

Abstract
The t(14;18) chromosomal translocation typically involves breakage at the bcl-2 major breakpoint region (MBR) to cause human follicular lymphoma. A theory to explain the striking propensity of the MBR breaks at three CpG clusters within the 175-bp MBR region invoked activation-induced deaminase (AID). In a test of that theory, we used here minichromosomal substrates in human pre-B cell lines. Consistent with the essential elements of the theory, we found that the MBR breakage process is indeed highly dependent on DNA methylation at the CpG sites and highly dependent on the AID enzyme to create lesions at peak locations within the MBR. Interestingly, breakage of the phosphodiester bonds at the AID-initiated MBR lesions is RAG dependent, but, unexpectedly, most are also dependent on Artemis. We found that Artemis is capable of nicking small heteroduplex structures and is even able to nick single-base mismatches. This raises the possibility that activated Artemis, derived from the unjoined D to JH DNA ends at the IgH locus on chromosome 14, nicks AID-generated TG mismatches at methyl CpG sites, and this would explain why the breaks at the chromosome 18 MBR occur within the same time window as those on chromosome 14.
INTRODUCTION
Chromosomal translocations consist of an initial chromosome breakage phase, and a DNA end-joining phase (1). The joining phase is attributable primarily to NHEJ (or alternative end joining) (2, 3). However, there has been much less progress in understanding the breakage phase. It is clear that proximity of the involved DNA must occur at some point during the process (4, 5). Neoplastic translocations and chromosomal rearrangements can be nonreciprocal or reciprocal. Human lymphomas and lymphoid leukemias typically have reciprocal translocations (6).
Some lymphoid translocations merely involve the RAG complex pairing an antigen receptor locus with an off-target site that is similar in DNA sequence to the physiologic RAG recombination signal sequence (RSS) (7). This is often the case in T-cell acute lymphoblastic lymphoma, where the only other requirement is that the off-target site has histone modifications that are suitable for the RAG complex (8).
However, the most common human lymphoid translocations do not involve simple mispairing by the RAG complex between one physiologic antigen receptor RSS and an off-target RSS-like sequence. Most human B cell lymphoma translocations have a propensity to occur at the CpG sites within short zones of 20 to 600 bp, which can be called translocation fragile zones (1). We have proposed a model in which activation-induced deaminase (AID) acts at the methylated form of these CpG sites to deaminate the methyl C (meC) to yield a T. In this model, the resulting TG mismatch is repaired relatively slowly compared to the more typical UG mismatch and would be vulnerable to any enzyme that nicks at mismatches or small bubbles, such as the RAG complex. It is not yet clear why the CpGs in translocation fragile zones are more vulnerable than CpGs outside the zone, but a propensity for single-stranded character within the fragile zones is one possibility since AID only acts on single-stranded DNA (9).
Genes that confer a proliferative advantage (oncogenes) can break at any of multiple translocation fragile zones, and the various fragile zones for a given oncogene can be distributed over considerable distances from each other and from the oncogene (e.g., bcl-1, bcl-2, MALT1, and CRLF2) (1, 10, 11). bcl-2 translocations occur at CpG sites within the major breakpoint region (MBR) (50%), intermediate cluster region (13%), or minor cluster region (5%), but these translocation fragile zones are distributed over 31 kb. Of the translocations at CpG-type breaks within the fragile zones near oncogenes, the partner locus in most cases is the immunoglobulin heavy chain (IgH) locus on human chromosome 14, and the breaks at these sites are usually D-to-JH recombination events in which the D and JH joining failed to complete (Fig. 1). Clearly, the RAG complex is needed to initiate these chromosome 14 breaks, and hence, these translocations occur at the pro-B or pre-B stage of development (1).
Fig 1.

Diagram of bcl-2 translocation. The bcl-2 translocations occur between the IgH locus on chromosome 14, mostly during DH-to-JH recombination, and the bcl-2 locus on chromosome 18, mostly downstream of the coding portion of the bcl-2 gene. About 50% of patient translocation breaks occur within the 175-bp major breakpoint region (MBR), which is shown as the red starburst square at the bottom left and expanded to a sequence level above it. The MBR breakpoint distribution from follicular lymphoma patients show highly significant focusing on three CpG peaks (see DNA sequence in the middle left of figure). The side of the break closest to the bcl-2 gene (telomeric side) joins to the JH segment of IgH locus on chromosome 14 and becomes the derivative 14 (der 14). The centromeric side of the bcl-2 locus usually joins to the DH segment and becomes derivative 18 (der 18). In the DNA sequence of the MBR (middle left), the triangles on top of the sequence represent the breakpoints sequenced from the derivative chromosome 14, and the triangles on the bottom of the sequence represent breakpoints sequenced from the derivative chromosome 18. The bottom panel shows the approximate distribution propensity of translocation breakpoints occurring around the MBR region of the bcl-2 gene on chromosome 18 in human patients. The density of lines in the starburst square represents the number of breakpoints at the MBR region relative to breakpoint outside of the MBR region (shown as the vertical bars to the right of the starburst).
AID expression is very high in germinal center B cells. However, it is now clear that AID mRNA, AID enzymatic activity, and some levels of AID-induced class switch recombination are present at a lower level in mouse and human pro-B and pre-B cells (12–28). Our proposed model invoked concurrent expression of AID and the RAG complex (1). AID was needed to initiate TG mismatch sites. The RAG complex was needed to create the DH and JH breaks, at a minimum. We proposed the additional possible role for the RAG complex in converting the TG lesions into double-strand breaks (DSBs). CpG sites are palindromic; hence, if the meCs on both strands at that site are deaminated by AID, then a 5′-TG/5′-TG 2-bp mismatch site would be created. We noted that action by TDG or MBD4 glycosylases, followed by APE1, could also create a DSB (1).
Here, we test the key mechanistic elements of the model that we proposed to explain the CpG propensity of human B cell translocations. First, we found that, indeed, the CpG sites must be methylated in order to be a focus of breakage. Second, AID is indeed required for the CpG-focused breakage. Third, the RAG complex does appear to contribute to some of the conversion of AID-initiated TG mismatches to DSBs. Fourth, surprisingly, Artemis also appears to contribute significantly to the conversion of AID-initiated TG mismatches to DSBs. In light of this support for the major essential elements of our model and now additional findings, we can propose a more detailed mechanistic model for how the CpG-focused breaks are likely to occur.
MATERIALS AND METHODS
Oligonucleotides.
The oligonucleotide primers used in the present study were the first-round forward primer XC111(5′-AAAATAGGCGTATCACGAGGCCC-3′), the first-round reverse primer XC108 (5′-AATGTTCTTTACGATGCCATTGGG-3′), the second-round forward primer XC139 (5′-GTACTAGTCAGCACAGGATTGGATATTCC-3′), and the second-round reverse primer XC110 (5′-CCATTTTAGCTTCCTTAGCTCCTG-3′). The positions of each oligonucleotide DNA on the plasmid substrate are detailed in Fig. S1 in the supplemental material.
The following oligomers were used to form substrates for the Artemis:DNA-dependent protein kinase catalytic subunit (DNA-PKcs) nuclease activity assay: 6-nucleotide (nt) bubble, SCR245 (5′-GACCTGAGGGCGAGCCCGTTGCACAGTGCTTAACAG-3′) and SCR246 (5′-CTGTTAAGCACTGTGTTTTTTGCTCGCCCTCAGGTC-3′); 3-nt bubble, SCR247 (5′-GACCTGAGGGCGAGCCGTCACAGTGCTTAACAG-3′) and SCR248 (5′-CTGTTAAGCACTGTGTTTGCTCGCCCTCAGGTC-3′); and 1-nt bubble, SCR257 (5′-GACCTGAGGGCGAGCTCACAGTGCTTAACAG-3′) and SCR258 (5′-CTGTTAAGCACTGTGTGCTCGCCCTCAGGTC-3′). These oligomers were purified by 12% denaturing polyacrylamide gel electrophoresis (PAGE).
