

Abstract
During viral infection or cellular stress, cap-dependent translation is shut down. Proteins that are synthesized under these conditions use alternative mechanisms to initiate translation. This study demonstrates that at least two alternative translation initiation routes, internal ribosome entry site (IRES) initiation and ribosome shunting, rely on ribosomal protein S25 (RPS25). This suggests that they share a mechanism for initiation that is not employed by cap-dependent translation, since cap-dependent translation is not affected by the loss of RPS25. Furthermore, we demonstrate that viruses that utilize an IRES or a ribosome shunt, such as hepatitis C virus, poliovirus, or adenovirus, have impaired amplification in cells depleted of RPS25. In contrast, viral amplification of a virus that relies solely on cap-dependent translation, herpes simplex virus, is not hindered. We present a model that explains how RPS25 can be a nexus for multiple alternative translation initiation pathways.
INTRODUCTION
The predominant translation initiation pathway for cellular mRNAs is cap dependent, which requires recognition of the mRNA 5′ cap structure by the cap binding protein eukaryotic initiation factor 4E (eIF4E). eIF4E interacts with a complex of eukaryotic initiation factors to recruit the ribosome to the 5′ end of the mRNA (1). However, some viral and cellular mRNAs contain an internal ribosome entry site (IRES) in their 5′ untranslated region (UTR) that recruits the ribosome in a cap-independent manner. This allows translation of key regulatory proteins under conditions where cap-dependent translation is downregulated, such as during cell stress (2). In fact, 5 to 10% of cellular mRNAs have been predicted to contain IRES elements. IRESs are enriched in genes that encode proteins regulating growth, differentiation, and responses to stress (3, 4). Viruses have coopted this pathway by inhibiting cap-dependent translation and using an IRES to recruit host ribosomes to synthesize viral proteins (5).
The mechanism of IRES-mediated translation is not well understood; however, our best understanding has come from studying viral IRESs. Viral IRESs have been characterized functionally according to the type and number of initiation factors required (6). The picornaviral IRESs require several canonical initiation factors and noncanonical IRES trans-acting factors (ITAFs) (6, 7). The hepatitis C virus (HCV) IRES can bind directly to the 40S ribosomal subunit but requires additional factors to initiate protein synthesis (8–10). The most streamlined IRESs are found in the intergenic region (IGR) of the Dicistroviridae virus family; they recruit the 40S and 60S subunits to form functional 80S complexes in the absence of any initiation factors (11–13).
The IGR IRES binds to the intersubunit surface of the 40S subunit and occupies the peptidyl (P) and exit (E) sites, whereas the HCV IRES binds to the solvent side of the 40S subunit with only the finger-like domain IIb occupying the E site (14–16). While the binding of these two IRESs to the 40S subunit is quite different, they do share some similarities with respect to mechanism. They both induce a conformational change in the ribosome upon binding (10, 15), and they are both dependent on the nonessential ribosomal protein S25 (RPS25) for efficient IRES-mediated translation (17–19). RPS25 is a 15-kDa protein located in the head domain of the 40S ribosomal subunit in the E site contacting helix 41 of the 18S rRNA and situated between ribosomal proteins S5 and S18 (20–22). Consistent with RPS25 being required for full HCV and IGR IRES activities, the E site, where RPS25 resides, is the only commonly shared contact on the 40S subunit (aside from the decoding center) for these IRESs.
Another alternate mechanism of initiating translation during stress that is used by cells and hijacked by viruses is ribosome shunting. Unlike IRES-mediated translation, the 40S ribosomal subunit is recruited to the 5′ end of the mRNA through a cap-dependent mechanism and commences scanning of the mRNA 5′ UTR, sometimes translating a short open reading frame. The 40S subunit is then transferred from a shunt donor region to a shunt acceptor region, bypassing (without scanning) regions of the transcript to initiate translation at a downstream AUG. Ribosome shunting has been best characterized for cauliflower mosaic virus (CaMV), Sendai virus, rice tungro bacilliform virus, duck hepatitis B virus, and adenovirus (Ad) (23–27). The HSP70 and cIAP2 cellular mRNAs have also been shown to utilize a ribosome shunting mechanism (28, 29). While the mechanism for how this shunting event occurs is not well understood, some studies have shown that the ribosome is tethered to the mRNA either through base pairing with the 18S rRNA or by binding to eIF3 (28, 30). In the work presented here, we present data that suggest that ribosome shunting may share at least some features with IRES-mediated translation.
To determine whether RPS25 is essential in mammalian cells and examine the requirement of RPS25 for translation, we generated a cell line in which RPS25 is stably knocked down. Consistent with our previous findings in Saccharomyces cerevisiae (17), mammalian cells that have undetectable levels of RPS25 have only slight decreases in cell growth and global translation in rich medium. RPS25 depletion led to decreased IRES activity in representatives of different types of viral IRESs as well as in cellular IRESs. Importantly, the decrease in translation of these viral RNAs when RPS25 was knocked down corresponded to a decrease in viral amplification. In addition, ribosome shunting by the adenovirus tripartite leader was also defective in RPS25-deficient cells. These data demonstrate that RPS25 is required for both IRES-mediated translation and ribosome shunting and suggest that both methods of initiating translation may share a mechanism.
MATERIALS AND METHODS
Cell culture.
HeLa cells (Ambion) were cultured in complete medium (high-glucose Dulbecco's modified Eagle's medium [DMEM] supplemented with 10% [vol/vol] fetal bovine serum [FBS], 1% [vol/vol] l-glutamine, and 1% [vol/vol] penicillin-streptomycin). Huh7.5 cells were cultured in complete medium supplemented with 1% nonessential amino acids. All cells were maintained at 37°C and 5% CO2.
Plasmids.
To generate pLVTHMshS25 short hairpin RNA (shRNA), the pLVTHM vector (plasmid 12247; Addgene) was digested with ClaI and MluI. The RPS25 shRNA insert was generated by annealing and phosphorylating (T4 kinase; Promega) the complementary P7 oligonucleotides (see Table S2 in the supplemental material). The shRNA insert was inserted into the restricted pLVTHM vector and verified by sequencing.
The cricket paralysis virus (CrPV), HCV, encephalomyocarditis virus (EMCV), poliovirus (PV), and enterovirus 71 (EV71) plasmids were previously described (17, 31). To construct the classic swine fever virus (CSFV) IRES dual-luciferase reporter, the CSFV IRES plus 69 bases of the coding region were amplified from the pXLcsfv1-442 plasmid using primer set P1 (see Table S1 in the supplemental material) (32) and cloned into the EcoRI-NcoI-digested pΔEMCV plasmid (33). The p53 short IRES (34) (nucleotides 64 to 197 [numbering based on the reference sequence reported under GenBank accession number NM_000546.4]) was amplified from HeLa cDNA using primer set P2 (see Table S1 in the supplemental material) and cloned into the EcoRI-NcoI-restricted pΔEMCV plasmid. To verify that only the full-length dual-luciferase transcript was produced by the pΔEMCV-CSFV and pΔEMCV-p53 reporter plasmids, a Northern analysis against the firefly luciferase gene was performed on poly(A) isolated RNA from HeLa cells transfected with the indicated reporter plasmid. A single product ensures that firefly luciferase activity originated from IRES-mediated translation (see Fig. S2a in the supplemental material). In addition, small interfering RNA (siRNA) knockdown of the first cistron confirmed that the firefly activity observed resulted from the dicistronic reporter and not from a small monocistronic transcript (see Fig. S2b in the supplemental material). The other cellular IRESs Apaf-1, Bag-1, c-myc, KMI2, L-myc, MNT, MTG8α, myb, and Set7 are derived from the pRF dual-luciferase plasmid and were described and verified previously (4, 35–39).
To create the hS25 rescue plasmid, the RPS25 coding region (bases 64 to 441 based on the reference sequence reported under GenBank accession number NM_001028) modified with six synonymous mutations in the siRNA recognition site (nucleotides 283 to 301) (see Fig. 2A) was synthesized by assembly PCR and cloned into the NheI and BamHI sites of the dual-luciferase plasmid pSRT222, replacing the entire dual-luciferase cassette with the RPS25 coding region (17). Long oligonucleotides were designed by using the assembly PCR Oligo Maker, a freely accessible program (http://publish.yorku.ca/∼pjohnson/AssemblyPCRoligomaker.html), and assembly PCR was carried out as described previously (40). Briefly, a PCR with primer set P3 (see Table S1 in the supplemental material) was used to assemble the long oligomers of DNA (one cycle at 94°C for 4 min and then 8 cycles of 94°C for 60 s, 54°C for 2 min, and 72°C for 3 min, followed by a final single cycle at 72°C for 5 min). A 2-μl aliquot of this reaction mixture was added to the second-stage PCR mixture with 20-mer flanking primers (primer set P4 [see Table S1 in the supplemental material]) encoding NheI and BamHI sites on their termini to facilitate cloning into pSRT222 (which was denatured at 94°C for 5 min and then for 24 cycles of 94°C for 30 s, 54°C for 2 min, and 72°C for 90 s, followed by a final extension cycle at 72°C for 5 min). All cloning was verified by sequencing.
