

Abstract
Correct reprogramming of epigenetic marks in the donor nuclei is crucial for successful cloning by nuclear transfer. Specific epigenetic modifications, such as repressive histone lysine methylation marks, are known to be very stable and difficult to reprogram. The discovery of histone lysine demethylases has opened up opportunities to study the effects of removing repressive histone lysine methylation marks in donor cells prior to nuclear transfer. In this study, we generated mouse embryonic stem (ES) cells for the inducible expression of JMJD2B (also known as KDM4B), a demethylase that primarily removes the histone-3 lysine-9 trimethylation (H3K9me3) mark. Induction of jmjd2b in the ES cells decreased total levels of H3K9me3 by 63%. When these cells were used for nuclear transfer, H3K9me3 levels were normalized within minutes following fusion with an enucleated oocyte. This transient reduction of H3K9me3 levels improved in vitro development into cloned embryos by 30%.
INTRODUCTION
Despite sharing the same genetic information, cell types within an individual are morphologically and functionally diverse. Diversity arises from a complex set of epigenetic modifications, such as DNA methylation and histone modifications, that control cell-specific gene expression profiles and determine the cellular phenotype. Correct setting of these modifications is critical during embryonic development (1–6). Reprogramming of epigenetic marks occurs first during gametogenesis and later during fertilization and subsequent embryonic differentiation (3, 5). Reprogramming during gametogenesis involves extensive demethylation and sex-specific remethylation at imprinted loci of primordial germ cells and gametes (3). Following fertilization, reprogramming of epigenetic marks is essential for resolving the early parental asymmetry in histone modifications, DNA methylation, and chromatin proteins to allow correct embryonic gene activation (7–9). Nuclear transfer (NT) cloning is almost a reversal of this process. It requires that the genome of a single differentiated cell with all its epigenetic modifications, which, unlike the gametes, are not formatted to initiate development, be reprogrammed from a restricted cell lineage-appropriate gene expression profile to a totipotent state (2, 4). Live cloned offspring have been produced from a range of mammalian species, demonstrating that somatic donor nuclei can be reprogrammed back to the embryonic state (10, 11). However, the efficiency of this process remains low, and various molecular, cellular, and developmental abnormalities have been detected in clones. Incorrect reprogramming of the epigenetic donor cell marks has been proposed to be the main cause of this low efficiency (1, 2, 4, 6).
To facilitate nuclear reprogramming, epigenetic modifications in donor cells have been modified by treating them with pharmacological histone deacetylase and DNA methyltransferase inhibitors (12–15). These agents increase global histone acetylation and reduce DNA methylation, respectively, resulting in a more open, transcriptionally permissive chromatin that generally reprograms better. Histone lysine methylation marks play a key role in controlling gene expression profiles and directing cell lineage specification. Some of these marks can persist through multiple cell divisions (16–18). In particular, lysine methylation marks associated with gene repression are thought to be responsible for restricting the reprogrammability of the genome (19–22). The discovery of enzymes that remove histone methylation (23) allows more targeted approaches to investigate the role of these epigenetic modifications during NT-induced reprogramming. The histone lysine demethylase JMJD2B, which demethylates the trimethyl modification of H3K9 (24), is such an enzyme. It provides a molecular tool to manipulate and investigate the role of the thermodynamically very stable histone-3 lysine-9 trimethylation (H3K9me3) modification previously shown to resist reprogramming following NT and potentially limiting the efficiency of nuclear reprogramming (22). Overexpression of a truncated, yet fully active form of the JMJD2B demethylase in NIH 3T3 cells was shown to result in a decline in H3K9me3 levels (24). Due to their nonreprogrammable karyotypic abnormalities and mutations, NIH 3T3 cells do not support development of NT embryos beyond the 2-cell stage (F. Oback, unpublished observation). Hence, we generated embryonic stem (ES) cells engineered for the inducible expression of the truncated form of JMJD2B. We demonstrate that reduced H3K9me3 levels in induced donor cells are quickly restored after fusion with an enucleated oocyte. Nevertheless, such ES cell donors reprogrammed into cloned embryos significantly better. These results confirm that repressive H3K9me3 marks are implicated in restricting cloning efficiency and thus validate this novel experimental strategy to examine mechanistic aspects of nuclear reprogramming.
MATERIALS AND METHODS
Vector construction.
To generate the jmjd2b-egfp Flp recombinase-mediated DNA insertion (Flp-In) vectors for functional and mutant jmjd2b, plasmids pI-jmjd2b-T2-GFP and pI-jmjd2b-T2-H189A-GFP (24), which contain the 1- to 424-amino-acid truncated functional or H189A mutated inactive jmjd2b fused to the enhanced green fluorescent protein (EGFP) gene, were digested with HindIII, and the overhangs were filled in with the Klenow fragment to isolate a 2-kb fragment encoding the respective jmjd2b-egfp fusion constructs. The EGFP Flp-In vector (pBS31-EGFP), kindly provided by C. Beard (25), was digested with EcoRI to excise the EGFP marker gene. The resulting Flp-In vector fragment was blunted, dephosphorylated, and then ligated to the jmjd2b-egfp fragments. The newly generated Flp-In vectors for functional (F) and mutant (M) jmjd2b-egfp were validated by restriction enzyme digestion and sequencing analysis.
Cell culture.
Parental KH2 ES cells (25) (obtained from Open Biosystems) and all ES cell lines derived from KH2 cells were cultured in Dulbecco's modified Eagle's medium–nutrient mixture F-12 (DMEM–F12; Life Technologies) containing 20% fetal bovine serum (Life Technologies), 100 μM β-mercaptoethanol (Sigma), 1× minimal essential medium nonessential amino acids (Life Technologies), 2,000 U/ml of human leukemia inhibitory factor (GenScript), and 0.4 μM PD0325901 (Stemgent) on 0.1% gelatin-coated dishes or were seeded onto dishes containing mitomycin C (Sigma)-inactivated STO fibroblast feeders.
Generation of ES cells for the inducible expression of jmjd2b-egfp.
KH2 ES cells (approximately 5 million cells) were transfected by Nucleofection using a mouse ES Nucleofection kit according to the manufacturer's guidelines (Amaxa AG). The jmjd2b-egfp Flp-In expression plasmids (5 μg) were each cotransfected into KH2 cells with different amounts of FLPo (26) expression plasmid pPGKFLPobpA, obtained from Addgene (plasmid 13793), at plasmid ratios of 3:1, 1:1, and 1:3. To isolate cell clones with correct Flp-In based on the restoration of a nonfunctional hygromycin selection marker at the homing site, cells were selected with 140 μg/ml hygromycin B (Life Technologies) at 48 h after transfection. Resulting hygromycin-resistant clones were picked approximately 10 days later and expanded in hygromycin-free ES cell medium. Picked clones were analyzed for correct targeting by PCR using standard conditions. Successful Flp-In of jmjd2b-egfp was detected with primer pair P2/P3, and primers P1 and P2 were used to identify the original nonrecombined Col1A1 locus. The possible integration of the FLPo recombinase was tested using primer pair P4/P5. Primer sequences are shown in Table 1.
Table 1.
Primer sequences
| Primer | Sequence |
|---|---|
| P1 | CTCCCTTTAGGGTTCCGATT |
| P2 | AGCAATCGCGCATATGAAAT |
| P3 | GTTTTCCCAGTCACGACGTT |
| P4 | CACCATCATCCCTTACAACG |
| P5 | TTCAGCAGCTGGTACTCCTG |
| P6 | AGAAGACACCGGGACCGATC |
| P7 | TGAATTCATCCATGGTGGGG |
| P8 | AGAAGCCTTCCTGTTCTCAG |
| P9 | TGTACTGACTGGCTGTAGGG |
| GAPDH f | TGCACCACCAACTGCTTAG |
| GAPDH r | GATGCAGGGATGATGTTC |
| β-actin f | CAGAAGGACTCCTATGTGGG |
| β-actin r | TTGGCCTTAGGGTTCAGGG |
| HPRT fa | GAAATGTCAGTTGCTGCGTC |
| HPRT ra | GCCAACACTGCTGAAACATG |
| GusB fb | ATAAGACGCATCAGAAGCCG |
| GusB rb | ACTCCTCACTGAACATGCGA |
Induction of transgene expression.
