

Abstract
Huntington's disease (HD) is a neurodegenerative disease caused by the expansion of a CAG repeat in the Huntingtin (HTT) gene. Abnormal regulation of the cyclic AMP (cAMP)/protein kinase A (PKA) pathway occurs during HD progression. Here we found that lower PKA activity was associated with proteasome impairment in the striatum for two HD mouse models (R6/2 and N171-82Q) and in mutant HTT (mHTT)-expressing striatal cells. Because PKA regulatory subunits (PKA-Rs) are proteasome substrates, the mHTT-evoked proteasome impairment caused accumulation of PKA-Rs and subsequently inhibited PKA activity. Conversely, activation of PKA enhanced the phosphorylation of Rpt6 (a component of the proteasome), rescued the impaired proteasome activity, and reduced mHTT aggregates. The dominant-negative Rpt6 mutant (Rpt6S120A) blocked the ability of a cAMP-elevating reagent to enhance proteasome activity, whereas the phosphomimetic Rpt6 mutant (Rpt6S120D) increased proteasome activity, reduced HTT aggregates, and ameliorated motor impairment. Collectively, our data demonstrated that positive feedback regulation between PKA and the proteasome is critical for HD pathogenesis.
INTRODUCTION
Huntington's disease (HD) is an autosomal dominant neurodegenerative disease caused by expansion of a CAG repeat in the Huntingtin (HTT) gene. The normal HTT gene has 35 or fewer CAG repeats in its N-terminal region, whereas expansion of the repeat beyond 35 units confers toxicity to the Huntingtin protein (HTT) and evokes HD pathogenesis (1). The major clinical presentations of HD include chorea, cognitive decline, psychiatric symptoms, and a systemic metabolic defect (2). Selective neuronal loss occurs in multiple brain regions, particularly in the striatum and cortex. Since a great number of cellular machineries (including transcription, vesicle transport, molecular chaperones, and the ubiquitin-proteasome system [UPS]) are compromised by the expression of mutant HTT (mHTT) (3), effective removal of mHTT has become one of the most challenging aspects of drug development for HD.
The UPS, which degrades misfolded and/or damaged proteins in eukaryotes, is the major regulatory proteolytic pathway. It regulates many essential cellular processes, including transcription, translation, DNA repair, chromatin remodeling, cell survival, signal transduction, protein trafficking, and synaptic plasticity (4). A recent study demonstrated that AAA-ATPase subunits are critical for cell-type-specific regulation of UPS activity (5). Interestingly, UPS activity is under metabolic control. Posttranslational modifications, such as O-GlcNacylation and phosphorylation of 19S subunits, affect the proteolytic activity of the UPS (6). In addition, impaired functioning of the UPS is believed to contribute to the pathogenic processes of many neurodegenerative diseases, including Alzheimer's disease, Parkinson's disease (PD), and HD. Recent studies revealed that the UPS might be impaired in HD mouse models and patients (7, 8). Intriguingly, although the degradation of polyubiquitin-tagged proteins decreased, the ubiquitination process was not affected in cells expressing mHTT (9). The results of those studies suggest that regulation of the UPS is complex and might occur in multiple steps.
Previous studies demonstrated that components involved in regulating the cyclic AMP (cAMP)/protein kinase A (PKA) pathway are abnormally regulated during HD progression (10–12). In the present study, we report that the mHTT-evoked suppression of proteasome activity was associated with accumulation of PKA regulatory subunits and reduced PKA activity in the striatum for two HD mouse models (R6/2 and N181-82Q) and in striatal cells expressing mHTT. Activation of PKA increased proteasome activity via phosphorylation of Rpt6 (a component of the 19S complex) at the serine 120 residue (S120), which in turn led to enhanced proteasome activity, suppressed aggregate formation, and improved motor function in R6/2 mice. The results of our study suggest that feedback regulation between PKA and the proteasome occurs during HD disease progression and significantly contributes to HD pathogenesis. In addition, both the UPS and PKA are promising drug targets in HD.
MATERIALS AND METHODS
Reagents.
All reagents were purchased from Sigma (St. Louis, MO) except where otherwise specified. Dulbecco's modified Eagle's medium (DMEM) and fetal bovine serum (FBS) were purchased from Invitrogen (Carlsbad, CA).
Animals and CGS21680 (CGS) administration.
Male R6/2 mice (13), N171-82Q mice (14), and littermate controls were originally obtained from Jackson Laboratories (Bar Harbor, ME) and mated to female control mice (B6CBAFI/J). Offspring were identified by a PCR genotyping technique with genomic DNA extracted from tail tissues using primers located in the transgene (5′-CCGCTCAGGTTCTGCTTTTA-3′ and 5′-GGCTGAGGAAGCTGAGGAG-3′ for R6/2 and A5′-TGGCGACCCTGGAAAAGCTG-3′ and 5′-GGCTGAGGAAGCTGAGGAG-3′ for N171-82Q) to determine the numbers of CAG repeats, which were approximately 190 and 82 for R6/2 and N171-82Q mice, respectively. In total, 36 R6/2 transgenic mice and 18 wild-type (WT) littermate controls and 6 N171-82Q mice and 6 WT littermate controls were used in this study. Animals were housed at the Institute of Biomedical Sciences Animal Care Facility, Academia Sinica, under a 12-h light/dark cycle. A daily intraperitoneal injection of CGS (an agonist of the A2A adenosine receptor, A2AR; 2.5 μg/g body weight) or vehicle was given to mice between 1300 and 1800 h from the age of 7 weeks for 5 weeks as described earlier (15). Animal experiments were performed under protocols approved by the Academia Sinica Institutional Animal Care and Utilization Committee (Taipei, Taiwan).
Cell culture and transfection.
The ST14A rat striatal progenitor cell line (16) was a generous gift from E. Cattaneo (University of Milano, Milan, Italy), and was cultured in high-glucose DMEM containing 10% FBS, 2 mM l-glutamine, and 1% penicillin-streptomycin (Invitrogen GibcoBRL, Carlsbad, CA) at 33°C under 5% CO2. Cells were transfected using Lipofectamine 2000 (Invitrogen) based on the manufacturer's protocol and cultured for another 72 h in the presence of the indicated reagents. Primary striatal neurons were prepared from R6/2 and control mice on embryonic day 18 as described previously (17) and grown in Neurobasal medium supplemented with B27 (Invitrogen, San Diego, CA), penicillin (100 units/ml), streptomycin (100 μg/ml), glutamate (12.5 μM), and l-glutamine (0.5 mM). At 7 days in vitro (DIV), cells were transfected using Optifect (Invitrogen), following the manufacturer's instructions.