Preparation of oligonucleotide DNA substrates.
The double-stranded oligonucleotides containing 6-nt, 3-nt, and 1-nt bubble structures were prepared by annealing a [γ-32P]ATP end-labeled top strand (SCR245, SCR247, and SCR257, respectively) with the corresponding cold bottom strand (SCR246, SCR248, and SCR258, respectively) at a 1:1 ratio with slow annealing in 10 mM Tris (pH 8.0) and 100 mM NaCl.
Artemis:DNA-PKcs cleavage assay.
The 5′-labeled substrate (25 nM) was incubated in the presence or absence of Artemis (100 nM) or Artemis (100 mM) plus DNA-PKcs (50 nM) complex in 25 mM Tris (pH 8.0), 10 mM KCl, 10 mM MgCl2, 1 mM dithiothreitol, 0.25 mM ATP, and 50 μg of bovine serum albumin/ml in a total reaction volume of 10 μl. Reaction mixtures were incubated for 1 h at 37°C and then denatured for 5 min at 100°C in an equal volume of denaturing gel loading dye (98% formamide, 10 mM EDTA, 0.025% of bromphenol blue, 0.025% of xylene cyanol FF). Reaction mixtures were resolved by 12% denaturing PAGE, and the gel image was obtained with PhosphorImager SI445 (Amersham Biosciences). The details of protein purification were described previously (33).
Plasmid construction.
The plasmid substrate used in the present study, pXC46, was modified based on plasmid pSCR71, which includes a 311-bp human MBR sequence and a 12/23RSS signal and coding sequences (29). In plasmid pXC46, a T-to-C point mutation at nt 215 and a G-to-C point mutation at nt 272 were each created at HhaI sites (see the supplemental material).
In vitro plasmid DNA methylation.
The substrate was methylated in vitro by using HhaI methyltransferase (New England BioLabs), under the conditions recommended by the manufacturer. After methylation, DNA was cleaned by phenol-chloroform extraction. The methylation status of each plasmid DNA was confirmed by HhaI restriction enzyme digestion.
Cell culture and transfection.
Nalm-6 wild-type (wt) cells and the derived knockout cell lines (RAG1−/− and Artemis−/−) were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum. For each experimental condition, 2 × 106 cells were used for each transfection. The DNA mix for each transfection included 1 μg of methylated or unmethylated substrate plasmid DNA, 3 μg of plasmid pKY70 (which expresses wt AID) or pKY155 (which expresses mutant AID with no deamination activity), and 0.5 μg of green fluorescent protein (GFP) expression plasmid pMAXGFP (to monitor the transfection efficiency) in RPMI 1640. Cells mixed with DNA were shocked with an Amaxa Nucleofactor II device (Lonza).
Creation of RAG1 KO in Nalm-6 cells.
The detailed method of targeted disruption of genes in Nalm-6 cells was described previously (30). Briefly, the first 1,980 bp on the RAG1 gene open reading frame was first replaced with either the puromycin resistance gene or a hygromycin resistance gene in each allele, followed by Cre recombination to generate Rag1−/− Nalm-6 cells.
In vitro V(D)J recombination assay.
Transient cellular V(D)J assays were performed as described previously. The recombination substrate, pGG49, retaining signal joint upon V(D)J recombination was tested in three cell lines: Reh, Nalm-6, and RAG1−/− Nalm-6.
Recombination event analysis.
At 48 h after transfection with pXC46, pXC42, or pXC25, episomal DNA was harvested by the rapid alkaline method. The recovered DNA was resuspended in 20 μl of Tris-EDTA (pH 8.0) and used as a template for nested PCR to amplify recombinant junctions between the MBR region and the 23RSS region. Multiple transfections and PCRs were repeated for each experimental condition, 16 to 32 junctions were sequenced for each PCR, and the identical junctions from the same PCR were omitted and only counted once. Four to twelve different transfections or PCRs were performed for each experimental condition, and the numbers were averaged. The PCR products were subcloned in pGEM-T Easy vector (Promega; catalog no. A1360), and junction sequences were revealed by Li-Cor DNA analyzer sequencing (model 4200; Li-Cor, Lincoln, NE).
Statistical analysis.
For comparison of the MBR break sites for pXC46 under various conditions, we compared the number of events within a break zone extending from the CpG sites of peaks 1 and 2 upstream toward the primer for a distance of 30 bp (Fig. 2). The P values shown were calculated using a Student t test comparison between the Fig. 2B, line 1, values for peak 1 and all of the conditions for peak 1 in lines 2 to 6; the same applies to peak 2 comparisons of line 1 with lines 2 to 6. More specifically for Fig. 2B, the percentage of events within the peak box was divided by the total number of sequenced recombinants for each transfection, and then the mean was calculated for multiple transfections (usually 4 to 12 transfections).
Fig 2.

Summary of breakpoint locations for substrate pXC46. (A) Schematic graph of the pXC46 plasmid. The small squares represent three peaks in the MBR region. Red squares represent the methylated peaks. The triangles represent 12RSS and 23RSS. The arrows represent the relative positions of primers for nested PCR assays. Given that there is NHEJ nucleolytic resection after DSBs occur, a 30-bp zone of interest was delineated at each peak region, designated by two vertical dashed lines. A 30-bp zone of interest is also demarcated for V(D)J recombination breaks at the 12/23RSS coding region. (B) The middle columns of the table show percentages of breakpoints occurring at peak 1, 2, or 3. The right part of the table shows percentages of deletions that have a left side boundary among the MBR peaks and a right side boundary near the 23 coding end (within 30 bp of the 23RSS heptamer). Each data number in this figure is the sum of 4 to 12 independent experiments. The P values connected by solid red lines were calculated using a Student t test comparison between the line 1 values for peak 1 and all of the conditions for peak 1 in lines 2 to 6. The same applies to peak 2 comparisons of line 1 with lines 2 to 6. *, P values connected with the dashed red line represent chi-square test comparisons of methylated peak 1 or peak 2 with the nonmethylated peak 3 when all of the factors are present (line 1). Note that when peaks 1 and 2 are not the focus of breakage, due to omission of one factor (e.g., no CpG methylation, use of mutant AID, or knockout of Artemis), then it is not surprising that the percentage of breaks at all other locations rise, as we see for peak 3 in lines 2, 3, and 5.
Comparisons between pXC46 peaks 1, 2 and 3 in Fig. 2B were done using the chi-square test (see the legend to Fig. 2B). An alternative method for comparing break locations between different experimental conditions also used the chi-square test (see the legend to Fig. 3B).
Fig 3.