Fig 2.
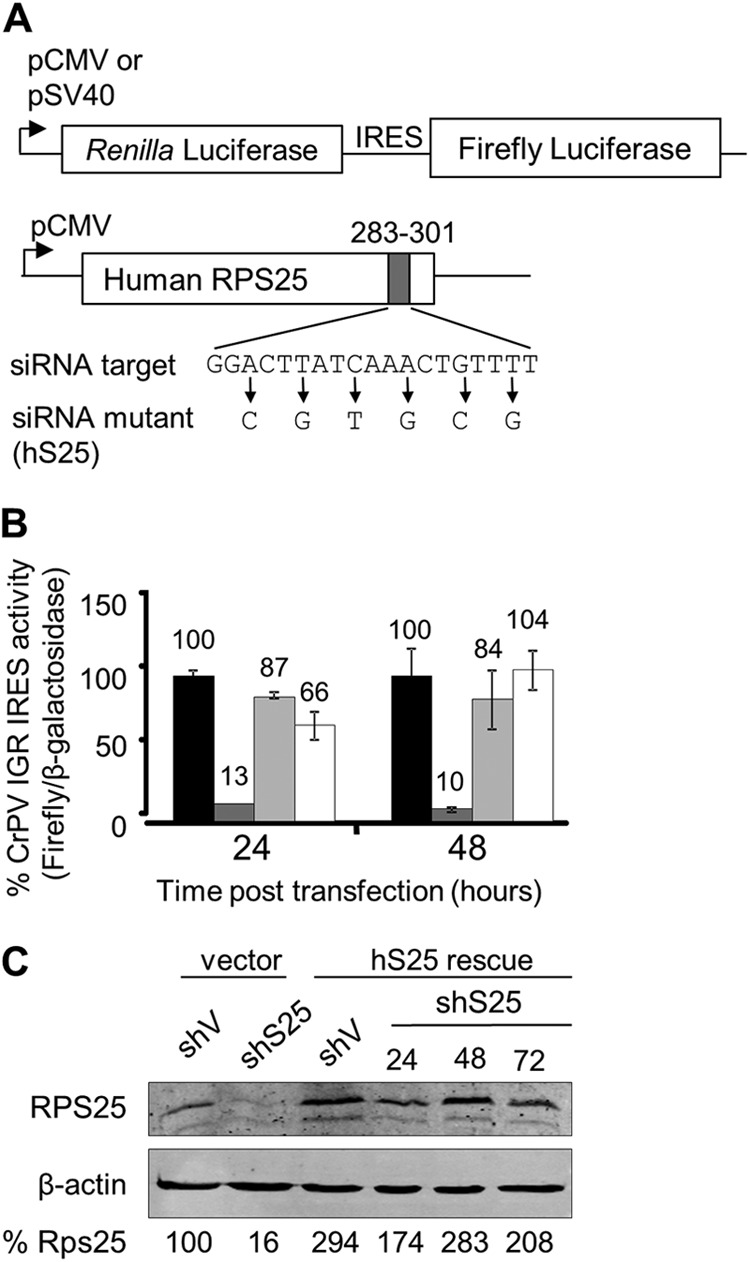
HeLashS25 cells exhibit a specific defect in IRES-mediated translation, which can be complemented by exogenous expression of the shRNA-resistant RPS25, hS25. (A) Schematic representation of the dual-luciferase reporter and the hS25 rescue construct. Transcription of the bicistronic dual-luciferase reporters was from the simian virus 40 promoter (pSV40) or cytomegalovirus promoter (pCMV). Cap-dependent translation drives expression of the Renilla luciferase, while the firefly luciferase is under the control of the CrPV IGR IRES located in the intercistronic region. A diagram of the hS25 rescue plasmid indicates the synonymous mutations made to confer resistance to the siRNA. (B) CrPV IGR IRES activity in cells cotransfected with the dual-luciferase reporter and either the hS25 plasmid or empty pcDNA3 vector. IRES activity (firefly luciferase) was normalized to the cap-dependent translation of β-galactosidase and expressed as a percentage of the activity in HeLashV cells with the empty vector (black), HeLashS25 cells with the empty vector (dark gray), HeLashV cells with hS25 (light gray), and HeLashS25 cells with hS25 (white). Standard errors for 3 experiments are shown. (C) RPS25 Western analysis of cells 24, 48, and 72 h following transfection with the hS25 rescue plasmid.
Lentiviral vectors.
Virus was generated by cotransfection of pLVTHMshS25, the psPAX2 packaging plasmid (plasmid 12260; Addgene), and pMG2.G, a vesicular stomatitis virus G protein (VSV-G) envelope plasmid (plasmid 12259; Addgene), into HEK293T cells. After 24 h, supernatant was collected every 12 h for 2 days. The virus supernatant was filter sterilized by using a 0.2-μm filter and applied directly onto the HeLa cells.
Proliferation assay.
A total of 3 × 104 cells were seeded into 6-well plates, and medium was replaced with either 1% or 10% serum after 24 h. Viable cells were counted at 1, 2, 3, and 4 days by removing them from the plate with trypsin, staining them with trypan blue, and manually counting the cells by using a hemocytometer. The cells were fed with the indicated media every 24 h. Each assay was performed in triplicate.
Global protein synthesis rate.
To pulse-label cells, 1 × 105 HeLashS25 or HeLashV vector control cells were grown to 70% confluence in 12-well plates and were then incubated in DMEM supplemented with dialyzed FBS lacking methionine and cysteine to starve the cells of the sulfur-containing amino acids for 15 min. The cells were radiolabeled for 20 min in the same medium supplemented with 0.1 mCi 35S protein labeling mix (PerkinElmer, Waltham, MA). Trichloroacetic acid (TCA) precipitation was performed as described previously (17). Briefly, cells were lysed with E1 lysis buffer (50 mM HEPES [pH 7.0], 250 mM NaCl, 0.1% NP-40) for 30 min on ice. Twenty microliters of the lysate was mixed with 100 μl bovine serum albumin (BSA)-NaN3 (1 mg/ml BSA, 0.2% NaN3) and 1 ml of 10% TCA to precipitate the proteins. Precipitates were filtered over a glass microfiber filter and washed with 10% TCA, followed by 100% ethanol. The radioactivity of the precipitates on the filter was measured with a Wallac 1409 liquid scintillation counter (PerkinElmer).
Transfections.
The day before transfection, 5 × 104 HeLa cells were seeded into 24-well plates. Once the cells reached 90% confluence, cotransfection of pcDNA3.1/His B/LacZ (Invitrogen) with either an IRES-containing dual-luciferase reporter or a shunting reporter was done by using Lipofectamine 2000 (Invitrogen), according to the manufacturer's recommendations, using 0.4 μg DNA per well. Cells were harvested for luciferase and β-galactosidase assays after 24 h. Double-stranded RPS25 siRNAs, 5′-GGACUUAUCAAACUGGUUUtt-3′ and 5′-AAACCAGUUUGAUAAG-UCCtt-3′ (siRNA identification number 142220; Ambion; the TT DNA overhang is in lowercase), were used to knock down RPS25 in Huh7 cells. The Dicector DS scrambled negative-control duplex was used as a negative control (Integrated DNA Technologies [IDT]). siRNA complexes were prepared in opti-MEM (Invitrogen Life Technologies) with 5 μl siPORT NeoFX transfection agent (Ambion) to a final siRNA concentration of 0.375 μM, according to the manufacturer's specifications. siRNA complexes were plated and overlaid with 2 × 105 Huh7.13 cells in antibiotic-free medium. The transfection medium was replaced with complete medium after 24 h.
Viral infections and virus titer assays.
HeLashS25 or HeLashV cells were infected with poliovirus (Mahoney strain) at a multiplicity of infection (MOI) of 0.1 in CPBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM potassium phosphate [pH 7.4], 0.1 mg/ml CaCl2, 0.1 mg/ml MgCl2). Infections were carried out for 30 min at 37°C and 5% CO2, by rolling the plates every 10 min. The virus was removed, and complete medium was added. At 6 h postinfection, the medium was removed, and the cells were scraped into phosphate-buffered saline (PBS) (137 mM NaCl, 2.7 mM KCl, 10 mM sodium phosphate dibasic, 2 mM potassium phosphate [pH 7.4]). Virus was isolated by 3 freeze-thaw cycles, the debris was then pelleted, and the PFUs in the supernatant was determined. Briefly, 10-fold serial dilutions of the virus were used to inoculate HeLa cells. The inoculums were removed after 30 min, and 2 ml of agarose (1% agarose, 1× 199 medium, 10% FBS, 12 mM HEPES, 0.2% NaHCO3, 1% penicillin-streptomycin, 1% l-glutamine) was overlaid onto the HeLa cells. After 36 h, cells were fixed with 10% TCA and stained with 1% crystal violet, and plaques were counted.