Immunohistochemistry.
For immunostaining and quantitation of mHTT aggregates, coronal serial sections (20 μm) containing the striatum (interaural, 5.34 mm/bregma, 1.54 mm; to interaural, 3.7 mm/bregma, −0.1 mm) were immunohistochemically stained as described previously (18). Briefly, brain sections were blocked with normal goat serum for 1 h and stained with an anti-HTT antibody (EM48, 1: 500) at 4°C overnight, followed by incubation with goat anti-mouse IgG conjugated to Alexa Fluor 568 for 2 h at room temperature. Nuclei were stained with Hoechst 33258 stain. Patterns of immunostaining were analyzed with a laser confocal microscope (LSM510; Carl Zeiss MicroImaging). Twenty images of serial Z-axis sections (1 μm per section) were recorded and then merged using the LSM 5 Image Examiner software program (Carl Zeiss MicroImaging). Sizes of the mHTT aggregates were analyzed using the ImageJ software program (http://rsbweb.nih.gov/ij/) (Research Services Branch of the National Institute of Mental Health, Bethesda, MD). Five different brain sections of each animal were assessed.
Western blot assays.
Equal amounts of protein were separated by SDS-PAGE using 10% polyacrylamide gels according to the method of Laemmli (19). The resolved proteins were electroblotted onto Immobilon polyvinylidene difluoride (PVDF) membranes (Millipore, Bedford, MA). Membranes were blocked with 5% skim milk or 3% bovine serum albumin (BSA) in phosphate-buffered saline (PBS) and incubated with an anti-Htt antibody (EM48, 1:500; Chemicon International, Temecula, CA), an antiubiquitin antibody (1:2,000; DakoCytomation, Glostrup, Denmark), an anti-Rpt6 antibody (1:1,000; Santa Cruz Biotechnology, Santa Cruz, CA), an anti-phospho-(Ser/Thr)-PKA substrate antibody (1:1,000; Cell Signaling), an anti-GluR1 antibody (1:1,000; Chemicon International), an anti-PKA-R2β antibody (1:1,000; BD), or an antiactin antibody (1:5,000; Chemicon International) at 4°C overnight, followed by the corresponding secondary antibody for 1 h at room temperature (RT). Immunoreactive bands were detected by enhanced chemiluminescence (ECL; Pierce, Rockford, IL) and recorded using Kodak XAR-5 film (Rochester, NY).
Filter retardation assay.
Sodium dodecyl sulfate (SDS)-insoluble mutant Htt aggregates were detected and quantified using a filter retardation assay as described previously (20). Blots were blocked with 5% skim milk in PBS and incubated with an anti-Htt antibody (1:100) or an anti-Rpt6 antibody (1:1,000) at 4°C overnight, followed by the corresponding secondary antibody for 1 h at RT. Immunoreactive bands were detected by ECL (Pierce) and recorded using Kodak XAR-5 film.
Preparation of synaptosome-enriched fractions.
Synaptosome-enriched fractions were prepared as detailed elsewhere (21). Briefly, striatal tissues were resuspended in ice-cold homogenization buffer (0.32 M sucrose, 15 mM Tris-HCl [pH 8.0], 60 mM KCl, 15 mM NaCl, 5 mM EDTA, 1 mM EGTA, 1× PhosSTOP phosphatase inhibitor cocktail (Roche, Basel, Switzerland), 10 μM phenylmethylsulfonyl fluoride [PMSF], and 2 mM ATP) and homogenized by 15 Dounce strokes. The homogenate was centrifuged at 1,300 × g for 10 min to remove unbroken tissue clumps and nuclei. The supernatant was centrifuged at 10,000 × g for 10 min to obtain the synaptosome-enriched fraction. The protein concentration was measured using the Bio-Rad protein assay reagent (Hercules, CA,).
Constructs.
The pcDNA3.1-Htt-(Q)25-hrGFP and pcDNA3.1-Htt-(Q)109-hrGFP constructs, encoding an N-terminal fragment of Htt with the indicated number of poly(Q) residues fused to humanized renilla green fluorescent protein (hrGFP), were created as described earlier (10). Expression constructs of Rpt6-myc and Rpt6S120A-myc (with Rpt6 phosphorylation eliminated) were gifts from Jan-Jong Hung (National Cheng Kung University, Tainan, Taiwan) (22). A phosphomimetic mutant, Rpt6S120D, was created by a two-step PCR technique as described previously (23) using the primers 5′-AAGCTTATGGCGCTTGACGGA-3′, 5′-TGCAGAGTGTAGTCGTCATT-3′, 5′-AATGACGACTACACTCTGCA-3′, and 5′-AAGCTTTTGCTTCCATAATTTCTT-3′ and standard molecular biological techniques. Mutation of S120D was confirmed by DNA sequencing. The complementary (c)DNA encoding full-length mouse PKA-R2β was generated by PCR amplification using the primers 5′-CACGGGCAGGATGAGCATCGAG-3′ and 5′-TGCAGTGGGCTCAACAATATCCATGTTTG-3′ from mouse brain cDNA and cloned into the pcDNA3.1 vector (Invitrogen) by TA cloning, according to the manufacturer's instructions.
cAMP assay.
The intracellular cAMP content was determined as described before with slight modifications (24). ST14A cells (3 × 105 per 35-mm plate) were washed twice with Ca2+-free Locke's solution (150 mM NaCl, 5.6 mM KCl, 5 mM glucose, 1 mM MgCl2, and 10 mM HEPES [pH 7.4]) containing 0.5 mM isobutylmethylxanthine (IBMX) (Sigma) and were then treated with the indicated reagent(s) for 20 min at 25°C. The cAMP content was assayed using the 125I-cAMP assay system (Amersham Biosciences, Little Chalfont, Buckinghamshire, United Kingdom).
PKA activity assay.
Tissues or cells were lysed using ice-cold lysis buffer (20 mM Tris-HCl [pH 8.0], 0.5% [vol/vol] Nonidet P-40, 150 mM NaCl, 1 mM EDTA, 1× PhosSTOP phosphatase inhibitor cocktail, and 10 μM PMSF). PKA activity was determined using a commercially available kit (Millipore) according to the manufacturer's instructions.