Breakpoint distributions around the bcl-2 MBR. (A) Breakpoint distributions for the substrate pXC46. Each inverted triangle represents one breakpoint. The region of sequence in this plot includes (from left to right) 285 bp from human MBR gene (chromosome 18 reverse strand: bp 60793336 to 60793620). Peak 1 (bp 60793548 to 60793551) and peak 2 (bp 60793493 to 60793495) are labeled in a small rectangle with solid lines and are methylated by HhaI methyltransferase except in the group for which methylation is absent. Each line represents one experimental condition, as labeled under each line of sequence. The positions of peaks 1, 2, and 3 are marked above the figure, and the 30-bp zone of interest of each peak is also shown in a large rectangle with dashed lines. The breakpoints plotted are collected from all of the experiments for each group. (B) Comparisons based on normalization. As an alternative to the Student t test, we also compared the conditions of lines 1 to 6 of panel 1 by normalizing the total number of events in lines 2 to 6 to the number in line 1. We divided the left-side break event locations into the peak 1, 2, and 3 zones, as well as the additional zones of events between peaks 1 and 2 (P2-1), between peaks 2 and 3 (P3-2), and upstream of peak 3 (N-P3). For each zone, the normalized number of events for lines 1 to 6 are shown from left to right. Many events are detected to the left of peak 3 because the AT-rich region favors paused and short PCR products, but the differences between conditions are not substantial. Only peaks 1 and 2, when all factors are present (line 1), are significantly higher than all of the other conditions (lines 2 to 6), and this was tested using the chi-square test on the normalized number of events within each peak box versus the normalized total. The significance (P < 0.05) is designated by an asterisk. Chi-square analysis was not run on the other zones. For the P2-1 zone, the large value of the line 4 condition (no RAG1) is likely because AID is present, and most of the breaks in this zone are near a WGCW site, which is a preferred AID motif (35).
Statistical analysis of motif proximity.
Methods for conducting the statistical analysis were described previously (1). Only the methylated motif (the HhaI site) plus 1 bp upstream are analyzed for breakpoint localization propensity; hence, nGCGC is the motif analyzed. Because of the PCR pause at the AT-rich zone upstream of peak 3 in the pXC46 substrate, we modified the reference sequence for motif propensity analysis. The modified reference sequence starts 30 bp lateral to peak 3 and therefore includes only 207 bp of the MBR (i.e., bp 60793417 to bp 60793620). Each motif test was analyzed with three tests (binomial distribution, Student t test, and Mann-Whitney U test). These statistical analyses have various strengths and limitations. In order to ensure the fairest evaluation of the data, only the groups that have all three P values lower than 0.05 are considered statistically significant at each analyzed motif.
RESULTS
Experimental system using minichromosomes.
Within the bcl-2 MBR, breaks found in follicular lymphoma patients are centered on the CpG sites within the MBR with high statistical significance (1). In a previous study, we showed that the MBR had increased fragility on a similar minichromosome substrate that carries a 12RSS (equivalent to a DH site) and 23RSS (equivalent to a JH site), in addition to the MBR (29, 31). However, in that system, breakage within the MBR was relatively uniform across the 175-bp zone and failed to show the peak predilection for the CpG clusters within the MBR. This suggested that the overall fragility of the MBR zone in the genome can be reproduced on the minichromosome but that requirements to recapitulate the localization of the breaks to peaks 1, 2, and 3 within the MBR remain undefined.
Here we have devised methods to methylate the CpG sites at some of the peaks within the MBR. This allowed us to test the contribution of methylation status to the break localization at or near that peak within the 175-bp MBR. We proposed that AID, CpG methylation, RAGs, and Artemis are essential for the fragility in the MBR region to be targeted to the three peaks. Minichromosomal translocation substrates with DNA methylation can be cotransfected with an AID expression vector (pKY70) into Nalm-6 cells, which produce the wt RAG complex and express a very low level of endogenous AID mRNA, to create the condition with all of the essential elements present. We have created a clone of Nalm-6 cells with homozygous deletion of RAG1 alleles (see Fig. S2 in the supplemental material) and a clone with homozygous deletion of Artemis alleles (30). These knockout human cell lines and a mutant AID expression vector allow testing for the contribution of AID, RAG, and Artemis in the breakage in MBR peaks in this pre-B cell line.
To capture DNA breaks with various junctions for analysis, the relevant size ranges of products from 25 cycles of PCR were excised from the gel and then amplified for cloning and sequencing (see Fig. S3 in the supplemental material). We examined the locations of the breaks relative to the MBR and relative to the three peaks within it, when various proteins were present or absent. Because either side of the MBR can recombine with either DH or JH in patient bcl-2 translocations and because this was verified in our earlier studies, we positioned the MBR on the initial substrate, pXC46, as shown in Fig. S1A in the supplemental material (29, 32). Substrates with the MBR in an opposite orientation relative to that in pXC46 were also tested (see Fig. S1B and C in the supplemental material).
The PCR products we analyzed consisted of a population of deletion recombinants. The junctions we detected likely involve two breaks: one on the “left” or MBR side of the recombination zone (the same side as the forward primers) and one on the right side of the recombination zone (the same side as the reverse primers). The sequence between the two breaks in each recombination event was deleted (see Fig. S4 in the supplemental material).
Our procedures are designed to capture recombination between the MBR zone and the 12/23RSS sites (right side of the recombination zone). However, the level and temporal coordination of AID and the RAGs may not be similar to the precise conditions prevailing when bcl-2 translocations occur in patients. RAGs are endogenously expressed in Nalm-6 and thus can act before AID expression. Therefore, we can only capture the recombinant junctions between the MBR and the 12/23RSS in a small fraction of the recombinant plasmids (see below). That is, many deletions within the recombination zone have a break within the MBR on the left side, but the break on the right side of the recombination zone is not near the RSSs. Although many breaks on the right side are RAG dependent, many occur in the absence of both RAG1 and AID (see below), and we assume that these are the background breakages generated by the transfection process or DNA preparation. Because of this, our analysis focuses primarily on the location of breaks within the MBR region, although we include information for the 12/23 region throughout.
Fragile site character of the bcl-2 major breakpoint region is reproduced with methylated CpG substrates in the presence of AID, RAGs, and Artemis.
We were interested in the role of CpG methylation in the localization of breaks within the MBR. A substrate, pXC46, was generated to introduce a single nucleotide point mutation into both peak 1 (by changing the 3′ G of peak 1 GCGG to a “c,” resulting in GCGc) and peak 2 (by changing the T at the 3′ edge of GCGT to a “c,” resulting in GCGc) to create HhaI (GCGC) sites that could be methylated by HhaI methylase. No mutations or changes were made at peak 3 (see Fig. S5A in the supplemental material).
We have proposed that AID, RAGs, and CpG methylation are essential for the fragility associated with the CpG sites in the MBR within wt cells (1). Here, we first test whether recombination products can be detected when all of the proposed essential elements are present. We cotransfected methylated pXC46 (me-pXC46), on which MBR peaks 1 and 2 are methylated, with the AID expression vector (pKY70) into wt Nalm-6 cells. As mentioned above, Nalm-6 cells express RAGs and Artemis. To identify recombinant products and exclude the unrecombined products, we PCR amplified across the recombination zone and gel purified potential recombinant products away from the full-length (unrecombined) band (see Fig. S3 in the supplemental material). Using this gel-purified material, we then performed a second round of PCR using internal primers (Fig. 2A; see also Fig. S1 and S3 in the supplemental material).
As mentioned above, our primary focus was on the positions of the left-side breaks (the MBR side). In particular, are the left-side breaks in proximity to the methylated peaks of the MBR? To include events in which nucleolytic resection has occurred from the break site, we included breakpoints within a zone of up to 30 bp leftward (upstream [i.e., toward the sequencing primer]) from each of the MBR peaks. To show the distribution of breakage on the left side of the recombination zone, we plotted the location of all of the breakage events (Fig. 3A). Each line represents events from each experimental condition. The 30-bp zone for each peak is identified by the boxed areas (Fig. 3A, blue boxes are methylated peaks and gray boxes are unmethylated).