ES cells were cultured on 0.1% gelatin-coated dishes in the presence of 1 μg/ml doxycycline (Sigma) for 48 h. The induced cells were then trypsinized, resuspended in phosphate-buffered saline (PBS), and analyzed for EGFP fluorescence on a FACScalibur apparatus (Becton and Dickinson). To assess the switch off of jmjd2b-egfp expression, induced ES cells were reseeded in medium without doxycycline and analyzed for EGFP fluorescence after 24 h of culture.
RNA and cDNA isolation.
For total RNA extraction, jmjd2b-egfp ES cells were seeded onto gelatin-coated 6-well plates without feeders and cultured for 48 h in the presence or absence of 1 μg/ml of doxycycline for 48 h. For each treatment, total RNA equivalent to cells of one 6-well plate was isolated using a Qiagen RNeasy minikit. cDNA was synthesized from 2 μg of total RNA using SuperScript III first-strand synthesis SuperMix (Life Technologies), precipitated, and resuspended in 100 μl of RNase-free water.
qRT-PCR.
Endogenous and transgene-derived jmjd2b expression levels were determined by quantitative reverse transcriptase PCR (qRT-PCR) using the Syber Premix ExTaq (TaKaRa). Transgene-derived jmjd2b was amplified using primers P6 and P7, while primers P8 and P9 were used to amplify the endogenous demethylase. All primer sequences are shown in Table 1, and target sequences were amplified with a Corbett Rotorgene 6000 system with the following cycling conditions: 3 min of denaturation and 40 cycles of 10 s at 95°C and 25 s at 60°C. Each PCR was first validated for its efficiency with external standard curves from serial 5-log dilutions for each gene in duplicate. For transcript quantification, either 1 μl or 0.5 μl of cDNA synthesized using the SuperScript III first-strand synthesis SuperMix was used. The comparative quantification in the Rotorgene software was used to determine the amplification efficiency of each reaction (29). Transcript levels of endogenous and transgene-derived jmjd2b were determined relative to the geometric mean of the four housekeeping genes for glyceraldehyde-3-phosphate dehydrogenase (GAPDH), β-actin (ACTB), hypoxanthine-guanine phosphoribosyltransferase (HPRT), and glucuronidase beta (GUSB) while normalizing for amplification efficiency (a) and the expression level of the gene of interest (goi), which is equal to , where CT is the threshold cycle (30). For both primer combinations P6/P7 and P8/P9, differences in amplicon sizes (190 bp and 189 bp, respectively), the G+C contents of the amplicons (57.4% and 56.6%, respectively), and the amplification efficiencies for the reactions (range, 1.74 to 1.8) were within the compulsory limits for calculating relative levels and accounted for by the equation.
Immunostaining.
ES cells were seeded onto sterilized gelatin-coated (0.1%) coverslips without feeders and cultured in the presence or absence of 1 μg/ml of doxycycline for 48 h. Cells were then fixed and permeabilized simultaneously in 4% (wt/vol) paraformaldehyde–1% (wt/vol) Triton X-100 in PBS, blocked with 2.5% (wt/vol) bovine serum albumin in PBS, all at room temperature, and incubated overnight at 4°C with the primary antibody specific for H3K9me1 (rabbit polyclonal, 1:1,000), H3K9me2 (rabbit polyclonal, 1:1,000), H3K9me3 (rabbit polyclonal, 1:1,000), H3K27me3 (rabbit polyclonal, 1:1,000), and GFP (1:1,000; catalog no. A11120; Life Technologies). The histone methylation antibodies have been previously described and validated in detail (31). Following binding of the primary antibody, cells were washed in PBS–0.05% Tween 20 and incubated for 1 h at room temperature with 5 μg/ml of Hoechst 33342 (to visualize DNA) and the respective secondary antibodies: Alexa Fluor 568–goat anti-rabbit antibody (1:1,000; catalog no. A11011; Life Technologies) and Alexa Fluor 488–donkey anti-mouse antibody (1:1,000; catalog no. A21202; Life Technologies). After another washing step, cells were mounted with Dako fluorescent mounting medium (catalog no. S3023; Dako) onto glass slides, and immunostained cells were visualized on an AX-70 Olympus fluorescence microscope. Images were taken and processed using a Spot RT-KE slider charge-coupled-device camera and associated software (Diagnostics Instruments Inc.). A similar protocol for immunofluorescence staining was carried out on NT reconstructs, with the difference being that these were first fixed in 4% paraformaldehyde (30 min at 4°C) and then permeabilized in 0.1% Triton X-100 (5 min at room temperature) before blocking and immunostaining. Quantification of global H3K9me3 levels in NT reconstructs was conducted in ImageJ software and normalized on the nuclear area. For each reconstruct, the region of interest (ROI) around the nucleus (marked D [donor nucleus] or PN [pronucleus] in Fig. 7) was outlined. The mean background intensity, measured at a random location within the cytoplast (marked C in Fig. 7), was subtracted from the mean ROI intensity. This background-corrected mean intensity was then divided by the area to determine normalized pixel intensity.
Fig 7.

H3K9me3 in NT reconstructs generated from induced and noninduced F-jmjd2b ES cells. NT reconstructs were fixed and costained for DNA (H33342) and antibodies specific for EGFP and H3K9me3 prior to fusion of the donor cells with enucleated oocytes (A), 10 min after fusion with the oocyte (B), 1 h after fusion (C), and 8 h after fusion (D). Incorporation of the donor cells into the enucleated oocyte cytoplasm was monitored under a microscope. DNA images are merged images of phase-contrast and H33342-stained images. C, cytoplast; D, donor nucleus; PN, pronucleus; PB, polar body.
Western blot detection of modified histones.
Histones were extracted using an EpiQuik total histone extraction kit (catalog no. OP-0006-100; Epigentek) according to the manufacturer's guidelines. For induced cell populations, histones were extracted from cells verified by flow cytometry for greater than 80% induction. Histone extracts (10 to 15 μg per lane) were separated on a 15% SDS-polyacrylamide gel and transferred onto a nitrocellulose membrane, and relevant methylated histones were detected with 1:3,000 or 1:6,000 dilutions of the primary antibodies specified above. Following incubation with a secondary goat anti-rabbit antibody conjugated with horseradish peroxidase (1:5,000 or 1:10,000; catalog no. G21234; Life Technologies), the modified histones were visualized with enhanced chemiluminescence. Signal intensity was normalized for the histone H4 signal (Ponceau S stain) and quantified using Quantity One software (Bio-Rad Laboratories Inc.).
Nuclear transfer.
Zona-free NT was carried out according to a modified protocol based on the method described by Ribas et al. (32). Donor jmjd2b ES cells were cultured in the presence or absence of doxycycline for 48 h. On the day of NT, cells were treated with 1.65 μM nocodazole for 2 to 3 h to arrest them at metaphase, following which they were shaken off the plate. Metaphase II-arrested oocytes were collected from 10- to 12-week-old B6C3 (C3H × C57BL/6) female mice 14 to 17 h after human chorionic gonadotropin stimulation, and the cumulus cells were dispersed with bovine testicular hyaluronidase. The zona pellucida was removed with Tyrode's acidic solution, pH 2.5, with 1% polyvinylpyrrolidone and zona-free oocytes enucleated under differential interference contrast. Metaphase-arrested ES cell donors were attached to enucleated oocytes using 10 μg/ml lectin (L-9017; Sigma). Couplets were automatically aligned and fused in a parallel plate chamber in 0.2 mM mannitol fusion buffer (33). Progression of fusion was observed under a stereomicroscope and scored according to donor cell morphology: from round donor cell (not fused) to progressively flattening donor cell (fusing) and absence of donor cell (fused). Until fusion had occurred, which was observed under the stereomicroscope, all solutions contained nocodazole to keep the donors arrested in metaphase. At at least 1 h postfusion, NT reconstructs were activated with 10 mM SrCl2 in Ca-free M16 medium (M7292; Sigma-Aldrich). After 6 h, polar body extrusion was scored and all reconstructs with one polar body were cultured in M16 medium drops at 37.5°C under 5% CO2 in air for 4 days. Embryo development was measured as the number of compacted morulae or blastocysts relative to the number of NT reconstructs placed into culture. On day 4, compacted morulae and blastocysts were either fixed for immunofluorescence or transferred into the oviduct of day 0.5 pseudopregnant females as previously described (34).
