

Abstract
Using affinity purifications coupled with mass spectrometry and yeast two-hybrid assays, we show the Saccharomyces cerevisiae translation initiation factor complex eukaryotic translation initiation factor 2B (eIF2B) and the very-long-chain fatty acid (VLCFA) synthesis keto-reductase enzyme YBR159W physically interact. The data show that the interaction is specifically between YBR159W and eIF2B and not between other members of the translation initiation or VLCFA pathways. A ybr159wΔ null strain has a slow-growth phenotype and a reduced translation rate but a normal GCN4 response to amino acid starvation. Although YBR159W localizes to the endoplasmic reticulum membrane, subcellular fractionation experiments show that a fraction of eIF2B cofractionates with lipid membranes in a YBR159W-independent manner. We show that a ybr159wΔ yeast strain and other strains with null mutations in the VLCFA pathway cause eIF2B to appear as numerous foci throughout the cytoplasm.
INTRODUCTION
In eukaryotic translation initiation, the eukaryotic translation initiation factor 2 (eIF2) bound with GTP is required to interact with the initiator Meti-tRNA to form the ternary complex. After start codon recognition, eIF2-GTP is hydrolyzed to GDP, and eIF2-GDP dissociates from the translation initiation complex (1, 2). eIF2-GDP must exchange GDP with GTP before it can initiate another round of translation (Fig. 1A). The initiation factor eIF2B is an essential guanine nucleotide exchange factor (GEF) responsible for exchanging GDP for GTP on eIF2 (3). It is the only known target of eIF2B. This exchange reaction is one of the rate-limiting steps in translation initiation and is the target of numerous signaling pathways in yeast, as well as higher eukaryotes (4–10). Although the majority of eukaryotic GEFs are monomeric, eIF2B is unique among GEFs in that it is composed of multiple subunits. In Saccharomyces cerevisiae, eIF2B is composed of the five subunits GCD1, GCD2, GCN3, GCD6, and GCD7. The GCD6 subunit is necessary and sufficient for catalytic activity, although at a significantly reduced rate compared to the eIF2B complex (11–13). Coexpression of GCD6 with GCD1 yields similar GEF activity as the eIF2B holoenzyme (13). Of the other three subunits, previous studies show GCD2 and GCD7 to be involved in the stability of the complex and regulatory activity (13–15). GCN3 is required for eIF2B's role in the GCN4 stress response pathway (16, 17). With the exception of GCN3, all of the yeast eIF2B genes are essential (3).
Fig 1.
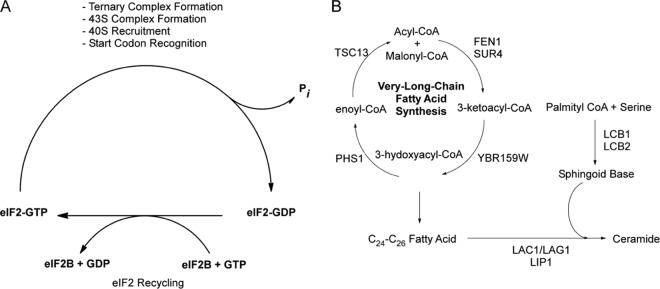
eIF2B and the VLCFA functional pathways. (A) Diagram showing the GEF pathway of eIF2B that is required for recharging eIF2 with GTP to begin a new round of translation initiation. (B) Diagram showing the cyclical VLCFA elongase pathway and the genes required for the catalytic steps. Also depicted is the pathway utilizing VLCFAs by the ceramide synthase complex LAC1/LAG1 and LIP1 to make ceramide. Ceramide is later modified to generate various sphingolipids.
Recent studies show that a significant fraction of yeast eIF2B resides in distinct foci in the cytoplasm known as “2B bodies” (18, 19). Green fluorescent protein (GFP) fluorescence microscopy shows the bodies contain both eIF2B and eIF2. The initiation factor eIF2 appears to shuttle in and out of the 2B bodies (18). The shuttling occurs quickly during logarithmic growth and slower following disruptions of translation initiation. The 2B bodies are thought to be the sites of eIF2B's GEF activity.
In eukaryotes, two distinct complexes are responsible for the synthesis of fatty acids (FA) (20, 21). The cytoplasmic fatty acid synthase (FAS) complex elongates fatty acids from 2 to 18 carbons in length in a four-reaction cycle. A second fatty acid synthesis complex, the elongase complex, is responsible for the elongation of fatty acids from 18 to 26 carbons (Fig. 1B) (22). The longer-chain fatty acids are known as very-long-chain fatty acids (VLCFAs). In S. cerevisiae, VLCFAs make up 1 to 5% of total fatty acids (22, 23) and the predominant VLCFA is 26 carbons long (24). The VLCFAs are crucial for the formation of lipid rafts in yeast (25). Although the FAS and elongase complexes share very similar catalytic steps, different sets of enzymes catalyze the elongation reactions in the two pathways (Fig. 1B). The elongase complex's enzymes localize to the endoplasmic reticulum (ER) membrane (26, 27). The complex receives fatty acids from the cytoplasmic FAS complex and elongates them to VLCFAs (28). Previous studies have shown that YBR159W, also known as IFA38, is a keto-acyl reductase required for the second step in the yeast elongase's pathway (Fig. 1B) (29, 30). A ybr159wΔ null yeast strain has a slow-growth phenotype and altered VLCFA lipid composition (30). Although both FEN1 and SUR4 catalyze the first enzymatic step in the elongase pathway, they are not redundant and are responsible for different-chain-length precursor fatty acids. FEN1 prefers 20-carbon-long precursors, while SUR4 has a broader range of chain length specificity but is required to convert 24-carbon-long VLCFAs to their final 26-carbon-long form (31). The elongase enzymes TSC13 and PHS1 are both essential (32, 33). In yeast, newly synthesized VLCFAs are predominantly incorporated first into ceramide and eventually into sphingolipids (24). LIP1 is a component of the ceramide synthase complex required for the formation of ceramide from a VLCFA and a sphingoid base (34). Each sphingolipid contains one 24- to 26-carbon-long VLCFA in addition to the long-chain base and head group (35).
The ER in budding yeast is composed of the classical membrane network connected to the nuclear envelope as well as a network of tubules known as the cortical ER. The cortical ER extends throughout the cell and encases the inner face of the entire plasma membrane (36). In microscopy, the cortical ER can often be mistaken as the plasma membrane itself (36). Although the bulk of yeast's cortical ER lies under the plasma membrane, in most metazoan cells, including mammalian cells, the ER is continuous with the nuclear envelope and forms a network of tubules throughout the cytoplasm that closely align with microtubules (37).
Using protein affinity purifications coupled with mass spectrometry and yeast two-hybrid analysis, we provide direct evidence for an interaction between the S. cerevisiae eIF2B complex and YBR159W. We found that in wild-type (WT) cells eIF2B colocalizes with lipid membranes and that this membrane colocalization is not altered in a ybr159wΔ strain. Our experiments show that a ybr159wΔ mutation causes eIF2B to appear as numerous foci. Although ybr159wΔ null cells have a lower rate of translation, the appearance of numerous eIF2B foci does not appear to correlate with the cell's translation rate. Other VLCFA mutant strains showing multiple eIF2B foci have WT translation rates. Overall, we demonstrate here a novel interaction between the essential yeast translation initiation factor and the fatty acid synthesis enzyme YBR159W.
MATERIALS AND METHODS
Strains and media.
All yeast medium, growth, and genetic manipulation experiments were performed according to standard techniques (38). To create the ybr159wΔ strain AL401, the kanamycin resistance cassette from plasmid pFa6a-kanmx6 was first amplified with primers CGTACGCTGCAGGTCGAC and ATCGATGAATTCGAGCTCG. Using the PCR double-fusion approach (38), the primers CGGATTTGGAAGTCCTTTATAG, GTCGACCTGCAGCGTACGCATTTCTTAAGCTGCACCG, CGAGCTCGAATTCATCGATTAGAATTATCGTTCTCG, and GGACTTGGTCCTTCCACC were used to expand the YBR159W genomic regions flanking the kanmx6 cassette. The YBR159W disruption cassette was transformed into strain BY4741, and transformants were selected on yeast extract-peptone-dextrose (YPD) plus 300 mM G418 plates and screened using Western blotting and anti-YBR159W polyclonal antibodies. Candidate BY4741 ybr159wΔ strains were crossed with the HIS+ strain H1511 and sporulated to create the ybr159wΔ null strain AL401. An isogenic WT HIS+ control strain AL400 was selected from the same sporulation. The lip1Δ strain RH5994 was kindly provided by the Howard Riezman laboratory (34). The gcn3Δ, fen1Δ, and sur4Δ deletion strains were obtained from the MATa yeast deletion collection (39). The fen1Δ and sur4Δ deletion strains from the MATa yeast deletion collection were mated with the HIS+ strain H1511 and sporulated to create the fen1Δ and sur4Δ strains, AL413 and AL414, respectively. The tandem affinity purification (TAP)-tagged strains were obtained from the yeast TAP-tagged library (40). The GFP-tagged strains were obtained from a GFP-tagged yeast library (41). To make the ybr159wΔ, GCD7-GFP strain, we mated the ybr159wΔ strain AL401 with the GCD7-GFP strain AL429 from the GFP-tagged yeast library and sporulated the diploids to obtain the ybr159wΔ, GCD7-GFP strain AL403. The yeast two-hybrid activation-domain strains derived from parent strain PJ69-4a, the binding-domain parent strain PJ69-4α, and yeast two-hybrid plasmids were obtained from the Yeast Resource Center (University of Washington) (42). Using the protocol previously described by the Yeast Resource Center (42), the AL408 (YBR159W-BD), AL409 (GCD1-BD), AL410 (GCD2-BD), AL411 (GCD6-BD), and AL412 (GCD7-BD) strains expressing yeast two-hybrid binding-domain tagged alleles were generated from parent strain PJ69-4α. Briefly, initial forward and reverse primers were used to PCR the target gene from yeast genomic DNA. The PCR product and the common forward and reverse two-hybrid primers were used for a second round of PCR to extend the flanking sequences. The second PCR product and the PvuII- and NcoI-linearized pOBD2 plasmid were cotransformed into yeast strain PJ69-4α and plated on synthetic complete medium lacking Trp (SC-Trp) to select for recombinants fusing the target gene to the GAL4-binding domain. Tables 1, 2, and 3 list all of the strains, plasmids, and primers used in the present study.
Table 1.