We found that 15% of the recombination events occur at or near (within 30 bp upstream) peak 1, and 13% of the recombination events occurred at or near peak 2 in Nalm-6 cells with AID expression vector cotransfection (Fig. 2B, line 1). At peak 3, where there is no CpG methylation on the minichromosome, we found no propensity for breakage, with only 6% of recombination events occurring there (Fig. 2B, line 1), and the differences between peak 3 versus peaks 1 and 2 are significant (P = 0.005 and P = 0.02, respectively, as determined by chi-square analysis). Note that there is a propensity for PCR pausing at an AT-rich zone to the left of peak 3 in pXC46, which raises the background at peak 3, but consideration of this would suggest an even greater difference when the unmethylated peak 3 was compared to the methylated peaks 1 and 2. These findings indicate that breakage in MBR peaks can occur when HhaI sites are methylated on the minichromosome when AID, RAGs, and Artemis are present. This substrate also reflects the differential breakage propensity for peaks within the MBR at methylated peaks 1 and 2 and not at the unmethylated peak 3. This difference may be due to the methylation status or due to the intrinsic sequence or position difference, which we further analyzed in the next section.
Importantly, substrates with the MBR in the reversed orientation relative to that in pXC46 were also tested (see Fig. S1B and C, S6, and S7 in the supplemental material); these are substrates pXC42 and pXC25. Analysis of these substrates is complicated by the fact that PCR products had a greater tendency to terminate just upstream of peak 1 (see the methods section on recombination event analysis in the supplemental material). Therefore, each peak was analyzed from PCR products generated using a different internal primer (see Fig. S1B and C in the supplemental material). Because of this the tabulation of breaks was done for the individual peaks from molecules amplified from the relevant peak in different PCRs rather than for all three peaks in the entire region from molecules amplified in the same PCR (as it was for pXC46). This manner of tabulation for pXC42 makes the percentage of breaks at each peak larger because it does not include events that occur in the other two peaks in the denominator, as was done for the tabulation with pXC46. The pXC42 substrate, like pXC46, was methylated only at peaks 1 and 2 but not at peak 3 (see Fig. S5B in the supplemental material). This substrate showed the same basic results as pXC46. Namely, breaks are common in or upstream of peaks 1 and 2 and much less so at peak 3 (see Fig. S6 in the supplemental material).
pXC25 is similar to pXC42 except that CpG methylation was done using the HpaII methylase, which methylates the natural sequence at peak 3. Methylation at peak 1 using HpaII methylase requires a single nucleotide mutation of the natural peak 1 sequence (see Fig. S5B in the supplemental material). Using this substrate, ample breaks were detected at the methylated peaks 1 and 3 and much less so at the unmethylated peak 2 (see Fig. S7 in the supplemental material). Findings using all three substrates are consistent with localization of breaks to methylated MBR peaks, when AID and RAGs are present in wt Nalm-6 cells.
CpG methylation is important for the fragile character at the MBR.
To further test whether the methylation status at the peaks is important for the localization of breaks to the peaks, as we had proposed in our original model (1), the substrate, pXC46, with no methylation was cotransfected into wt Nalm-6 cells with AID expression vector. The recombination events were amplified and sequenced as described above. Without methylation, the breakage percentage drops significantly from 15 to 3% (Student t test, P = 0.001) at peak 1 and from 13 to 2% (P = 0.0009) at peak 2 (Fig. 2B, line 2). Hence, CpG methylation status appears to be quite important for fragility at peaks 1 and 2. Also, very little difference was observed among peaks 1, 2, and 3 when there is no methylation.
To test the background level of breakage frequency, the unmethylated substrate, pXC46, was transfected alone into Nalm-6 cells. The breakage percentage remains low in both peaks 1 and 2 (Fig. 2B, line 6 compared to line 1).
In addition to knowing that breakage events appear to focus in the peaks with CpG methylation, it is also important to know how the breakage events distribute within the recombination zone. In order to compare the results between Fig. 2B, lines 1 and 2, for event distribution, we normalized the total number of events detected throughout the recombination zone in each experiment to the total number in line 1 of Fig. 2B. The estimated number of events in each of six zones across the left-side of the recombination zone was calculated based on the percentage of the events observed in the specific zone (Fig. 3B). This comparison illustrates that peaks 1 and 2 in line 1 (all factors present) are significantly more often sites of breakage than peaks 1 and 2 in line 2 (all factors minus CpG methylation at peaks 1 and 2) (Fig. 3B).
The lack of difference in recombination events in peaks 1, 2, and 3 when all three peaks are not methylated on pXC46 would indicate that breakage at these three peaks is not sequence or position dependent (Fig. 2B, line 2). Therefore, the differential breakage rate in peak 3 compared to peaks 1 and 2 is a true reflection of the essential role of CpG methylation in breakage in these peaks. Corresponding analysis of pXC42 and pXC25 also showed a strong dependence of breakage on CpG methylation in two ways: (i) between unmethylated and methylated substrates and (ii) between unmethylated and methylated peaks within the same substrate (see Fig. S6 and S7 in the supplemental material). Therefore, the importance of CpG methylation for the fragility to be focused at the specific peaks in the MBR is clearly evident.
AID enzymatic activity is important for the fragility at the MBR.
Another critical element of our original model was that AID was responsible for the localization of breaks at the peaks within the MBR (1). To test this, the methylated recombination substrate me-pXC46 was cotransfected with an active-site point mutant of AID (pKY155 expresses AID with an E58Q mutation), which produces equivalent amounts of protein in mammalian cells but is catalytically inactive.
The breakage reduced from 15 to 4% (P = 0.009) at peak 1 and from 13 to 5% (P = 0.03) at peak 2 (Fig. 2B, line 3) when mutant AID was expressed in the cells. Therefore, the results indicate that AID is important for fragility at specific CpG methylated peaks within the MBR. The same marked AID dependence was observed for substrates pXC42 and pXC25 (see Fig. S6 and S7 in the supplemental material).
RAG activity is important for MBR breakage.
In contrast to the roles of CpG methylation and AID, the role of the RAG complex in the t(14;18) translocation could be at more than one step. At a minimum, the RAG complex is clearly essential to create the chromosome 14 breaks at the DH and JH segments of the IgH locus and then inadvertently releases these two DNA ends, thereby allowing them to participate in the translocation. Any further participation of the RAG complex in the chromosome 18 MBR breakage process, after AID has created a TG mismatch from the meCG site, is unclear.
In order to evaluate whether the RAG complex is needed to convert the AID-initiated lesions into DSBs, we created a Nalm-6 RAG1−/− clone with homozygous deletion of RAG1 in the same human pre-B cell line, Nalm-6, as we used above (see Fig. S2 in the supplemental material). We confirmed that the Nalm-6 RAG1−/− has no detectable V(D)J recombination activity using the extrachromosomal V(D)J recombination assay that we have described previously (see Table S1 in the supplemental material).
The me-pXC46 substrate was cotransfected with the AID expression vector into Nalm-6 RAG1−/− cells for comparison to the wt Nalm-6. The absence of RAG1 results in a significant reduction in breakage at peak 1 from 15 to 8% (P = 0.04) (Fig. 2B, line 4) and at peak 2 from 13 to 5% (P = 0.03) (Fig. 2B, line 4). The breakage at peak 3 remains the same as unmethylated pXC46 in wt Nalm-6 cells with AID expression. These results indicate that a significant number of MBR breaks at peaks 1 and 2 are RAG dependent.
