate of Erk1/2 phosphorylation of eIF4G(S1232) is affected by the phosphorylation state of S1239 (Fig. 4). First, Ck2-α inhibition (with CX4945) in TPA-stimulated HEK293 cells showed an inhibitory effect on endogenous eIF4G(S1232) phosphorylation without diminishing Pkc/Ras downstream signals to Erk1/2 (Fig. 4A). Second, tests with the 1222 IDL fragments bearing a Ser, Ala, or Glu residue in position 1239 revealed effects on the efficiency of S1232 phosphorylation (Fig. 4B). Serum-starved HEK293 cells transfected with the S1239, S1239A, or S1239E constructs were stimulated with TPA for the indicated intervals, and their lysates were analyzed by Flag-IP and immunoblotting (Fig. 4B). Substitution of S1239A reduced and delayed TPA-induced S1232 phosphorylation (4B and C). This effect was partially compensated for by the S1239 phospho-mimic (Fig. 4B). The degree of S1232 phosphorylation inversely correlated with eIF4B/-3A binding to the 1222 fragment, indicating that the phosphorylation of S1232, likely under the influence of S1239 phosphorylation status, physiologically controls eIF4B access to its binding site in the distal IDL (Fig. 4B). Accordingly, eIF4B binding to the wt 1222 IDL was reduced with increasing S1232 phosphorylation, and eIF4B dissociation was substantially slowed with the S1232A mutant (Fig. 4B). At 2 h after TPA addition, all eIF4B/-3A association with the 1222 fragment had ceased, in correlation with the enhanced S1232 phosphorylation state of all S1239 variants, even S1239A (Fig. 4B).
Fig 4.
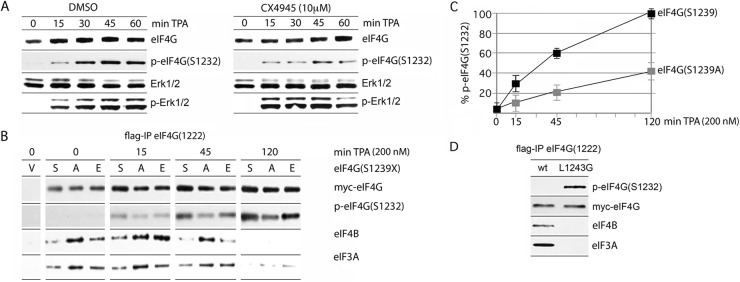
Effect of eIF4G(S1239) phosphorylation on S1232 phosphorylation and binding with eIF3A/-4B. (A) HEK293 cells were mock treated (DMSO) or treated with 10 μM CX4945 (2 h) and lysed after the indicated intervals of TPA (200 nM) stimulation. (B) Kinetics of TPA-stimulated eIF4G-IDL fragment 1222(S1232) phosphorylation and binding with eIF3A/-4B. 1222(S1239A) and 1222(S1239E) mutants were compared to the wt 1222 fragment. HEK293 cells were transfected with the indicated fragments (16 h), serum starved (24 h), and treated with 200 nM TPA as shown. Lysates were subjected to IP/immunoblotting with the indicated antibodies. The test was repeated three times; the results of a representative assay are shown. (C) Quantitative assessment of S1232 phosphorylation in the 1222 wt versus 1222(S1239A) fragment. Protein bands were quantified as described in Materials and Methods, adjusted for the loading control (myc-eIF4G), and plotted as the percent phosphorylation versus the time of TPA stimulation. The data represent average values from three independent experiments and standard error. (D) Constitutive eIF4G(S1232) phosphorylation upon L1243G substitution. HEK293 cells were transfected (16 h) with the 1222 wt fragment or a 1222(L1243G) mutant and serum starved (24 h). Lysates were subjected to IP/immunoblotting with the indicated antibodies. The experiment was repeated three times; the results of a representative assay are shown.