Strains used in this study
| Strain | Genotype | Source or reference |
|---|---|---|
| AL400 | MATaura3 leu2 HIS+ | This study |
| AL401 | MATaura3 leu2 HIS+ YBR159W::KanR | This study |
| AL402 | MATaura3 leu2 HIS+ YBR159W::KanR [YCp-YBR159W] | This study |
| AL403 | MATaura3 leu2 HIS+ YBR159W::KanR GCD7-GFP | This study |
| AL404 | MATaura3 leu2 HIS+ YBR159W::KanR GCD7-GFP[YCp-YBR159W] | This study |
| AL405 | MATaleu2 ura3 met15 GCD1-GFP[YCp-YBR159W-dsRed] | This study |
| AL406 | MATaleu2 ura3 met15 GCD6-GFP[YCp-YBR159W-dsRed] | This study |
| AL407 | MATaleu2 ura3 met15 GCD7-GFP[YCp-YBR159W-dsRed] | This study |
| AL408 | MATαtrp1-901 leu2-3,112 ura3-52 his3-200 gal4Δ gal80Δ LYS2::GAL1-HIS3 GAL2-ADE2 met2::GAL7-lacZ[YBR159W-GAL4DBD] | This study |
| AL409 | MATαtrp1-901 leu2-3,112 ura3-52 his3-200 gal4Δ gal80Δ LYS2::GAL1-HIS3 GAL2-ADE2 met2::GAL7-lacZ[GCD1-GAL4DBD] | This study |
| AL410 | MATα trp1-901 leu2-3,112 ura3-52 his3-200 gal4Δ gal80Δ LYS2::GAL1-HIS3 GAL2-ADE2 met2::GAL7-lacZ[GCD2-GAL4DBD] | This study |
| AL411 | MATα trp1-901 leu2-3,112 ura3-52 his3-200 gal4Δ gal80Δ LYS2::GAL1-HIS3 GAL2-ADE2 met2::GAL7-lacZ[GCD6-GAL4DBD] | This study |
| AL412 | MATα trp1-901 leu2-3,112 ura3-52 his3-200 gal4Δ gal80Δ LYS2::GAL1-HIS3 GAL2-ADE2 met2::GAL7-lacZ[GCD7-GAL4DBD] | This study |
| AL413 | MATaleu2 ura3 met15 HIS+ FEN1::KanR | This study |
| AL414 | MATaleu2 ura3 met15 HIS+ SUR4::KanR | This study |
| AL415 | MATaura3 leu2 HIS+[p180] | This study |
| AL416 | MATaura3 leu2 HIS+ YBR159W::KanR [p180] | This study |
| AL417 | MATaura3 leu2 HIS+ YBR159W::KanR [YCp-YBR159W] [p180] | This study |
| AL418 | MATaleu2 ura3 met15 FEN1::KanR [p180] | This study |
| AL419 | MATaleu2 ura3 met15 SUR4::KanR [p180] | This study |
| AL420 | MATα ura3-52 gcd1-101[p180] | This study |
| AL421 | MATα ura3-52 trp1-63 leu2-3 leu2-112 GAL2+ gcn2Δ[p180] | This study |
| AL422 | MATaleu2 ura3 met15 FEN1-GFP[YCp-YBR159W-dsRed] | This study |
| AL423 | MATaleu2 ura3 met15 DPM1-GFP[YCp-YBR159W-dsRed] | This study |
| AL424 | MATaleu2 ura3 met15 his3 GCN3::KanR | Deletion library (39) |
| AL425 | MATaleu2 ura3 met15 YBR159W-GFP | GFP library (41) |
| AL426 | MATaleu2 ura3 met15 FEN1-GFP | GFP library (41) |
| AL427 | MATaleu2 ura3 met15 GCD1-GFP | GFP library (41) |
| AL428 | MATaleu2 ura3 met15 GCD6-GFP | GFP library (41) |
| AL429 | MATaleu2 ura3 met15 GCD7-GFP | GFP library (41) |
| AL430 | MATaleu2 ura3 met15 GCD2-TAP | TAP library (40) |
| AL431 | MATaleu2 ura3 met15 GCD7-TAP | TAP library (40) |
| AL432 | MATaleu2 ura3 met15 YBR159W-TAP | TAP library (40) |
| AL433 | MATaleu2 ura3 met15 FEN1-TAP | TAP library (40) |
| AL434 | MATaleu2 ura3 met15 SUR4-TAP | TAP library (40) |
| AL435 | MATaleu2 ura3 met15 TSC13-TAP | TAP library (40) |
| AL436 | MATaleu2 ura3 met15 his3 FEN1::KanR | Deletion library (39) |
| AL436 | MATaleu2 ura3 met15 DPM1-GFP | GFP library (41) |
| AL437 | MATaleu2 ura3 met15 his3 SUR4::KanR | Deletion library (39) |
| F98 | MATα ura3-52 gcd1-101 | A. Hinnebusch |
| H1511 | MATα ura3-52 trp1-63 leu2-3 leu2-112 GAL2+ | A. Hinnebusch |
| H2557 | MATα ura3-52 trp1-63 leu2-3 leu2-112 GAL2+ gcn2Δ | A. Hinnebusch |
| pAD(GCD7) | MATatrp1-901 leu2-3,112 ura3-52 his3-200 gal4Δ gal80Δ LYS2::GAL1-HIS3 GAL2-ADE2 met2::GAL7-lacZ[GCD7-AD] | Yeast Resource Center (42) |
| PJ69-4a | MATatrp1-901 leu2-3,112 ura3-52 his3-200 gal4Δ gal80Δ LYS2::GAL1-HIS3 GAL2-ADE2 met2::GAL7-lacZ | Yeast Resource Center (42) |
| PJ69-4α | MATα trp1-901 leu2-3,112 ura3-52 his3-200 gal4Δ gal80Δ LYS2::GAL1-HIS3 GAL2-ADE2 met2::GAL7-lacZ | Yeast Resource Center (42) |
| pOAD(GCD1) | MATatrp1-901 leu2-3,112 ura3-52 his3-200 gal4Δ gal80Δ LYS2::GAL1-HIS3 GAL2-ADE2 met2::GAL7-lacZ[GCD1-AD] | Yeast Resource Center (42) |
| pOAD(GCD2) | MATatrp1-901 leu2-3,112 ura3-52 his3-200 gal4Δ gal80Δ LYS2::GAL1-HIS3 GAL2-ADE2 met2::GAL7-lacZ[GCD2-AD] | Yeast Resource Center (42) |
| pOAD(GCD6) | MATatrp1-901 leu2-3,112 ura3-52 his3-200 gal4Δ gal80Δ LYS2::GAL1-HIS3 GAL2-ADE2 met2::GAL7-lacZ[GCD6-AD] | Yeast Resource Center (42) |
| pOAD(GCN3) | MATatrp1-901 leu2-3,112 ura3-52 his3-200 gal4Δ gal80Δ LYS2::GAL1-HIS3 GAL2-ADE2 met2::GAL7-lacZ[GCN3-AD] | Yeast Resource Center (42) |
| pOAD(SUI2) | MATatrp1-901 leu2-3,112 ura3-52 his3-200 gal4Δ gal80Δ LYS2::GAL1-HIS3 GAL2-ADE2 met2::GAL7-lacZ[SUI2-AD] | Yeast Resource Center (42) |
| pOAD(TDH1) | MATatrp1-901 leu2-3,112 ura3-52 his3-200 gal4Δ gal80Δ LYS2::GAL1-HIS3 GAL2-ADE2 met2::GAL7-lacZ[TDH1-AD] | Yeast Resource Center (42) |
| pOAD(YBR159W) | MATatrp1-901 leu2-3,112 ura3-52 his3-200 gal4Δ gal80Δ LYS2::GAL1-HIS3 GAL2-ADE2 met2::GAL7-lacZ[YBR159W-AD] | Yeast Resource Center (42) |
| RH5994 | MATα leu2 ura3 trp1 bar1 LIP1::HIS3 | H. Riezman |
Table 2.
Plasmids used in this study
| Plasmid | Backbone | Notes | Source (reference) |
|---|---|---|---|
| pOBD2 | pOBD2 | ampR TRP1 CEN4 ORI GAL4-DBD | Yeast Resource Center (42) |
| YCp-YBR159W | pAG415GAL-ccdB | ampR LEU2 CEN ORI YBR159W 5′ UTR-YBR159W | This study |
| YCp-YBR159W-dsRed | pAG415GPD-ccdB-dsRed | ampR LEU2 CEN ORI PGPD-YBR159W-dsRed | This study |
| p180 | YCp50 | ampR URA3 CEN ORI GCN4 5′ UTR-LacZ | A. Hinnebusch |
| pFa6a-kanmx6 | pFa6a-kanmx6 | ampR KanR2 | Addgene |
| pENTR | pENTR | KanR | Invitrogen |
| pENTR-YBR159W 5′ UTR-YBR159W | pENTR | KanR YBR159W 5′ UTR-YBR159W | This study |
| pENTR-YBR159W | pENTR | KanR YBR159W | This study |
| pAG415GAL-ccdB | pAG415GAL-ccdB | ampR LEU2 CEN ORI ccdB | Addgene |
| pAG415GPD-ccdB-dsRed | pAG415GPD-ccdB-dsRed | ampR LEU2 CEN ORI ccdB-dsRed | Addgene |
Table 3.
Primers used in this study
| Primer | Orientationa | Sequence (5′–3′) |
|---|---|---|
| Genomic YBR159W deletion primers | ||
| YBR159W deletion primer | F | CGTACGCTGCAGGTCGAC |
| R | ATCGATGAATTCGAGCTCG | |
| 5′ Homology extension primer | F | CGGATTTGGAAGTCCTTTATAG |
| R | GTCGACCTGCAGCGTACGCATTTCTTAAGCTGCACCG | |
| 3′ Homology extension primer | F | CGAGCTCGAATTCATCGATTAGAATTATCGTTCTCG |
| R | GGACTTGGTCCTTCCACC | |
| Yeast two-hybrid primers | ||
| Two-hybrid common primer | F | CTATCTATTCGATGATGAAGATACCCCACCAAACCCAAAAAAAGAGATCGAATTCCAGCTGACCACCATG |
| R | GTACCGTTAAGGGCCCCTAGGCAGCTGGACGTCTCTAGATACTTAGCATCTATGACTTTTTGGGGCGTTC | |
| Two-hybrid YBR159W | F | AATTCCAGCTGACCACCATGACTTTTATGCAACAGCTTCAAGAGGCTGG |
| R | GATCCCCGGGAATTGCCATGCTATTCCTTTTTAACCTGTCTTGCGGCTTTTTTTAAGG | |
| Two-hybrid GCD1 | F | AATTCCAGCTGACCACCATGTCAATTCAGGCTTTTGTCTTTTGCGGTAAAGG |
| R | GATCCCCGGGAATTGCCATGTTAACGCTCAAATAATCCGTCATCTTCGTACTCGTAC | |
| Two-hybrid GCD2 | F | AATTCCAGCTGACCACCATGAGCGAATCGGAAGCCAAATCTAGGTCG |
| R | GATCCCCGGGAATTGCCATGTTATGCGGAACCTTTGTACTCTCTTAAAATAACAGGGAC | |
| Two-hybrid GCD6 | F | AATTCCAGCTGACCACCATGGCTGGAAAAAAGGGACAAAAGAAAAGTGGACTAG |
| R | GATCCCCGGGAATTGCCATGTTATTCCTCTTCTGAGGAAGATTCTTCGTCAGCATTC | |
| Two-hybrid GCD7 | F | AATTCCAGCTGACCACCATGTCCTCTCAAGCATTCACTTCAGTACATCCG |
| R | GATCCCCGGGAATTGCCATGTCACGCCTTATTTTTATCCAAATGCACATCAATTTGC | |
| YBR159W ORF + 600-bp upstream primers | ||
| 600-bp upstream YBR159W primer | F | CACCATGGTTTTTGTGACTTTACCTATAAATAGTACACAAC |
| YBR159W ORF primer | R | CTATTCCTTTTTAACCTGTCTTGCGGCTTTTTTTAAGGC |
| pAG415GAL-ccdB promoter remover 1 | GGGAGCTCCATACTGATTAGTACACTAGTGG | |
| pAG415GAL-ccdB promoter remover 2 | CCACTAGTGTACTAATCAGTATGGAGCTCCC |
F, forward; R, reverse.
Plasmids.
The plasmid pOBD2 used in generating yeast two-hybrid binding-domain strains has been previously described (43). To create a plasmid expressing an endogenous level of YBR159W, we used PCR to amplify the YBR159W gene along with 600 bp of the genomic region upstream of the gene's start codon and the YBR159's stop codon using the primers CACCATGGTTTTTGTGACTTTACCTATAAATAGTACACAAC and CTATTCCTTTTTAACCTGTCTTGCGGCTTTTTTTAAGGC. The PCR product was cloned into the pENTR entry vector using Directional TOPO Cloning (Invitrogen) to create pENTR-YBR159W 5′ untranslated region (UTR)-YBR159W. The YBR159W cassette was transferred to the pAG415GAL-ccdB yeast destination vector using LR Clonase recombination (Invitrogen) (44). To eliminate possible promoter interference, the vector's endogenous GAL promoter was deleted using the restriction enzymes SacI and SpeI and replaced with the primer insert GGGAGCTCCATACTGATTAGTACACTAGTGG and CCACTAGTGTACTAATCAGTATGGAGCTCCC to create the YBR159W expression plasmid YCp-YBR159W. To create a plasmid expressing RFP-tagged YBR159W, the YBR159W open reading frame (ORF) without the stop codon was amplified by PCR and cloned into the pENTR vector creating pENTR-YBR159W. The YBR159W ORF insert was transferred by recombinational cloning into the pAG415GPD-ccdB-dsRed vector (Addgene) to create the final expression plasmid YCp-YBR159W-dsRed. All plasmids used in the present study are listed in Table 2.