The results with pXC42 were similar to those for pXC46 in demonstrating a substantial RAG dependence of the breaks (see Fig. S6 in the supplemental material). The results using pXC25 appear to show that breaks at peak 3 are RAG dependent while breaks at peak 1 of pXC25 are not (see Fig. S7 in the supplemental material). This suggests that there may be more than one pathway to achieve double-strand breakage of AID-initiated lesions (see Discussion).
Artemis is important for detection of a subset of the fragile events.
Artemis, when activated by DNA-PKcs, can nick at a wide range of single- to double-stranded transitions. We wondered whether Artemis could provide another pathway by which TG mismatches generated by AID could be converted to DSBs. To test this, the methylated minichromosomal substrates were cotransfected with the AID expression vector, pKY70, into a Nalm-6 Artemis−/− cell line that harbors homozygous deletion of Artemis.
We found that the absence of Artemis results in a significant decrease in breakage at both peaks 1 and 2 within the MBR when me-pXC46 was cotransfected with AID expression vector into the knockout cells. Breakage at peak 1 decreases from 15 to 8% (P = 0.04), and breakage at peak 2 decreases from 13 to 5% (P = 0.02) (Fig. 2B, line 5).
The results with pXC42 and pXC25 also show a very strong dependence of breakage at methylated MBR peaks on Artemis (see Fig. S6 and S7 in the supplemental material). These results compelled us to consider the possibility that Artemis participates in the breakage process at the MBR breaks.
The Artemis:DNA-PKcs complex can nick mismatches and small unpaired regions.
We previously showed that the Artemis:DNA-PKcs complex could nick 10-bp DNA bubble structures (33). If the purified Artemis:DNA-PKcs complex were to act at TG or TGTG double-mismatch sites, then it would need to act at DNA bubble structures that are substantially smaller than any that we had previously studied. To test this, we incubated highly purified Artemis:DNA-PKcs with DNA containing a small heteroduplex structure (bubble). We found that nicking occurs efficiently at 6-bp heteroduplexes and detectably at 3-bp heteroduplexes (Fig. 4). A low level of nicking is also detectable when there is a 1-bp mismatch. These findings suggest that activated Artemis:DNA-PKcs can participate in converting mismatches or larger heteroduplexes into DSBs (see below).
Fig 4.

Artemis:DNA-PKcs complex nicking at bubble structures. Oligonucleotide DNA substrates (with 6-bp, 3-bp, or 1-bp bubble structure in the middle and each arm 15 bp long) were incubated in the absence or presence of Artemis and DNA-PKcs. The diagrams above the gel show the DNA structure and size of the bubble of each substrate. The asterisk indicates the position of the 5′ end labeling. The products of the 6- and 3-bp bubble substrates are indicated by a bracket. The product of the 1-bp bubble substrate was darkened to give better illustration and is more apparent in the right panel.
DISCUSSION
We previously discovered that most human B cell lymphoma translocation breakpoints (outside of the IgH locus) occur in proximity to CpG sites, and we theorized that this could be explained with a model invoking initial AID-initiated lesions (1). Because these B cell translocations occur during V(D)J recombination (the IgH breaks at chromosome 14 require RAGs), our model proposed concurrent expression of both AID and the RAG complex as requirements. Recent studies have documented AID mRNA and AID enzymatic activity in mouse and human pro-B and pre-B cells (12–28). Therefore, AID may potentially play a role in translocations in these B cells. Here we have tested and demonstrated the two central elements, CpG methylation and AID presence, of the model. We found that CpG methylation is important for directing breakage in proximity to the MBR peaks. We also found that the MBR focusing at the peaks requires AID enzymatic activity. The requirement of RAGs and Artemis was also demonstrated here.
Role of CpG methylation at the fragile site.
The studies here are the first to demonstrate that DNA methylation can affect the location of a DNA breakage (recombination) process. The peak localization analysis described above in Results is based on position, irrespective of the sequence. One can also do an independent analysis based purely on motif sequence, regardless of position, and we have done this (Table 1). This motif analysis shows that the CpG motif is the preferred site of breakage when it is methylated and when AID, RAGs and Artemis are present in the cells (Table 1).
Table 1.
Proximity of breaks to the methylated CpG motif
| Condition | Percentage at motif |
Avg distance (bp) to motif |
Pa |
||||
|---|---|---|---|---|---|---|---|
| Actual | Random | Actual | Random | At motif (binomial distribution) | Proximity to motif (Student t test) | Proximity to motif (Mann-Whitney U test) | |
| HhaI me-pXC46 + AID in wt Nalm-6 cells | 10.3 | 5.8 | 19.96 | 28.9 | 0.045012 | 0.0003079 | 2.58286E–05 |
| pXC46 + AID in wt Nalm-6 cells | 14.3 | 5.8 | 31.82 | 28.9 | 0.075120 | 0.63004453 | 0.366175446 |
| HhaI me-pXC46 +muAID in wt Nalm-6 cells | 0.0 | 5.8 | 34.67 | 28.9 | 1 | 0.25076711 | 0.117219833 |
| HhaI me-pXC46 + AID in RAG1−/− Nalm-6 cells | 3.1 | 5.8 | 23.28 | 28.9 | 0.850663 | 0.647175498 | 0.109348552 |
| HhaI me-pXC46 + AID in Artemis−/− Nalm-6 cells | 0.0 | 5.8 | 32.68 | 28.9 | 1 | 0.404722487 | 0.225724957 |
Statistical measures of nGCGC are compared in the pXC46 substrate when enzyme factors are present or absent. GCGC is used rather than CpG because these are the specific sites methylated at peaks 1 and 2 (rather than all CpG sites in the zone). In order to include events in the binomial test for which there was some NHEJ nucleolytic resection during the joining process, we included one extra base pair on the 5′ side of the CCGG or GCGC (designated as “n”). P values are calculated such that “random” is defined as a uniform breakpoint distribution from the most 5′ to the most 3′ breakpoint. Each motif test was analyzed based on three tests (binomial distribution, Student t test, and Mann-Whitney U test). Shaded P values indicate significance in the three tests, and only the shaded group is considered statistically significant for the motif propensity.
We demonstrated previously that the CpG sites within the bcl-2 MBR in primary pre-B cells from healthy normal bone marrow donors are methylated at a high percentage in a typically random manner (1, 34). Therefore, the CpG sites are in a sufficient state to give rise to TG mismatches upon AID action, rather than the UG mismatches that would arise from AID action at unmethylated cytosines. The same partially methylated state is also observed in another common human pre-B cell translocation zone, the major translocation cluster of the CCND1 gene in the bcl-1 t(11;14) translocation (1, 35).
The TG mismatches that would be generated by AID action at meCG sites are repaired back to the CG state at a rate that is 1,000- to 2,500-fold slower than are UG mismatches (6). All Cs are presumably subject to deamination by AID, as biochemical and cellular studies have shown (36–38). However, the rapid repair of UG mismatches relative to TG mismatches results in a marked predilection for human B cell translocations to occur at the meCG sites (35). We have proposed that this is why meCG sites are markedly overrepresented at sites of human B cell chromosomal translocation.
Three pathways for creating double-strand breaks at the CpG sites of the MBR.