Statistical analysis.
Values for all quantitative expression analyses are provided as the averages of several replicates ± standard errors of the means (SEMs). Significance of differences for Western analysis was determined via two-tailed paired t tests on log expressions. For the quantification of fluorescent signals of immunostained NT reconstructs, significance was determined by two-tailed paired t tests, and for qRT-PCR results, significance was determined by two-tailed unpaired t tests. For comparisons of Flp-In efficiencies and in vitro and in vivo development of reconstructs derived from induced and noninduced jmjd2b ES cells, the significance of differences was determined using the two-tailed Fisher exact test for independence in 2-by-2 tables.
RESULTS
Generation of jmjd2b-inducible ES cells.
The acceptor cell line KH2 (25) has been designed for efficient site-specific integration of transgenes and was used for the generation of ES cells that can be induced to express the histone demethylase JMJD2B. KH2 ES cells carry an expression cassette for the M2 reverse tetracycline transactivator (M2-rtTA) at the Rosa26 locus and an Flp recombination target (FRT) homing site at the collagen type 1 alpha 1 (Col1a1) locus for insertion of a gene of interest that can be induced according to the Tet-On system (Clontech Laboratories). For the targeted insertion, we generated Flp-In vectors for two previously characterized jmjd2b-egfp fusion constructs (24). The first construct consisted of a truncated (amino acids 1 to 424) but fully functional JMJD2B (F-JMJD2B) that mainly contains the catalytic domains fused to EGFP. The second construct is truncated JMJD2B containing an H189A point mutation (M-JMJD2B) that abrogates all enzymatic activity. Following cotransfection of Flp-In and FLPo recombinase constructs, cells with targeted insertions can be identified by hygromycin selection (25). Of the isolated resistant cell clones, only 50 to 78% could be further expanded, and all displayed the morphological phenotype of the parental KH2 ES cells (data not shown).
To confirm correct Flp-In, cell clones were screened via PCR with primers detecting the presence of the original locus and recombined locus (Fig. 1). The genotyping results for all isolated ES cell clones that could be sufficiently expanded are summarized in Table 2. Across all treatments, between 40 and 100% of hygromycin-resistant cell clones had undergone Flp recombination and contained the jmjd2b-egfp fusion construct at the Col1al locus. The use of a 3-fold excess of FLPo over the Flp-In vector compared to a 3-fold excess of the Flp-In vector over FLPo resulted in significantly increased Flp-In efficiency (85% and 100% versus 40%; P = 0.019 and P = 0.0001, respectively). In spite of the stringent selection, some background hygromycin-resistant clones that were negative for jmjd2b Flp-In (6 to 60% across the different groups) and clones which were negative for both the original and jmjd2b Flp-In (0 to 9% across the different groups) were also obtained. In addition, hygromycin-resistant clones were screened by PCR for potential nontargeted integration of the FLPo construct, which was detected in a small number of hygromycin-resistant clones (0 to 18%). For subsequent analyses, we selected one F-jmjd2b cell clone and one M-jmjd2b ES cell clone that contained the correct Flp-In jmjd2b-egfp and no random FLPo insertions and showed in vitro growth rates typical of mouse ES cells.
Fig 1.

Schematic representation of the parental and modified Col1A1 locus jmjd2b-egfp Flp-In. The parental locus consists of a neomycin resistance gene flanked by FRT sites (triangles) and a promoterless hygromycin resistance gene that lacks an ATG start codon [Hygro (no ATG)]. Correct FLPo-mediated insertion of the jmjd2b vector replaces the neomycin cassette with the TetO-controlled jmjd2b-egfp gene and converts the nonfunctional Hygro (no ATG) into a functional selection cassette (Hygro) by providing a phosphoglycerate kinase promoter and ATG (PGK-ATG). Binding sites of primers (arrows) used for the characterization of ES clones with correct Flp-In (primers P1 to P3) and transgene-specific qRT-PCR (primers P6 and P7) and the resulting amplicon sizes are indicated.
Table 2.
Results for generation of jmjd2b Flp-In ES cell clones
| FLPo construct and plasmid ratio | No. of ES clones picked | No. of expandable ES clones | No. (%) of clones: |
Flp-In efficiencyb | |||||
|---|---|---|---|---|---|---|---|---|---|
| Positive for jmjd2b Flp-Ina | Positive for parental locusa | Negative for jmjd2b and parental locusa | Positive for jmjd2b and parental locusa | FLPo insertiona | Correctly targeted, no FLPo insertionc | ||||
| F-jmjd2b | |||||||||
| 3:1 | 42 | 33 | 28 (85)d | 2 (6) | 3 (9) | 0 | 6 (18) | 23 (82) | 85 |
| 1:1 | 34 | 22 | 18 (82)e | 2 (9) | 2 (9) | 0 | 2 (9) | 17 (94) | 82 |
| 1:3 | 19 | 10 | 4 (40) | 6 (60) | 0 | 0 | 0 | 4 (100) | 40 |
| M-jmjd2b: 3:1 | 46 | 35 | 35 (100)f | 2 (6) | 0 | 2 (6) | 4 (11) | 31 (89) | 100 |
Percentages indicate the proportions of expandable cell clones with the specified genotype.
Proportion of expandable cell clones with a targeted insertion of jmjd2b.
Percentages indicate the proportions of cell clones with the targeted jmjd2b insertion that are not compromised by a random insertion of the FLPo construct.
P = 0.019 compared with F-jmjd2b at the 1:3 plasmid ratio determined by the Fisher 2-by-2 test.
P = 0.054 compared with F-jmjd2b at the 1:3 plasmid ratio determined by the Fisher 2-by-2 test.
P = 0.0001 compared with F-jmjd2b at the 1:3 plasmid ratio determined by the Fisher 2-by-2 test.
Doxycycline-induced expression of jmjd2b-egfp.
Selected F- and M-jmjd2b ES cell clones were cultured in the presence of doxycycline to induce expression of the jmjd2b-egfp transgene. Due to the fusion with EGFP, cells expressing JMJD2B could be readily monitored and were analyzed by flow cytometry. Representative flow cytometry profiles of noninduced and induced cells are shown in Fig. 2. In the presence of doxycycline, up to 94% (average ± SEM, 86% ± 2%; n = 19) and 88% (average ± SEM, 81% ± 2%; n = 12) of F-jmjd2b and M-jmjd2b cells, respectively, displayed EGFP fluorescence, indicative of JMJD2B-EGFP expression and the doxycycline-dependent inducibility of both cell clones. We also examined the kinetics for switching off JMJD2B-EGFP expression and reanalyzed induced cells for green fluorescence 24 h after removal of doxycycline. This showed that the formerly induced cells had returned to a profile that was indistinguishable from that of noninduced cells, confirming the complete switch off of jmjd2b-egfp transgene expression.
Fig 2.

Flow cytometry analyses of jmjd2b-egfp induction. Green fluorescence generated by F-jmjd2b (A) and M-jmjd2b (B) ES cells was determined using the FL1 emission channel. The range of intensities for green fluorescent cells (M1) is indicated. The relative cell numbers are plotted as a function of variable intensities of green fluorescence from individual cells. Gray filled graph, noninduced cells; solid-line graph, induced cells; dotted-line graph, induced cells 24 h after removal of doxycycline.