Based on the proposed helicase complex structure (8), we hypothesized that eIF4G(S1232) phosphorylation may be codetermined by intramolecular α-helix stacking interactions in HEAT-2 (indicated in Fig. 2A). Phosphorylation of the adjacent S1239, generating a negative charge and electrostatic repulsion of the neighboring α2-helix (8), may favor Erk1/2 association with its docking motif in HEAT-2. To test this hypothesis, we analyzed S1232 phosphorylation of a mutant 1222(L1243G) fragment (Fig. D). L1243G substitution was implemented to discourage putative α1-, α2-helix stacking interactions between L1243 (α1) and L1270 (α2) (indicated in Fig. 2A). The 1222(L1243G) variant, but not the wt fragment, was constitutively phosphorylated in the absence of TPA stimulation, presumably due to inherent, low Erk1/2 activity in HEK293 cells (Fig. 4D). Constitutive S1232 phosphorylation in 1222(L1243G) negatively regulates the binding of mutant eIF4G with eIF4B and -3A (Fig. 4D), in agreement with a role for eIF4G(S1232) phosphorylation in dissociation of eIF4B from the distal IDL (Fig. 4B). Taken together, our findings support a possible conformational arrangement of HEAT-2 that discourages Erk1/2 binding and S1232 phosphorylation in the S1239 native state. Upon S1239 phosphorylation, Erk1/2 binding to eIF4G, the phosphorylation of S1232, and the dissociation of eIF4B/-3A from the distal IDL are enhanced.
S1232 phosphorylation controls protein-protein interactions in the C-terminal part of eIF4G.
Our assays suggest that eIF4G(S1232) phosphorylation by Erk1/2 controls eIF4B binding to a newly identified binding site in the distal IDL (Fig. 4B). Since protein-protein interactions in eIF4G HEAT-2/-3 are determined by a multitude of inter-relating and dynamic features, we conducted a broad investigation of the effect of S1232 and S1239 phospho status on eIF4G binding with eIF4B, -3A, and -4A and Mnk1 (Fig. 5). HEK293 cells were transfected with 1222 eIF4G fragments (either wt, an S1239A mutant, an S1232A mutant or an SS1232/9AA double mutant [Fig. 5]), and binding with eIF4A, -4B, and -3A and Mnk1 was analyzed by Flag-IP. The cells were serum starved (for 24 h), TPA stimulated, and analyzed for dynamic protein-protein interactions over a 2-h time interval.
Fig 5.

Time course of TPA-induced binding of eIF4A, eIF4B, eIF3A, and Mnk1 with wt and mutant 1222 eIF4G fragments. HEK293 cells were transfected with the indicated 1222 fragments (16 h), serum starved (24 h), and treated with 200 nM TPA for the intervals shown. Lysates were subjected to IP/immunoblotting with the indicated antibodies. The experiments were repeated three times, and the results of a representative series are shown. (A) TPA-induced binding of eIF4A, -4B, and -3A to wt 1222, 1222(S1239A), 1222(S1232A), or 1222(SS1239/2AA). Due to very weak binding of the 1222 fragment with eIF4A (Fig. 1D and E), we exposed filters for extended intervals for detection of signal. (B) Quantitative chemiluminescence detection of the immunoblot signals for wt and double mutant 1222 fragments. Average values of the relative binding are shown; standard errors were <12% from the average values for 0 and 15 min of TPA stimulation and <15% for the 45- and 120-min time points.
With wt 1222, TPA stimulation gradually enhanced Mnk1-eIF4G binding to reach ∼6-fold levels of the uninduced state by 2 h (Fig. 5B, left panel). Simultaneously, binding with eIF4A/-4B and eIF3A declined rapidly, and all but disappeared by 2 h (Fig. 5). TPA-induced Mnk1 binding with the S1239A and SS1232/9AA eIF4G mutants was less efficient relative to wt eIF4G or the S1232A mutant, and Mnk1-eIF4G binding was increased in the uninduced state with all eIF4G phosphosite mutants (compared to wt; Fig. 5A).