Proteasome activity assay.
The chymotrypsin-like activity of the proteasome was determined using a specific proteasome substrate (succinyl [suc]-Leu-Leu-Val-Tyr-7-amino-4-methyl coumarin [AMC]; Sigma-Aldrich, St. Louis, MO) as described earlier (20). Total lysates or the synaptosome-enriched fraction (10 μg) was incubated with the substrate (40 μM) in 100 μl of proteasome assay buffer (0.05 M Tris-HCl [pH 8.0], 0.5 mM EDTA, 1 mM ATP, and 1 mM dithiothreitol [DTT]) at 37°C for 60 min, where the relationship between the incubation time and product formation remained linear. The fluorescence of the released AMC was detected using a fluorescence microplate reader system (Molecular Devices, Sunnyvale, CA) at 380-nm excitation and 460-nm emission wavelengths.
Immunoprecipitation.
ST14A cells were transfected with the indicated plasmid(s) for 72 h. Cells were lysed using ice-cold lysis buffer (20 mM Tris-HCl [pH 8.0], 0.5% [vol/vol] Nonidet P-40, 150 mM NaCl, 1 mM EDTA, 1× PhosSTOP phosphatase inhibitor cocktail, and 10 μM PMSF). The anti-Rpt6 antibody (10 μl) was incubated with 50 μl ExactaCruz C IP matrix (Santa Cruz Biotechnology) for 1 h at 4°C on a rolling wheel, washed using ice-cold lysis buffer, and incubated with the cell lysate (1 mg) for 1 h at 4°C on a rolling wheel. After extensive washing, the resultant immunoprecipitation complexes were analyzed by SDS-PAGE and Western blot assays as described above.
AAV vector production.
The coding sequence of Rpt6S120A and Rpt6S120D was generated by PCR amplification using the primers 5′-GCGGCCGCATGGCGCTTGACGGACC-3′ and 5′-GCGGCCGCTTACTTCCATAATTTCTTG-3 from pcDNA3.1-Rpt6S120A and Rpt6S120D, respectively. The Rpt6 amplicon was then restriction digested with NotI and cloned into the adeno-associated viral (AAV) construct (pXX-UF1-CB-IRES-EGFP). AAV8 was generated by transient transfection of HEK293 cells with the target plasmid along with helper plasmids as detailed by Xiao et al. (25). Titers of AAVs were assessed by determining the number of packaged vector genomes using a quantitative PCR (qPCR) method as described elsewhere (26). Before the injection, all AAV vectors were adjusted to a titer of 1011/μl.
Stereotaxic injection of AAV.
Adult mice (5 weeks old) were anesthetized by an intraperitoneal injection of ketamine (500 mg/kg) and xylazine (100 mg/kg) and placed in a stereotaxic frame (Stoelting, Wood Dale, IL). AAVs were bilaterally injected into the striatum at 3 different locations (1 μl per location; 200 nl/min) per side using a 10-μl Hamilton syringe (Reno, NV). The respective target anterior-posterior (AP), medial-lateral (ML), and dorsoventral coordinates were 0.5, ±2, and −2.7 mm relative to the bregma. After the injection, the needle was left in place for 10 min and then withdrawn slowly. Seven weeks after the injection, animals were sacrificed and perfused with 4% (wt/vol) paraformaldehyde. After sucrose sedimentation, the brains were sectioned, and an immunohistochemical analysis was performed.
Rotarod performance.
Fore- and hindlimb motor coordination was assessed using a rotarod apparatus assay (model 7600; Ugo Basile, Comerio, Italy) at a constant speed of 12 rpm over a period of 2 min, and the latency to falling off the rotarod was recorded. All mice were tested 3 times per week. Each mouse received 3 trials for a maximum of 2 min for each trial.
Beam walk.
Motor coordination was assessed by measuring the ability of mice to traverse a narrow beam to reach an enclosed box. The beam consisted of a 1-m round piece of wood with a 17- or 11-mm diameter. The 17-mm-diameter round beam was used for training the first time, and the latency of a mouse in traversing the 11-mm-diameter beam was recorded once weekly.
Limb clasping.
Limb clasping during tail suspension is a special phenotype and has been used to study the disease progression in HD transgenic mice. In this study, mice were suspended by the tail for 30 s, and limb clasping was scored on a scale of 0 to 2, with 0 being normal, 1 meaning clasping by either the fore- or hindlimbs, and 2 meaning clasping by both the fore- and hindlimbs.
RESULTS
Aberrant regulation of proteasome activity in synaptosomes of HD mice was previously documented (8). Such deregulation of the activity of proteasomes in synapses might alter expression profiles of synaptic proteins and contribute to abnormal synaptic transmission observed in HD mice (27). Consistent with these findings, we found that chymotrypsin-like activity of the proteasome in the striatal synaptosome fraction of two HD mouse models (R6/2 and N171-82Q) was lower than that of wild-type (WT) mice (Fig. 1A and B).
Fig 1.
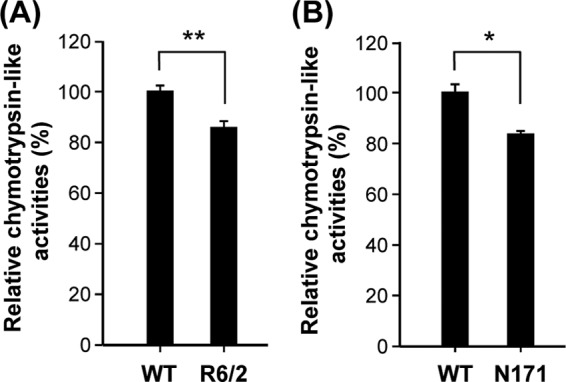
Mutant Huntingtin (Htt) suppressed proteasome activity in synapses of the striatum of R6/2 and N171-82Q mice. Chymotrypsin-like activities of the proteasome in the synaptic fraction of the striatum of 12-week-old R6/2 mice and littermate controls (A) or 16-week-old N171-82Q mice and littermate controls (B) were assessed as described in Materials and Methods. Data are presented as the means ± SEM in each group. ∗, P < 0.05; ∗∗, P < 0.01 (by one-way analysis of variance [ANOVA]).