Herpes simplex virus 1 (HSV1) strain F was a generous gift from Jacqueline Parker, University of Alabama (UAB). HeLashS25 or HeLashV cells were infected with HSV-1 at an MOI of 0.1 and incubated. One round of infection was carried out at 37°C and 5% CO2. After 24 h, the medium was removed, and the virus was harvested in sterile milk. The virus titer was determined as described above for poliovirus.
HeLashS25 or HeLashV cells were infected with adenovirus serotype 5 (Ad5) at an MOI of 0.1. Infections were carried out at 37°C and 5% CO2 for 30 h (one round of infection). The medium was removed, and virus was harvested by scraping into PBS. Virus was isolated by 3 freeze-thaw cycles, the debris was then pelleted, and the virus titer in the supernatant was determined as described above, except that 911 cells were used and plaques were counted at 8 days postinfection.
HCV infection was carried out by inoculating Huh7.13 cells with 1 ml of culture medium from the HCV producer cell line Huh7 JFH1 HCV (genotype 2a). Medium was replaced after 2 h, and the cells were incubated for 3 days at 37°C prior to analysis. Cells were lysed at 72 h postinfection, and a Western analysis was performed by using the NS5A antibody (Charles Rice, Rockefeller University).
Northern analysis.
Total RNA was harvested with TRIzol (Invitrogen), according to the manufacturer's protocol, from shRNA lentivirus-transduced cells. Five micrograms of RNA or 5 μl of RNA extracted from polysome fractions was separated on a denaturing agarose gel (0.8% agarose, 16% formaldehyde) in morpholinepropanesulfonic acid (MOPS) buffer (20 mM MOPS, 5 mM sodium acetate [NaOAc], 1 mM EDTA [pH 7.0]) and transferred onto a zeta-probe membrane (Bio-Rad). [32P]dCTP (PerkinElmer)-radiolabeled probes were generated with the Prime-a-gene kit (Promega), using PCR products amplified from a HeLa cDNA pool with primer sets P5 and P6 for RPS25 and β-actin (see Table S1 in the supplemental material). The p53 probe template was made by using the IRES sequence, by digesting pΔEMCV-p53 with EcoRI-NcoI.
Western analysis.
Cells were lysed in E1 lysis buffer with 0.1% SDS. Twenty micrograms of protein was separated by 12 or 15% SDS-PAGE. Proteins were transferred onto an Immobilon-P polyvinylidene difluoride membrane (Millipore Co., Milford, MA), using the Genie wet transfer system (Idea Scientific Company, Minneapolis, MN). Membranes were probed with the indicated antibody. A rabbit polyclonal RPS25 antibody was generated by using a His-tagged recombinant RPS25 protein by PrimmBiotech (Cambridge, MA) and used at a 1:200 dilution. The commercial antibodies used were β-actin antibody (catalog number sc-47778; Santa Cruz) at a 1:2,000 dilution and NS5A antibody (a generous gift from Charles Rice, Rockefeller University). Signals were detected by using a 1:5,000 dilution of a fluorochrome-conjugated secondary antibody for quantitative Western blotting using the Odyssey scanner and software (Li-Cor, Lincoln, NE).
Luciferase assay.
Cells from a 24-well plate were washed with PBS and lysed directly in the plate with 100 μl of 1× passive lysis buffer (Promega). In the adenovirus shunting experiments, 5 × 104 HeLa cells were transfected with the B202 shunting reporter and β-galactosidase reporter (pcDNA3.1/His B/LacZ; Invitrogen). The cells were allowed to recover from transfection for 24 h and were then infected with Ad5 at an MOI of 25. After one round of infection, the cells were processed for the luminescence assays. Four microliters of lysate was assayed by using an FB 12 luminometer (Berthold) according to the manufacturer's instructions for the dual-luciferase kit (Promega). All assays were performed three independent times in triplicate. The IRES activity (firefly luciferase) was normalized to the cap-dependent translation of β-galactosidase (see blow) and expressed as a percentage of the activity in HeLashV cells with the empty vector.
β-Galactosidase assay.
Cells from a 24-well plate were washed with PBS and lysed directly in the plate with 100 μl of lysis buffer (100 mM potassium phosphate [pH 7.8], 0.2% Triton X-100 [Applied Biosystems]). The β-galactosidase activity of each lysate was measured by using the Galacto-Light Plus kit according to the manufacturer's instructions (Applied Biosystems). Briefly, 20 μl of lysate was incubated with 200 μl of 1× chemiluminescent substrate in reaction buffer (100 mM NaPO4 [pH 8.0], 1 mM MgCl2). After incubation at room temperature for 1 h, 300 μl of Accelerator-II was added, and the luminescence was measured by using an FB 12 luminometer (Berthold). All assays were performed three independent times in triplicate.
Polysome analysis.
Approximately 2 × 107 cells were grown to 70% confluence. Cycloheximide (0.1-mg/ml final concentration) was added to the medium for 3 min at 37°C to arrest the ribosomes. The cells were washed with PBS containing 0.1 mg/ml cycloheximide and then lysed for 10 min on ice with 400 μl polysome extraction buffer (15 mM Tris-Cl [pH 7.4], 15 mM MgCl2, 0.3 M NaCl, 0.1 mg/ml cycloheximide, 1 mg/ml heparin, 1% Triton X-100). The lysates were cleared by centrifugation at 13,200 × g for 10 min. Five hundred microliters of the supernatant was layered onto 20 to 50% sucrose gradients, which contained polysome extraction buffer with no Triton X-100. The gradients were sedimented at 151,263 × g for 190 min in an SW41 rotor at 4°C. An ISCO UA-5 fraction collection system (Teledyne) was used to collect 14 fractions, which were immediately mixed with 1 volume of 8 M guanidine HCl.
Total RNA were precipitated from polysome fractions by ethanol precipitation and dissolved in 25 μl of H2O based on common protocols (3). Briefly, guanidine-containing samples were vortexed for 20 s. Three milliliters of 100% ethanol was added, and the fraction was vortexed again. The faction was incubated overnight at −20°C to allow for complete precipitation of the RNA. The fractions were centrifuged at 14,462 × g in an SS-34 rotor for 30 min at 4°C. The RNA pellet was washed with 75% ethanol. The pellet was resuspended in 400 μl 1× Tris-EDTA (TE) (pH 8.0), transferred into a microcentrifuge tube containing 0.1 volumes of 3 M NaOAc (pH 5.3) and 2.5 volumes of ethanol, and incubated at −20°C to precipitate RNA. The RNA pellet was washed with 75% ethanol and resuspended in 25 μl of H2O. One-fifth (5 μl) of the total RNA from each fraction was separated on a denaturing RNA gel and probed for a specific mRNA, as indicated above for the Northern analysis.
RESULTS
RPS25 is not essential for mammalian cells in culture.
We demonstrated previously that RPS25 was not an essential gene in yeast and that transient knockdown had no apparent effect on mammalian cells (17). To determine if RPS25 is essential in mammalian cells, we generated stable HeLa cells lines that have RPS25 stably knocked down by transducing them with a lentivirus that either expressed an shRNA against RPS25 (HeLashS25) or not (HeLashV). Stably transduced cells were isolated by cell sorting based on expression of the green fluorescent protein (GFP) from the lentiviral vector (Fig. 1A). RPS25 mRNA and protein expression levels were undetectable in HeLashS25 cells (Fig. 1B), demonstrating that RPS25 is efficiently knocked down. The HeLashS25 cell line was further characterized to determine if there were any gross defects due to the prolonged knockdown of RPS25. The HeLashS25 cells were morphologically similar to the control HeLashV cells (Fig. 1A), and there was no significant defect in growth rate except at the highest serum concentration (Fig. 1C). Cap-dependent translation initiation was not reduced in the HeLashS25 cells when examined by measuring the activity of two different reporters (Fig. 1D). There was a 17% decrease in the global protein synthesis rate of the HeLashS25 cells cultured in 10% serum (Fig. 1E). These findings are consistent with our previous findings in yeast and offer a possible explanation as to why HeLashS25 cells have a slight growth defect at 10% serum in Fig. 1C (17). Taken together, these results suggest that RPS25 is not essential in mammalian cells but may confer a slight growth advantage to rapidly growing cells.
Fig 1.