Antibodies.
The anti-YBR159W polyclonal antibodies were generated by inoculation of a rabbit with the synthetic peptide CETVKAENKKSGTRG (Covance). The peptide was covalently bound to cyanogen bromide beads (Sigma-Aldrich) to affinity purify anti-YBR159W from rabbit whole blood. Polyclonal antibodies to yeast SUI2 were kindly provided by Tom Dever. Polyclonal antibodies to yeast GCD6 and GCD1 were kindly provided by Alan Hinnebusch. The mouse anti-DPM1 was obtained from Molecular Probes. Antibodies to yeast TDH1, TDH2, and TDH3 were obtained from Millipore. The antibody to yeast RPL32 was kindly provided by Jonathan Warner.
Mass spectrometry and proteomics.
For yeast TAP experiments, TAP-tagged protein complexes were purified as previously described (45, 46). For each TAP strain, a 2-liter culture was grown to an optical density at 660 nm (OD660) of 1 to 2 in YPD. The purified TAP complexes were reduced with 1/10 volume of 50 mM dithiothreitol (DTT) at 65°C for 5 min, and cysteines were alkylated with 1/10 volume of 100 mM iodoacetamide at 30°C for 30 min. The proteins were digested overnight at 37°C with modified sequencing grade trypsin at an ∼25:1 substrate/enzyme ratio (Promega, Madison, WI). Proteins were identified using multidimensional protein identification technology (MudPIT) and an LTQ linear ion trap mass spectrometer (Thermo Fisher) (47, 48). A fritless, microcapillary (100-μm-inner-diameter) column was packed sequentially with 12 cm of 5-μm C18 reversed-phase packing material (Synergi 4μ Hydro RP80a; Phenomenex) and 3 cm of 5-μm strong cation exchange packing material (Partisphere SCX; Whatman). The entire trypsin-digested samples were loaded onto the biphasic column equilibrated in 0.1% formic acid–2% acetonitrile, which was then placed in-line with an LTQ linear ion trap mass spectrometer. An automated six-cycle multidimensional chromatographic separation was performed using buffer A (0.1% formic acid, 5% acetonitrile), buffer B (0.1% formic acid, 80% acetonitrile), and buffer C (0.1% formic acid, 5% acetonitrile, 500 mM ammonium acetate) at a flow rate of 300 nl/min. Cycles 1 to 6 consisted of 3 min of buffer A, 2 min of 0 to 100% buffer C, and 5 min of buffer A, followed by a 60-min linear gradient to 60% buffer B. In cycles 1 to 6, the percentage of buffer C was increased incrementally from 0, 15, 30, 50, 70, and 100% in each cycle. During the linear gradient, the eluting peptides were analyzed by one full mass spectrometry (MS) scan (300 to 2,000 m/z), followed by five tandem MS (MS/MS) scans on the five most abundant ions detected in the full MS scan while operating under dynamic exclusion.
For proteomic analysis of membrane float experiment's membrane and cytoplasmic fractions, a modified MudPIT protocol was utilized. Purified yeast protein and subcellular complexes were processed and analyzed essentially as described above except a 12-step MudPIT experiments was used with the salt pulses of 0, 25, 50, 75, 100, 150, 200, 250, 300, 500, and 750 mM and 1 M ammonium acetate. Eluting peptides were analyzed using an LTQ-OrbitrapXL mass spectrometer with preview mode and monoisotopic precursor selection enabled. The top 10 precursor ions based on intensity were fragmented using CID in the ion trap using 35% normalized collision energy. Dynamic exclusion was enabled for 180 s with repeat count of 1 at 30-s duration, list size of 500, mass tolerance of 10 ppm. MS data were analyzed as previously described (49).
GFP affinity purification.
Two liters of the GCD7-GFP ybr159wΔ strain AL403, the GCD7-GFP strain AL429, and the untagged ybr159wΔ strain AL401 were grown to an OD660 of 1 to 2 in YPD. Yeast cells were harvested by centrifuging at 1,500 × g for 5 min and resuspended in 10 ml of ice-cold NP-40 lysis buffer (6 mM Na2HPO4, 4 mM NaH2PO4, 1% NP-40, 150 mM NaCl, 2 mM EDTA, 50 mM NaF, 4 μg of leupeptin/ml, 0.1 mM Na3VO4). Cells were lysed for 10 min with glass beads in NP-40 lysis buffer. The lysates were centrifuged at 500 × g for 5 min. The cleared supernatant was brought up to 25 ml with ice-cold lysis buffer. A 500-μl bed volume of protein A/G-agarose beads (Thermo Scientific) and 50 μg of anti-GFP antibody (ThermoFisher) were added simultaneously and allowed to incubate for 1 h at room temperature. The beads were centrifuged at 300 × g for 5 min, transferred to a Poly Prep chromatography column (Bio-Rad), and washed at 4°C with 50 column volumes of wash buffer (10 mM Tris [pH 8.0], 150 mM NaCl, 0.1% NP-40). Protein digestion was carried out directly on the agarose beads. The beads were suspended in 1 ml of digestion buffer (10 mM Tris [pH 8.0], 150 mM NaCl, 0.5 mM EDTA, 1 mM DTT) and transferred to a 1.5-ml microcentrifuge tube. The resuspended beads were trypsin digested as described for yeast TAP complexes. After digestion the beads were centrifuged at 13,000 × g for 1 min, and the supernatant was transferred to a fresh microcentrifuge tube. MudPIT was performed identical as described for the TAP purifications. MS data was analyzed as previously described using Cn scoring filters of 1.5 (+1), 3.5 (+2), and 3.5 (+3) (49).
Fatty acid profiling.
The protocol for extracting lipids from yeast cells was adapted from Ejsing et al. (50). Each yeast strain was grown to an OD660 of 1.0 to 1.5 in YPD medium at 30°C. A 50-mg portion of wet weight yeast cells was incubated in 200 μl of phosphate-buffered saline (PBS) with 100 μg of lyticase (Sigma)/ml for 1 h at 37°C. Next, 990 μl of chloroform-methanol (17:1 [vol/vol]) was added, followed by incubation for 2 h at 37°C. The lower organic layer was collected and vacuum evaporated. Next, 990 μl of chloroform-methanol (2:1 [vol/vol]) was added to the upper aqueous layer and incubated for 2 h at 37°C. The lower layer was collected and pooled with the evaporated fraction taken from the first extraction and vacuum evaporated. The sample was solubilized with 100 μl of chloroform-methanol (1:2 [vol/vol]) and mixed 1:1 with 0.4 mM methylamine in methanol. Samples were directly injected into an ESI-LTQ OrbitrapXL at 2 μl/min and precursor ions were scanned using the Orbitrap analyzer at a resolution of 30,000 in negative ion mode. Using published inositolphosphoceramide (IPC) precursor m/z values, precursor ion peaks were identified using a mass tolerance of 10 ppm (51, 52). Using IPC structure data at the LIPID MAPS Lipidomics Gateway (52), the following theoretical precursor [M-H]− ion m/z values were used to identify the IPC ions in the high-resolution scan (sphingolipid species are identified by lipid class followed by numbers indicating carbons in FA moiety:double bonds in FA moiety;hydroxyl groups in FA moiety): IPC 46:0;4, 980.717); IPC 44:0;4, 952.686; IPC 42:0;4, 924.655; IPC 40:0;4, 896.623; IPC 38:0;4, 868.592. To validate the identity of these IPC ions, the IPC precursor ions were fragmented by CID in the linear ion trap. The observed m/z values of the MS/MS fragment ions for each IPC precursor was compared to predicted [ceramide phosphate-H2O]− and [ceramide phosphate]− m/z values at a mass tolerance of 0.1 Da. The following theoretical m/z [ceramide phosphate-H2O]− and [ceramide phosphate]− fragment ion values were used to validate the IPC lipids: IPC 38:0;4, 688.53, 706.54; IPC 40:0;4, 716.56, 734.57; IPC 42:0;4, 744.59, 762.60; IPC 46:0;4, 800.65, 818.66. In addition, to validate the identification of IPC 44:0;4, the fragmentation spectrum of the precursor with m/z 952.68 at a mass tolerance of 0.1 Da was compared to the previously published values for the fragment ions [ceramide phosphate-H2O]−, m/z 772.62, and [ceramide phosphate]−, m/z 790.63 (51). To compare the observed abundance for each IPC species between strains, the precursor ion signal intensity for each identified IPC species was normalized to the signal intensity of the m/z 835.53 base peak corresponding to the phosphatidylinositol (PI) species PI 16:1-18:0 and PI 16:0-18:1, where the numbers indicate carbons in first FA moiety:double bonds in first FA moiety-carbons in second FA moiety:double bonds in second FA moiety.
Growth rate analysis.
Yeast strains were grown overnight at 30°C in YPD. Relative cell number was measured as the OD660 by using a Beckman DU 530 spectrophotometer. Cells were diluted in 50 ml of fresh YPD to ∼0.05 OD660 unit/ml. Individual strains were grown at 30°C, and an OD660 measurement was obtained every 2 h. The formula for used for converting OD660 readings to cell numbers was y = 1.1564x3 − 0.6815x2 + 1.3996x, with y as cell number/ml and x as the OD660 value (38). Cell doubling time was determined by plotting the growth curve for each strain and measuring the maximum rate of cell growth during logarithmic growth.
Yeast two-hybrid assay.
Mating type A strains containing AD-tagged alleles and mating type α strains containing BD-tagged alleles have been previously described (53). The A and α strains were allowed to mate in liquid YPD at 30°C overnight. The relative cell number was determined by measuring the OD660, and 4 μl of a solution containing 107 cells/ml was plated onto SC-Leu-Trp-His–1.5 mM 3-aminotriazole (3-AT) agar plates. The plates were scanned after 48 h.
Membrane flotation.
Membrane flotation of yeast extracts was performed as previously described (54). A 50-ml culture of each yeast strain was grown to an OD660 of 1.0 to 1.5 in YPD medium at 30°C. The cells were lysed with glass beads in ice-cold breaking buffer (30 mM Tris [pH 7.0], 1 mM EDTA). The lysate was cleared by centrifugation at 500 × g for 3 min. Lysate corresponding to 10 OD660 units of cells in 222 μl was mixed with 1,778 μl of ice-cold 90% sucrose (wt/vol)–10 mM Tris (pH 7.0) solution. Then, 2 ml of lysate-sucrose solution was transferred to the bottom of a 14- by 89-mm ultracentrifuge tube (Beckman, catalog no. 344059) and layered with 6 ml of 65% sucrose–10 mM Tris (pH 7.0) and then 3 ml of 10% sucrose–10 mM Tris (pH 7.0). The tubes were centrifuged in a Beckman SW-41 rotor at 24,000 rpm for 18 h. Individual 1.5-ml fractions were collected from the top of the gradient, and the proteins were trichloroacetic acid (TCA) precipitated. Ten percent of each fraction was used for SDS-PAGE and Western blotting.
Membrane flotation fractions affinity purifications.
For each TAP strain, a 1-liter culture was grown to an OD660 of ∼1 in YPD, and the cells were split into six fractions. Each cell fraction was separated using the membrane flotation gradients as described above. The 10 to 65% sucrose interface layer and a 80% sucrose layer from each gradient were collected and pooled. TAP purification was performed as previously described up to TEV protease cleavage (45, 46).
GCN4-LacZ induction.