Among substrates of the proteasome, we were interested in the regulatory subunits of PKA (PKA-Rs) because cAMP signaling is deregulated in HD (11, 12, 15). The PKA holoenzyme consists of two PKA-Rs and two catalytic subunits and is catalytically inactive in the absence of cAMP. In the presence of cAMP, cAMP-bound PKA-Rs disassociate from the catalytic subunits and are degraded by the proteasome (4). Accumulation of PKA-Rs promotes the formation of PKA holoenzyme, reduces the amounts of free PKA catalytic subunits, and therefore impairs PKA activity. Since PKA-Rs are substrates of the proteasome (28), we hypothesized that lower proteasome activity in cells expressing mHTT causes accumulation of PKA-Rs, which inhibits PKA activity. To confirm that PKA-Rs are substrates of the UPS, a striatal progenitor cell line (ST14A) was treated with an inhibitor (MG132) of the proteasome. As shown in Fig. 2A, MG132 dose-dependently enhanced the expression of a regulatory subunit (PKA-R2β) of PKA in ST14A cells. Levels of PKA-R2β in the striatal synaptosome fractions of two HD mouse models (R6/2 and N171-82Q) were also higher than those observed in WT mice (Fig. 2B and C). Similarly, the level of PKA-R2β in ST14A cells expressing CAG-expanded mHTT (Q109-HTTEx1-GFP [109Q]) was also higher than that detected in cells expressing the control construct (Q25-HTTEx1-GFP [25Q]) (Fig. 2D). To assess whether expression levels of PKA-Rs are critical for PKA activity, an expression construct for PKA-R2β was transfected into ST14A cells. As predicted, overexpression of PKA-R2β markedly decreased the activity of PKA (Fig. 2E). Consistent with this observation, the activity of PKA in the striatal synaptosomes of two HD mouse models (R6/2 and N171-82Q), which contained higher levels of PKA-R2β (Fig. 2B and C), were lower than those observed in their littermate controls (Fig. 3).
Fig 2.
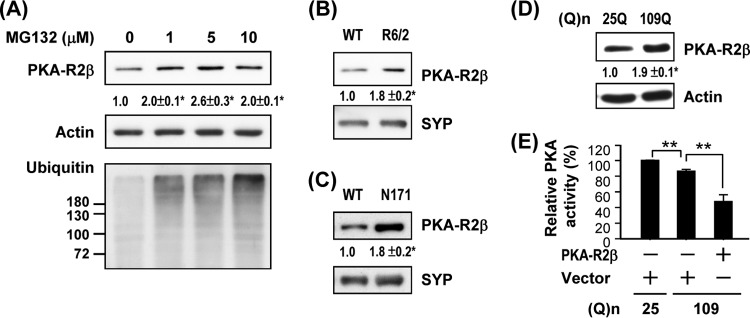
Impairment of proteasome activity caused accumulation of PKA regulatory subunits and suppression of PKA activity. (A) ST14A cells were treated with vehicle or MG132 for 6 h. Lysates collected for the indicated condition were used to determine the expression of PKA-R2β by a Western blot analysis. Data points represent the means ± SEM for three independent experiments. Synaptic fractions of the striatum of 12-week-old R6/2 mice and littermate controls (B) or 16-week-old N171-82Q mice and littermate controls (C) were used to determine the expression of PKA-R2β by a Western blot analysis. (D) ST14A cells were transfected with pcDNA3.1-Htt-(Q)25-hrGFP or pcDNA3.1-Htt-(Q)109-hrGFP for 72 h. Lysates collected for the indicated condition were used to determine the expression of PKA-R2β by a Western blot analysis. (E) ST14A cells were cotransfected with pcDNA3.1-Htt-(Q)25-hrGFP or pcDNA3.1-Htt-(Q)109-hrGFP and pcDNA3.1 or pcDNA3.1-PKAR2β for 72 h. Lysate collected for the indicated condition was used to determine PKA activity. Data are presented as the means ± SEM for 3 experiments. ∗, P < 0.05; ∗∗, P < 0.01 (by one-way ANOVA).
Fig 3.
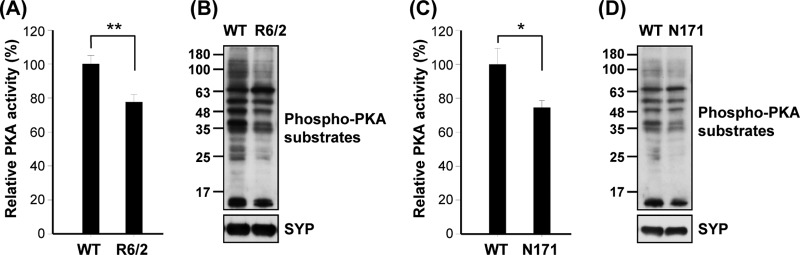
Mutant Huntingtin (Htt) downregulated PKA activity in vivo. PKA activities in the synaptic fraction of the striatum of 12-week-old R6/2 mice and littermate controls (A) or 16-week-old N171-82Q mice and littermate controls (C) were assessed as described in Materials and Methods. Data are presented as the means ± SEM for each group. ∗, P < 0.05; ∗∗, P < 0.01 (by one-way ANOVA). Synaptic fractions of the striatum of 12-week-old R6/2 mice and littermate controls (B) or 16-week-old N171-82Q mice and littermate controls (D) were used to determine the expression of phospho-PKA substrates by a Western blot analysis.