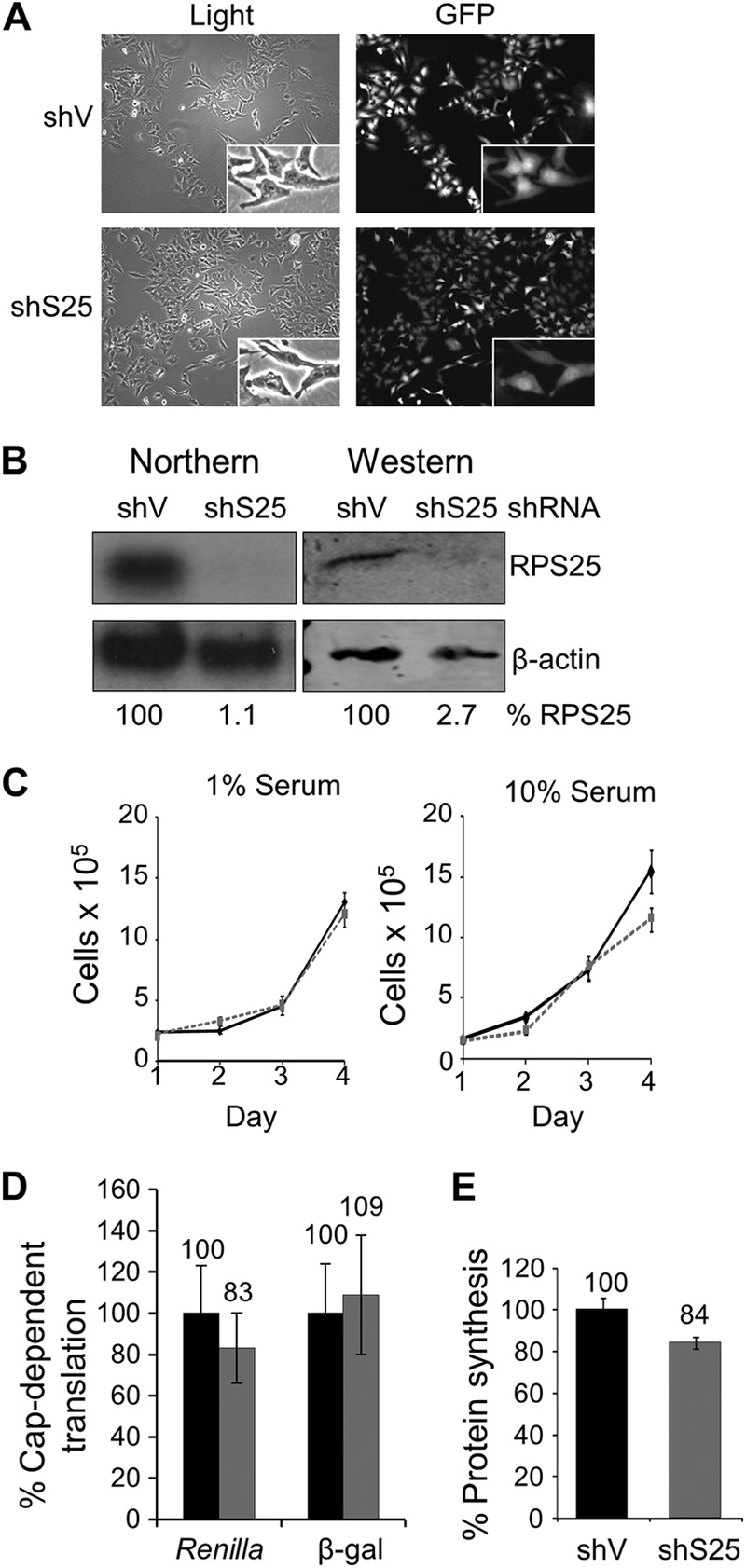
A stable knockdown of RPS25 in HeLa cells is viable, and the cells do not have significant defects in growth or global translation. (A) Light (left) and florescence (right) microscopy of HeLashV and HeLashS25 cells at a ×100 magnification. (B) Northern and quantitative Western analyses of RPS25 RNA and protein in HeLashV and HeLashS25 cells. (C) Proliferation assay for HeLashV (black) and HeLashS25 (gray) cells in 1% or 10% serum. Standard errors for 3 experiments are shown. (D) Relative cap-dependent translation levels in HeLashV (black) and HeLashS25 (gray) cells. Cells were transfected with plasmids encoding one of two reporter proteins, Renilla luciferase or β-galactosidase (β-gal), and the enzyme activity was measured after 48 h. (E) Protein synthesis rate in 10% serum determined by [35S]methionine incorporation into HeLashV and HeLashS25 cells. The counts per minute were measured for HeLashV and HeLashS25 cells and expressed relative to HeLashV cells at 100%. Standard errors for 4 experiments are shown.
IRES-mediated translation is defective in HeLashS25 cells.
We previously demonstrated that RPS25 is required for translation initiation by the CrPV IGR IRES in human cell lines following a transient knockdown of RPS25 (17). The dicistronic reporter assay is used to measure IRES activity. Briefly, the first cistron, Renilla luciferase, is translated in a cap-dependent manner, and the second cistron, firefly luciferase, is translated only if there is a functional IRES located upstream (Fig. 2A). The CrPV IGR IRES activity in the RPS25-depleted cells was 10 to 13% of the wild-type activity (Fig. 2B, compare black and dark gray bars), which is a more pronounced defect in IRES activity than what was observed previously during transient knockdown of RPS25 (17). To verify that the HeLashS25 cell line did not accumulate additional confounding mutations that could potentially affect IRES activity, we tested whether IRES activity could be rescued by expression of shRNA-resistant RPS25 (Fig. 2A). Since the RPS25 shRNA targeted the coding region of RPS25, an shRNA-resistant RPS25 expression plasmid, hS25, was generated, with synonymous mutations in the siRNA recognition motif (Fig. 2A). Transient expression of hS25 in HeLashS25 cells resulted in an increase in RPS25 protein expression levels after 24 h that was maintained for at least 72 h, demonstrating that hS25 from the rescue plasmid was resistant to the shRNA (Fig. 2C). Expression of hS25 resulted in a partial restoration of CrPV IGR IRES activity at 24 h posttransfection and a complete restoration by 48 h (Fig. 2B, white bars). The fact that exogenous expression of an shRNA-resistant RPS25 rescued IRES activity confirms that RPS25 limitation is responsible for the reduction in IRES-mediated translation in HeLashS25 cells from the CrPV IGR IRES.
Viral IRESs that are structurally and functionally different rely on RPS25.
To determine whether RPS25 knockdown affects the activity of other viral IRESs, the IRES activity for a range of viral IRESs was tested with the HeLashS25 cell line (Fig. 3A). HCV and classic swine fever virus (CSFV) are both members of the Flaviviridae virus family and have similar IRES elements (41). The activity of the HCV IRES was decreased to 25% in HeLashS25 cells, in agreement with results obtained previously after a transient knockdown of RPS25 in HeLa cells (17). The CSFV IRES activity was reduced to 44% in HeLashS25 cells, demonstrating that other flaviviral IRESs also use RPS25-driven translation (Fig. 3A).
Fig 3.
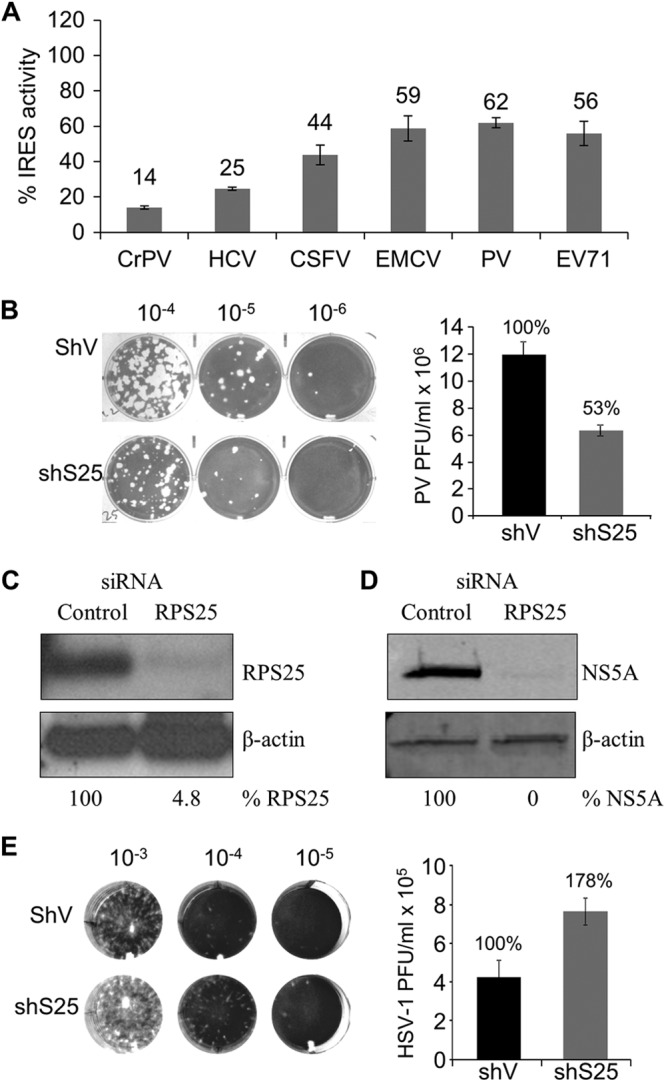
Viral IRESs that are structurally and functionally different rely on RPS25. (A) Normalized activity of several viral IRESs in HeLashS25 cells expressed as a percentage of the activity for each IRES in the control cells. Raw luciferase and β-galactosidase values are presented in Table S2 in the supplemental material. (B) Representative poliovirus plaque assay images and quantification of titers after one round of infection in HeLashV or HeLashS25 cells. (C) Northern analysis of RPS25 mRNA levels in Huh7.13 cells 72 h after siRNA knockdown. Quantifications of RPS25 levels are indicated. (D) Replication efficiency of HCV (JHF1 strain) in control and RPS25 knockdown Huh7.13 cells was assessed by a quantitative Western analysis for the HCV NS5A protein normalized to β-actin at 72 h postinfection. (E) Representative herpes simplex virus 1 plaque assay images and quantification of virus titers following one round of infection in HeLashV or HeLashS25 cells. Standard errors for 3 experiments are shown.