The yeast reporter plasmid p180 containing the GCN4 5′ UTR coupled to a lacZ reporter has been previously described (17). Yeast strains transformed with p180 were grown overnight at 30°C in SC-uracil (SC-ura). Cultures were diluted 1:10 and allowed to continue growing for 2 h in SC-ura-His. Cells were spun down and split into two tubes containing 10 ml of SC-ura-His medium. A 1 M 3-AT solution was added to the starvation tube to a final concentration of 10 mM. The cells continued to grow for 4 h at 30°C. β-Galactosidase assays were performed as previously described (55). The cells were centrifuged at 1,500 × g for 5 min and lysed with glass beads in 1 ml of ice-cold breaking buffer (100 mM Tris [pH 8.0], 1 mM DTT, 20% glycerol). A 20-μl portion of whole-cell extract was added to 900 μl of Z buffer (16.1 g of Na2HPO4·7H2O/liter, 5.5 g of NaH2PO4·H2O/liter, 0.75 g of KCl/liter, 0.246 g of MgSO4·7H2O/liter, 2.7 ml of β-mercaptoethanol/liter [pH 7.0]), followed by incubation at 28°C for 5 min. The reaction was initiated by adding 200 μl of 4 mg of ONPG (o-nitrophenyl-β-d-galactopyranoside)/ml in Z buffer, followed by incubation at 28°C. After the reaction turned a pale yellow color, 0.5 ml of 1 M Na2CO3 was added. LacZ expression was determined by measuring the absorbance at 420 nm using a Beckman DU 530 spectrophotometer. The protein concentration of the extracts was determined by using the Bio-Rad DC protein assay. LacZ specific activity was determined according to the following formula: [(OD420 × 1.7)/(0.0045 × protein concentration (mg/ml) × extract volume (ml) × time (min)] (38). Values were normalized to the WT.
[35S]methionine incorporation.
Overnight cultures of yeast grown in YPD were diluted 1:10 in 10 ml of SC-Met and grown for 3 h at 30°C. The OD660 of the culture was measured to determine the cell numbers. For labeling, [35S]methionine (MP Biomedicals) was added to 5 ml of the cell culture to a final concentration of 10 μCi/ml. Samples were incubated with shaking for 30 min at 30°C. Labeling was stopped by the addition of 1/10 volume 100% TCA and heating to 100°C for 30 min. TCA precipitates were collected on GFC filters (Whatman) and then washed sequentially with 5 ml each of 10% TCA and 95% ethanol. Filters were then placed in 5 ml of EcoLume scintillation fluid (MP Biomedicals), and [35S]methionine incorporation was measured using a Beckman LS 6500 scintillation counter. Values were reported as the counts per minute/OD660 unit.
Microscopy.
Epifluorescence microscopy was performed using live yeast cells grown in SC medium to an OD660 of 1.0 to 1.5 at 30°C. Cells were mounted on slides and visualized using a Zeiss Axiophot bright-field microscope with a ×63/1.40 Plan-Apochromat oil differential interference contrast (DIC) lens. Images were analyzed with MetaMorph imaging software (Molecular Devices). Live yeast cells imaged using confocal microscopy were grown in SC medium to an OD660 of 1.0 to 1.5 at 30°C. Cells were visualized with a Zeiss LSM 510 META inverted confocal microscope using a ×63/1.40 Plan-Apochromat oil immersion lens. Microscopic images used for quantitative analysis were analyzed using ImageJ imaging software (56). To quantify the percentage of 2B bodies that colocalized with the ER, a 2B body was judged to be colocalized with the ER only if the 2B body signal overlapped with an area of YBR159W at least half as bright as the brightest YBR159W signal seen in the cell. Cells were pooled into groups of ∼25 cells to calculate a standard deviation for the percentage of 2B bodies colocalized with the ER. The bright regions of the ER were subtracted from the total area of the cell minus the nuclear area to determine the fraction of the cell taken up by the ER. The compound 3,3′-dihexyloxacarbocyanine iodide [DiOC6(3)] was used to stain and image the membranes of the WT strain AL400 and the ybr159wΔ strain AL401 as previously described (57). Yeast cells were incubated in media containing 2.5 μg of DiOC6(3)/ml for 10 min before imaging.
Polysome profiling.
Polysome analysis was performed as previously described (58). Yeast strains were grown in YPD to an OD660 of ∼1. Cells were lysed with glass beads in ice-cold breaking buffer (10 mM Tris [pH 7.0], 100 mM NaCl, 30 mM MgCl2, 50 μg of cycloheximide/ml, 200 μg of heparin/ml). The crude lysate was cleared by centrifugation at 500 × g for 3 min, and 20 OD660 units of cells were loaded on top of a 7 to 47% continuous sucrose gradient (wt/vol) cast in 50 mM Tris (pH 7.0), 50 mM NH4Cl, 12 mM MgCl2, and 50 μg of cycloheximide/ml in a 14-by-89-mm ultracentrifuge tube (Beckman). Gradients were centrifuged in a Beckman SW-41 rotor at 14,000 rpm for 18 h at 4°C. An absorbance profile at 254 nm was collected from the gradients as previously described (16). Fractions (1 ml) were used for Western blotting. Monosome and polysome peak areas were determined by using ImageJ software (56). A moving baseline for each profile was established by connecting the minima between each peak, and the area under each peak above this line was calculated. The polysome peak areas were summed and compared to the monosome peak area.
Subcellular fractionation.
WT yeast strain AL400 was grown to an OD660 of 1.0 to 1.5 in YPD medium at 30°C. To isolate subcellular fractions, 45 OD660 units of cells were split into three samples: control, puromycin treatment, and EDTA treatment. The control sample was lysed using glass beads in 750 μl of ice-cold control buffer (10 mM Tris [pH 8.0], 100 mM NaCl, 1 mM EDTA, 10 mM KCl, 30 mM MgCl2). The puromycin and EDTA treatment samples were lysed using glass beads in 750 μl of ice-cold ribosome dissociation buffer (10 mM Tris [pH 8.0], 100 mM NaCl, 1 mM EDTA). The control sample was diluted in 750 μl of control buffer. The puromycin treated sample was diluted with 750 μl of ribosome dissociation buffer containing 2 mM puromycin to a final concentration of 1 mM. The EDTA-treated sample was diluted in ribosome dissociation buffer (20 mM EDTA) to a final concentration of 10 mM EDTA. Lysates were gently mixed at room temperature for 30 min to facilitate dissociation of ribosomes from the ER. Lysates were centrifuged at 900 × g for 5 min, and the supernatant was centrifuged at 11,000 × g for 20 min. The soluble fraction was recovered from the supernatant. The pellets were resuspended in either control buffer (10 mM Tris [pH 8.0], 100 mM NaCl, 1 mM EDTA, 10 mM KCl, 30 mM MgCl2), puromycin solution (1 mM puromycin, 10 mM Tris [pH 8.0], 100 mM NaCl, 1 mM EDTA), or EDTA solution (10 mM EDTA, 10 mM Tris [pH 8.0], 100 mM NaCl) and centrifuged at 11,000 × g for 20 min. The pellets were resuspended in 1.5 ml of resuspension buffer (1 mM puromycin, 10 mM Tris [pH 8.0], 100 mM NaCl, 1 mM EDTA, 1% SDS), and 15 μl of each fraction was used for Western blotting.
RESULTS
In a tandem affinity purification proteomics screen of S. cerevisiae translation initiation factors, followed by liquid MS analysis, we discovered that all five subunits of eIF2B copurified with the VLCFA enzyme YBR159W (A. J. Link et al., unpublished data) (Fig. 2A). Subsequent liquid chromatography (LC)-MS/MS analysis of TAP-YBR159W affinity purification showed YBR159W copurified with all five subunits of the eIF2B complex and several members of the VLCFA synthesis pathway. In the present study, additional TAP experiments examined whether other members of the VLCFA synthesis pathway also interact with eIF2B subunits. With the exception of YBR159W, our data showed that other members of the VLCFA synthesis pathway did not interact with eIF2B (Fig. 2A). To rule out the possibility that the YBR159W-eIF2B interaction was due to an artifact of the TAP-tagged strains, we performed a GFP affinity purification using the GCD7-GFP strain AL429. LC-MS/MS analysis identified YBR159W copurifying with all five subunits of eIF2B (see Fig. 5E). Next, we utilized yeast two-hybrid analysis to identify interactions between eIF2B subunits and YBR159W. The activation-domain tagged strains pOAD(YBR159W), pOAD(GCD1), pOAD(GCD2), pOAD(GCN3), pOAD(GCD6), pOAD(GCD7), pOAD(SUI2), and pOAD(TDH1) were mated with binding-domain tagged strains AL408 (YBR159W), AL409 (GCD1), AL410 (GCD2), AL411 (GCD6) and AL412 (GCD7). The positive interactions between different subunits of the eIF2B complex validated the experiment's ability to detect previously described interactions (Fig. 2B). The two-hybrid analysis showed that YBR159W positively interacted with both the GCD6 and GCD7 subunits of eIF2B (Fig. 2B).
Fig 2.

YBR159W's interaction with eIF2B is unique among VLCFA genes. (A) MS analysis of the affinity-purified TAP-GCD2, TAP-YBR159W, and other TAP-tagged VLCFA protein complexes. Listed are unique peptide identifications with the percent coverage of identified peptides in the protein in parentheses. A “–” indicates that no peptides were identified for the gene. (B) Yeast two-hybrid (Y2H) analysis of interactions between YBR159W and eIF2B subunits GCD6 and GCD7. Shown is both the assay plate used for scoring the Y2H interactions and a table of the interactions tested at each spot. Shading on the table corresponds to a positive interaction on the plate.
Fig 5.

Translation assays on the ybr159wΔ strain. (A) Translation efficiency as measured by [35S]methionine incorporation. Values are counts per minute per OD660 unit of cells. The results shown are from at least three replicates. (B) Polysome profiling of the ybr159wΔ and other VLCFA null strains. At least three replicates were performed for each strain. Although the example ybr159wΔ plot does not show a 40S ribosome peak, all other replicates of the strain showed a 40S peak similar to the WT. (C) Assay for GCN4 pathway competence by GCN4-LacZ induction. The results are expressed as the LacZ expression per mg of protein per min normalized to the WT starvation condition. Starvation conditions were induced by 10 mM 3-AT in synthetic complete minus histidine media for 4 h. The gcd1-100 strain has a constitutively derepressed GCN4 pathway and constant GCN4 protein translation, while the gcn2Δ strain is incapable of derepression of GCN4 and cannot produce significant amounts of GCN4 protein. (D) Ratio of monosome to polysome peak areas for the polysome profiles. P values were generated using the Student t test from at least three individual replicates. (E) GFP pulldown of eIF2B complexes in a ybr159wΔ background. After pulldown LC-MS/MS was performed to identify the proteins. An untagged ybr159wΔ strain and GCD7-GFP tagged strain were used as controls. Displayed are unique peptide hits and the percentage of coverage as described in Fig. 2A. (F) Western blot analysis of WT, ybr159wΔ, and GFP-GCD7 strains. The yeast strains in panel D and the WT strain AL400 were used. Equivalent amounts of whole-cell extracts were loaded on the SDS-PAGE gel. The Western signals for GCD6 and TDH1 to -3 were determined by densitometry. The ratio of the anti-GCD6 to the anti-TDH1-3 signal is shown for each strain. The anti-TDH1-3 antibody does not distinguish between the three GAPDH (glyceraldehyde-3-phosphate dehydrogenase) gene duplications in yeast.
The GFP-tagged YBR159W strain AL425 showed the YBR159W protein localizes to the ER membrane using epifluorescence microscopy (Fig. 3A). DPM1 encodes the enzyme dolichol phosphate mannose synthase that adds a mannose moiety to dolichyl phosphate on the cytosolic side of the endoplasmic reticulum (59, 60). DPM1 is an ER membrane protein unrelated to VLCFA synthesis or utilization (60). Confocal microscopy using the FEN1-GFP YBR159W-RFP strain AL422 and the DPM1-GFP YBR159W-RFP strain AL423 confirmed that RFP-tagged YBR159W expressed from a low-copy-number plasmid colocalizes with the VLCFA protein FEN1 and ER protein DPM1 (Fig. 3B).
Fig 3.

Cellular analysis of YBR159W. (A) Live cell epifluorescence imaging of endogenously tagged YBR159W-GFP indicates YBR159W localizes mainly to the ER membrane. (B) Live cell confocal microscopy showing the colocalization of YBR159W with the VLCFA pathway enzyme FEN1 and ER membrane protein DPM1. YBR159W is expressed on a low-copy-number plasmid and tagged with DsRed. FEN1 and DPM1 are endogenously expressed and tagged with GFP. (C) Deletion of YBR159W results in a very slow growth rate.