In addition, levels of transgene-derived jmjd2b expression were determined by qRT-PCR with the transgene-specific primer pair P6/P7 (Fig. 1). Expression levels relative to the geometric mean of four housekeeping genes were significantly higher for induced F-jmjd2b cells than M-jmjd2b cells (1.58 ± 0.11 and 1.06 ± 0.07, respectively; P = 0.015) (Fig. 3A), essentially reflecting the difference in induction rates. Compared to noninduced control cells, this corresponds to 1,230- and 558-fold increases in expression of F-jmjd2b and M-jmjd2b, respectively. We next measured the expression of the endogenous jmjd2b gene to determine how it compares to the induced expression of the transgene-derived jmjd2b. Using a primer pair (P8 and P9) specific for an endogenous jmjd2b sequence that is deleted in the truncated transgene version, relative expression levels of the endogenous gene were determined in comparison to the geometric mean of the same four housekeeping genes. Expression levels of the endogenous gene were 0.037 ± 0.002 and 0.043 ± 0.001 in noninduced F- and M-jmjd2b cells, respectively. Under inducing conditions, the endogenous jmjd2b transcript levels remained unchanged (0.037 ± 0.002 for F-jmjd2b and 0.033 ± 0.001 for induced M-jmjd2b cells) and were not affected by the overexpression of the respective jmjd2b transgene (Fig. 3B). Thus, relative to the natural expression levels of the endogenous jmjd2b gene, transgene expression levels are more than an order of magnitude greater, resulting in substantially increased total jmjd2b expression in induced F- and M-jmjd2b ES cells.
Fig 3.
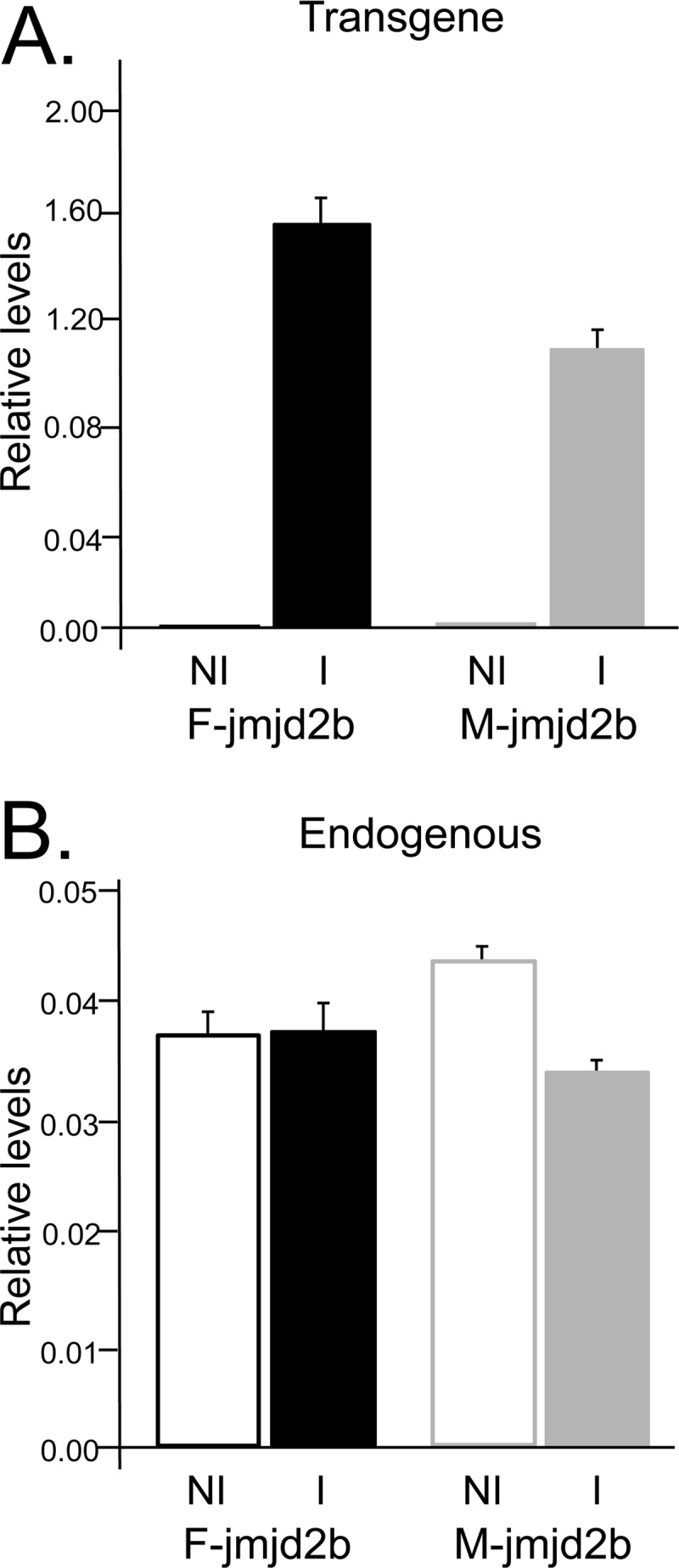
jmjd2b expression levels. Shown are the relative expression levels of transgene-derived jmjd2b (A) and endogenous jmjd2b (B) compared to the geometric mean of four housekeeping genes. Results for induced (I) and noninduced (NI) F-jmjd2b and M-jmjd2b ES cells are averages of 3 to 6 replicates, with error bars depicting the SEMs.
Induction of F-JMJD2B reduces H3K9me3 in ES cells.
We then evaluated the enzymatic activity of transgene-encoded JMJD2B. Using qualitative immunofluorescence, we determined the methylation status of H3K9 in induced and noninduced F- and M-jmjd2b ES cells using antibodies specific for each of the three different methyl modifications H3K9me1, H3K9me2, and H3K9me3. In contrast to noninduced cells, induced cells expressing F-JMJD2B were green fluorescent, which correlated with markedly reduced H3K9me3 levels (Fig. 4). The observed change in methylation upon induction of F-JMJD2B was specific for H3K9me3, as F-JMJD2B expression had no apparent effect on H3K9me2 and H3K9me1. Cells expressing M-JMJD2B showed no effect on any of the methylated states of H3K9. To quantify the reduction in H3K9me3 levels, we performed Western analyses of bulk histone extracts prepared from induced and noninduced F- and M-jmjd2b ES cells (Fig. 5A). Consistent with the immunofluorescence results, signal quantification of the immunoblots showed a selective and significant decrease of H3K9me3 levels of approximately 63% ± 2.4% (n = 3 repeats; P < 0.01) in induced F-jmjd2b ES cells compared to noninduced F-jmjd2b cells and induced or noninduced M-jmjd2b cells (Fig. 5B). Furthermore, the Western blotting quantification indicated a 28% ± 9.5% increase in H3K9me1 levels upon induction of F-jmjd2b, which was close to reaching significance (n = 3 repeats; P = 0.07) and not observed in induced M-jmjd2b cells. H3K9me2 levels remained unchanged upon induction of F- or M-jmjd2b. In addition, the two lines of ES cells were also analyzed for the unrelated, repressive trimethyl modification of H3K27. Immunofluorescence with H3K27me3-specific antibodies did not detect any significant changes in this mark with either induced F-jmjd2b ES cells or induced M-jmjd2b ES cells (Fig. 6).
Fig 4.

Immunofluorescence analysis of histone modifications in induced (I) and noninduced (NI) F- and M-jmjd2b ES cells. Cells were costained for DNA and with antibodies specific for EGFP and the indicated histone modification. The arrowheads indicate an induced cell next to two noninduced cells, highlighting the specific reduction of H3K9me3 in induced cells.
Fig 5.

JMJD2B-dependent changes in levels of H3K9 methylation states. Histone extracts from induced (I) and noninduced (NI) F- and M-jmjd2b cells were quantified for the indicated histone modifications by Western analysis. (A) Representative immunoblots for the indicated H3K9 methylation mark. For each modification, the top panel (W) is the immunoblot and the bottom panel (P) is the respective Ponceau S-stained blot. The H4 band that was used for normalization is indicated in the Ponceau S-stained blot. (B) Results represent H3K9 methylation levels in induced cells as a percentage of the levels in noninduced cells. F, F-jmjd2b cells; M, M-jmjd2b cells; error bars, SEMs determined from 3 repeats; asterisks, significant changes (**, P < 0.01; *, P = 0.074, determined by two-tailed paired t test on log expressions).