Both the Dicistroviridae and Flaviviridae IRESs recruit the 40S ribosomal subunit in the absence of any initiation factors directly to the start codon. To examine whether other types of IRESs that require some subset of initiation factors to recruit the 40S subunits also use RPS25, the activity of various picornaviral IRESs were determined when RPS25 was knocked down. The encephalomyocarditis virus (EMCV) IRES recruits the ribosome directly to the AUG start codon (42). In contrast, the IRES elements in poliovirus (PV) and enterovirus 71 (EV71) recruit the ribosome upstream of the start codon, and the ribosome scans down to the start codon (43, 44). Interestingly, both types of picornaviral IRESs are equally compromised in HeLashS25 cells (Fig. 3A), indicating that RPS25 has a role in picornaviral IRESs that is independent of ribosome scanning.
RPS25 knockdown reduces amplification of IRES-containing viruses.
Many viruses, such as HCV and PV, rely solely on IRES-mediated translation to generate viral proteins, which suggests that depletion of RPS25 would inhibit viral amplification. Therefore, the amplifications of PV and HCV were assayed in RPS25-depleted cells. Both HeLashS25 and control cells (HeLashV) were infected with PV, and the amount of virus produced from a single round of replication was determined by a plaque assay (Fig. 3B). There was a 47% reduction in the PV titer in RPS25 knockdown cells, demonstrating that the decrease in PV IRES activity from the knockdown of RPS25 results in a decrease in PV amplification (Fig. 3B). Taken together, these data suggest that RPS25 is not essential for PV IRES activity but rather enhances IRES activity.
HCV replicates efficiently in Huh7 cell lines but not in HeLa cells (45). Therefore, to determine whether RPS25 knockdown affects HCV amplification, an Huh7.13 cell line was transiently transfected with an siRNA that targets RPS25. The RPS25 knockdown was over 95% effective (Fig. 3C). These cells were infected with HCV at a low MOI (multiplicity of infection), such that amplification of the virus is required in order to detect viral proteins by Western analysis (see Fig. S1 in the supplemental material) (46). In the presence of a nontargeting control siRNA, there was robust expression of the nonstructural protein NS5A 72 h after infection (Fig. 3D). However, in contrast, HCV NS5A was not detected in RPS25 knockdown Huh7.13 cells, demonstrating that RPS25 is required for amplification of HCV in cell culture (Fig. 3D).
We hypothesize that the reduction in viral amplification is due to a decrease in viral protein production from a decrease in IRES-mediated translation when RPS25 levels are reduced. Accordingly, a virus that does not use an RPS25-dependent IRES should not have a defect in replication in the HeLashS25 cell line. To test this hypothesis, we determined the effect of RPS25 knockdown on a DNA virus, herpes simplex virus 1 (HSV-1) strain F. HSV-1 uses a cap-dependent mechanism to translate viral proteins (47) and therefore should be unaffected by RPS25 depletion. Viral titers were determined after a single round of infection in HeLashS25 or HeLashV cells (Fig. 3E). Unlike HCV and PV, we observed a reproducible 1.5- to 2-fold increase in HSV-1 titers in HeLashS25 cells. Taken together, these results demonstrate that the reduced virus titers are due to impaired translation of IRESs and not due to a decrease in cell fitness.
RPS25 aids in the translation of cellular IRESs.
Thus far, we have shown that a diverse group of viral IRESs use RPS25 to initiate protein synthesis. To examine whether RPS25 is also used by cellular IRESs, several cellular IRESs were assayed for RPS25 dependence in HeLashS25 and HeLashV cells using a bicistronic reporter assay. The IRES activity was determined for all dicistronic reporters in HeLashV and HeLashS25 cells (see Table S3 in the supplemental material for raw values). IRES activity is reported relative to the IRES activity observed in HeLashV cells (Fig. 4A). All of the cellular IRESs demonstrated a dependence on RPS25 (Fig. 4A, gray bars). Importantly, IRES activity was rescued by hS25 (Fig. 4A, white bars).
Fig 4.

RPS25 aids in the translation of cellular IRESs. (A) Cellular IRES activity was measured 48 h after cotransfection with the bicistronic IRES reporter and the monocistronic β-galactosidase reporter (gray bars) or also with the hS25 rescue plasmid (white bars) and expressed as a percentage of the activity in HeLashV cells (wild-type [WT] cells) (dotted line) for each IRES. Raw luciferase and β-galactosidase values are presented in Table S3 in the supplemental material. (B) Polysome analysis of the HeLashV and HeLashS25 cells. P/M, polysome-to-monosome ratio. (C) RNA isolated from the HeLashV and HeLashS25 polysome fractions was separated on a denaturing agarose gel. 18S and 28S rRNAs are indicated on the ethidium bromide-stained gel. The RNA was probed by Northern analysis for p53 and β-actin mRNAs.
To examine the translational efficiency of an endogenous IRES-containing mRNA, a polysome analysis was performed on lysates from HeLashS25 and HeLashV cells. The polysome profiles demonstrate that the global translation profiles and the polysome-to-monosome (P/M) ratios are similar for both HeLashS25 and HeLashV cells, indicating that there are no differences in global translation (Fig. 4B). Furthermore, the sizes of the 40S peaks are equivalent, suggesting that there is no defect in ribosomal subunit production. This result is consistent with our previous finding that there is no significant defect in rRNA biogenesis in yeast harboring a deletion in RPS25 (17). Lastly, there is no increase in free ribosomal subunits, which indicates that there is no defect in translation initiation or subunit joining.
To visualize the relative translation efficiencies of specific messages, Northern analysis was performed on RNA extracted from the polysome fractions. The β-actin mRNA was associated with high-molecular-weight polysomes in both cell lines, demonstrating that cap-dependent translation was unaffected by the loss of RPS25 (Fig. 4C). The slight shift in β-actin mRNA in the polysome fractions was not consistently observed, nor was it observed for other cap-dependent mRNAs (see Fig. S3 in the supplemental material). In contrast, a subset of the p53 mRNA accumulated in the 40S peak in the HeLashS25 cells, indicating a block in initiation (Fig. 4C). This suggests that the endogenous p53 IRES can bind to 40S subunits but is blocked at a downstream step. Many IRES-containing cellular RNAs can also be translated by both cap-dependent and cap-independent mechanisms. Consistent with this, there appears to be a population of p53 mRNAs that are translated through a cap-dependent mechanism and remain associated with the polysomes in the absence of RPS25.
RPS25 is used for ribosome shunting by adenovirus.
We next asked if another alternative mechanism of translation initiation is RPS25 dependent. Ribosome shunting is a process in which the ribosome is recruited to the mRNA in a cap-dependent manner and the 40S ribosome bypasses a region of secondary structure in the 5′ UTR without scanning through it. Instead, the ribosome is shunted to a downstream site to initiate protein synthesis. This strategy was described previously for cauliflower mosaic virus and adenovirus and is also used in other viral and cellular mRNAs (25, 27–29, 48). A shunting luciferase reporter was used to establish whether ribosome shunting was specifically impaired in RPS25-deficient cells. The Ad-hp-Luc (adenovirus-hairpin-luciferase) shunting reporter contains the tripartite leader shunting sequence from the 5′ UTR of adenovirus with a thermodynamically stable stem-loop engineered immediately downstream to eliminate scanning through the 5′ UTR (Fig. 5A) (27). In HeLashS25 cells, shunting by the adenovirus tripartite leader exhibited a modest reduction (26%) (Fig. 5C). However, shunting is known to be upregulated during infection (27); therefore, we tested the efficiency of shunting in the absence of RPS25 following infection in HeLashS25 cells. In infected HeLashV cells, shunting increased by 3.7-fold over that in mock-infected cells. In contrast, shunting in infected HeLashS25 cells increased by only 1.9-fold. Therefore, shunting was reduced by 60% in infected HeLashS25 cells compared to infected control cells (Fig. 5B), suggesting a role for RPS25 in ribosome shunting. Next, we evaluated if RPS25 was necessary for adenovirus replication by measuring the titer of adenovirus type 5 (Ad5) produced following one round of infection. The Ad5 titer was more than 5-fold lower in HeLashS25 cells, indicating that RPS25 is important for the replication of adenovirus (Fig. 5C).
Fig 5.