Because proteasome activity can be regulated by PKA (29), we attempted to identify the downstream target of PKA that mediates activation of the proteasome. Among subunits of the 26S proteasome, we were particularly interested in Rpt6 (a subunit of the 19S proteasome) because phosphorylation of Rpt6 at Ser120 by PKA enhances the activity of the proteasome (29). To evaluate the involvement of PKA-mediated phosphorylation of Rpt6 in regulating proteasome activity, ST14A cells were transfected with Q25-HTTEx1-GFP or Q109-HTTEx1-GFP and treated with an agonist (CGS) of the A2A adenosine receptor (A2AR) and/or an antagonist [8-(3-chlorostyryl)-caffeine (CSC)] of the A2AR. The A2AR is highly enriched in the striatum and enhances striatal cAMP after stimulation (15). In ST14A cells, treatment with CGS elevated the cellular level of cAMP, which was blocked by CSC, demonstrating the involvement of the A2AR in this process (see Fig. S1 in the supplemental material). Rpt6 was first immunoprecipitated by an anti-Rpt6 antibody. The immunoprecipitated complexes were then analyzed by Western blotting using an anti-phospho-PKA substrate antibody. As shown in Fig. 4A, expression of mHTT decreased the phosphorylation of Rpt6 by PKA. This finding was consistent with the observation that ST14A cells expressing Q109-HTTEx1-GFP had lower PKA activity than those expressing Q25-HTTEx1-GFP (see Fig. S2). To demonstrate that the PKA-mediated phosphorylation of Rpt6 is functionally important, we mutated the PKA-phosphorylated Ser residue (Ser120) of Rpt6 into Ala. The expression of mutant Rpt6 (Rpt6S120D), which mimics the phosphorylated form of Rpt6, markedly increased the proteasome activity (Fig. 4B). To confirm whether phosphorylation of Rpt6 at Ser120 enhances the formation of 26S proteasome complexes, Rpt6 mutants (Rpt6S120D or Rpt6S120A) were transiently expressed in ST14A cells, immunoprecipitated, and analyzed using Western blotting. As shown in Fig. 4C, only the phosphorylated form of Rpt6 (Rpt6S120D) was able to pull down the α6 component of the 20S proteasome, indicating appropriate complex formation between the 19S and 20S particles. Thus, phosphorylation of Rpt6 at Ser120 was critical for the formation of 26S proteasome complexes in ST14A cells.
Fig 4.
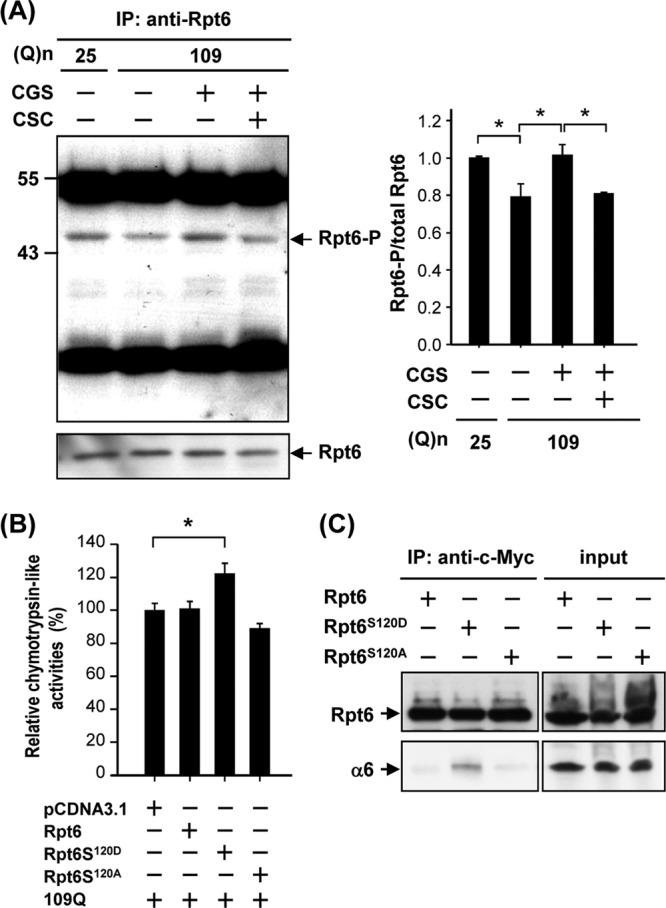
Activation of PKA via the A2A adenosine receptor (A2AR) increased phosphorylation of Rpt6 in ST14A cells. ST14A cells were transfected with pcDNA3.1-Htt-(Q)25-hrGFP or pcDNA3.1-Htt-(Q)109-hrGFP for 24 h and then treated with the desired reagent(s) for another 48 h. Rpt6 was immunoprecipitated using an anti-Rpt6 antibody, and the immunoprecipitated complexes were analyzed by Western blotting analyses, using an anti-phospho-(Ser/Thr) PKA substrate antibody and anti-Rpt6 antibody. The level of phosphorylated Rpt6 was quantified and is shown in the right panel, and Rpt6 was used as an internal control. Data are presented as the means ± SE for 3 experiments. ∗, P < 0.05 (by one-way ANOVA). (B) ST14A cells were transfected with the indicated constructs for 72 h. Chymotrypsin-like activity of proteasomes was assessed as described in Materials and Methods. (C) ST14A cells were transfected with the indicated construct for 72 h. Cell lysates were immunoprecipitated by an anti-c-Myc antibody conjugated with protein G beads. The resultant complexes were subjected to a Western blot analysis using anti-c-Myc and anti-α6 antibodies. Representative images of 3 independent experiments are shown.
To further test whether the cAMP/PKA pathway is involved in the impairment of the proteasome in HD, we treated ST14A cells with an activator (forskolin [FK]) of adenylyl cyclases or a cell-permeative cAMP analogue (dibutyryl cAMP [db-cAMP]) to activate the cAMP/PKA-dependent pathway and assess proteasome activity. In line with our hypothesis, the two cAMP-elevating reagents (FK and db-cAMP) effectively enhanced proteasome activity in the presence of Q109-HTTEx1-GFP (Fig. 5A). Because an inhibitor (H89) of PKA suppressed the enhancing effect of FK and db-cAMP, the regulation mentioned above was determined to be PKA dependent. Consistent with the results of treatment with FK and db-cAMP, CGS enhanced proteasome activity (Fig. 5B), normalized the elevated levels of high-molecular-mass ubiquitin (Fig. 5C), and reduced the formation of mutant HTT aggregates (Fig. 5D) in ST14A cells expressing Q109-HTTEx1-GFP. These effects of CGS were blocked by an A2AR-selective antagonist (CSC) and by an inhibitor (H89) of PKA. Therefore, PKA appears to mediate actions of the A2AR in regulating proteasome activity and subsequently suppressing the formation of HTT aggregates. We noted that a proteasome inhibitor (MG132), which greatly suppresses UPS activity and facilitates aggregate formation, completely eliminated the effects of CGS (Fig. 5B to D), further supporting our hypothesis that PKA functions by modulating the activity of the UPS.
Fig 5.