The S1239A, S1232A, and SS1232/9AA substitutions significantly protracted dissociation of eIF4A/-4B and eIF3A from eIF4G HEAT-2 (Fig. 5). Accordingly, binding of eIF4A/-4B/-3A with eIF4G HEAT-2 in the uninduced state was substantially enhanced, and eIF4A/-4B/-3A dissociation was delayed with the SS1232/9AA double mutant (Fig. 5). Binding of eIF3A with the SS1232/9AA eIF4G mutant was much more sensitive to TPA stimulation than the association of eIF4B with the same variant (compare the relative binding slopes in Fig. 5B). We stipulate that this is due to the changing phosphorylation status of eIF4B and/or eIF3 components upon activation of the Pkc/Ras/Erk cascade. For example, it has been shown that phosphorylation of eIF4B(S422) by the Erk1/2 MAPK-activated protein kinase (Rsk) controls the association of eIF4B with eIF3 (18). Our observations indicate that coordinated phosphorylation of S1239 and S1232 at the IDL-HEAT-2 junction affects dynamic protein-protein interactions with eIF4A/-4B/-3A and Mnk1 in the C-terminal portion of eIF4G.
Binding of eIF4A/-4B and eIF3 with intact eIF4G compared to HEAT-1.
The eIF4G/-4A/-4B helicase complex can assemble with eIF4G HEAT-1 or, alternatively, with HEAT-2/the distal IDL alone. To investigate the relative influence of HEAT-2 on eIF4G helicase complex assembly upon Pkc/Ras/Erk activation, we tested binding of full-size eIF4G with eIF4A/-4B compared to HEAT-1 alone. HEK293 cell lines with Tet-inducible expression of myc-eIF4G(197-1600)-Flag (13) (Fig. 6A) or tagged eIF4G(682-1132), corresponding to HEAT-1 (Fig. 6B) were TPA stimulated, and the association of eIF4G with eIF4A, eIF4B, eIF3A, rpS6, and Mnk1 was assessed (Fig. 6). As expected, intact eIF4G exhibited S1232 phosphorylation in a TPA-dependent manner (Fig. 6A), whereas HEAT-1, lacking S1232, did not (Fig. 6B). In accordance with previously published data (13), Mnk1 and p-rpS6 coimmunoprecipitation with eIF4G was significantly enhanced by TPA stimulation (Fig. 6A). We observed modest, but reproducible, dissociation of eIF4B from full-length eIF4G by ∼4-h TPA stimulation (Fig. 6A). This did not occur with coimmunoprecipitation of the HEAT-1 fragment, suggesting that eIF4A/-4B associations with HEAT-1/the proximal IDL (8) are not influenced by TPA-induced signals. Thus, the differences in eIF4B coimmunoprecipitation with full-length eIF4G versus HEAT-1 upon TPA induction may be due to Pkc/Ras/Erk-mediated signals to eIF4G(S1232) resulting in eIF4B detachment from distal IDL/HEAT-2 (Fig. 6B). To test our hypothesis that eIF4A/-4B/-3A complex formation with eIF4G is codetermined by Erk/Rsk signal transduction to eIF4B (see above) (Fig. 5B), we compared coimmunoprecipitation of total eIF4B and p-eIF4B(S406) to eIF4G(682-1132) (Fig. 6B). Despite equal binding of total eIF4B with or without TPA stimulation, the proportion of p-eIF4B(S406) that coimmunoprecipitated with eIF4G(682-1132) increased with prolonged TPA stimulation (Fig. 6B). This suggests that multiple, coordinated translation factor phosphorylation events control the formation of the eIF4G/-4A/-4B helicase complex and its association with eIF3.
Fig 6.