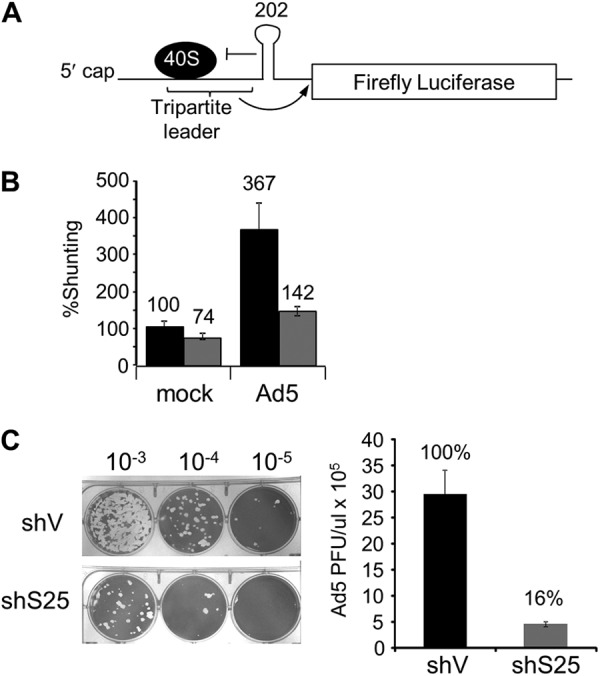
RPS25 is used for ribosome shunting during adenovirus infection. (A) Diagram of the Ad-hp-Luc adenovirus shunting reporter (35). A stable stem-loop at the 3′ end of the tripartite shunting sequence inhibits normal scanning of the 40S ribosome, allowing only shunting to proceed, as indicated by the arrow. (B) The relative shunting rate was determined in HeLashV (black bars) and HeLashS25 (gray bars) cells cotransfected with the Ad-hp-Luc shunting reporter and β-galactosidase reporter as a control for cap-dependent translation. After one round of Ad5 infection, ribosome shunting activity (firefly luciferase) was normalized to β-galactosidase activity and expressed as a percentage of the shunting activity in the mock-infected HeLashV cells. (C) Representative plaque assay images and titers following one round of infection with Ad5 in HeLa cells. Three independent replicates of each assay were performed, and error bars indicate standard errors for three experiments.
DISCUSSION
IRES-mediated translation and ribosome shunting are employed by cells, and exploited by viruses, as ways to regulate protein synthesis for specific mRNAs. Based on the known diversity of IRES elements, it was suggested that there are multiple strategies for IRES-mediated translation initiation (6). In addition, it appears that there are equally diverse mechanisms of ribosome shunting (49). However, this study strongly suggests that there is a more unified mechanism among IRESs and between IRESs and ribosome shunting. Our results demonstrate that RPS25 is essential for the efficient translation of multiple viral and cellular IRESs in mammalian cells, indicating that these IRESs share a mechanism. Furthermore, we showed that RPS25 is also used for ribosome shunting by the adenovirus tripartite leader, which suggests that RPS25 performs a function that is common to both IRESs and ribosome shunts. The biological significance of RPS25 in these mechanisms of translation initiation was demonstrated by depletion of RPS25, which resulted in reduced amplification of viruses that utilize an IRES or ribosome shunt to express viral proteins. In contrast, amplification of HSV-1, a virus that depends solely on cap-dependent translation, is definitely not hindered and appears to be enhanced when RPS25 is knocked down. This demonstrates that cells deficient in RPS25 are not less fit for viral amplification. This agrees with our findings that RPS25 does not affect cap-dependent translation or hinder global translation rates significantly. Taken together, these results support a model whereby the activities of IRESs and ribosome shunts are augmented by RPS25 during translation initiation.
The data presented here demonstrate that there is a specific inhibition of IRES-mediated initiation and ribosome shunting in mammalian cells that have RPS25 knocked down. Based on our current knowledge of these mechanisms of initiation, it is not obvious why both ribosome shunts and IRESs would share a mechanism. Therefore, we propose a model in which IRESs and ribosomal shunts use various mechanisms to recruit the 40S subunit but rely on RPS25 for a downstream step in initiation, such as mRNA loading into the mRNA binding channel of the 40S ribosomal subunit, start codon recognition, or 60S subunit joining (Fig. 6).
Fig 6.

Model for how RPS25 plays a common role in initiation by IRESs and ribosome shunts. (A) Cap-dependent translation does not require RPS25 for any of the steps in initiation. (B) HCV or picornaviral IRESs bind to the 40S subunit independently of RPS25. Next, RPS25 likely functions in a downstream step following 40S subunit recruitment, such as loading of the mRNA into the mRNA binding channel of the 40S subunit, start codon recognition, or 60S subunit joining. (C) The CrPV IGR IRES depends on RPS25 for binding to the 40S subunit (17). (D) In ribosomal shunting, the 40S subunit is recruited in a cap-dependent manner. RPS25 may be involved in transferring the ribosome from the donor to the acceptor site on the mRNA during ribosome shunting or in the events following shunting. Tan, 40S ribosome subunit; green, RPS25; black lines, IRES; black rectangles, coding region; blue, cap binding complex (eIF4F [composed of eIF4E, eIF4G, and eIF4A]); purple, eIF3; brown, ternary complex; red, eIF1A; yellow, eIF1.
The reason for placing the function of RPS25 downstream of 40S subunit recruitment is due to the fact that CrPV IGR, HCV, and picornaviral IRESs and ribosome shunts all use various means to recruit the 40S ribosome subunit to the mRNA. Specifically, the CrPV IGR IRES binds to the intersubunit surface of the 40S and depends on RPS25 for 40S recruitment (17). The HCV IRES binds to the solvent side of the 40S subunit. Circumstantial evidence that RPS25 is not involved in recruitment of the HCV IRES to the ribosome was gained from previous studies that observed no defect in HCV IRES affinity for the 40S subunit when domain IIb, which interacts with the E site, where RPS25 resides, was deleted (16, 50). The poliovirus IRES recruits the 40S subunit through interactions with the C terminus of eIF4G (51, 52). Ribosome shunts recruit the 40S subunit initially to the mRNA though a cap-dependent mechanism, but the transfer of the 40S from the donor site to the acceptor site was reported previously to involve tethering of the 40S subunit to the mRNA through either interactions with eIF3 or complementarities to the 18S rRNA (28, 53, 54). Lastly, the p53 endogenous mRNA appears to be able to associate with 40S ribosomes that lack RPS25 (Fig. 4C). Taken together, these findings suggest that RPS25 plays a role downstream of 40S subunit recruitment.
Following 40S subunit recruitment, the mRNA needs to be loaded into the mRNA channel on the 40S ribosome, the start codon must be positioned into the P site of the ribosome, and the 60S subunit then joins. IRESs or ribosomal shunts could be dependent upon RPS25 for any one of these downstream steps, and currently, there are no direct data that address this. The mRNA binding channel of an empty 40S subunit is in a “closed” conformation; therefore, the channel must be “opened” to allow loading of the mRNA. Prior to cap-dependent initiation, eIF1 and eIF1A bind to the 40S subunit and induce a conformational change in the 40S subunit to open the latch of the mRNA entry channel in order for the mRNA to be loaded onto the 40S subunit (55). Interestingly, binding of the CrPV IGR and the HCV IRES to the 40S subunit induces similar conformational changes to the 40S ribosomal subunit, as was observed following eIF1 and eIF1A ribosome binding (15, 16, 55). It is possible that RPS25 acts directly or indirectly to trigger the opening of the entry latch of the mRNA channel in the 40S ribosomal subunit. It is notable that some initiation events, such as those utilizing the HCV or CrPV IGR IRESs, do not require eIF1 and eIF1A. Presumably, they are able to open the mRNA entry channel by a different mechanism. For ribosome shunting, the 40S subunit is initially recruited in a cap-dependent mechanism that requires eIF1 and eIF1A to open the mRNA binding channel as part of the preinitiation complex. However, some ribosome shunts involve translation of a short upstream open reading frame (uORF) prior to shunting. Since eIF1 and eIF1A are released upon 60S joining, it is unlikely that eIF1 and eIF1A would remain associated with the 40S subunit following translation of a short uORF (56–58). Therefore, IRESs and ribosome shunts face the common problem of inducing the conformational change in the 40S subunit in order to open the mRNA binding channel to load the mRNA or the start codon into the entry channel of the 40S ribosome.
While clearly any interaction between the 40S and the IRES could potentially result in conformational changes in the 40S subunit, the CrPV IGR IRES and HCV IRES bind to opposite surfaces of the 40S subunit, and their only common point of interaction on the 40S subunit is in the E site, where RPS25 resides. Furthermore, an HCV IRES mutant that lacks domain IIb, which contacts the E site, fails to induce the conformational change in the ribosome to open the mRNA channel (16), suggesting that IRES interactions with the E site are critical for inducing a conformational change to open the mRNA channel. Taken together, these results suggest that a contact in the E site, where RPS25 resides, may be important for inducing the conformational change.