Activation of PKA via cAMP elevation rescued impairment of proteasome activity and reduced mutant Huntingtin (Htt) aggregates. (A) ST14A cells were transfected with pcDNA3.1-Htt-(Q)25-hrGFP or pcDNA3.1-Htt-(Q)109-hrGFP for 68 h and then treated with the desired reagent(s) (FK, 10 μM; db-cAMP, 100 μM; H89, 10 μM) for another 4 h. Chymotrypsin-like activity of proteasomes was assessed as described in Materials and Methods. Data were normalized to that of control Q25-expressing cells and are presented as the means ± SE for 3 experiments. ∗, P < 0.05 (by one-way ANOVA). (B) ST14A cells were transfected with pcDNA3.1-Htt-(Q)25-hrGFP or pcDNA3.1-Htt-(Q)109-hrGFP for 24 h and then treated with the desired reagent(s) (CGS, 10 μM; CSC, 20 μM; H89, 10 μM; MG132, 1 μM) for another 48 h. Chymotrypsin-like activity of proteasomes was assessed as described in Materials and Methods. Data were normalized to that for control Q25-expressing cells and are presented as the means ± SE for 3 experiments. ∗, P < 0.05 (by one-way ANOVA). (C) ST14A cells were transfected with pcDNA3.1-Htt-(Q)25-hrGFP or pcDNA3.1-Htt-(Q)109-hrGFP for 24 h and then treated with the desired reagent(s) for another 48 h. Lysates (30 μg per lane) collected from the indicated condition were subjected to a Western blot analysis using an antiubiquitin antibody. (D) ST14A cell lysates (30 μg per lane) collected from the indicated condition were subjected to a filter retardation assay. Insoluble Htt aggregates retained on the filter were detected using an anti-Htt antibody. The level of actin in the lysates was assessed using a Western blot analysis and served as an internal control. Representative images of 3 independent experiments are shown. (E) Primary striatal neurons were prepared from E18 R6/2 or wild-type WT mice. Striatal neurons were cotransfected with Snap25-DsRed and Snap25-pZsProSensor-1 at 7 days in vitro (DIV) and then treated with vehicle or 10 μM CGS for 72 h. Representative images of 3 independent experiments are shown. Scale bar = 20 μm.
To further assess the synaptic proteasome activity in striatal neurons, we created a fusion protein (SNAP25-pZsProSensor-1) comprising a synaptic protein (SNAP25) (30) and a fluorescent proteasome sensor (pZsProSensor-1), which encodes a ZsGreen protein fused to the degradation domain of ornithine decarboxylase and is sensitive to proteasome-mediated cleavage. The level of SNAP25-pZsProSensor-1 in neuronal processes, which was observed as a green fluorescent signal, inversely reflected synaptic proteasome activity. A control fusion protein (SNAP25-DsRed) was also created to normalize the transfection efficiency. As shown in Fig. 5E, ZsGreen signals were detected in the soma of both WT and R6/2 neurons. However, strong green fluorescent signals were observed in neuronal processes of primary striatal neurons harvested from R6/2 but not WT mice. Most importantly, CGS treatment reduced the accumulation of ZsGreen signals (Fig. 5E) in the neuronal processes of R6/2 neurons.
Similar to the findings obtained in ST14A and primary striatal neurons (Fig. 5), we demonstrated that chronic treatment of R6/2 mice with CGS for 5 weeks significantly enhanced the activity of the proteasome (Fig. 6A) and reduced the number of mutant HTT aggregates in the synaptosome fraction (Fig. 6B) of R6/2 mice. In addition, the level of a known downstream synaptic target of the proteasome (the AMPA glutamate receptor subunit, GluR1) (31) was higher in the synaptosome fraction of R6/2 mice than in that of WT mice. In agreement with its ability to increase proteasome activity, chronic CGS treatment normalized the abnormal upregulation of GluR1 (Fig. 6C).
Fig 6.

Chronic CGS21680 (CGS) treatment enhanced proteasome activity and reduced Huntingtin (Htt) aggregates in synapses of the striatum of R6/2 mice. R6/2 mice were injected daily with CGS (2.5 μg/g body weight) or vehicle for 5 weeks from the age of 7 weeks. (A) Chymotrypsin-like activities of the proteasome in the synaptic fraction of the striatum of the indicated 12-week-old mice were assessed as described in Materials and Methods. Data are presented as the mean ± SEM for each group. ∗∗, specific comparison to R6/2 control mice (P < 0.01; by one-way ANOVA). (B and D) Striatal synaptic fractions (10 μg per lane) were subjected to a Western blot analysis using an anti-HTT, antisynaptophysin (anti-SYP), or anti-GluR1 antibody, as indicated. (C) Striatal synaptic fractions (10 μg per well) were subjected to a filter retardation assay. Insoluble HTT aggregates retained on the filter were detected using an anti-HTT antibody. The level of synaptophysin in the lysates was assessed using Western blot analyses and served as an internal control. Representative images of 3 independent experiments are shown. (D) Striatal synaptic fractions (10 μg per lane) were subjected to a Western blot analysis using an antisynaptophysin or anti-GluR1 antibody.
The results described above collectively suggest that reagents that activate the cAMP/PKA pathway increase synaptic proteasome activity. To evaluate the involvement of PKA-mediated phosphorylation of Rpt6 in regulating proteasome activity, ST14A cells were transfected with Rpt6 or Rpt6S120A and treated with CGS. The expression of the resultant mutant, Rpt6S120A, but not WT Rpt6 in ST14A cells blocked the enhancing effect of CGS on proteasome activity (Fig. 7A) and the suppressive effect of CGS on mHTT aggregate formation (Fig. 7B and C). These data suggest that stimulation of the A2AR activates PKA, which phosphorylates Rpt6 at Ser120, consequently enhancing proteasome activity and reducing mHTT aggregation. In contrast, expression of the Rpt6S120D mutant, which mimics the phosphorylated form of Rpt6, markedly suppressed aggregate formation by Q109-HTTEx1-GFP, as assessed using a filter assay (Fig. 7D) and a fluorescence microscopic assay (Fig. 7E).
Fig 7.