Fig 6.

Analysis of H3K27me3 in jmjd2b ES cells. H3K27me3 levels in induced (I) and noninduced (NI) F- and M-jmjd2b ES cells were analyzed by immunofluorescence (A) and Western blot quantification (B). For the detection by immunofluorescence, cells were costained for DNA and antibodies specific for EGFP and H3K27me3. For the Western blot quantification, shown are the representative immunoblot (W) and the Ponceau S-stained blot (P). The H4 band that was used for normalization is indicated in the Ponceau S-stained blot. The quantification of induced H3K27me3 levels by Western analysis is presented as a percentage of the levels for noninduced cells. Error bars, SEMs determined from 2 repeats.
Reduced H3K9me3 levels are rapidly restored after nuclear transfer.
Next we examined how the reduced H3K9me3 levels from the induced F-jmjd2b donor cells change after NT and subsequent in vitro embryo development under noninducing conditions. NT reconstructs generated from induced and noninduced F-jmjd2b ES cells were fixed at various time points after NT and analyzed by immunofluorescence for the presence of H3K9me3 marks (Fig. 7 and 8). After electrofusion, incorporation of donor cell nuclei into the enucleated oocyte (cytoplast) was monitored under a stereomicroscope. Following the attachment of induced F-jmjd2b donor cells to cytoplasts, just prior to NT, the donor cells still showed substantially reduced H3K9me3 levels (51% of noninduced H3K9me3; P < 0.05) compared to noninduced donor cells (induced, n = 11; noninduced, n = 12) (Fig. 7A and 8, couplets). However, within 10 min after fusion of the donor cells and entry into the oocyte cytoplasm, the initial difference in H3K9me3 signal between induced and noninduced groups (induced, n = 13; noninduced, n = 14) had disappeared (Fig. 7B and 8, postfusion 10 min). At the same time, the EGFP signal that previously marked the induced donor cell was no longer visible in the reconstruct. This status remained unchanged 1 h after fusion (induced, n = 9; noninduced, n = 8) (Fig. 7C and 8, postfusion 1 h), with no difference in the H3K9me3 signal being detected between induced and noninduced embryos. Similar results were obtained with NT reconstructs at 2, 4, and 6 h postfusion (data not shown). At 8 h postfusion, both the newly formed pronucleus and the extruded polar body displayed similar H3K9me3 intensities in NT reconstructs derived from induced versus noninduced donor cells (induced, n = 9; noninduced, n = 8) (Fig. 7D and 8, postfusion 8 h). In addition, some NT embryos were analyzed at the 2-cell, 4-cell, and blastocyst stages. No significant differences were detected between the H3K9me3 signals from the induced and noninduced treatment groups at any of those developmental stages (data not shown). These results suggest that the initial reduction in H3K9me3 levels in induced donor cells is restored within minutes after NT.
Fig 8.

Quantification of H3K9me3 in NT reconstructs generated from induced and noninduced F-jmjd2b ES cells. Shown is the normalized pixel intensity for noninduced (NI) and induced (I) F-jmjd2b ES donor cells attached to enucleated oocytes (or cytoplast marked C in Fig. 7) prior to fusion (couplets; noninduced, n = 12; induced, n = 11) and NT reconstructs 10 min (noninduced, n = 14; induced, n = 13), 1 h (noninduced, n = 8; induced, n = 9), and 8 h (noninduced, n = 8; induced, n = 9) after fusion of the donor nuclei (marked D in Fig. 7) and cytoplast. Note that due to differences in the exposure times for different developmental reconstruct stages, only values of induced versus noninduced of the same reconstruct stage are directly comparable. Error bars depict SEMs. RU, relative units; *, significant (P < 0.05) difference determined by two-tailed paired t test.
Reduced H3K9me3 levels improve in vitro development of cloned embryos.
To evaluate whether ES cells with reduced H3K9me3 levels would improve donor cell reprogrammability into cloned embryos, we determined the in vitro development of NT reconstructs into blastocysts. NT was performed under noninducing conditions using either induced or noninduced F-jmjd2b ES cells. Prior to each NT run, cells were validated for induction of F- or M-jmjd2b by monitoring EGFP fluorescence. The average induction rate ± SEM for F-jmjd2b was 80% ± 0.04% (range, 60 to 94%; n = 9). For M-jmjd2b, the average induction rate ± SEM was very similar at 77% ± 4% (range, 71 to 85%, n = 3). Despite the rapid restoration of H3K9me3 levels following NT (Fig. 7 and 8), the initial donor chromatin modification significantly improved the in vitro development of NT reconstructs (Table 3). Reconstructs generated with induced F-jmjd2b donor cells developed into 38% more blastocysts (induced versus noninduced, 33% versus 24%; P = 0.041) and showed a 30% higher overall development (induced versus noninduced, 48% versus 37%; P = 0.018) than NT reconstructs derived from noninduced F-jmjd2b ES cells. In contrast, no significant differences of in vitro development were detected in control experiments using induced and noninduced M-jmjd2b donor cells.
Table 3.
In vitro development of NT reconstructs
| Cell line | No. of independent NT expts | No. of NT reconstructsa | No. (% ± SEM) |
||
|---|---|---|---|---|---|
| Morulae | Blastocysts | Total developmentb | |||
| F-jmjd2b, noninduced | 12 | 240 | 31 (13 ± 4) | 58 (24 ± 6) | 89 (37 ± 7) |
| F-jmjd2b, induced | 12 | 223 | 34 (15 ± 4) | 74 (33 ± 5)c | 108 (48 ± 5)d |
| M-jmjd2b, noninduced | 3 | 76 | 11 (14 ± 4) | 36 (47 ± 11) | 47 (62 ± 15) |
| M-jmjd2b, induced | 3 | 76 | 7 (9 ± 0.4) | 37 (49 ± 17) | 44 (58 ± 17) |
All data relate to reconstructs that extruded a single polar body and that were cultured in vitro until day 4.
Morulae and blastocysts.
P = 0.041 compared with noninduced F-jmjd2b cells determined by the Fisher 2-by-2 test.
P = 0.018 compared with noninduced F-jmjd2b cells determined by the Fisher 2-by-2 test.
In vivo development of NT embryos generated with jmjd2b ES cells.
To assess the in vivo developmental potential of cloned embryos generated from induced F-jmjd2b ES cells compared to that of embryos derived from noninduced cells, some embryos were transferred into pseudopregnant females. For most transfers, pregnancies were established and maintained until day 10.5 of gestation, and fetuses recovered on that day appeared to be normal and fully developed (data not shown). However, surgical retrieval of the uterus on day 13.5 revealed only implantation sites, suggesting that cloned fetuses failed between days 10.5 and 13.5 and were reabsorbed. No significant differences in the number of implantation sites were noted between induced and noninduced jmjd2b ES donor cells (Table 4).
Table 4.
In vivo development of NT embryos generated from jmjd2b-egfp ES cells
| Cell line | No. of transfers | No. of embryos transferred | No. (%) of embryos that resulted in: |
|
|---|---|---|---|---|
| Implantation sites | Offspring at term | |||
| F-jmjd2b, noninduced | 3 | 28 | 13 (46) | 0 (0) |
| F-jmjd2b, induced | 6 | 54 | 15 (28) | 0 (0) |
| M-jmjd2b, noninduced | 2 | 23 | 11 (48) | 0 (0) |
| M-jmjd2b, induced | 2 | 27 | 11 (41) | 0 (0) |
DISCUSSION
Histone lysine methylation marks play a key role in controlling gene expression profiles and directing cell lineage specification. Some of these marks are quite stable and can persist through multiple cell divisions (16–18). In particular, the presence of lysine methylation marks that are associated with gene repression is widely seen to be responsible for restricting the reprogramming potential of the genome (19–22). H3K9me3 is one of the well-studied repressive lysine methylation marks and is implicated in maintaining silencing at heterochromatic regions, imprinting related gene repression and cell-specific identity (12, 35–41). Its role in consolidating stable repression of these regions suggests that H3K9me3 marks are potential candidates involved in obstructing the reprogramming of the genome following NT into a pluripotent ground state.