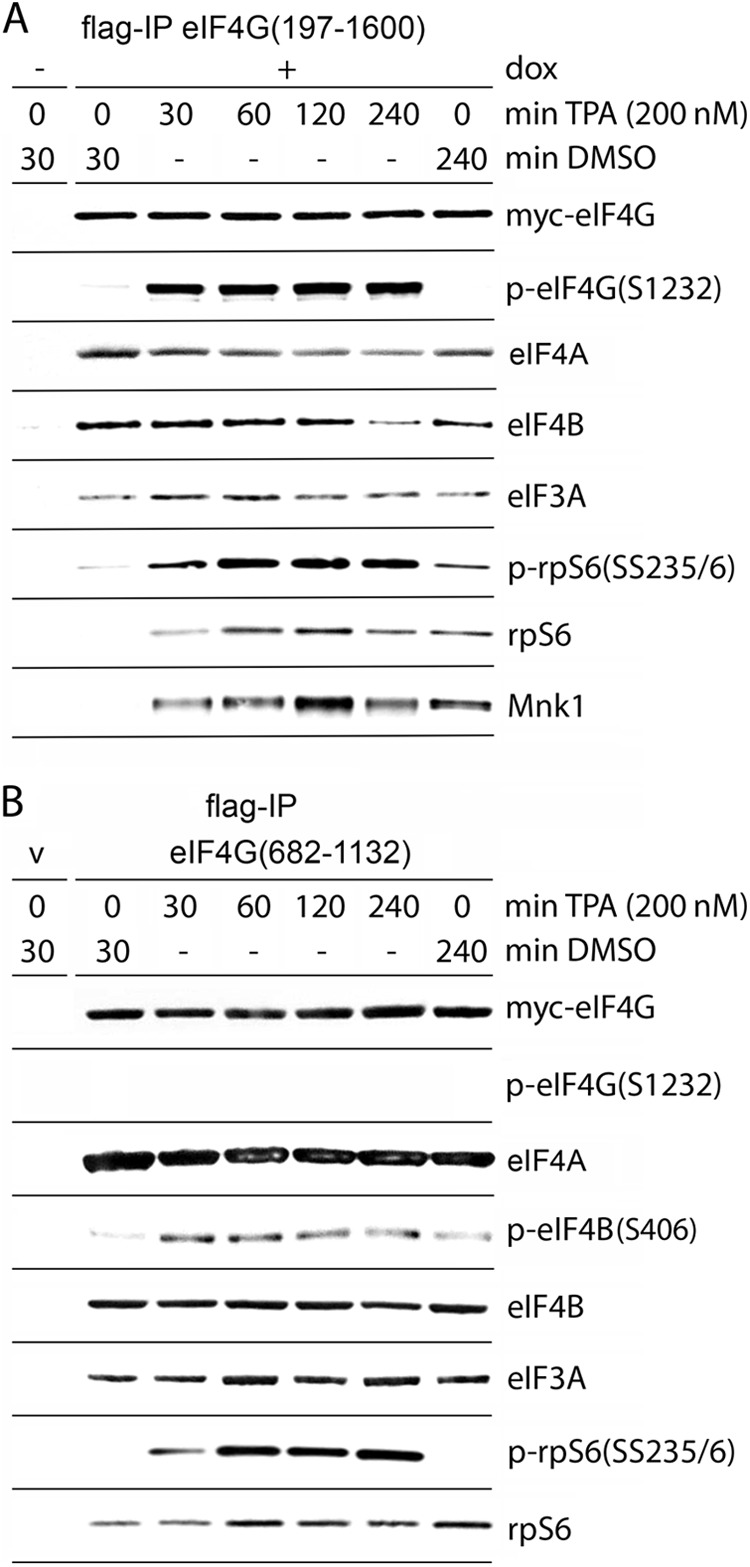
eIF4G/-4A/-4B helicase complex formation and/or association with eIF3 and the 40S ribosomal subunit with full-length eIF4G or with HEAT-1. (A and B) Dox-inducible HEK293 cells expressing myc-eIF4G(197-1600)-Flag (A) or HEK293 cells transfected (16 h) with myc-eIF4G(682-1132)-Flag (B) were serum starved (24 h) and treated for the intervals shown with DMSO or 200 nM TPA. Lysates were subjected to Flag-IP/immunoblotting with the indicated antibodies. The experiments were repeated three times, and the results of representative assays are shown.