Our original model had proposed that the RAG complex might do more than merely cause the breaks at the DH and JH segments at the IgH locus on human chromosome 14 (1). We proposed that the RAG complex might additionally nick one or both strands at the AID-generated TG mismatch. The data here are consistent with this as one possibility, but we now recognize this may be one of three concurrent pathways by which the DSBs at the TG mismatches might arise, and these three are as follows.
First, all cells have the two glycosylases, TDG and MBD4, which are able to act at TG mismatches to create an abasic site at which APE1 can nick 5′ of the abasic position. One possible scenario is that AID acts to create a TG at a methylated CpG site. Then, given the palindromic nature of CpG sites, the meC on the antiparallel strand can be deaminated by AID. The second AID deamination would be even more likely than the initial one because the initial TG mismatch would favor local DNA breathing, thereby providing the single-stranded DNA character required by AID to attack the antiparallel strand (39). With two TG mismatches across from one another, a DSB would result when TDG/MBD4 and APE1 act (Fig. 5, lower left).
Fig 5.
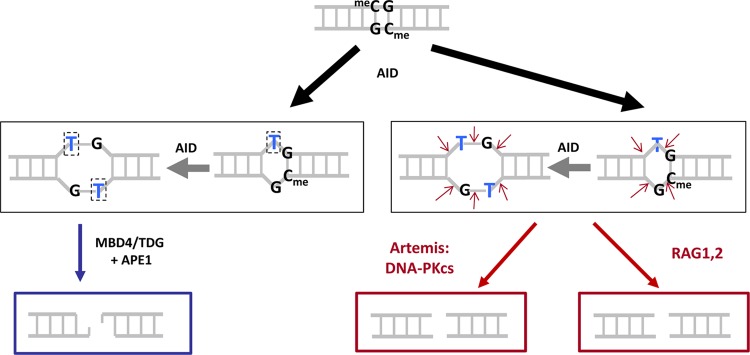
Three mechanisms for generating CpG-type DSBs. Deamination by AID at a methylcytosine within a CpG creates a TG mismatch (black arrow). A second deamination (gray arrow) by AID could occur on the antiparallel strand—an event that would be favored by breathing due to the initial TG mismatch—and this would generate a TG/TG double mismatch or 2-bp bubble structure (middle row). Once the single- or double-mismatch structures are generated, these could be converted to DSBs by any of three mechanisms. (Left) The MBD4 or TDG glycosylases can remove the thymines to generate abasic sites at the boxed Ts, and APE1 could cut 5′ of each of these abasic sites. (Middle) Artemis:DNA-PKcs, upon activation, could nick both strands to create a DSB. (Right) The RAG complex could nick both strands to create a DSB.
A second pathway again would begin with a single AID-generated TG mismatch (at a meCG sites) (Fig. 5, lower right). At this point, the RAG complex could nick both DNA strands by way of its structure-specific nuclease activity (1, 40), which allows it to nick any single- to double-strand transition, including small bubble structures. This is the pathway that we suggested in our original model and which remains plausible (1).
A third pathway again would start with the AID-generated TG mismatch (Fig. 5, lower center). As we have shown in the present study, activated Artemis is able to nick at small DNA bubble structures. Artemis is present in all cells, and activated Artemis (i.e., the Artemis:DNA-PKcs complex) would be present in the nucleus because of the unjoined DH and JH DNA ends at chromosome 14. Hence, Artemis would create a DSB at the bubble structure.
Relative contribution of the three pathways for double-strand break formation.
It is difficult to be certain of the relative contributions of the above three pathways in our experimental system or in patient translocations. When the RAG complex is not present, Artemis will not be as efficiently activated because no breaks at the DH and JH sites will arise. In our system, adventitious breaks due to DNA damage during the transfection process are a likely cause of breaks that are not located at the DH (12RSS) and JH (23RSS) sites, and such breaks also would activate Artemis. When the RAG complex is absent, there is a reduction in most MBR peak-localized breaks. This would seem to indicate that most of the breakage of phosphodiester bonds at the TG mismatches at the MBR peaks are done by RAGs or Artemis.
For pXC42 and pXC25, when Artemis is absent, we see a larger reduction in peak-localized MBR breaks relative to the reduction in the RAG1 knockout, even though we see no obvious change in the total level of breaks on the MBR (left) side of the recombination zone in our substrates (see Fig. S6 and S7 in the supplemental material). This suggests that Artemis is playing a role in the DSB formation at the AID-generated TG mismatches rather than merely carrying out its known roles in hairpin opening or NHEJ end processing (30, 41). Neither hairpin opening nor an NHEJ role would be expected to influence the location of the break within the MBR region, although this is difficult to prove. Therefore, we believe that the larger effect of the Artemis knockout on MBR peak localization (relative to the RAG1-null mutant) suggests that the Artemis pathway for converting the TG mismatches to DSBs may be the most important one of the three pathways for DSB formation (Fig. 5).
Model for simultaneity of the double-strand DNA breaks at chromosomes 14 and 18 in patient translocations.
We have provided evidence here for the two key aspects of our earlier model; namely, a role for CpG methylation and a role for AID action at these CpG sites. Like all other models for the bcl-2 translocation, our earlier model assumed that the chromosome 14 event (failed DH-to-JH joining) and the DSB event at the MBR were independent and had nothing to do with each other causally (1). Based on that earlier model, many DSBs at the MBR (perhaps hundreds or more) would occur before one such event would become entangled with the failed DH-to-JH event on chromosome 14. If this were true, then one might expect to see evidence of NHEJ due to DSBs at the MBR. NHEJ is usually accompanied by 1 to 10 bp of nucleolytic resection, and these microdeletions would be expected to accumulate if the MBR were prone to DSBs at AID-initiated lesions. Such an accumulation would be expected regardless of the rare coincidental simultaneous failed DH-to-JH joining.
We saw no evidence of NHEJ (i.e., 1- to 10-bp microdeletions) at the MBR after introducing exogenous AID in human pre-B cell lines expressing endogenous RAGs (see Table S2 in the supplemental material). Such cell lines have already recombined their DH and JH segments, and so the rare failed DH-to-JH joinings would even be even more rare, and independent DSBs at the MBR would be unaffected by this, according to our earlier model (1). The lack of NHEJ microdeletions within the genomic MBR caused us to consider the possibility that the MBR break is actually dependent on the failed DH-to-JH joining. Our observation here of reduced DSBs in the MBR, when Artemis is lacking, supports this possibility (Fig. 2 and 3; see also Fig. S6 and S7 in the supplemental material).
Our findings now permit a more detailed model proposing that the breaks at the MBR depend on the failed DH-to-JH joining (Fig. 6). Such a dependence would explain why the MBR breaks occur in the same time window as the failed DH-to-JH joining. In the first step of this model, the RAG complex would create hairpin DNA ends at DH and JH, which would activate the Artemis:DNA-PKcs complex. Second, activated Artemis:DNA-PKcs would nick at both strands of the AID-generated TG mismatches and create the DSB at chromosome 18 (Fig. 6). Therefore, the chromosome 14 break would activate the Artemis:DNA-PKcs complex so that it can cause the break at the AID-initiated TG lesions on chromosome 18. Without the action of Artemis:DNA-PKcs, the TG lesions would be repaired back to the original sequence, and this would explain why we do not see any evidence of numerous cycles of break/NHEJ at the MBR region (see Table S2 in the supplemental material). Hence, the new model provides an explanation not only for the mechanism of the DSBs at the MBR but also for the timing between chromosomes 14 and 18. More experiments will be needed to definitively establish the extent to which this new model applies to patient translocations.
Fig 6.