Development of ES cells for the conditional overexpression of the H3K9me3-specific histone demethylase JMJD2B in this study provided a new tool to directly assess the effect of the stable, repressive H3K9me3 mark on nuclear reprogramming. However, strong overexpression of the transgene-encoded JMJD2B could not achieve complete removal of H3K9me3 but resulted in a relatively modest, 63% decline of H3K9me3 levels. Similar results were observed in analogous experiments using fibroblasts, where the overexpression of JMJD2B resulted in a reduction of H3K9me3 levels of between 57 and 78% (24).
In addition to the expected JMJD2B-mediated decrease of H3K9me3 levels, our Western blotting quantifications indicated that this may be correlated with a concurrent increase in H3K9me1 by approximately 28%. While this small increase in H3K9me1 did not reach significance in our study, it is consistent with a previous report showing that a JMJD2B-mediated decrease in H3K9me3 in mouse fibroblasts was associated with a 50% increase in H3K9me1 (24). Based on this finding, the authors had proposed that JMJD2B-mediated demethylation may convert H3K9me3 into H3K9me1. Deletion of the H3K9-specific Suv39h histone methylases generated a similar phenotype characterized by an increase in H3K9me1 and a decrease in H3K9me3 (40, 41), providing further support for the conversion of the repressive H3K9me3 mark into the activating H3K9me1 modifications. Apart from the repressive H3K9me3 mark, JMJD2B has been shown to also demethylate H3K36me2, a mark mainly associated with transcriptional activation (24). However, its demethylase activity for H3K36me2 is much weaker than that for its main target, H3K9me3, effecting a decrease in overall levels of H3K36me2 by only 10% (24).
The reduced H3K9me3 levels first established in induced donor cells did not persist in reconstructed embryos and were rapidly, within minutes of NT, restored to levels indistinguishable from those for noninduced controls. Thus, the onset of reprogramming must start immediately with the introduction of the nucleus into the cytoplasm of the enucleated oocyte. The prompt restoration of H3K9me3 marks indicates the presence of a fully active chromatin remodeling machinery in the reconstructs. Indeed, proteome analysis has revealed that metaphase-arrested oocytes are loaded with histone-modifying factors and enzymes, including H3K9me3-specific methyltransferases (42). That this reprogramming machinery is also fast acting is evident from the reprogramming during normal fertilization. The sperm chromatin, which is complexed with protamines and lacks histones, rapidly acquires acetylated histones immediately upon entry into the oocyte cytoplasm (43). An elegant study by Liu et al. more specifically demonstrated that the cytoplasm of metaphase-arrested oocytes contains strong histone methyltransferase activity causing the rapid change of the histone methylation profile, similar to what we have observed (44). When unmethylated male mouse pronuclei were transplanted into enucleated metaphase-arrested II oocytes, intense H3K9 methylation of the male pronuclei was already established at the earliest time point examined (after 3 h). The rapid change in methylation status that we observed in our study would suggest that the normalization of H3K9me3 levels is most likely the result of remethylation of H3K9 sites, which can occur faster than the alternative incorporation of new, already methylated histones.
Concomitant with the normalization of H3K9me3, there was a complete loss of JMJD2B-EGFP signal in the reconstructs. A similar rapid loss of the immunosignal of donor nuclei-associated chromatin factors after NT has previously been shown to be caused by their rapid dissociation from the condensing donor chromatin and dilution in the oocyte cytoplasm (45).
In spite of the rapid restoration of H3K9me3, the reduced H3K9me3 levels in induced donor cells did have a significant effect on the in vitro development of NT reconstructs. Compared to NT with noninduced controls, development to blastocysts improved by 38% and overall development (blastocysts and morulae) increased by 30%. While we have focused our study on H3K9me3, the truncated JMJD2B has minor demethylase activity for H3K36me2 (24). Therefore, it is conceivable that our approach may have introduced changes in H3K36me2 levels that are more persistent and partly responsible for the enhanced reprogramming.
NT is a single-cell reprogramming assay, and with an induction of only 80% of the donor cells, we may underestimate the improvements by 20%. Even taking this into account, the observed improvement in the development of NT embryos was relatively modest. This suggests that a greater reduction or total removal of H3K9me3 might be required to more substantially increase reprogramming efficiencies. In addition, our approach targeted only a subset of specific histone modifications. All other repressive histone modifications and DNA methylation were not manipulated and are likely to present additional impediments for nuclear reprogramming, as shown by several other studies. The use of ES donor cells containing a functional disruption of the histone methyltransferase G9a, an enzyme with specificity for mono- and dimethylation of H3K9 (46) and implicated in DNA methylation by recruiting DNA methyltransferases, was correlated with slightly improved nuclear reprogramming into cloned blastocysts (47). In a different reprogramming assay based on ES cell fusion-mediated reprogramming of somatic nuclei, similar positive effects were achieved with the knockdown of G9a or the overexpression of the H3K9-specific histone demethylase JHDM2A (48). Moreover, beneficial effects on reprogramming have not only been described for the modification of repressive epigenetic marks. Overexpression of the histone demethylases JHDM1A and JHDM1B to reduce the level of H3K36me2, a mark that is correlated with transcriptional activity and also reduced by the truncated JMJD2B (24), resulted in an increased efficiency for the reprogramming of somatic cells into induced pluripotent stem cells (49).
It is intriguing that the very short-lived change in H3K9me3 levels can result in improved reprogramming. This finding suggests that early reprogramming events may affect key developmental genes with the potential to trigger a cascading effect resulting in downstream consequences after the initial difference in H3K9me3 has already disappeared. Indeed, rapid reprogramming events following natural fertilization and erasure of preexisting marks in donor nuclei within minutes after NT due to the dissociation of chromatin-associated factors provide evidence for a very rapid onset of epigenetic reprogramming (44, 45). The transient knockdown of Xist, the gene responsible for X chromosome inactivation, in cloned mouse embryos exemplifies how a short-lived effect can generate long-lasting consequences. Following injection of Xist small interfering RNA into cloned embryos, Xist expression was decreased compared to that of controls in morulae-stage embryos but returned to normal levels at the blastocyst stage only 24 h later. However, this short-term repression of Xist expression resulted in reactivation of a number of X-linked genes in cloned blastocysts and greatly improved survival of cloned embryos to term (17).
Considering the implication of H3K9me3 in the regulation of embryonic, imprinted, and heterochromatin-associated genes, the enhanced reprogramming due to a very transient decrease in overall H3K9me3 levels could be the result of initially facilitating the reprogramming of these regions. Subsequent consolidation of the changes and amplification through associated enhanced gene activities may then be responsible for attaining an enduring effect.
While we were able to generate cloned blastocysts with jmjd2b-egfp ES cells, we failed to produce any live cloned mice. Although the proportion of implantation sites obtained was lower following transfer of induced F-jmjd2b cells, these results were not significant. Hence, we cannot conclude that induction of jmjd2b in donor cells improved in vivo development of cloned embryos to term. Furthermore, ES cells are known to accumulate epigenetic abnormalities with prolonged culture (4, 50), which was necessary to introduce the targeted insertions of the jmjd2b transgenes. Thus, the jmjd2b ES cells that we used might have been compromised and no longer had the potential to support the development of live offspring. Considering the substantial technical difficulties of cloning mice, achieved by only a very small number of groups, our lack of success may also be attributable to these inherent technical difficulties (11, 51).