We constructed the ybr159wΔ yeast strain AL401 to examine the null phenotype. The mutant strain had a slow-growth phenotype (Fig. 3C) and was temperature sensitive at 37°C (data not shown). To show that the slow-growth phenotype was due to the deletion of ybr159wΔ and not a second site mutation in the strain, the ybr159wΔ null yeast strain was complemented in strain AL402 expressing YBR159W from the low-copy-number plasmid YCp-YBR159W (Fig. 3C). Our results agreed with previous studies using an unrelated ybr159wΔ null strain (29).
Previous work has shown that disruption of VLCFA utilization in yeast causes abnormal formation of lipid membranes (61). The compound 3,3′-dihexyloxacarbocyanine iodide [DiOC6(3)] is a lipophilic dye used to label a variety of lipid membranes (57). We used DiOC6(3) to stain membranes of WT strain AL400, ybr159wΔ strain AL401, and the VLCFA mutant strains AL413 (fen1Δ), and AL414 (sur4Δ), and the ceramide synthase mutant strain RH5994 (lip1Δ). The ybr159wΔ, fen1Δ, sur4Δ, and lip1Δ null strains all displayed disrupted lipid membranes using epifluorescence imaging (Fig. 4). This supported previous work showing that VLCFAs are important for proper membrane formation (61).
Fig 4.

Null mutations of genes in the VLCFA pathway disrupt lipid membranes. The lipophilic dye DiOC6(3) was used to label membranes in live yeast cells. Dye was applied to cells in suspension 10 min before plating on a microscope slide and imaging. Included are the VLCFA and ceramide synthase fen1Δ, sur4Δ, and lip1Δ mutants as controls. A total of 100% of these mutants showed abnormal membranes (n = 246) versus 1.1% for WT (n = 89) and 9.7% for the rescue (n = 93).
To determine whether YBR159W has a role in translation, we examined whether the ybr159wΔ strain AL401 causes a defect in protein synthesis. We used [35S]methionine incorporation to quantify the global translation rate. The [35S]methionine incorporation experiments showed that the ybr159wΔ strain has a reduced translation rate (Fig. 5A). The ceramide synthase mutant lip1Δ strain RH5994 also showed a reduction in the rate of translation. The lip1Δ strain had a slow growth rate similar to that of the ybr159wΔ strain. However, the VLCFA mutant strains AL413 (fen1Δ) and AL414 (sur4Δ) showed no reduction in translation or growth rates (Fig. 5A).
Next, we performed polyribosome profiling to examine the distribution of 40S, 60S, 80S, and polyribosomes in the ybr159wΔ strain AL401. Compared to the WT strain, we observed the polysome profiles for the ybr159wΔ strain showed an increase in the 80S monosome peak and a decrease in polysome peaks (Fig. 5B). As expected, the complemented ybr159wΔ strain AL402 showed a polysome profile similar to that of the WT. To normalize and quantify the observed differences in the peak areas, the ratio of the 80S monosome to polysome peak areas was calculated. The monosome/polysome ratio significantly increased for the ybr159wΔ strain compared to the WT and complemented strains (Fig. 5D). Polysome profiles of the lip1Δ strain RH5994 showed defects similar to those of the ybr159wΔ strain (Fig. 5B and D). Polysome profiling of the fen1Δ strain AL413 and sur4Δ strain AL414 showed no noticeable differences from WT strain AL400 (Fig. 5B and D). These polysome distributions were consistent with the reduced global translation rates seen previously in the [35S]methionine labeling experiments.
We next examined ybr159wΔ's effect on eIF2B's activity. We used a GCN4-lacZ expression assay to examine GCN4 expression during the starvation response (62). Strains AL400 (HIS+ control strain), AL401 (ybr159wΔ), AL402 (ybr159wΔ + YCp-YBR159W), AL413 (fen1Δ), AL414 (sur4Δ), RH5994 (lip1Δ), H2557 (gcn2Δ), and F98 (gcd1) were transformed with the GCN4-lacZ reporter plasmid p180. Our results showed that the ybr159wΔ, fen1Δ, sur4Δ, and lip1Δ null strains did not affect the induction of GCN4 during amino acid starvation (Fig. 5C). This suggested that eIF2B's role in the regulation of GCN4 response is not affected by the ybr159wΔ null or other VLCFA pathway mutation.
We next tested whether the ybr159wΔ mutation affected the composition of the eIF2B complex. Using the ybr159wΔ GCD7-GFP tagged strain AL403, the untagged ybr159wΔ strain AL401, and the GCD7-GFP strain AL429, we performed GFP affinity purifications and LC-MS/MS analysis of the affinity-purified complexes. All five subunits of eIF2B were identified in the ybr159wΔ GCD7-GFP strain and the GCD7-GFP strain (Fig. 5E). No subunits of eIF2B were identified in the untagged ybr159wΔ control strain AL401. These results suggested that the composition of eIF2B is not dependent upon the presence of YBR159W.
Although the composition of eIF2B appeared to be independent of YBR159W, the consistently lower number of identified peptides for each eIF2B subunit from the MS data for the GCD7-GFP, ybr159wΔ null strain compared to the GCD7-GFP strain suggested that the cellular abundance of eIF2B was lower in the ybr159wΔ null background (Fig. 5E). To determine whether the cellular abundance of eIF2B is lower in a ybr159wΔ null strain, Western analysis was performed on the yeast strains used in the GCD7-GFP affinity purification of eIF2B complexes. Lack of signal for YBR159W in the ybr159wΔ strains confirmed the expected null genotype (Fig. 5F). In concordance with the MS results, the GCD7-GFP, ybr159wΔ strain had a lower abundance of eIF2B compared to the GCD7-GFP strain (Fig. 5E). To validate this observation in untagged strains, Western analysis was also performed using the WT strain AL400 and the untagged ybr159wΔ null strain AL401. The ybr159wΔ null strain again showed lower abundance of eIF2B compared to the WT strain (Fig. 5F)
We next tested whether eIF2B played a role in VLCFA synthesis. Previous studies had shown a ybr159wΔ null strain had an altered VLCFA lipid composition (30). Since four of the five subunits of eIF2B are essential, we used a gcn3Δ strain AL424 to test for VLCFA defects. The WT strain AL400, the ybr159wΔ strain AL401, the ybr159wΔ rescue strain AL402, the sur4Δ strain AL414, and the lip1Δ strain RH5994 were used as positive and negative controls. To profile the VLCFAs, lipids were extracted from yeast cells and directly infused into an ESI-LTQ-OrbitrapXL mass spectrometer while scanning at high resolution in negative-ion mode. Several IPCs, a class of VLCFA-containing sphingolipid, were identified using previously published m/z values at a 10-ppm mass accuracy (50, 52). We validated the identification of the IPC species using either previously observed fragmentation spectrum or expected m/z values for the IPC's [ceramide phosphate-H2O]− and [ceramide phosphate]− fragment ions (Fig. 6) (51). The IPC 44:0;4 and IPC 46:0;4 sphingolipids contain full-length VLCFAs and are the most abundant yeast sphingolipid species (24). Compared to the WT strain, the gcn3Δ, ybr159wΔ, and other VLCFA and ceramide synthase mutant strains all showed a reduction in the IPC 44:0;4 and IPC 46:0;4 sphingolipids containing full-length VLCFAs (Fig. 7A). The IPC sphingolipid species IPC 38:0;4, IPC 40:0;4, and IPC 42:0;4 contain shorter-chain fatty acids and are typically only detected in VLCFA biosynthesis mutant strains (24). As previously observed, the sur4Δ strain had elevated shorter-chain fatty acid-containing IPC species 38:0;4, 40:0;4, and 42:0;4 (36). We observed that IPC 38:0;4 and IPC 42:0;4 were also elevated in the ybr159wΔ strain. The gcn3Δ strain showed no significant changes in the shorter-chain fatty acid sphinglipid's IPC 38:0;4, 40:0;4, and 42:0;4 levels (Fig. 7B). The lip1Δ strain contained barely perceptible levels of any IPC, supporting its requirement for ceramide synthesis (34).
Fig 6.

Validation and identification of IPCs. (A) MS precursor and MS/MS fragmentation spectrum for IPC 44:0;4 from WT yeast. The observed precursor ion m/z 952.681 represents the expected ion IPC 44:0;4 (952.686). The observed m/z 835.529 corresponds to the two PIs, PI 16:0-18:1 and PI 16:1-18:0, used to normalize the relative abundance of each IPC species. In the lower MS/MS spectra of the 952.681 precursor ion, the first and second most abundant peaks correspond to the expected IPC 44:0;4 fragment ions [ceramide phosphate-H2O]− at m/z 772.62 and [ceramide phosphate]− at m/z 790.63. (B) Theoretical fragmentation database for IPCs 38:0;4, 40:0;4, 42:0;4, and 46:0;4. Shown are the theoretical m/z values for fragment ions [ceramide phosphate-H2O]− and [ceramide phosphate]− for IPCs 38:0;4, 40:0;4, 42:0;4, and 46:0;4. (C) MS/MS fragmentation spectra for IPCs 38:0;4, 40:0;4, 42:0;4, and 46:0;4. The observed precursor m/z values “Pre” of 868.586, 896.617, 924.648, and 980.712 correspond to the expected m/z values of IPC 38:0;4 (868.592), IPC 40:0;4 (896.623), IPC 42:0;4 (924.655), and IPC 46:0;4 (980.717), respectively. In the MS/MS spectra, the peaks corresponding to the expected theoretical IPC fragment ions [ceramide phosphate-H2O]− and [ceramide phosphate]− are marked with an asterisk. In each case, the peak corresponding to the expected [ceramide phosphate-H2O]− fragment ion was the most intense ion in the MS/MS spectrum.
Fig 7.

Fatty acid profiling of WT and mutant yeast strains. (A) Longer-chain fatty acid-containing sphingolipid species. IPC species with 44 and 46 carbon-containing acyl chains are shown. The VLCFA and ceramide mutants sur4Δ and lip1Δ synthase are included as controls. (B) Shorter-chain fatty acid-containing sphingolipid species. Three IPC species with 38, 40, and 42 carbon-containing acyl chains are shown. For both panels A and B, the data represent the percentage of signal of each lipid species normalized to the signals of the PI 16:0-18:1 and PI 16:1-18:0 ions.
We next looked at cellular localization of eIF2B and YBR159W using strains with subunits of eIF2B endogenously tagged with GFP and the YBR159W-RFP expression plasmid YCp-YBR159W-dsRed. To show that the RFP-tagged YBR159W allele was functional, the plasmid YCp-YBR159W-dsRed complemented the ybr159wΔ null strain AL401 (data not shown). Confocal microscopy of the dual-fluorescentce-labeled strains was used to look for colocalization between eIF2B and YBR159W (Fig. 8). As observed in previous studies and Fig. 2, YBR159W localized to membranes corresponding to the ER (Fig. 8). Using strains with different eIF2B subunits tagged with GFP, we observed eIF2B localized as one to two large foci (Fig. 8). In addition, GFP-tagged eIF2B is seen dispersed throughout the cytoplasm (data not shown). Surprisingly, the confocal microscopy images did not convincingly show the majority of YBR159W signal colocalizing with eIF2B subunits. Because we observed that the 2B bodies localize near the ER membrane-bound YBR159W, we performed a statistical analysis to test if eIF2B and YBR159W colocalize. We examined 221 individual 2B bodies from 140 dually labeled cells by pooling results from the YCp-YBR159W-dsRed transformed GCD1-GFP, GCD6-GFP, and GCD7-GFP strains (AL405, AL406, and AL407). We found that 60.1% ± 6.6% of 2B bodies examined showed partial colocalization with a bright area of YBR159W signal. Based on the area of the cell taken up by bright areas of YBR159W signal, it would be expected that only 30.7% ± 6.9% of 2B bodies would colocalize with YBR159W signal if the two signals were independent of each other. Student t test (P = 2.9 × 10−8) shows this difference to be significant.
Fig 8.

eIF2B and YBR159W localization in live cells. Confocal microscopy of live yeast cells showing localization of eIF2B subunits in relation to the localization of YBR159W. eIF2B subunits are endogenously tagged with GFP, while YBR159W-RFP is expressed on a centromeric plasmid.