ATP/ADP binding negatively regulates eIF4A–HEAT-2 interactions, whereas eIF4B binding to the distal IDL is nucleotide independent.
eIF4A interactions with eIF4G HEAT-1/-2 (and other accessory proteins) are continuously rearranged during the ATP-binding/hydrolysis cycle of the helicase in vitro (8). To investigate possible effects of nucleotides on eIF4A/-4B association with IDL-HEAT-2 in vivo, we transfected HEK293 cells with eIF4G fragment 1222 (to test the binding of eIF4B to the distal IDL/HEAT-2) or fragments 1192 and 1245 (to probe for eIF4A binding to HEAT-2). Cell lysates were subjected to Flag-IP overnight. For the last hour of incubation, the IP reactions were treated with 0.3 to 1 mM ADP/ATP or a 1 mM concentration of the nonhydrolyzable ATP analog adenosine 5′-[β,γ-imido]triphosphate (pNppA) and probed for HEAT-2-eIF4A/-4B and HEAT-3-Mnk1 associations (Fig. 7). Both ADP and ATP significantly decreased the affinity of eIF4A for HEAT-2 in a concentration-dependent manner (ATP) but failed to influence Mnk1 binding to HEAT-3 (Fig. 7A). The eIF4A helicase possesses ATPase activity, and the moderate decrease in binding to the eIF4G(1245) fragment may be due to hydrolysis of ATP to ADP. To control for this possibility, we use the nonhydrolyzable ATP analog pNppA in our assay (Fig. 7B). As observed before, ADP (1 mM) treatment had the strongest inhibitory effect on eIF4A/-4G binding. ATP or pNppA (both at 1 mM) had similar, moderate effects in comparison (Fig. 7B). These findings suggest that ATP hydrolysis is not a significant factor in controlling the association of eIF4A with eIF4G in our assay.
The 1245 fragment has no affinity for eIF4B (Fig. 1D and E). Therefore, to analyze the nucleotide dependence of eIF4B-binding to IDL/HEAT-2, we compared the 1222 wt fragment with a 1222(SS1232/9AA) double mutation that exhibited substantially increased binding affinity to eIF4A/-4B/-3A (Fig. 5B). IP of lysates from transfected HEK293 cells, followed by incubation with ADP or ATP, revealed that ATP/ADP prevented the binding of eIF4A, but not of eIF4B, with the distal IDL-HEAT-2 in eIF4G (Fig. 7C).
DISCUSSION
The C-terminal part of eIF4G in higher eukaryotes, and its interactions with the eIF4A helicase and with Mnk1, likely serve modulatory roles in protein synthesis (19). In this report, we show that Mnk1 binding to eIF4G is strongly regulated by the flexible IDL and its conformational arrangement within eIF4G. Our studies revealed two inhibitory regions within the IDL (aa 1162 to 1177 and aa 1222 to 1245) that restrict Mnk1 association with HEAT-3 and eIF4A binding to HEAT-2. The role of the IDL in forming eIF4G/-4A/-4B helicase complexes and controlling eIF4G-Mnk1 interaction is underlined by the identification of a new eIF4B-binding site (within the distal inhibitory region of aa 1222 to 1245).
The eIF4G HEAT-2 domain (exemplified by fragment 1245 in our study) contains a complete eIF4A-binding site, enabling very efficient eIF4A binding. This association is precluded when adding the distal IDL (aa 1222 to 1245), harboring a binding site for eIF4B. Extending the IDL further (fragments 1192 and 1177) exposed possible intramolecular interactions between IDL in the range of aa 1177 to 1192 with the HEAT-3 domain (13). This “closed” conformation might restrict the access of eIF4B to its binding site (aa 1222 to 1245), stimulate HEAT-2-eIF4A association, and enable Mnk1 binding to HEAT-3. Our assays show that within the aa 1222 to 1245 segment of the IDL, two phosphorylation sites at S1232 and S1239 are substrates of Erk1/2 and Ck2-α, respectively.