Clearly, the activities of some IRESs are more affected by knockdown of RPS25 than others. This may suggest that different IRESs use different mechanisms that are more or less dependent on RPS25. Alternatively, there may be more of a sliding scale for RPS25 dependence for different IRESs. The picornaviral IRESs were less dependent upon RPS25 than CrPV IGR or HCV IRESs. It is interesting that the minimum complement of initiation factors required to form 48S complexes at the start codon of a picornaviral IRES does not include eIF1 or eIF1A (52, 59). However, if eIF1 and eIF1A are added, they enhance the translation efficiency of picornaviral IRESs (60). This suggests that picornaviral IRESs may have two modes of initiation: (i) a ribosome bound by eIF1 and eIF1A is recruited to the IRES in an open conformation, or (ii) a ribosome is recruited in a closed conformation, and IRES binding triggers RPS25 to induce an open conformation. Cap-dependent translation is unaffected by the loss of RPS25 because the ribosome is recruited to the 5′ end of the mRNA only as a 43S preinitiation complex, which inevitably has eIF1 and eIF1A associated with the 40S subunit. Thus, the capped mRNA never needs to rely on RPS25 to open the mRNA channel on the 40S subunit. While we have presented no data to support the role of RPS25 in opening the mRNA channel and recognize that it may have other downstream roles, we find this model to be the most consistent with all the available published data. Future studies that define the role of RPS25 in IRES-mediated translation will reveal why there is a range of effects on various IRESs when RPS25 is knocked down.
Importantly, RPS25 contains an N-terminal extension that contacts the decoding center (20). Terminal extensions are common in ribosomal proteins and function to communicate changes between distant regions of the ribosome. eIF1 and eIF1A are also known to be important for scanning of the ribosomal subunit; however, some IRESs that we tested are known to use ribosome scanning (PV and EV71), while others do not (CrPV IGR and HCV). Therefore, RPS25 is less likely to be involved in scanning.
Knockdown of RPS25 has a dramatic effect on IRES-mediated translation and ribosome shunting. However, its effect on global translation is less obvious. Several lines of evidence indicate that cap-dependent translation is unaffected by the depletion of RPS25: (i) the activities of two reporters, Renilla luciferase and β-galactosidase, which utilize cap-dependent translation, are not inhibited by the depletion of RPS25; (ii) there is no defect in global translation initiation based on polysome analysis; and (iii) amplification of HSV-1, which relies solely on cap-dependent translation for viral protein synthesis, is not inhibited when RPS25 is knocked down. Generally, under rich-medium conditions, initiation is the rate-limiting step of translation. Under these conditions, there is a slight but reproducible decrease in global translation rates in cells lacking RPS25 (Fig. 1E) (17). However, at low serum concentrations, there is no difference in cell proliferation, suggesting that depletion of RPS25 imparts only a slight growth advantage to cells that are dividing rapidly. Importantly, this slight decrease in translation does not render the cells less fit for viral amplification, as evidenced by the efficient amplification of HSV-1. Our previous studies in yeast also suggest that ribosomes lacking RPS25 are functional and display only minor or no significant defects in initiation, ribosome biogenesis, stop codon recognition, programmed ribosomal frameshifting, or correct incorporation of amino acids (17). While the role of RPS25 in global translation remains unknown, there exists the possibility that it plays a role in either elongation or termination but not cap-dependent initiation. Taken together, these results suggest that RPS25 is essential for cells or viruses only if they require IRES-mediated translation or ribosome shunting. Therefore, since viral RNAs and cellular mRNAs that use alternative mechanisms of translation converge on RPS25, this suggests that RPS25 is a good target for a broad-spectrum antiviral or anticancer therapeutic agent.
Supplementary Material
ACKNOWLEDGMENTS
We thank David Bedwell for careful reading of the manuscript. We thank Peter Sarnow (Stanford School of Medicine) for the HCV plasmid, Jacqueline N. Parker (University of Alabama at Birmingham) for HSV-1, and Charles Rice (Rockefeller University) for the NS5A antibody.
This work is supported by the National Institutes of Health (grants R01GM084547 and 3R01GM084547-01A1S1 to S.R.T.) and a UAB Cancer Center HIV-Associated Malignancy pilot research grant (UAB Comprehensive Cancer Center core support grant P30 CA13148) to S.R.T.
We declare that we have no conflict of interest.
Footnotes
Published ahead of print 28 December 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00879-12.
REFERENCES
- 1.Jackson RJ, Hellen CU, Pestova TV. 2010. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 11:113–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spriggs KA, Stoneley M, Bushell M, Willis AE. 2008. Re-programming of translation following cell stress allows IRES-mediated translation to predominate. Biol. Cell 100:27–38 [DOI] [PubMed] [Google Scholar]
- 3.Johannes G, Carter MS, Eisen MB, Brown PO, Sarnow P. 1999. Identification of eukaryotic mRNAs that are translated at reduced cap binding complex eIF4F concentrations using a cDNA microarray. Proc. Natl. Acad. Sci. U. S. A. 96:13118–13123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bushell M, Stoneley M, Kong YW, Hamilton TL, Spriggs KA, Dobbyn HC, Qin X, Sarnow P, Willis AE. 2006. Polypyrimidine tract binding protein regulates IRES-mediated gene expression during apoptosis. Mol. Cell 23:401–412 [DOI] [PubMed] [Google Scholar]
- 5.Bushell M, Sarnow P. 2002. Hijacking the translation apparatus by RNA viruses. J. Cell Biol. 158:395–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kieft JS. 2008. Viral IRES RNA structures and ribosome interactions. Trends Biochem. Sci. 33:274–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fitzgerald KD, Semler BL. 2009. Bridging IRES elements in mRNAs to the eukaryotic translation apparatus. Biochim. Biophys. Acta 1789:518–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pestova TV, Shatsky IN, Fletcher SP, Jackson RJ, Hellen CU. 1998. A prokaryotic-like mode of cytoplasmic eukaryotic ribosome binding to the initiation codon during internal translation initiation of hepatitis C and classical swine fever virus RNAs. Genes Dev. 12:67–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Otto GA, Puglisi JD. 2004. The pathway of HCV IRES-mediated translation initiation. Cell 119:369–380 [DOI] [PubMed] [Google Scholar]
- 10.Fraser CS, Doudna JA. 2007. Structural and mechanistic insights into hepatitis C viral translation initiation. Nat. Rev. Microbiol. 5:29–38 [DOI] [PubMed] [Google Scholar]
- 11.Pestova TV, Lomakin IB, Hellen CU. 2004. Position of the CrPV IRES on the 40S subunit and factor dependence of IRES/80S ribosome assembly. EMBO Rep. 5:906–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deniz N, Lenarcic EM, Landry DM, Thompson SR. 2009. Translation initiation factors are not required for Dicistroviridae IRES function in vivo. RNA 15:932–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jan E, Sarnow P. 2002. Factorless ribosome assembly on the internal ribosome entry site of cricket paralysis virus. J. Mol. Biol. 324:889–902 [DOI] [PubMed] [Google Scholar]
- 14.Schuler M, Connell SR, Lescoute A, Giesebrecht J, Dabrowski M, Schroeer B, Mielke T, Penczek PA, Westhof E, Spahn CM. 2006. Structure of the ribosome-bound cricket paralysis virus IRES RNA. Nat. Struct. Mol. Biol. 13:1092–1096 [DOI] [PubMed] [Google Scholar]
- 15.Spahn CM, Jan E, Mulder A, Grassucci RA, Sarnow P, Frank J. 2004. Cryo-EM visualization of a viral internal ribosome entry site bound to human ribosomes: the IRES functions as an RNA-based translation factor. Cell 118:465–475 [DOI] [PubMed] [Google Scholar]
- 16.Spahn CM, Kieft JS, Grassucci RA, Penczek PA, Zhou K, Doudna JA, Frank J. 2001. Hepatitis C virus IRES RNA-induced changes in the conformation of the 40s ribosomal subunit. Science 291:1959–1962 [DOI] [PubMed] [Google Scholar]
- 17.Landry DM, Hertz MI, Thompson SR. 2009. RPS25 is essential for translation initiation by the Dicistroviridae and hepatitis C viral IRESs. Genes Dev. 23:2753–2764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muhs M, Yamamoto H, Ismer J, Takaku H, Nashimoto M, Uchiumi T, Nakashima N, Mielke T, Hildebrand PW, Nierhaus KH, Spahn CM. 2011. Structural basis for the binding of IRES RNAs to the head of the ribosomal 40S subunit. Nucleic Acids Res. 39:5264–5275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hertz MI, Thompson SR. 2011. In vivo functional analysis of the Dicistroviridae intergenic region internal ribosome entry sites. Nucleic Acids Res. 39:7276–7288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Armache JP, Jarasch A, Anger AM, Villa E, Becker T, Bhushan S, Jossinet F, Habeck M, Dindar G, Franckenberg S, Marquez V, Mielke T, Thomm M, Berninghausen O, Beatrix B, Soding J, Westhof E, Wilson DN, Beckmann R. 2010. Localization of eukaryote-specific ribosomal proteins in a 5.5-A cryo-EM map of the 80S eukaryotic ribosome. Proc. Natl. Acad. Sci. U. S. A. 107:19754–19759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ben-Shem A, Jenner L, Yusupova G, Yusupov M. 2010. Crystal structure of the eukaryotic ribosome. Science 330:1203–1209 [DOI] [PubMed] [Google Scholar]