Phosphorylation of Rpt6 at Ser120 is important in the PKA-mediated rescue effect on ST14A cells. ST14A cells were cotransfected with pcDNA3.1-Htt-(Q)109-hrGFP and pcDNA3-Rpt6-myc or pcDNA3.1-Rpt6S120A-myc for 24 h and then treated with the desired reagent(s) for another 48 h. Lysates collected for the indicated condition were used to perform the proteasome activity assay (A) and filter retardation assay (B and C). Data points represent the means ± SEM for 3 independent experiments. ST14A cells were cotransfected with pcDNA3.1-Htt-(Q)109-hrGFP and pcDNA3.1-Rpt6S120D-myc for 72 h. Lysates (30 μg per lane) collected for the indicated condition were subjected to a filter retardation assay (D). Insoluble Htt aggregates retained on the filter were detected using an anti-Htt antibody. The level of actin in the lysate was assessed using Western blot analysis and served as an internal control. A representative image of 3 independent experiments is shown. (E) Expression of hrGFP is shown in green. Nuclei were visualized using H33258 (blue). Representative pictures of 3 independent experiments are shown. Percentages of Htt aggregates containing cells were quantified using fluorescence microscopy. Data points represent the means ± SEM for 3 independent experiments. For each experiment, at least 130 transfected cells were scored.
To evaluate the in vivo role of Rpt6S120D, an AAV carrying Rpt6S120D or GFP was injected into the striatum of 5-week-old WT and R6/2 mice. The motor functions of these animals were analyzed 2 weeks after infection. Compared to findings for R6/2 mice injected with a control virus expressing GFP, expression of Rpt6S120D in the striatum of HD mice significantly attenuated the progressive deterioration of motor coordination observed in these mice to a near-normal level, as measured using rotarod performance (Fig. 8B), beam walk latency (Fig. 8C), and foot-clasping tests (Fig. 8D). Brain sections were collected to determine aggregate formation using immunofluorescence staining. Similar to what was observed in ST14A cells, expression of Rpt6S120D markedly reduced aggregate formation (Fig. 8E). These results collectively demonstrate that phosphorylation of Rpt6 at Ser120 by PKA enhances proteasome activity and delays the formation of HTT aggregates.
Fig 8.

Expression of phosphomimetic Rpt6 (Rpt6S120D) rescued motor function and reduced mutant Huntingtin (Htt) aggregates in R6/2 mice. Adeno-associated viruses (AAVs) carrying Rpt6S120D, Rpt6S120A, or green fluorescent protein (GFP) were injected into the striatum of 5-week-old wild-type (WT) and R6/2 mice. Body weight (A), beam walk (B), rotarod performance (C), and foot clasping (D) were measured weekly 2 weeks after the AAV injection. Data are presented as the means ± SEM for each group. ∗∗∗, ∗∗, and ∗, specific comparison to GFP-HD mice (P < 0.001, P < 0.01, and P < 0.05, respectively [by one-way ANOVA]) (n = 7 to 11 in each group). (E) Brain sections were harvested for immunocytochemical staining. Levels of mutant Htt aggregates (red) in the striatum were assessed using an anti-Htt antibody. Cells infected with the indicated virus are marked by expression of the reporter gene (GFP, green). Nuclei were visualized using H33258 (blue). Representative pictures of 3 independent experiments are shown. Scale bar = 20 μm. Data are presented as the means ± SEM for each group. ∗, specific comparison to GFP-HD mice (P < 0.01 [by Student's t test]).
DISCUSSION
The cAMP/PKA pathway has long been recognized as fundamentally important to HD pathogenesis and for drug development in HD (11, 12, 15, 20). In the present study, we showed that the activity of PKA in the striatum of two HD mouse models was lower than that observed in their littermate controls (Fig. 3), indicating that impaired PKA activity is an authentic pathogenic event in HD. Given the importance of PKA-mediated processes, including neuronal survival and plasticity (32), a wide variety of neuronal functions are expected to be affected. In particular, we demonstrated herein that low PKA activity in striatal cells expressing mHTT led to low phosphorylation of Rpt6. Because PKA-mediated phosphorylation of Rpt6 enhances the assembly and activity of the proteasome (29, 33), suppression of PKA activity further impaired the function of the proteasome (Fig. 5A and B). Conversely, low activity of the UPS caused accumulation of proteasome substrates, including PKA regulatory subunits (Fig. 2B and C), thus further reducing PKA activity. This novel feedback regulation between PKA and proteasome activity occurred during HD progression and likely contributed to HD pathogenesis, because activation of PKA using cAMP-inducing reagents or expression of a phosphorylated form of the Rpt6 mutant (Rpt6S120D) increased proteasome activity, reduced aggregation of mHTT, and improved motor impairment in HD mice (Fig. 5 to 8). Reagents that activate the cAMP/PKA pathway in the striatum are therefore beneficial in HD mice, as was reported previously (11, 20, 34). This cross talk between PKA and the proteasome might be brain area specific or disease stage dependent. In the hippocampus of an HD mouse model (R6/1), PKA was upregulated before the onset of motor impairment because of the downregulation of phosphodiesterase 4, which might have contributed to defects in recognition memory and spatial memory (35). Cell-type-specific regulation of Rpt subunits and UPS activity, probably due to differential structural changes in the proteasome in different tissues (5), might also account for this brain-area-specific regulation of PKA in HD. Future studies are needed to further investigate the complex feedback regulation between PKA and the proteasome during HD progression because of their critical roles in HD pathogenesis and neuronal survival.
The proteasome is a major regulatory proteolytic pathway for recognizing and degrading abnormal and misfolded proteins in eukaryotes (36). It was recently proposed as an important drug target in HD because impairment of the UPS leads to the accumulation of toxic N-terminal fragments of mutant HTT (37). The role and regulation of the proteasome in HD appear to be complex. Results from studies performed in several laboratories indicate that impairment of the UPS by polyglutamine [poly(Q)]-expanded mHTT might not be global (8, 38). Acute expression of mHTT impaired HD mouse models harboring a proteasome reporter (ubiquitinG76V-GFP) (39). Surprisingly, mHTT aggregation restored the activity of the proteasome (40). Abnormal upregulation of cAMP signaling might contribute, at least partially, to the compensatory effect on the proteasome observed in HD. Results of a previous report indicated that the expression of phosphodiesterase, an enzyme responsible for cAMP degradation, is downregulated in HD mice (R6/1 and R6/2), therefore resulting in an elevation of cAMP levels in the striatum of HD mice (12). Consistent with this report, we found that R6/2 mice at the presymptomatic stage (7 weeks of age) had high cAMP content and enhanced PKA activity in the striatum compared to WT mice (see Fig. S3 in the supplemental material), which likely contributed to the increased activity of the proteasome observed in the present study. During HD progression, proteins that mediate cAMP synthesis (e.g., type V adenylyl cyclase and the Gαs protein) are gradually downregulated (15), causing a consequent decrease in PKA activity (Fig. 3). Such downregulation of PKA in the late disease stage in HD mice might lead to reduced phosphorylation of Rpt6, lower activity of the proteasome, and increased mutant HTT aggregation, as demonstrated in the present study (Fig. 3 to 6). Proteasome activity appears to be tightly regulated by multiple mechanisms during HD progression. The disease stage, mouse models used, and experimental approaches employed to assess the activity of the proteasome are critical in assessing the dynamic regulation of this system in HD. Our study highlights a new means of manipulating the function of the proteasome, a critical element in the pathogenesis of HD.