Model for the simultaneity of DSBs at chromosome 14 IgH and the bcl-2 MBR at chromosome 18. After RAG-generated DSBs at the IgH locus, unjoined DH and JH DNA ends activate the Artemis:DNA-PKcs complex. The activated Artemis:DNA-PKcs complexes can nick at TG mismatches. Initiation of repair at TG mismatches (by TDG or MBD4) is known to be 1,000- to 2,500-fold slower than at UG mismatches (by uracil glycosylase). The model here would explain how these long-lived TG mismatch lesions would be vulnerable to conversion to DSBs at the same time that free DH and JH ends exist at the IgH locus on chromosome 14. This also would explain why AID-initiated lesions would not accumulate as NHEJ deletional lesions within the MBR over time. This is because TG mismatches would eventually be corrected back to CG. The only instance when a TG mismatch would be converted to a DSB is under the rare circumstance when activated Artemis:DNA-PKcs at a DH or a JH end comes close to the TG lesion at chromosome 18.
Limitations of this study.
It is notable that many key aspects of the CpG-dependent, methylation-dependent, and AID-dependent localization of breakage can be recapitulated on replicating minichromosomes in a human pre-B cell line. There are many facets of this topic that we have not yet addressed or which are suboptimally examined using this system. First, we have only been able to CpG methylate a subset of the peaks in each substrate. For the methylated peaks, the localization is significant. However, perhaps because not all three peaks are methylated at once, we were not able to reproduce the sharp rises at the peaks and valleys between peaks that we noted for the distribution of the breaks in the patients. In the neoplasm of each patient, the methylation status cannot ever be known at the time of the translocation. In our analysis of normal primary human bone marrow pre-B cells, the pattern of DNA methylation for the three peaks and the lateral regions was heterogeneous (1), as it is for most genomic locations in primary human cells (34). The heterogeneity applies from one pre-B cell to the next, even from the same individual (1). It is possible that with appropriate combinations and ratios of differently methylated substrates, we could more precisely reproduce the fine structure of the peaks and valleys of the observed patient distribution.
There are several other suboptimal aspects of our approach. Among these is the use of transiently introduced substrates. We initially invested substantial time to use chromosomally integrated substrates in human pre-B cell lines. However, the DNA methylation status of the substrates always changed upon integration and did so in a highly variable manner, primarily with the loss of most or all of the methylation or the gain of new methylation. This does not occur with the extrachromosomal substrates (34, 42).
The PCR complexities that we experienced in the present study cannot be avoided using integrated substrates or analysis of the natural MBR genomic site in human cells. For detection of rare events, intrinsic system biases seem inescapable, but we have been careful to document them in our system.
An advantage of our system here is that the RAG expression is natural and consists of untagged full-length RAGs. Use of stably integrated substrates would almost certainly require either exogenous and/or fusion or tagged versions of the RAG proteins in order to limit the action of RAGs on the substrate until AID is introduced, and such a system would be unnatural in the level or sequence of the RAG proteins. Despite the acknowledged limitations of our approach, the observation of CpG and AID methylation dependence for breakage is a major step in the understanding of these translocations.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from NIH to M.R.L. Use of the USC Norris Cancer Center cores was supported by grant P30CA014089. X.C. was supported in part by grant T32CA09320 as a postdoctoral trainee.
Footnotes
Published ahead of print 21 December 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.01436-12.
REFERENCES
- 1. Tsai AG, Lu H, Raghavan SC, Muschen M, Hsieh CL, Lieber MR. 2008. Human chromosomal translocations at CpG sites and a theoretical basis for their lineage and stage specificity. Cell 135: 1130–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Boboila C, Alt FW, Schwer B. 2012. Classical and alternative end-joining pathways for repair of lymphocyte-specific and general DNA double-strand breaks. Adv. Immunol. 116: 1–49 [DOI] [PubMed] [Google Scholar]
- 3. Gostissa M, Alt FW, Chiarle R. 2011. Mechanisms that promote and suppress chromosomal translocations in lymphocytes. Annu. Rev. Immunol. 29: 319–350 [DOI] [PubMed] [Google Scholar]
- 4. Hakim O, Resch W, Yamane A, Klein I, Kieffer-Kwon KR, Jankovic M, Oliveira T, Bothmer A, Voss TC, Ansarah-Sobrinho C, Mathe E, Liang G, Cobell J, Nakahashi H, Robbiani DF, Nussenzweig A, Hager GL, Nussenzweig MC, Casellas R. 2012. DNA damage defines sites of recurrent chromosomal translocations in B lymphocytes. Nature 484: 69–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang Y, McCord RP, Ho YJ, Lajoie BR, Hildebrand DG, Simon AC, Becker MS, Alt FW, Dekker J. 2012. Spatial organization of the mouse genome and its role in recurrent chromosomal translocations. Cell 148: 908–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tsai AG, Lieber MR. 2010. Mechanisms of chromosomal rearrangement in the human genome. BMC Genomics 11(Suppl 1): S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mahowald GK, Baron JM, Sleckman BP. 2008. Collateral damage from antigen receptor gene diversification. Cell 135: 1009–1012 [DOI] [PubMed] [Google Scholar]
- 8. Shimazaki N, Tsai AG, Lieber MR. 2009. H3K4me3 Stimulates V(D)J RAG complex for both nicking and hairpinning in trans in addition to tethering in cis: implications for translocations. Mol. Cell 34: 535–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tsai AG, Engelhart AE, Hatmal MM, Houston SI, Hud NV, Haworth IS, Lieber MR. 2009. Conformational variants of duplex DNA correlated with cytosine-rich chromosomal fragile sites. J. Biol. Chem. 284: 7157–7164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tsai AG, Lu Z, Lieber MR. 2010. The t(14;18)(q32;q21)/IGH-MALT1 translocation in MALT lymphomas is a CpG-type translocation, but the t(11;18)(q21;q21)/API2-MALT1 translocation in MALT lymphomas is not. Blood 115: 3640–3642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tsai AG, Yoda A, Weinstock DM, Lieber MR. 2010. t(X;14)(p22;q32)/t(Y;14)(p11;q32) CRLF2-IGH translocations from human B-lineage ALLs involve CpG-type breaks at CRLF2, but CRLF2/P2RY8 intrachromosomal deletions do not. Blood 116: 1993–1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Edry E, Koralov SB, Rajewsky K, Melamed D. 2007. Spontaneous class switch recombination in B cell lymphopoiesis generates aberrant switch junctions and is increased after VDJ rearrangement. J. Immunol. 179: 6555–6560 [DOI] [PubMed] [Google Scholar]