- 22.Rabl J, Leibundgut M, Ataide SF, Haag A, Ban N. 2011. Crystal structure of the eukaryotic 40S ribosomal subunit in complex with initiation factor 1. Science 331:730–736 [DOI] [PubMed] [Google Scholar]
- 23.Curran J, Kolakofsky D. 1988. Scanning independent ribosomal initiation of the Sendai virus X protein. EMBO J. 7:2869–2874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pooggin MM, Ryabova LA, He X, Futterer J, Hohn T. 2006. Mechanism of ribosome shunting in Rice tungro bacilliform pararetrovirus. RNA 12:841–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Futterer J, Kiss-Laszlo Z, Hohn T. 1993. Nonlinear ribosome migration on cauliflower mosaic virus 35S RNA. Cell 73:789–802 [DOI] [PubMed] [Google Scholar]
- 26.Cao F, Tavis JE. 2011. RNA elements directing translation of the duck hepatitis B virus polymerase via ribosomal shunting. J. Virol. 85:6343–6352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yueh A, Schneider RJ. 1996. Selective translation initiation by ribosome jumping in adenovirus-infected and heat-shocked cells. Genes Dev. 10:1557–1567 [DOI] [PubMed] [Google Scholar]
- 28.Yueh A, Schneider RJ. 2000. Translation by ribosome shunting on adenovirus and hsp70 mRNAs facilitated by complementarity to 18S rRNA. Genes Dev. 14:414–421 [PMC free article] [PubMed] [Google Scholar]
- 29.Sherrill KW, Lloyd RE. 2008. Translation of cIAP2 mRNA is mediated exclusively by a stress-modulated ribosome shunt. Mol. Cell. Biol. 28:2011–2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Park HS, Browning KS, Hohn T, Ryabova LA. 2004. Eucaryotic initiation factor 4B controls eIF3-mediated ribosomal entry of viral reinitiation factor. EMBO J. 23:1381–1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thompson SR, Gulyas KD, Sarnow P. 2001. Internal initiation in Saccharomyces cerevisiae mediated by an initiator tRNA/eIF2-independent internal ribosome entry site element. Proc. Natl. Acad. Sci. U. S. A. 98:12972–12977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fletcher SP, Jackson RJ. 2002. Pestivirus internal ribosome entry site (IRES) structure and function: elements in the 5′ untranslated region important for IRES function. J. Virol. 76:5024–5033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carter MS, Sarnow P. 2000. Distinct mRNAs that encode La autoantigen are differentially expressed and contain internal ribosome entry sites. J. Biol. Chem. 275:28301–28307 [DOI] [PubMed] [Google Scholar]
- 34.Ray PS, Grover R, Das S. 2006. Two internal ribosome entry sites mediate the translation of p53 isoforms. EMBO Rep. 7:404–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stoneley M, Paulin FE, Le Quesne JP, Chappell SA, Willis AE. 1998. C-Myc 5′ untranslated region contains an internal ribosome entry segment. Oncogene 16:423–428 [DOI] [PubMed] [Google Scholar]
- 36.Coldwell MJ, deSchoolmeester ML, Fraser GA, Pickering BM, Packham G, Willis AE. 2001. The p36 isoform of BAG-1 is translated by internal ribosome entry following heat shock. Oncogene 20:4095–4100 [DOI] [PubMed] [Google Scholar]
- 37.Coldwell MJ, Mitchell SA, Stoneley M, MacFarlane M, Willis AE. 2000. Initiation of Apaf-1 translation by internal ribosome entry. Oncogene 19:899–905 [DOI] [PubMed] [Google Scholar]
- 38.Jopling CL, Spriggs KA, Mitchell SA, Stoneley M, Willis AE. 2004. L-Myc protein synthesis is initiated by internal ribosome entry. RNA 10:287–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mitchell SA, Spriggs KA, Bushell M, Evans JR, Stoneley M, Le Quesne JP, Spriggs RV, Willis AE. 2005. Identification of a motif that mediates polypyrimidine tract-binding protein-dependent internal ribosome entry. Genes Dev. 19:1556–1571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rydzanicz R, Zhao XS, Johnson PE. 2005. Assembly PCR oligo maker: a tool for designing oligodeoxynucleotides for constructing long DNA molecules for RNA production. Nucleic Acids Res. 33:W521–W525 doi:10.1093/nar/gki380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kolupaeva VG, Pestova TV, Hellen CU. 2000. An enzymatic footprinting analysis of the interaction of 40S ribosomal subunits with the internal ribosomal entry site of hepatitis C virus. J. Virol. 74:6242–6250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Plank TD, Kieft JS. 2012. The structures of nonprotein-coding RNAs that drive internal ribosome entry site function. Wiley Interdiscip. Rev. RNA 3:195–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pelletier J, Sonenberg N. 1988. Internal initiation of translation of eukaryotic mRNA directed by a sequence derived from poliovirus RNA. Nature 334:320–325 [DOI] [PubMed] [Google Scholar]
- 44.Thompson SR, Sarnow P. 2003. Enterovirus 71 contains a type I IRES element that functions when eukaryotic initiation factor eIF4G is cleaved. Virology 315:259–266 [DOI] [PubMed] [Google Scholar]
- 45.Cai Z, Zhang C, Chang KS, Jiang J, Ahn BC, Wakita T, Liang TJ, Luo G. 2005. Robust production of infectious hepatitis C virus (HCV) from stably HCV cDNA-transfected human hepatoma cells. J. Virol. 79:13963–13973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cun W, Jiang J, Luo G. 2010. The C-terminal alpha-helix domain of apolipoprotein E is required for interaction with nonstructural protein 5A and assembly of hepatitis C virus. J. Virol. 84:11532–11541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smith RW, Graham SV, Gray NK. 2008. Regulation of translation initiation by herpesviruses. Biochem. Soc. Trans. 36:701–707 [DOI] [PubMed] [Google Scholar]
- 48.Futterer J, Gordon K, Sanfacon H, Bonneville JM, Hohn T. 1990. Positive and negative control of translation by the leader sequence of cauliflower mosaic virus pregenomic 35S RNA. EMBO J. 9:1697–1707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morley SJ, Coldwell MJ. 2008. A cunning stunt: an alternative mechanism of eukaryotic translation initiation. Sci. Signal. 1:pe32 doi:10.1126/scisignal.125pe32. [DOI] [PubMed] [Google Scholar]
- 50.Kieft JS, Zhou K, Jubin R, Doudna JA. 2001. Mechanism of ribosome recruitment by hepatitis C IRES RNA. RNA 7:194–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lamphear BJ, Kirchweger R, Skern T, Rhoads RE. 1995. Mapping of functional domains in eukaryotic protein synthesis initiation factor 4G (eIF4G) with picornaviral proteases. Implications for cap-dependent and cap-independent translational initiation. J. Biol. Chem. 270:21975–21983 [DOI] [PubMed] [Google Scholar]
- 52.Pestova TV, Shatsky IN, Hellen CU. 1996. Functional dissection of eukaryotic initiation factor 4F: the 4A subunit and the central domain of the 4G subunit are sufficient to mediate internal entry of 43S preinitiation complexes. Mol. Cell. Biol. 16:6870–6878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Park HS, Himmelbach A, Browning KS, Hohn T, Ryabova LA. 2001. A plant viral “reinitiation” factor interacts with the host translational machinery. Cell 106:723–733 [DOI] [PubMed] [Google Scholar]
- 54.Xi Q, Cuesta R, Schneider RJ. 2004. Tethering of eIF4G to adenoviral mRNAs by viral 100k protein drives ribosome shunting. Genes Dev. 18:1997–2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Passmore LA, Schmeing TM, Maag D, Applefield DJ, Acker MG, Algire MA, Lorsch JR, Ramakrishnan V. 2007. The eukaryotic translation initiation factors eIF1 and eIF1A induce an open conformation of the 40S ribosome. Mol. Cell 26:41–50 [DOI] [PubMed] [Google Scholar]
- 56.Nanda JS, Cheung YN, Takacs JE, Martin-Marcos P, Saini AK, Hinnebusch AG, Lorsch JR. 2009. eIF1 controls multiple steps in start codon recognition during eukaryotic translation initiation. J. Mol. Biol. 394:268–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Olsen DS, Savner EM, Mathew A, Zhang F, Krishnamoorthy T, Phan L, Hinnebusch AG. 2003. Domains of eIF1A that mediate binding to eIF2, eIF3 and eIF5B and promote ternary complex recruitment in vivo. EMBO J. 22:193–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fringer JM, Acker MG, Fekete CA, Lorsch JR, Dever TE. 2007. Coupled release of eukaryotic translation initiation factors 5B and 1A from 80S ribosomes following subunit joining. Mol. Cell. Biol. 27:2384–2397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pestova TV, Hellen CU, Shatsky IN. 1996. Canonical eukaryotic initiation factors determine initiation of translation by internal ribosomal entry. Mol. Cell. Biol. 16:6859–6869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pestova TV, Borukhov SI, Hellen CU. 1998. Eukaryotic ribosomes require initiation factors 1 and 1A to locate initiation codons. Nature 394:854–859 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.