Ck2-α is a highly conserved, constitutively active serine/threonine protein kinase that participates in the regulation of multiple signaling networks including PI3k/Akt (20), Wnt (21) or the DNA-damage response (22). A unique feature of Ck2-α is that phosphorylation of its substrates can destabilize α-helices or other highly structured protein segments (23). Our data suggest that eIF4G(S1239) phosphorylation by Ck2-α may lead to partial unfolding of the α1-helix of HEAT-2, exposing an Erk1/2 binding site at aa 1245 to 1257, enabling Erk1/2 binding to eIF4G and stimulating phosphorylation of eIF4G(S1232) by Erk1/2. The functional consequence of phosphorylation of both sites is the dissociation of eIF4G HEAT-2 from the complex with eIF4A/-4B/-3 and the enhanced association of HEAT-3 with Mnk1.
The newly identified eIF4B/-3A binding site at the IDL/HEAT-2 junction (aa 1222 to 1245) of eIF4G could help to explain a previously proposed in vitro model for mRNA unwinding by the eIF4G/-4A/-4B(H) helicase complex (8). The HEAT-1 domain in eIF4G requires the closed, ATP-bound conformation of eIF4A for optimal binding, whereas HEAT-2 prefers the open, nucleotide-free conformation (8). Our data confirm those of Marintchev et al. (8), showing that eIF4A binding with HEAT-2 is also ATP/ADP-dependent in vivo. In contrast, eIF4B/-3A binding to the distal IDL is nucleotide and eIF4A independent. Thus, eIF4B/-3A binding to the aa 1222 to 1245 site in the IDL may facilitate the translocation of eIF4A from a complex with HEAT-2 to assemble with HEAT-1 during the unwinding cycle (Fig. 8).
Fig 8.
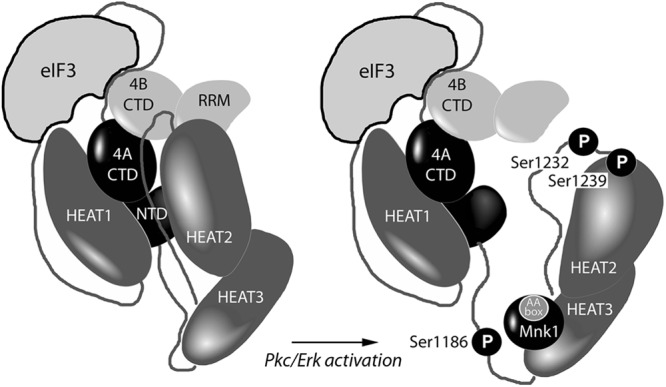
Model for proposed rearrangement of the eIF4G/-4A/-4B helicase complex upon Pkc/Erk activation. The mutual orientations of the eIF4G HEAT domains (dark gray) with the eIF4A N- and C-terminal domains (black), the eIF4B C-terminal and RRM domains (light gray), and eIF3 based on crystal structure and nuclear magnetic resonance chemical shift mapping data are depicted (8). Three investigated eIF4G phosphorylation sites and Mnk1 bound to AA box 1 of eIF4G HEAT-3 are shown.
The in vitro unwinding model of Marintchev et al. (8) proposed that, in the absence of mRNA, the function of HEAT-2 would be to lower the affinity of eIF4A for ATP and thus suppress idle ATPase activity in the eIF4G/-4A/-4B helicase complex. Phosphorylation of Ser1239 and S1232 occurs upon mitogenic stimulation, where mRNA 5′UTR scanning is active and the helicase complex is mostly bound with mRNAs and ribosomal subunits. During active unwinding/scanning, inhibiting the ATPase activity of eIF4A by sequestering it in a complex with HEAT-2 is no longer required, and SS1232/9-phosphorylated HEAT-2 dissociates from the complex with eIF4A/-4B (Fig. 8). This hypothesis is in agreement with the proposed function of another member of the eIF4G family: Dap5/p97 (with homology to aa 697 to 1600 in eIF4G). Dap5/p97 binds with eIF4A (24) but exclusively through a HEAT-1-like domain (25). Due to a lack of HEAT-2, tempering eIF4A ATPase activity, Dap5/p97 may exhibit constitutively high unwinding/scanning activities, e.g., required for the stimulation of translation initiation of specific mRNAs upon growth factor signaling (25) or in mitosis (26).