To observe the effects of the ybr159wΔ deletion on eIF2B localization, we performed live cell imaging using epifluorescence microscopy on the yeast strains ybr159wΔ GCD7-GFP (AL403), GCD7-GFP (AL429), and ybr159wΔ GCD7-GFP, [YCp-YBR159W] (AL404). Cells from the GCD7-GFP control strain were found to contain one to two large 2B bodies (Fig. 9A). In the ybr159wΔ strain AL403, eIF2B appeared as multiple foci (Fig. 9A). The ybr159wΔ phenotype of AL403 was rescued by expression of plasmid YCp-YBR159W in strain AL404 (Fig. 9A). Using these strains, we counted the number of cells containing one to two large 2B bodies compared to the number of cells having the multiple eIF2B foci or diffuse cytoplasmic localization. We found that the majority of ybr159wΔ cells had a multiple eIF2B focus phenotype (Table 4). For the GCD7-GFP WT control strain, no cells had the multiple eIF2B focus phenotype and a majority of cells had either one or two 2B bodies. The rescued ybr159wΔ GCD7-GFP, [YCp-YBR159W] strain AL404 did not have multiple eIF2B foci (Table 4). To show that the 2B body phenotype was independent of the GFP-tagged alleles, we performed immunofluorescence microscopy on untagged yeast strains using polyclonal antibody against the eIF2B subunit GCD6 (Fig. 9B). We observed the one to two large 2B body focus phenotype for the majority of the WT control AL400 cells, while the majority of the ybr159wΔ cells (AL401) displayed multiple eIF2B foci (Table 4). The ybr159wΔ [YCp-YBR159W] rescue strain AL402 showed a majority of cells had eIF2B present as either a single 2B focus or no detectable foci (Table 4). The VLCFA and ceramide synthase mutants AL413 (fen1Δ), AL414 (sur4Δ), and RH5994 (lip1Δ) were all found to have the multiple eIF2B focus phenotype (Table 4).
Fig 9.

eIF2B localization in the ybr159wΔ background. (A) Live cell fluorescence microscopy of endogenously tagged eIF2B subunit GCD7-GFP. Bright-field (BF) images are included for clarity. (B) Immunofluorescence microscopy of formaldehyde-fixed yeast cells. A polyclonal antibody against yeast eIF2B subunit GCD6 was used along with an Alexa Fluor 488-tagged secondary. Nuclei are stained with DAPI for clarity.
Table 4.
Statistics for eIF2B localization phenotypes in live yeast cells (group 1) and for eIF2B localization phenotypes via immunofluorescence analysis of fixed yeast cells (group 2)a
| Strain | No. of cells | Single 2B body (%) | Multiple foci (%) | No foci (%) |
|---|---|---|---|---|
| Group 1 | ||||
| WT | 173 | 50.9 | 0.0 | 49.1 |
| ybr159wΔmutant | 122 | 3.3 | 71.3 | 24.6 |
| Rescue | 72 | 44.4 | 0.0 | 55.6 |
| Group 2 | ||||
| WT | 105 | 63.8 | 7.6 | 28.6 |
| ybr159wΔmutant | 59 | 1.7 | 83.1 | 15.3 |
| Rescue | 111 | 35.1 | 15.3 | 49.5 |
| lip1Δmutant | 29 | 6.9 | 93.1 | 0.0 |
| fen1Δmutant | 96 | 4.2 | 63.5 | 32.3 |
| sur4Δmutant | 122 | 1.6 | 58.2 | 40.2 |
Because eIF2B is thought to be a soluble cytoplasmic protein and YBR159W has been shown to be an integral membrane protein in the ER (26), we performed membrane float experiments to determine whether a population of eIF2B complexes physically interacted with lipid membranes. The lack of signal from the Western blot for the control yeast GAPDH analogs TDH1, TDH2, and TDH3 in the membrane fraction showed that the fractionation was efficient at separating cytoplasmic proteins from membrane-associated proteins (Fig. 10A). A significant lipid membrane signal was seen for the ER proteins YBR159W and DPM1. A portion of the YBR159W and control ER membrane protein DPM1 signals was still present in the soluble fraction, indicating that the membrane-associated proteins do not appear to completely separate from the soluble fraction. The membrane float experiments showed that in WT AL400 cells, a significant fraction of the eIF2B subunit GCD6 localized to the lipid membrane fractions (Fig. 10A). The Western blot profiles of the membrane and soluble fractions for GCD6 showed the same pattern as the known ER membrane proteins YBR159W and DPM1 (Fig. 10A). Interestingly, the SUI2 component of eIF2 also showed a similar membrane association pattern. The data indicate a fraction of eIF2 complexes are associated with membranes in yeast cells.
Fig 10.

eIF2B and YBR159W localization using membrane flotation assays. (A) Western blot of membrane flotation assay fractions using protein extracts from WT yeast showing the localization of the eIF2B subunit GCD6 and YBR159W. Controls include the eIF2 subunit SUI2, the ER integral membrane protein DPM1, and the cytosolic protein GAPDH. The genes encoding TDH1 to TDH3 are the three GAPDH genes in yeast. The lanes represent 20% of fractions from the membrane flotation gradients. Labels show the location of the membrane-associated and soluble protein fractions. (B) MS analysis of affinity-purified TAP complexes from the membrane and soluble fractions of membrane flotation experiments. Unique peptides, the percent coverage, and “–” are all as described in Fig. 2A. (C) Western blot of membrane flotation assay fractions comparing WT and ybr159wΔ strains. The conditions are the same as in panel A. (D) Western blot of crude fractionation following EDTA or puromycin treatment. Abbreviations: WCE, whole-cell extract; Con, untreated control; EDTA, EDTA treatment; Puro, puromycin treatment; S, supernatant; P, pellet. ASC1 is a component of the small ribosomal subunit and RPL32 is a large ribosomal subunit protein. Lanes represent 15 μl of whole-cell extract (WCE) following fractionation, and pellets were resuspended in starting volume.
To validate our observation that eIF2B is membrane associated, we used whole-cell extracts prepared from TAP-tagged eIF2B and control strains and the membrane float separation experiment to collect fractions from the membrane-associated and soluble protein region of the density gradients. Next, we performed a modified TAP purification on each fraction and analyzed the affinity-purified complexes using LC-MS/MS. Our data showed that the interaction between eIF2B and YBR159W was still present in both the membrane-associated and soluble protein fractions (Fig. 10B). Because of the incomplete separation of membrane proteins in the assay, it is not known whether both soluble and membrane-associated eIF2Bs interact with YBR159W or whether only membrane-associated eIF2B interacts with YBR159W. To determine whether YBR159W was required for eIF2B's membrane association, we performed the membrane flotation assay and Western blot analyses using the ybr159wΔ strain AL401. We found that eIF2B associated with the membrane fraction in the ybr159wΔ strain at levels similar to those seen in the control strain (Fig. 10C). Overall, the membrane float experiments showed that a fraction of yeast eIF2B is associated with membranes but that the interaction is independent of YBR159W.
To determine whether the membrane association seen for eIF2B is possibly mediated by rough ER-bound ribosomes, we performed a subcellular fractionation experiment to isolate smooth membranes. Cell lysates from WT strain AL400 were treated with either elevated levels of EDTA or the ribosome-releasing antibiotic puromycin (63). After fractionation and Western blotting, ribosomal protein signal in the insoluble membrane fraction was significantly reduced in both the EDTA- and puromycin-treated cell extracts compared to untreated control extracts. However, the eIF2B signal in the rough or smooth membrane fraction did not noticeably change (Fig. 10D). The data indicate that the eIF2B-membrane association is independent of ribosomes.
DISCUSSION
Previous large-scale yeast interactions studies failed to show eIF2B interacting with the VLCFA pathway (64–66). We show using TAP-tagged and GFP-tagged affinity purifications, as well as yeast-two-hybrid analysis, that the VLCFA keto-reductase YBR159W interacts with the translation initiation factor complex eIF2B. Because our unpublished proteomic screen of translation factor interactions identified YBR159W interacting with eIF2B, we named the S. cerevisiae locus IFA38 for initiation factor-associated protein of 38 kDa (Link et al., unpublished). Affinity purification and LC-MS/MS experiments show that YBR159W copurifies with all five subunits of eIF2B and not in controls. No other member of the VLCFA pathway copurifies in the eIF2B affinity purifications. Interestingly, the TAP-tagged members of the VLCFA pathway do not seem to strongly interact with each other. Our Y2H data suggest that the eIF2B subunits GCD6 and GCD7 physically interact with YBR159W.
The interaction between the VLCFA synthesis pathway and the eIF2B translation initiation pathway presents a number of possibilities. Is one pathway regulating the other or vice versa? It can be hypothesized that the cell might need to regulate VLCFA synthesis if translation is disrupted. Alternatively, it might be advantageous to reduce translational activity if VLCFAs are being downregulated. Finally, the YBR159W-eIF2B complex could be involved in a novel function. A link between a translation initiation factor and lipid membranes is not totally unique. Experiments in human cells have shown an interaction between the translation initiation factor eIF4E and the Golgi apparatus (67).
To test the hypothesis that YBR159W and VLCFA synthesis play a role in translation, we used [35S]methionine incorporation and polysome profiling to assay translation activity in mutant strains. Both experiments show a reduction in the translation rate for the ybr159wΔ strain. However, a similar phenotype is seen for the slow-growing lip1Δ strain. The VLFCA mutant fen1Δ and sur4Δ strains have WT growth rates and do not share a translation defect with the slower-growing members of the pathway. It is not known whether the cause of the translation defect seen in the ybr159wΔ strain is directly related to its interaction with eIF2B or is an indirect consequence of slow growth or a VLCFA defect.
When GCN4 expression is examined using the GCN4-LacZ assay, the ybr159wΔ strain has WT levels of GCN4 induction. The GCN4-LacZ assay was normalized to protein concentration so the slow growth rate of the ybr159wΔ strain should not affect the results. The data indicate that the ybr159wΔ strain does not have a defect in the GCN4 pathway. We cannot rule out the possibility that the slow growth of the ybr159wΔ strain masks a subtle defect in eIF2B's GEF activity unrelated to the GCN4 pathway. Our affinity purification experiments of eIF2B in a ybr159wΔ deletion background showed that the eIF2B complex is intact. A Western blot of ybr159wΔ strains showed that the overall abundance of eIF2B was lower in the deletion background compared to WT. It is not clear whether the lower level of eIF2B is caused by the slow-growth phenotype of the ybr159wΔ null background or some other factor.
To test the hypothesis that eIF2B plays a role in VLCFA synthesis, several limitations arose that made answering the question problematic. Of the five yeast eIF2B subunits, only GCN3 is nonessential. The gcn3Δ strain did not show a defect in VLCFA production or utilization. Although the gcn3Δ strain showed a reduction in the sphingolipid species IPC 44:0;4 and IPC 46:0;4, it did not show a concomitant rise in shorter-chain fatty acid-containing IPC species indicative of a defect in VLCFA production. The presence of shorter-chain sphingolipids would indicate the cell is trying to compensate for a lack of VLCFAs. Therefore, we postulate the lower levels of IPC 44:0;4 and IPC 46:0;4 seen in the gcn3Δ strain are unrelated to a defect in VLCFA production. The VLCFA defect in the ybr159wΔ strain is modest, with only a small increase in the shorter-chain fatty acid-containing sphingolipids. The loss of IPC 46:0;4 is the strain's most striking characteristic. Previous work suggests that Ayr1p is able to perform 3-ketoacyl activity in the absence of YBR159W (30). The same study showed that ayr1 and ybr159w are synthetically lethal (30).