- 13. Feldhahn N, Henke N, Melchior K, Duy C, Soh BN, Klein F, von Levetzow G, Giebel B, Li A, Hofmann WK, Jumaa H, Muschen M. 2007. Activation-induced cytidine deaminase acts as a mutator in BCR-ABL1-transformed acute lymphoblastic leukemia cells. J. Exp. Med. 204: 1157–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Han JH, Akira S, Calame K, Beutler B, Selsing E, Imanishi-Kari T. 2007. Class switch recombination and somatic hypermutation in early mouse B cells are mediated by B cell and Toll-like receptors. Immunity 27: 64–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hasan M, Polic B, Bralic M, Jonjic S, Rajewsky K. 2002. Incomplete block of B cell development and immunoglobulin production in mice carrying the muMT mutation on the BALB/c background. Eur. J. Immunol. 32: 3463–3471 [DOI] [PubMed] [Google Scholar]
- 16. Hsu HC, Wu Y, Yang P, Wu Q, Job G, Chen J, Wang J, Accavitti-Loper MA, Grizzle WE, Carter RH, Mountz JD. 2007. Overexpression of activation-induced cytidine deaminase in B cells is associated with production of highly pathogenic autoantibodies. J. Immunol. 178: 5357–5365 [DOI] [PubMed] [Google Scholar]
- 17. Kitamura D, Roes J, Kuhn R, Rajewsky K. 1991. A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature 350: 423–426 [DOI] [PubMed] [Google Scholar]
- 18. Kuraoka M, Holl TM, Liao D, Womble M, Cain DW, Reynolds AE, Kelsoe G. 2011. Activation-induced cytidine deaminase mediates central tolerance in B cells. Proc. Natl. Acad. Sci. U. S. A. 108: 11560–11565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kuraoka M, Liao D, Yang K, Allgood SD, Levesque MC, Kelsoe G, Ueda Y. 2009. Activation-induced cytidine deaminase expression and activity in the absence of germinal centers: insights into hyper-IgM syndrome. J. Immunol. 183: 3237–3248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kuraoka M, McWilliams L, Kelsoe G. 2011. AID expression during B-cell development: searching for answers. Immunol. Res. 49: 3–13 [DOI] [PubMed] [Google Scholar]
- 21. Macpherson AJ, Lamarre A, McCoy K, Harriman GR, Odermatt B, Dougan G, Hengartner H, Zinkernagel RM. 2001. IgA production without mu or delta chain expression in developing B cells. Nat. Immunol. 2: 625–631 [DOI] [PubMed] [Google Scholar]
- 22. Mao C, Jiang L, Melo-Jorge M, Puthenveetil M, Zhang X, Carroll MC, Imanishi-Kari T. 2004. T cell-independent somatic hypermutation in murine B cells with an immature phenotype. Immunity 20: 133–144 [DOI] [PubMed] [Google Scholar]
- 23. Meffre E. 2011. The establishment of early B cell tolerance in humans: lessons from primary immunodeficiency diseases. Ann. N. Y. Acad. Sci. 1246: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Meyers G, Ng YS, Bannock JM, Lavoie A, Walter JE, Notarangelo LD, Kilic SS, Aksu G, Debre M, Rieux-Laucat F, Conley ME, Cunningham-Rundles C, Durandy A, Meffre E. 2011. Activation-induced cytidine deaminase (AID) is required for B-cell tolerance in humans. Proc. Natl. Acad. Sci. U. S. A. 108: 11554–11559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Milili M, Fougereau M, Guglielmi P, Schiff C. 1991. Early occurrence of immunoglobulin isotype switching in human fetal liver. Mol. Immunol. 28: 753–761 [DOI] [PubMed] [Google Scholar]
- 26. Muto T, Muramatsu M, Taniwaki M, Kinoshita K, Honjo T. 2000. Isolation, tissue distribution, and chromosomal localization of the human activation-induced cytidine deaminase (hAID) gene. Genomics. 68: 85–88 [DOI] [PubMed] [Google Scholar]
- 27. Qin H, Suzuki K, Nakata M, Chikuma S, Izumi N, THuong le, Maruya M, Fagarasan S, Busslinger M, Honjo T, Nagaoka H. 2011. Activation-induced cytidine deaminase expression in CD4+ T cells is associated with a unique IL-10-producing subset that increases with age. PLoS One 6: e29141 doi:10.1371/journal.pone.0029141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ueda Y, Liao D, Yang K, Patel A, Kelsoe G. 2007. T-independent activation-induced cytidine deaminase expression, class-switch recombination, and antibody production by immature/transitional 1 B cells. J. Immunol. 178: 3593–3601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Raghavan SC, Hsieh C-L, Lieber MR. 2005. Both V(D)J coding ends but neither signal end can recombine at the bcl-2 major breakpoint region, and the rejoining is ligase IV dependent. Mol. Cell. Biol. 25: 6475–6484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kurosawa A, Koyama H, Takayama S, Miki K, Ayusawa D, Fujii M, Iiizumi S, Adachi N. 2008. The requirement of Artemis in double-strand break repair depends on the type of DNA damage. DNA Cell Biol. 27: 55–61 [DOI] [PubMed] [Google Scholar]
- 31. Raghavan SC, Swanson PC, Wu X, Hsieh C-L, Lieber MR. 2004. A non-B-DNA structure at the bcl-2 major break point region is cleaved by the RAG complex. Nature 428: 88–93 [DOI] [PubMed] [Google Scholar]
- 32. Jaeger U, Bocskor S, Le T, Mitterbauer G, Bolz I, Chott A, Kneba A, Mannhalter C, Nadel B. 2000. Follicular lymphomas BCL-2/IgH junctions contain templated nucleotide insertions: novel insights into the mechanism of t(14;18) translocation. Blood 95: 3520–3529 [PubMed] [Google Scholar]
- 33. Ma Y, Schwarz K, Lieber MR. 2005. The Artemis:DNA-PKcs endonuclease can cleave gaps, flaps, and loops. DNA Repair 4: 845–851 [DOI] [PubMed] [Google Scholar]
- 34. Tsai AG, Chen DM, Lin M, Hsieh JC, Okitsu CY, Taghva A, Shibata D, Hsieh CL. 2012. Heterogeneity and randomness of DNA methylation patterns in human embryonic stem cells. DNA Cell Biol. 31: 893–907. [DOI] [PubMed] [Google Scholar]
- 35. Greisman HA, Lu Z, Tsai AG, Greiner TC, Yi HS, Lieber MR. 2012. IgH partner breakpoint sequences provide evidence that AID initiates t(11;14) and t(8;14) chromosomal breaks in mantle cell and Burkitt lymphomas. Blood 120: 2864–2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. DiNoia JM, Neuberger MS. 2007. Molecular mechanisms of antibody somatic hypermutation. Annu. Rev. Biochem. 76: 1–22 [DOI] [PubMed] [Google Scholar]
- 37. Pham P, Bransteitter R, Petruska J, Goodman MF. 2003. Processive AID-catalyzed cytosine deamination on single-stranded DNA stimulates somatic hypermutation. Nature 424: 103–107 [DOI] [PubMed] [Google Scholar]
- 38. Yu K, Huang FT, Lieber MR. 2004. DNA substrate length and surrounding sequence affect the activation induced deaminase activity at cytidine. J. Biol. Chem. 279: 6496–6500 [DOI] [PubMed] [Google Scholar]
- 39. Bransteitter R, Pham P, Scharff MD, Goodman MF. 2003. Activation-induced cytidine deaminase deaminates deoxycytidine on single-stranded DNA but requires the action of RNase. Proc. Natl. Acad. Sci. U. S. A. 100: 4102–4107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Raghavan SC, Swanson PC, Ma Y, Lieber MR. 2005. Double-strand break formation by the RAG complex at the Bcl-2 MBR and at other non-B DNA structures in vitro. Mol. Cell. Biol. 25: 5904–5919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Riballo E, Kuhne M, Rief N, Doherty A, Smith GCM, Recio M-J, Reis C, Dahm K, Fricke A, Kempler A, Parker AR, Jackson SP, Gennery A, Jeggo PA, Lobrich M. 2004. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to γ-H2AX foci. Mol. Cell 16: 715–724 [DOI] [PubMed] [Google Scholar]
- 42. Okitsu CY, Hsieh CL. 2007. DNA methylation dictates histone H3K4 methylation. Mol. Cell. Biol. 27: 2746–2757 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.