We recently reported that phorbol ester stimulation of cells results in phosphorylation of eIF4G(S1186) by Pkc-α and modulation of Mnk1 binding to eIF4G (13). In this report, we extend an important link between the Pkc/Ras/Erk signal transduction network and initiation of translation. We showed that TPA-dependent phosphorylation of eIF4G at S1232 by Erk1/2 caused dissociation of eIF4G HEAT2 from the complex with eIF4A/-4B/-3A and stimulated the association of HEAT-3 with Mnk1 (Fig. 8). Our results provide a mechanistic link between intracellular signal transduction and dynamic translation initiation complex formation coordinated by flexible eIF4G structure.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by Public Health Service grants CA140510 and CA124756 (M.G.).
We thank Mayya Shveygert (Duke University) for critical reading of the manuscript.
Footnotes
Published ahead of print 21 December 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.01441-12.
REFERENCES
- 1. Gingras AC, Raught B, Sonenberg N. 1999. eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu. Rev. Biochem. 68: 913–963 [DOI] [PubMed] [Google Scholar]
- 2. Parsyan A, Svitkin Y, Shahbazian D, Gkogkas C, Lasko P, Merrick WC, Sonenberg N. 2011. mRNA helicases: the tacticians of translational control. Nat. Rev. Mol. Cell. Biol. 12: 235–245 [DOI] [PubMed] [Google Scholar]
- 3. Caruthers JM, Johnson ER, McKay DB. 2000. Crystal structure of yeast initiation factor 4A, a DEAD-box RNA helicase. Proc. Natl. Acad. Sci. U. S. A. 97: 13080–13085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Richter-Cook NJ, Dever TE, Hensold JO, Merrick WC. 1998. Purification and characterization of a new eukaryotic protein translation factor: eukaryotic initiation factor 4H. J. Biol. Chem. 273: 7579–7587 [DOI] [PubMed] [Google Scholar]
- 5. Bellsolell L, Cho-Park PF, Poulin F, Sonenberg N, Burley SK. 2006. Two structurally atypical HEAT domains in the C-terminal portion of human eIF4G support binding to eIF4A and Mnk1. Structure 14: 913–923 [DOI] [PubMed] [Google Scholar]
- 6. Marcotrigiano J, Lomakin IB, Sonenberg N, Pestova TV, Hellen CU, Burley SK. 2001. A conserved HEAT domain within eIF4G directs assembly of the translation initiation machinery. Mol. Cell 7: 193–203 [DOI] [PubMed] [Google Scholar]
- 7. Imataka H, Sonenberg N. 1997. Human eukaryotic translation initiation factor 4G (eIF4G) possesses two separate and independent binding sites for eIF4A. Mol. Cell. Biol. 17: 6940–6947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Marintchev A, Edmonds KA, Marintcheva B, Hendrickson E, Oberer M, Suzuki C, Herdy B, Sonenberg N, Wagner G. 2009. Topology and regulation of the human eIF4A/4G/4H helicase complex in translation initiation. Cell 136: 447–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Oberer M, Marintchev A, Wagner G. 2005. Structural basis for the enhancement of eIF4A helicase activity by eIF4G. Genes Dev. 19: 2212–2223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Morino S, Imataka H, Svitkin YV, Pestova TV, Sonenberg N. 2000. Eukaryotic translation initiation factor 4E (eIF4E) binding site and the middle one-third of eIF4GI constitute the core domain for cap-dependent translation, and the C-terminal one-third functions as a modulatory region. Mol. Cell. Biol. 20: 468–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hsu PP, Kang SA, Rameseder J, Zhang Y, Ottina KA, Lim D, Peterson TR, Choi Y, Gray NS, Yaffe MB, Marto JA, Sabatini DM. 2011. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science 332: 1317–1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Raught B, Gingras AC, Gygi SP, Imataka H, Morino S, Gradi A, Aebersold R, Sonenberg N. 2000. Serum-stimulated, rapamycin-sensitive phosphorylation sites in the eukaryotic translation initiation factor 4GI. EMBO J. 19: 434–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dobrikov M, Dobrikova E, Shveygert M, Gromeier M. 2011. Phosphorylation of eukaryotic translation initiation factor 4G1 (eIF4G1) by protein kinase Cα regulates eIF4G1 binding to Mnk1. Mol. Cell. Biol. 31: 2947–2959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shveygert M, Kaiser C, Bradrick SS, Gromeier M. 2010. Regulation of eukaryotic initiation factor 4E (eIF4E) phosphorylation by mitogen-activated protein kinase occurs through modulation of Mnk1-eIF4G interaction. Mol. Cell. Biol. 30: 5160–5167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Al-Khouri AM, Ma Y, Togo SH, Williams S, Mustelin T. 2005. Cooperative phosphorylation of the tumor suppressor phosphatase and tensin homologue (PTEN) by casein kinases and glycogen synthase kinase 3beta. J. Biol. Chem. 280: 35195–35202 [DOI] [PubMed] [Google Scholar]
- 16. Brinkworth RI, Breinl RA, Kobe B. 2003. Structural basis and prediction of substrate specificity in protein serine/threonine kinases. Proc. Natl. Acad. Sci. U. S. A. 100: 74–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Litchfield DW. 2003. Protein kinase CK2: structure, regulation and role in cellular decisions of life and death. Biochem. J. 369: 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shahbazian D, Roux PP, Mieulet V, Cohen MS, Raught B, Taunton J, Hershey JW, Blenis J, Pende M, Sonenberg N. 2006. The mTOR/PI3K and MAPK pathways converge on eIF4B to control its phosphorylation and activity. EMBO J. 25: 2781–2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Marintchev A, Wagner G. 2004. Translation initiation: structures, mechanisms and evolution. Q. Rev. Biophysics 37: 197–284 [DOI] [PubMed] [Google Scholar]
- 20. Di Maira G, Salvi M, Arrigoni G, Marin O, Sarno S, Brustolon F, Pinna LA, Ruzzene M. 2005. Protein kinase CK2 phosphorylates and upregulates Akt/PKB. Cell Death Differ. 12: 668–677 [DOI] [PubMed] [Google Scholar]
- 21. Wang S, Jones KA. 2006. CK2 controls the recruitment of Wnt regulators to target genes in vivo. Curr. Biol. 16: 2239–2244 [DOI] [PubMed] [Google Scholar]
- 22. Keller DM, Lu H. 2002. p53 serine 392 phosphorylation increases after UV through induction of the assembly of the CK2.hSPT16.SSRP1 complex. J. Biol. Chem. 277: 50206–50213 [DOI] [PubMed] [Google Scholar]
- 23. Meggio F, Pinna LA. 2003. One-thousand-and-one substrates of protein kinase CK2? FASEB J. 17: 349–368 [DOI] [PubMed] [Google Scholar]
- 24. Imataka H, Olsen HS, Sonenberg N. 1997. A new translational regulator with homology to eukaryotic translation initiation factor 4G. EMBO J. 16: 817–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee SH, McCormick F. 2006. p97/DAP5 is a ribosome-associated factor that facilitates protein synthesis and cell proliferation by modulating the synthesis of cell cycle proteins. EMBO J. 25: 4008–4019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liberman N, Marash L, Kimchi A. 2009. The translation initiation factor DAP5 is a regulator of cell survival during mitosis. Cell Cycle 8: 204–209 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.