In summary, we provide evidence that repressive H3K9me3 marks are implicated in restricting genome reprogrammability by demonstrating improved in vitro reprogramming into cloned embryos following the targeted reduction of H3K9me marks in donor ES cells prior to NT. Considering that NIH 3T3 fibroblasts were previously shown to allow an almost complete removal of H3K9me3, it appears that the very distinctive characteristics of ES cells (52, 53) may have limited our ability to more substantially reduce H3K9me3 and improve nuclear reprogramming. This would suggest that the approach of a JMJD2B-mediated chromatin therapy may have a greater impact on enhancing the potential for successful nuclear reprogramming when applied in combination with somatic cells.
ACKNOWLEDGMENTS
We thank T. Jenuwein for donating plasmids pI-jmjd2b-T2-GFP and pI-jmjd2b-T2-H189A-GFP and anti-histone methylation antibodies, B. Fodor for sharing immunofluorescence and Western blotting protocols, S. Cole for assistance with flow cytometry analysis, B. Smith and G. Baildon for care of mice, R. Broadhurst for embryo transfer, and H. Henderson for assistance with statistical analysis.
J.A. was supported by a Bright Future Enterprise scholarship from the Tertiary Education Commission. This research was supported by funds from the New Zealand Ministry of Science and Innovation and the Royal Society of New Zealand Marsden Fund.
Footnotes
Published ahead of print 21 December 2012
REFERENCES
- 1. Armstrong L, Lako M, Dean W, Stojkovic M. 2006. Epigenetic modification is central to genome reprogramming in somatic cell nuclear transfer. Stem Cells 24: 805– 814 [DOI] [PubMed] [Google Scholar]
- 2. Fujita N, Wade PA. 2004. Nuclear transfer: epigenetics pays a visit. Nat. Cell Biol. 6: 920– 922 [DOI] [PubMed] [Google Scholar]
- 3. Morgan HD, Santos F, Green K, Dean W, Reik W. 2005. Epigenetic reprogramming in mammals. Hum. Mol. Genet. 14(Spec No. 1):R47–R58 [DOI] [PubMed] [Google Scholar]
- 4. Rideout WM, III, Eggan K, Jaenisch R. 2001. Nuclear cloning and epigenetic reprogramming of the genome. Science 293: 1093– 1098 [DOI] [PubMed] [Google Scholar]
- 5. Santos F, Dean W. 2004. Epigenetic reprogramming during early development in mammals. Reproduction 127: 643– 651 [DOI] [PubMed] [Google Scholar]
- 6. Shi W, Zakhartchenko V, Wolf E. 2003. Epigenetic reprogramming in mammalian nuclear transfer. Differentiation 71: 91– 113 [DOI] [PubMed] [Google Scholar]
- 7. Fussner E, Djuric U, Strauss M, Hotta A, Perez-Iratxeta C, Lanner F, Dilworth FJ, Ellis J, Bazett-Jones DP. 2011. Constitutive heterochromatin reorganization during somatic cell reprogramming. EMBO J. 30: 1778– 1789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Puschendorf M, Terranova R, Boutsma E, Mao X, Isono K-I, Brykczynska U, Kolb C, Otte AP, Koseki H, Orkin SH, van Lohuizen M, Peters AHFM. 2008. PRC1 and Suv39h specify parental asymmetry at constitutive heterochromatin in early mouse embryos. Nat. Genet. 40: 411– 420 [DOI] [PubMed] [Google Scholar]
- 9. Santos F, Peters AH, Otte AP, Reik W, Dean W. 2005. Dynamic chromatin modifications characterise the first cell cycle in mouse embryos. Dev. Biol. 280: 225– 236 [DOI] [PubMed] [Google Scholar]
- 10. Wilmut I, Beaujean N, de Sousa PA, Dinnyes A, King TJ, Paterson LA, Wells DN, Young LE. 2002. Somatic cell nuclear transfer. Nature 419: 583– 586 [DOI] [PubMed] [Google Scholar]
- 11. Yanagimachi R. 2002. Cloning: experience from the mouse and other animals. Mol. Cell. Endocrinol. 187: 241– 248 [DOI] [PubMed] [Google Scholar]
- 12. Delaval K, Govin J, Cerqueira F, Rousseaux S, Khochbin S, Feil R. 2007. Differential histone modifications mark mouse imprinting control regions during spermatogenesis. EMBO J. 26: 720– 729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Enright BP, Kubota C, Yang X, Tian XC. 2003. Epigenetic characteristics and development of embryos cloned from donor cells treated by trichostatin A or 5-aza-2′-deoxycytidine. Biol. Reprod. 69: 896– 901 [DOI] [PubMed] [Google Scholar]
- 14. Kishigami S, Mizutani E, Ohta H, Hikichi T, Thuan NV, Wakayama S, Bui H-T, Wakayama T. 2006. Significant improvement of mouse cloning technique by treatment with trichostatin A after somatic nuclear transfer. Biochem. Biophys. Res. Commun. 340: 183– 189 [DOI] [PubMed] [Google Scholar]
- 15. Lin L, Li Q, Zhang L, Zhao D, Dai Y, Li N. 2008. Aberrant epigenetic changes and gene expression in cloned cattle dying around birth. BMC Dev. Biol. 8: 14 doi:10.1186/1471-213X-8-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lachner M, Sengupta R, Schotta G, Jenuwein T. 2004. Trilogies of histone lysine methylation as epigenetic landmarks of the eukaryotic genome. Cold Spring Harbor Symp. Quant. Biol. 69:209– 218 [DOI] [PubMed] [Google Scholar]
- 17. Matoba S, Inoue K, Kohda T, Sugimoto M, Mizutani E, Ogonuki N, Nakamura T, Abe K, Nakano T, Ishino F, Ogura A. 2011. RNAi-mediated knockdown of Xist can rescue the impaired postimplantation development of cloned mouse embryos. Proc. Natl. Acad. Sci. U. S. A. 108: 20621– 20626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Reik W. 2007. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 447: 425– 432 [DOI] [PubMed] [Google Scholar]
- 19. Ang YS, Gaspar-Maia A, Lemischka IR, Bernstein E. 2011. Stem cells and reprogramming: breaking the epigenetic barrier? Trends Pharmacol. Sci. 32: 394– 401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Baxter J, Sauer S, Peters A, John R, Williams R, Caparros M-L, Arney K, Otte A, Jenuwein T, Merkenschlager M, Fisher AG. 2004. Histone hypomethylation is an indicator of epigenetic plasticity in quiescent lymphocytes. EMBO J. 23: 4462– 4472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pasque V, Jullien J, Miyamoto K, Halley-Stott RP, Gurdon JB. 2011. Epigenetic factors influencing resistance to nuclear reprogramming. Trends Genet. 27: 516– 525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Santos F, Zakhartchenko V, Stojkovic M, Peters A, Jenuwein T, Wolf E, Reik W, Dean W. 2003. Epigenetic marking correlates with developmental potential in cloned bovine preimplantation embryos. Curr. Biol. 13: 1116– 1121 [DOI] [PubMed] [Google Scholar]
- 23. Klose RJ, Kallin EM, Zhang Y. 2006. JmjC-domain-containing proteins and histone demethylation. Nat. Rev. Genet. 7: 715– 727 [DOI] [PubMed] [Google Scholar]
- 24. Fodor BD, Kubicek S, Yonezawa M, O'Sullivan RJ, Sengupta R, Perez-Burgos L, Opravil S, Mechtler K, Schotta G, Jenuwein T. 2006. Jmjd2b antagonizes H3K9 trimethylation at pericentric heterochromatin in mammalian cells. Genes Dev. 20: 1557– 1562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Beard C, Hochedlinger K, Plath K, Wutz A, Jaenisch R. 2006. Efficient method to generate single-copy transgenic mice by site-specific integration in embryonic stem cells. Genesis 44: 23– 28 [DOI] [PubMed] [Google Scholar]