A gcn3Δ null strain is unable to fully derepress GCN4 expression during amino acid starvation (68). GCN4 is a transcription factor involved in the expression of several hundred genes during a wide variety of cellular stresses (69). Although growth conditions for the gcn3Δ strain should not have activated a stress response, we suspected analysis of the lipid content of the gcn3Δ strain could prove problematic if the VLCFA pathway was a downstream target of the GCN4 transcription factor. We examined the effects of loss of GCN4 using expression data for gcn4Δ strains from the Gene Expression Omnibus (GEO) Database (70). Two separate data sets showed no significant changes in the expression of various VLCFA genes (data not shown; GEO accession no. GSE24057 [71] and GSE25582). We concluded that under the conditions used for the analysis of sphingolipids, the loss of GCN4 did not significantly alter VLCFA gene expression. We concluded that our gcn3Δ strain was not experiencing alterations in VLCFA gene expression due to the repression of GCN4. The lack of a direct translation defect in the ybr159wΔ strain and the lack of a VLCFA defect in the gcn3Δ strain suggest that there is no significant cross talk between the GEF and VLCFA pathways.
Membrane flotation and subcellular fractionation assays show eIF2B interacts with lipid membranes. Our data and previous studies showed YBR159W is an integral membrane protein that colocalizes with the ER membrane (26, 27). We interpret these findings to mean that the membranes eIF2B is interacting with are ER membranes. It is unknown whether ER-associated eIF2B is actively engaged in guanine nucleotide exchange. A number of conclusions can be made about this ER membrane-interacting eIF2B. First, the eIF2B-membrane interaction is not mediated by rough ER-bound ribosomes. Treatment of cell extracts with EDTA or puromycin greatly reduces the amount of ribosomes that fractionate with lipid membranes but does not reduce the portion of eIF2B that fractionates with membranes. This fits the prevailing theory that eIF2B's role in translation is independent of the ribosome (72). Second, YBR159W is not required for the interaction. The ybr159wΔ null strain does not affect eIF2B's interaction with the membrane showing that the interaction of eIF2B with ER membranes is YBR159W independent. This indicates that another factor(s) is possibly required.
Confocal microscopy shows that the majority of 2B bodies are in close proximity to YBR159Wp and ER membranes, supporting the model that 2B bodies and the ER interact. This could be taken to indicate that the eIF2B shown to interact with ER membranes resides in 2B bodies. A possible conflicting interpretation of the data is that YBR159W-RFP is being overexpressed and its localization is an artifact. The colocalization experiment used a RFP-tagged YBR159W allele expressed from a GPD promoter on a centromeric plasmid. Global protein expression analysis shows that the GPD promoter's target protein, TDH3, is expressed at roughly four times that of YBR159W (40). The fact that the RFP-tagged YBR159W localization agrees with endogenously expressed YBR159W-GFP localization leads us to believe that artifacts caused by the RFP tagged construct are not disrupting YBR159W's localization. In addition, the RFP-tagged allele complements a ybr159wΔ null strain. How and why eIF2B might be interacting with the ER membrane is unknown. A population of membrane-interacting 2B bodies might explain recent findings that 2B bodies can exist in a mobile or static state with mobile 2B bodies free in the cytoplasm and static 2B bodies being associated with membranes (73). Further work is needed to prove this hypothesis.
The observation that the ybr159wΔ null strain leads to multiple eIF2B foci is intriguing. This phenotype is also seen in other VLCFA mutants. The fact that these mutants all display disrupted lipid membranes lends itself to the theory that properly formed membranes are required for the integrity of 2B bodies. An intriguing question is whether the membrane disruption prevents the 2B bodies from forming properly or whether the 2B bodies are unable to be maintained once formed? For the first model, an as-yet-unknown factor in membranes required for 2B body formation could be disrupted and cause 2B bodies to form throughout the cell. We speculate that this membrane-associated factor could serve as a nucleating site for the formation of 2B bodies. The second model would predict that membrane disruption is affecting a factor needed for 2B body stability. Loss of this factor leads to 2B bodies dissociating into multiple smaller foci. A previous study showed VLCFAs were important for lipid raft formation (25). It is possible that lipid raft disruption in the VLCFA mutants causes the multiple eIF2B focus phenotype. Translation assays using the ybr159wΔ strain suggested the disruption of 2B bodies into multiple foci does not affect translation. The translation activity of yeast cells does not appear to be affected by the change from a single 2B body to multiple eIF2B foci.
Our work sheds light on the recently discovered 2B body. The data show a relationship between eIF2B localization and an ER membrane-bound protein. We discovered eIF2B's membrane colocalization while examining its interaction with YBR159W. Our data show that YBR159W is not necessary for 2B's colocalization to the membrane. The primary mediator of eIF2B's membrane association is unknown. It remains to be determined whether the translation defect seen in the ybr159wΔ strain is the cause of the strain's slow growth or vice versa. Further experiments are required to determine the functional role of YBR159W interacting with eIF2B.
ACKNOWLEDGMENTS
C.M.B. was supported by National Institutes of Health (NIH) training grant T32 AI007611, and P.S. and A.J.L. were supported by NIH grant GM64779. The experiments, data analysis, and presentation of fluorescence microscope images were performed in part through the use of the VUMC Cell Imaging Shared Resource, which is supported by NIH grants CA68485, DK20593, DK58404, HD15052, and DK59637.
We thank Tom Dever, Alan Hinnebusch, Howard Riezman, and Jonathan Warner for reagents and yeast strains.
Footnotes
Published ahead of print 21 December 2012
REFERENCES
- 1.Preiss T, Hentze WM. 2003. Starting the protein synthesis machine: eukaryotic translation initiation. Bioessays 25:1201–1211 [DOI] [PubMed] [Google Scholar]
- 2.Sonenberg NH, Mathews JM. 2000. Translational control of gene expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 3.Pavitt GD. 2005. eIF2B, a mediator of general and gene-specific translational control. Biochem. Soc. Trans. 33:1487–1492 [DOI] [PubMed] [Google Scholar]
- 4.Rowlands AG, Panniers R, Henshaw EC. 1988. The catalytic mechanism of guanine nucleotide exchange factor action and competitive inhibition by phosphorylated eukaryotic initiation factor 2. J. Biol. Chem. 263:5526–5533 [PubMed] [Google Scholar]
- 5.Zhan K, Narasimhan J, Wek RC. 2004. Differential activation of eIF2 kinases in response to cellular stresses in Schizosaccharomyces pombe. Genetics 168:1867–1875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schneider RJ, Mohr I. 2003. Translation initiation and viral tricks. Trends Biochem. Sci. 28:130–136 [DOI] [PubMed] [Google Scholar]
- 7.Harding HP, Zhang Y, Ron D. 1999. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397:271–274 [DOI] [PubMed] [Google Scholar]
- 8.Wek RC, Ramirez M, Jackson BM, Hinnebusch AG. 1990. Identification of positive-acting domains in GCN2 protein kinase required for translational activation of GCN4 expression. Mol. Cell. Biol. 10:2820–2831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olsen DS, Jordan B, Chen D, Wek RC, Cavener DR. 1998. Isolation of the gene encoding the Drosophila melanogaster homolog of the Saccharomyces cerevisiae GCN2 eIF-2α kinase. Genetics 149:1495–1509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sood R, Porter AC, Olsen DA, Cavener DR, Wek RC. 2000. A mammalian homologue of GCN2 protein kinase important for translational control by phosphorylation of eukaryotic initiation factor-2α. Genetics 154:787–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fabian JR, Kimball SR, Heinzinger NK, Jefferson LS. 1997. Subunit assembly and guanine nucleotide exchange activity of eukaryotic initiation factor-2B expressed in Sf9 cells. J. Biol. Chem. 272:12359–12365 [DOI] [PubMed] [Google Scholar]
- 12.Gomez E, Pavitt GD. 2000. Identification of domains and residues within the epsilon subunit of eukaryotic translation initiation factor 2B (eIF2Bε) required for guanine nucleotide exchange reveals a novel activation function promoted by eIF2B complex formation. Mol. Cell. Biol. 20:3965–3976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pavitt GD, Ramaiah KV, Kimball SR, Hinnebusch AG. 1998. eIF2 independently binds two distinct eIF2B subcomplexes that catalyze and regulate guanine-nucleotide exchange. Genes Dev. 12:514–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bushman JL, Asuru AI, Matts RL, Hinnebusch AG. 1993. Evidence that GCD6 and GCD7, translational regulators of GCN4, are subunits of the guanine nucleotide exchange factor for eIF-2 in Saccharomyces cerevisiae. Mol. Cell. Biol. 13:1920–1932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pavitt GD, Yang W, Hinnebusch AG. 1997. Homologous segments in three subunits of the guanine nucleotide exchange factor eIF2B mediate translational regulation by phosphorylation of eIF2. Mol. Cell. Biol. 17:1298–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kubica N, Jefferson LS, Kimball SR. 2006. Eukaryotic initiation factor 2B and its role in alterations in mRNA translation that occur under a number of pathophysiological and physiological conditions. Prog. Nucleic Acid Res. Mol. Biol. 81:271–296 [DOI] [PubMed] [Google Scholar]
- 17.Hinnebusch AG. 1985. A hierarchy of trans-acting factors modulates translation of an activator of amino acid biosynthetic genes in Saccharomyces cerevisiae. Mol. Cell. Biol. 5:2349–2360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Campbell SG, Hoyle NP, Ashe MP. 2005. Dynamic cycling of eIF2 through a large eIF2B-containing cytoplasmic body: implications for translation control. J. Cell Biol. 170:925–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Campbell SG, Ashe MP. 2006. Localization of the translational guanine nucleotide exchange factor eIF2B: a common theme for GEFs? Cell Cycle 5:678–680 [DOI] [PubMed] [Google Scholar]
- 20.Rossler H, Rieck C, Delong T, Hoja U, Schweizer E. 2003. Functional differentiation and selective inactivation of multiple Saccharomyces cerevisiae genes involved in very-long-chain fatty acid synthesis. Mol. Genet. Genomics 269:290–298 [DOI] [PubMed] [Google Scholar]
- 21.Stoops JK, Wakil SJ. 1978. The isolation of the two subunits of yeast fatty acid synthetase. Biochem. Biophys. Res. Commun. 84:225–231 [DOI] [PubMed] [Google Scholar]
- 22.Welch JW, Burlingame AL. 1973. Very long-chain fatty acids in yeast. J. Bacteriol. 115:464–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dittrich F, Zajonc D, Huhne K, Hoja U, Ekici A, Greiner E, Klein H, Hofmann J, Bessoule JJ, Sperling P, Schweizer E. 1998. Fatty acid elongation in yeast: biochemical characteristics of the enzyme system and isolation of elongation-defective mutants. Eur. J. Biochem. 252:477–485 [DOI] [PubMed] [Google Scholar]
- 24.Dickson RC, Sumanasekera C, Lester RL. 2006. Functions and metabolism of sphingolipids in Saccharomyces cerevisiae. Prog. Lipid Res. 45:447–465 [DOI] [PubMed] [Google Scholar]
- 25.Gaigg B, Toulmay A, Schneiter R. 2006. Very long-chain fatty acid-containing lipids rather than sphingolipids per se are required for raft association and stable surface transport of newly synthesized plasma membrane ATPase in yeast. J. Biol. Chem. 281:34135–34145 [DOI] [PubMed] [Google Scholar]
- 26.Klein HP. 1957. Some observations on a cell free lipid synthesizing system from Saccharomyces cerevisiae. J. Bacteriol. 73:530–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abraham S, Chaikoff IL, Bortz WM, Klein HP, Den H. 1961. Particle involvement in fatty acid synthesis in liver and yeast systems. Nature 192:1287–1288 [DOI] [PubMed] [Google Scholar]
- 28.Tehlivets O, Scheuringer K, Kohlwein SD. 2007. Fatty acid synthesis and elongation in yeast. Biochim. Biophys. Acta 1771:255–270 [DOI] [PubMed] [Google Scholar]
- 29.Beaudoin F, Gable K, Sayanova O, Dunn T, Napier JA. 2002. A Saccharomyces cerevisiae gene required for heterologous fatty acid elongase activity encodes a microsomal beta-keto-reductase. J. Biol. Chem. 277:11481–11488 [DOI] [PubMed] [Google Scholar]
- 30.Han G, Gable K, Kohlwein SD, Beaudoin F, Napier JA, Dunn TM. 2002. The Saccharomyces cerevisiae YBR159w gene encodes the 3-ketoreductase of the microsomal fatty acid elongase. J. Biol. Chem. 277:35440–35449 [DOI] [PubMed] [Google Scholar]
- 31.Oh CS, Toke DA, Mandala S, Martin CE. 1997. ELO2 and ELO3, homologues of the Saccharomyces cerevisiae ELO1 gene, function in fatty acid elongation and are required for sphingolipid formation. J. Biol. Chem. 272:17376–17384 [DOI] [PubMed] [Google Scholar]
- 32.Tuller G, Prein B, Jandrositz A, Daum G, Kohlwein SD. 1999. Deletion of six open reading frames from the left arm of chromosome IV of Saccharomyces cerevisiae. Yeast 15:1275–1285 [DOI] [PubMed] [Google Scholar]
- 33.Schuldiner M, Collins SR, Thompson NJ, Denic V, Bhamidipati A, Punna T, Ihmels J, Andrews B, Boone C, Greenblatt JF, Weissman JS, Krogan NJ. 2005. Exploration of the function and organization of the yeast early secretory pathway through an epistatic mini-array profile. Cell 123:507–519 [DOI] [PubMed] [Google Scholar]
- 34.Vallee B, Riezman H. 2005. Lip1p: a novel subunit of acyl-CoA ceramide synthase. EMBO J. 24:730–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dickson RC. 2008. Thematic review series: sphingolipids: new insights into sphingolipid metabolism and function in budding yeast. J. Lipid Res. 49:909–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Preuss D, Mulholland J, Kaiser CA, Orlean P, Albright C, Rose MD, Robbins PW, Botstein D. 1991. Structure of the yeast endoplasmic reticulum: localization of ER proteins using immunofluorescence and immunoelectron microscopy. Yeast 7:891–911 [DOI] [PubMed] [Google Scholar]
- 37.Lowe M, Barr FA. 2007. Inheritance and biogenesis of organelles in the secretory pathway. Nat. Rev. Mol. Cell Biol. 8:429–439 [DOI] [PubMed] [Google Scholar]
- 38.David Amberg C, DJB, Jeffrey Strathern N. 2005. Methods in yeast genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 39.Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, Chu AM, Connelly C, Davis K, Dietrich F, Dow SW, El Bakkoury M, Foury F, Friend SH, Gentalen E, Giaever G, Hegemann JH, Jones T, Laub M, Liao H, Liebundguth N, Lockhart DJ, Lucau-Danila A, Lussier M, M'Rabet N, Menard P, Mittmann M, Pai C, Rebischung C, Revuelta JL, Riles L, Roberts CJ, Ross-MacDonald P, Scherens B, Snyder M, Sookhai-Mahadeo S, Storms RK, Veronneau S, Voet M, Volckaert G, Ward TR, Wysocki R, Yen GS, Yu K, Zimmermann K, Philippsen P, Johnston M, Davis RW. 1999. Functional characterization of the Saccharomyces cerevisiae genome by gene deletion and parallel analysis. Science 285:901–906 [DOI] [PubMed] [Google Scholar]
- 40.Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O'Shea EK, Weissman JS. 2003. Global analysis of protein expression in yeast. Nature 425:737–741 [DOI] [PubMed] [Google Scholar]
- 41.Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O'Shea EK. 2003. Global analysis of protein localization in budding yeast. Nature 425:686–691 [DOI] [PubMed] [Google Scholar]
- 42.James P, Halladay J, Craig EA. 1996. Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics 144:1425–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hudson JR, JR, Dawson EP, Rushing KL, Jackson CH, Lockshon D, Conover D, Lanciault C, Harris JR, Simmons SJ, Rothstein R, Fields S. 1997. The complete set of predicted genes from Saccharomyces cerevisiae in a readily usable form. Genome Res. 7:1169–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alberti S, Gitler AD, Lindquist S. 2007. A suite of Gateway cloning vectors for high-throughput genetic analysis in Saccharomyces cerevisiae. Yeast 24:913–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Powell DW, Weaver CM, Jennings JL, McAfee KJ, He Y, Weil PA, Link AJ. 2004. Cluster analysis of mass spectrometry data reveals a novel component of SAGA. Mol. Cell. Biol. 24:7249–7259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sanders SL, Jennings J, Canutescu A, Link AJ, Weil PA. 2002. Proteomics of the eukaryotic transcription machinery: identification of proteins associated with components of yeast TFIID by multidimensional mass spectrometry. Mol. Cell. Biol. 22:4723–4738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Link AJ, Eng J, Schieltz DM, Carmack E, Mize GJ, Morris DR, Garvik BM, Yates JR., III 1999. Direct analysis of protein complexes using mass spectrometry. Nat. Biotechnol. 17:676–682 [DOI] [PubMed] [Google Scholar]
- 48.Link AJ, Fleischer TC, Weaver CM, Gerbasi VR, Jennings JL. 2005. Purifying protein complexes for mass spectrometry: applications to protein translation. Methods 35:274–290 [DOI] [PubMed] [Google Scholar]
- 49.McAfee KJ, Duncan DT, Assink M, Link AJ. 2006. Analyzing proteomes and protein function using graphical comparative analysis of tandem mass spectrometry results. Mol. Cell. Proteomics 5:1497–1513 [DOI] [PubMed] [Google Scholar]
- 50.Ejsing CS, Sampaio JL, Surendranath V, Duchoslav E, Ekroos K, Klemm RW, Simons K, Shevchenko A. 2009. Global analysis of the yeast lipidome by quantitative shotgun mass spectrometry. Proc. Natl. Acad. Sci. U. S. A. 106:2136–2141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ejsing CS, Moehring T, Bahr U, Duchoslav E, Karas M, Simons K, Shevchenko A. 2006. Collision-induced dissociation pathways of yeast sphingolipids and their molecular profiling in total lipid extracts: a study by quadrupole TOF and linear ion trap-orbitrap mass spectrometry. J. Mass Spectrom. 41:372–389 [DOI] [PubMed] [Google Scholar]
- 52.Sud M, Fahy E, Cotter D, Brown A, Dennis EA, Glass CK, Merrill AH, Jr, Murphy RC, Raetz CR, Russell DW, Subramaniam S. 2007. LMSD: LIPID MAPS structure database. Nucleic Acids Res. 35:D527–D532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fields S, Song O. 1989. A novel genetic system to detect protein-protein interactions. Nature 340:245–246 [DOI] [PubMed] [Google Scholar]
- 54.Bergmann JE, Fusco PJ. 1988. The M protein of vesicular stomatitis virus associates specifically with the basolateral membranes of polarized epithelial cells independently of the G protein. J. Cell Biol. 107:1707–1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rose M, Botstein D. 1983. Construction and use of gene fusions to lacZ (β-galactosidase) that are expressed in yeast. Methods Enzymol. 101:167–180 [DOI] [PubMed] [Google Scholar]
- 56.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH image to ImageJ: 25 years of image analysis. Nat. Methods 9:671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Terasaki M, Song J, Wong JR, Weiss MJ, Chen LB. 1984. Localization of endoplasmic reticulum in living and glutaraldehyde-fixed cells with fluorescent dyes. Cell 38:101–108 [DOI] [PubMed] [Google Scholar]
- 58.Gerbasi VR, Weaver CM, Hill S, Friedman DB, Link AJ. 2004. Yeast Asc1p and mammalian RACK1 are functionally orthologous core 40S ribosomal proteins that repress gene expression. Mol. Cell. Biol. 24:8276–8287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Orlean P. 1990. Dolichol phosphate mannose synthase is required in vivo for glycosyl phosphatidylinositol membrane anchoring, O mannosylation, and N glycosylation of protein in Saccharomyces cerevisiae. Mol. Cell. Biol. 10:5796–5805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Orlean P, Albright C, Robbins PW. 1988. Cloning and sequencing of the yeast gene for dolichol phosphate mannose synthase, an essential protein. J. Biol. Chem. 263:17499–17507 [PubMed] [Google Scholar]
- 61.Schneiter R, Brugger B, Amann CM, Prestwich GD, Epand RF, Zellnig G, Wieland FT, Epand RM. 2004. Identification and biophysical characterization of a very-long-chain-fatty-acid-substituted phosphatidylinositol in yeast subcellular membranes. Biochem. J. 381:941–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hinnebusch AG. 1994. Translational control of GCN4: an in vivo barometer of initiation-factor activity. Trends Biochem. Sci. 19:409–414 [DOI] [PubMed] [Google Scholar]
- 63.Adelman MR, Sabatini DD, Blobel G. 1973. Ribosome-membrane interaction: nondestructive disassembly of rat liver rough microsomes into ribosomal and membranous components. J. Cell Biol. 56:206–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gavin AC, Bosche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick JM, Michon AM, Cruciat CM, Remor M, Hofert C, Schelder M, Brajenovic M, Ruffner H, Merino A, Klein K, Hudak M, Dickson D, Rudi T, Gnau V, Bauch A, Bastuck S, Huhse B, Leutwein C, Heurtier MA, Copley RR, Edelmann A, Querfurth E, Rybin V, Drewes G, Raida M, Bouwmeester T, Bork P, Seraphin B, Kuster B, Neubauer G, Superti-Furga G. 2002. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 415:141–147 [DOI] [PubMed] [Google Scholar]
- 65.Krogan NJ, Cagney G, Yu H, Zhong G, Guo X, Ignatchenko A, Li J, Pu S, Datta N, Tikuisis AP, Punna T, Peregrin-Alvarez JM, Shales M, Zhang X, Davey M, Robinson MD, Paccanaro A, Bray JE, Sheung A, Beattie B, Richards DP, Canadien V, Lalev A, Mena F, Wong P, Starostine A, Canete MM, Vlasblom J, Wu S, Orsi C, Collins SR, Chandran S, Haw R, Rilstone JJ, Gandi K, Thompson NJ, Musso G, St. Onge P, Ghanny S, Lam MH, Butland G, Altaf-Ul AM, Kanaya S, Shilatifard A, O'Shea E, Weissman JS, Ingles CJ, Hughes TR, Parkinson J, Gerstein M, Wodak SJ, Emili A, Greenblatt JF. 2006. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 440:637–643 [DOI] [PubMed] [Google Scholar]
- 66.Miller JP, Lo RS, Ben-Hur A, Desmarais C, Stagljar I, Noble WS, Fields S. 2005. Large-scale identification of yeast integral membrane protein interactions. Proc. Natl. Acad. Sci. U. S. A. 102:12123–12128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Willett M, Brocard M, Davide A, Morley SJ. 2011. Translation initiation factors and active sites of protein synthesis colocalize at the leading edge of migrating fibroblasts. Biochem. J. 438:217–227 [DOI] [PubMed] [Google Scholar]
- 68.Hannig EM, Hinnebusch AG. 1988. Molecular analysis of GCN3, a translational activator of GCN4: evidence for posttranslational control of GCN3 regulatory function. Mol. Cell. Biol. 8:4808–4820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Natarajan K, Meyer MR, Jackson BM, Slade D, Roberts C, Hinnebusch AG, Marton MJ. 2001. Transcriptional profiling shows that Gcn4p is a master regulator of gene expression during amino acid starvation in yeast. Mol. Cell. Biol. 21:4347–4368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Barrett T, Troup DB, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Muertter RN, Holko M, Ayanbule O, Yefanov A, Soboleva A. 2011. NCBI GEO: archive for functional genomics data sets: 10 years on. Nucleic Acids Res. 39:D1005–D1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fendt SM, Oliveira AP, Christen S, Picotti P, Dechant RC, Sauer U. 2010. Unraveling condition-dependent networks of transcription factors that control metabolic pathway activity in yeast. Mol. Systems Biol. 6:432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Merrick WC, Hershey JWB. 1996. The pathway and mechanism of eukaryotic protein synthesis, p 31–69. In Hershey JWB, Mathews MB, Sonenberg N. (ed), Translation control. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 73.Taylor EJ, Campbell SG, Griffiths CD, Reid PJ, Slaven JW, Harrison RJ, Sims PF, Pavitt GD, Delneri D, Ashe MP. 2010. Fusel alcohols regulate translation initiation by inhibiting eIF2B to reduce ternary complex in a mechanism that may involve altering the integrity and dynamics of the eIF2B body. Mol. Biol. Cell 21:2202–2216 [DOI] [PMC free article] [PubMed] [Google Scholar]