- 26. Raymond CS, Soriano P. 2007. High-efficiency FLP and PhiC31 site-specific recombination in mammalian cells. PLoS One 2: e162 doi:10.1371/journal.pone.0000162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Peters J, Wroe SF, Wells CA, Miller HJ, Bodle D, Beechey CV, Williamson CM, Kelsey G. 1999. A cluster of oppositely imprinted transcripts at the Gnas locus in the distal imprinting region of mouse chromosome 2. Proc. Natl. Acad. Sci. U. S. A. 96:3830– 3835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tiedt R, Hao-Shen H, Sobas MA, Looser R, Dirnhofer S, Schwaller J, Skoda RC. 2008. Ratio of mutant JAK2-V617F to wild-type Jak2 determines the MPD phenotypes in transgenic mice. Blood 111: 3931– 3940 [DOI] [PubMed] [Google Scholar]
- 29. Tichopad A, Dilger M, Schwarz G, Pfaffl MW. 2003. Standardized determination of real-time PCR efficiency from a single reaction set-up. Nucleic Acids Res. 31: e122 doi:10.1093/nar/gng122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Smith C, Berg D, Beaumont S, Standley NT, Wells DN, Pfeffer PL. 2007. Simultaneous gene quantitation of multiple genes in individual bovine nuclear transfer blastocysts. Reproduction 133:231– 242 [DOI] [PubMed] [Google Scholar]
- 31. Perez-Burgos L, Peters AHFM, Opravil S, Kauer M, Mechtler K, Jenuwein T. 2003. Generation and characterization of methyl-lysine histone antibodies. Methods Enzymol. 376: 234– 254 [DOI] [PubMed] [Google Scholar]
- 32. Ribas R, Oback B, Ritchie W, Chebotareva T, Ferrier P, Clarke C, Taylor J, Gallagher EJ, Mauricio AC, Sousa M, Wilmut I. 2005. Development of a zona-free method of nuclear transfer in the mouse. Cloning Stem Cells 7: 126– 138 [DOI] [PubMed] [Google Scholar]
- 33. Gaynor P, Wells DN, Oback B. 2005. Couplet alignment and improved electrofusion by dielectrophoresis for a zona-free high-throughput cloned embryo production system. Med. Biol. Eng. Comput. 43: 150– 154 [DOI] [PubMed] [Google Scholar]
- 34. Nagy A, Gertsenstein M, Vintersten K, Behringer R. 2003. Manipulating the mouse embryo: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 35. Ait-Si-Ali S, Fritsch GVL, Yahi H, Sekhri R, Naguibneva I, Robin P, Cabon F, Polesskaya A, Harel-Bellan A. 2004. A Suv39h-dependent mechanism for silencing S-phase genes in differentiating but not in cycling cells. EMBO J. 23: 605– 615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lehnertz B, Ueda Y, Derijck AAHA, Braunschweig U, Perez-Burgos L, Kubicek S, Chen T, Li E, Jenuwein T, Peters AHFM. 2003. Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr. Biol. 13: 1192– 1200 [DOI] [PubMed] [Google Scholar]
- 37. McEwen K, Ferguson-Smith A. 2010. Distinguishing epigenetic marks of developmental and imprinting regulation. Epigenetics Chromatin 3: 2 doi:10.1186/1756-8935-3-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim T-K, Koche RP, Lee W, Mendenhall E, O'Donovan A, Presser A, Russ C, Xie X, Meissner A, Wernig M, Jaenisch R, Nusbaum C, Lander ES, Bernstein BE. 2007. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448:553– 560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nielsen SJ, Schneider R, Bauer UM, Bannister AJ, Morrison A, O'Carroll D, Firestein R, Cleary M, Jenuwein T, Herrera RE, Kouzarides T. 2001. Rb targets histone H3 methylation and HP1 to promoters. Nature 412: 561– 565 [DOI] [PubMed] [Google Scholar]
- 40. Peters AH, O'Carroll D, Scherthan H, Mechtler K, Sauer S, Schofer C, Weipoltshammer K, Pagani M, Lachner M, Kohlmaier A, Opravil S, Doyle M, Sibilia M, Jenuwein T. 2001. Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell 107: 323– 337 [DOI] [PubMed] [Google Scholar]
- 41. Peters AHFM, Kubicek S, Mechtler K, O'Sullivan RJ, Derijck AAHA, Perez-Burgos L, Kohlmaier A, Opravil S, Tachibana M, Shinkai Y, Martens JHA, Jenuwein T. 2003. Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Mol. Cell 12: 1577– 1589 [DOI] [PubMed] [Google Scholar]
- 42. Wang S, Kou Z, Jing Z, Zhang Y, Guo X, Dong M, Wilmut I, Gao S. 2010. Proteome of mouse oocytes at different developmental stages. Proc. Natl. Acad. Sci. U. S. A. 107: 17639– 17644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Adenot PG, Mercier Y, Renard JP, Thompson EM. 1997. Differential H4 acetylation of paternal and maternal chromatin precedes DNA replication and differential transcriptional activity in pronuclei of 1-cell mouse embryos. Development 124: 4615– 4625 [DOI] [PubMed] [Google Scholar]
- 44. Liu H, Kim J-M, Aoki F. 2004. Regulation of histone H3 lysine 9 methylation in oocytes and early pre-implantation embryos. Development 131: 2269– 2280 [DOI] [PubMed] [Google Scholar]
- 45. Gao T, Zheng J, Xing F, Fang H, Sun F, Yan A, Gong X, Ding H, Tang F, Sheng HZ. 2007. Nuclear reprogramming: the strategy used in normal development is also used in somatic cell nuclear transfer and parthenogenesis. Cell Res. 17: 135– 150 [DOI] [PubMed] [Google Scholar]
- 46. Rice JC, Briggs SD, Ueberheide B, Barber CM, Shabanowitz J, Hunt DF, Shinkai Y, Allis CD. 2003. Histone methyltransferases direct different degrees of methylation to define distinct chromatin domains. Mol. Cell 12: 1591– 1598 [DOI] [PubMed] [Google Scholar]
- 47. Epsztejn-Litman S, Feldman N, Abu-Remaileh M, Shufaro Y, Gerson A, Ueda J, Deplus R, Fuks F, Shinkai Y, Cedar H, Bergman Y. 2008. De novo DNA methylation promoted by G9a prevents reprogramming of embryonically silenced genes. Nat. Struct. Mol. Biol. 15: 1176– 1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ma DK, Chiang C-HJ, Ponnusamy K, Ming G-L, Song H. 2008. G9a and Jhdm2a regulate embryonic stem cell fusion-induced reprogramming of adult neural stem cells. Stem Cells 26: 2131– 2141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang T, Chen K, Zeng X, Yang J, Wu Y, Shi X, Qin B, Zeng L, Esteban MA, Pan G, Pei D. 2011. The histone demethylases Jhdm1a/1b enhance somatic cell reprogramming in a vitamin-C-dependent manner. Cell Stem Cell 9: 575– 587 [DOI] [PubMed] [Google Scholar]
- 50. Eggan K, Akutsu H, Loring J, Jackson-Grusby L, Klemm M, Rideout WM, III, Yanagimachi R, Jaenisch R. 2001. Hybrid vigor, fetal overgrowth, and viability of mice derived by nuclear cloning and tetraploid embryo complementation. Proc. Natl. Acad. Sci. U. S. A. 98: 6209– 6214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yang X, Smith SL, Tian XC, Lewin HA, Renard J-P, Wakayama T. 2007. Nuclear reprogramming of cloned embryos and its implications for therapeutic cloning. Nat. Genet. 39: 295– 302 [DOI] [PubMed] [Google Scholar]
- 52. Efroni S, Duttagupta R, Cheng J, Dehghani H, Hoeppner DJ, Dash C, Bazett-Jones DP, Le Grice S, McKay RDG, Buetow KH, Gingeras TR, Misteli T, Meshorer E. 2008. Global transcription in pluripotent embryonic stem cells. Cell Stem Cell 2: 437– 447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Meshorer E, Misteli T. 2006. Chromatin in pluripotent embryonic stem cells and differentiation. Nat. Rev. Mol. Cell Biol. 7: 540– 546 [DOI] [PubMed] [Google Scholar]