Expression levels of different proteasome components and the subcellular localization of the proteasome play important roles in the pathogenesis of HD. In addition, the structure of the 26S proteasome may vary greatly among different cell types and various pathophysiological conditions in which proteasome activity is dynamically regulated (5, 38). Several earlier studies demonstrated that PKA directly phosphorylates several subunits (including Rpt6) of the UPS and subsequently enhances its activity, at least partially, by promoting proteasome assembly (29, 33). Data generated in the present study show that a mutant (Rpt6S120A) of Rpt6, a major downstream target of PKA, which cannot undergo phosphorylation by PKA, ablated the beneficial effect of a cAMP-elevating reagent (CGS) on the formation of aggregates (Fig. 7C). Conversely, a phosphomimetic mutant of Rpt6 (Rpt6S120D) markedly suppressed mutant HTT aggregation and ameliorated the impaired motor function observed in HD mice (15) (Fig. 7 and 8). To the best of our knowledge, the present study is the first report to demonstrate that phosphorylation of Rpt6 at Ser120 plays a critical role in the function of a beneficial agent that activates the cAMP/PKA pathway in HD. In addition to the PKA-mediated phosphorylation of Rpt6 at the Ser120 residue, Rpt6 can also be phosphorylated by Ca2+/calmodulin-dependent protein kinase IIα (CaMKIIα) in a neuronal activity-dependent manner, which leads to promotion of spine outgrowth and modulation of synaptic strength (41–43). Given that dysregulated neuronal morphology and altered synaptic strength occur in HD (44–46), the proteasome-mediated regulation of synaptic remodeling might be impaired in HD and is likely to play a critical role in HD pathogenesis. Collectively, these findings suggest that enhanced phosphorylation of Rpt6 at Ser120 by PKA and CaMKIIα provides a potential route to identify new therapeutic agents for treating HD.
We noted that mHTT-evoked changes in proteasome activity in either cultured striatal cells or brains of HD mice were moderate (∼20%) (Fig. 1, 5, and 6). As discussed above, such small changes in proteasome activity may lead to cumulative morphological and physiological alterations in striatal neurons, especially at synapses, and worsened mHTT-evoked toxicity. Consistent with our findings for HD mice and striatal cells expressing an N-terminal fragment of mHTT (Q109), the chymotrypsin-like proteolytic proteasome activity in a striatal cell line expressing a full-length mHTT (Q111) was also moderate (∼15%) (9). In our study, we showed that cAMP-elevating reagents (i.e., CGS, FK, and db-cAMP) functioned to restore mHTT-damaged proteasome activity and therefore enhanced proteasome activity by ∼20%. Phosphorylation of Rpt6 by PKA is likely to elevate proteasome activity by enhancing proteasome assembly (Fig. 4C) (33). Our findings suggest that rescue of impaired proteasome activity by cAMP-elevating reagents or Rpt6S120D markedly reduced mHTT aggregates (Fig. 5 to 7), normalized levels of synaptic proteins (e.g., GluR1) (Fig. 6C), and ameliorated motor deficits of HD mice (Fig. 8) (15). The mHTT-evoked impairment of proteasome activity, although small, apparently causes cumulative pathogenic changes and exacerbates the progression of HD. In addition to HD, impairment of the proteasome was found in various neuronal degenerative diseases and brain injuries (4). For example, several recent studies showed that decreased UPS activity might contribute to PD pathogenesis (47). In particular, reduced proteasome activity was observed in degenerated brain areas (such as the nigra pars compacta) but not in other brain areas that are spared from degeneration in patients with PD (47). Suppression of proteasome activity, as detected here in striatal synaptic preparations of HD mice (Fig. 1), might impair the induction of a plastic response to neuronal activation (such as persistent presynaptic silencing) via the accumulation of synaptic proteins (e.g., GluR1 and Rim 1) (31, 48, 49). Such alterations in the properties of synaptic transduction might result in altered synaptic communication between the cortex and striatum, as previously reported (27, 50).
In the present study, we demonstrated that chronic treatment with CGS (an A2AR-selective agonist) normalized the reduced proteasome activity (Fig. 5 and 6) and increased levels of GluR1 in striatal synaptosomes of HD mice (Fig. 6C). Note that progressive degeneration of the corticostriatal pathway was proposed to cause the major symptoms of late-stage HD (51). Consistent with our finding that CGS treatment normalized expression levels of synaptic proteins (Fig. 6C), Cepeda and colleagues showed that CGS normalized the deregulated excitatory postsynaptic currents in striatal medium-sized spiny neurons of R6/2 mice (50). Thus, CGS rescues the corticostriatal synaptic disconnection and progressive motor impairment observed in HD mice (15, 50). These findings together with several earlier reports suggest that activation of the A2AR, a cAMP-elevating receptor, is a therapeutic option in HD and elicits beneficial effects on several key HD symptoms (including formation of intranuclear inclusions, impaired motor coordination, brain atrophy, and urea cycle deficiency) (15, 52–54). Consistent with the possible beneficial role of the A2AR in HD, Mievis and colleagues demonstrated that elimination of the A2AR exacerbated HD progression in a mouse model of HD (55). Future investigations are required to evaluate the therapeutic potential of A2AR drugs (56).
In conclusion, our data reveal novel feedback regulation between cAMP/PKA and proteasome activity in the striatum. Based on their ability to regulate proteasome activity, A2AR drugs and cAMP-inducing reagents might be considered potential drug candidates for HD and other diseases caused or exacerbated by deregulation of the proteasome.
Supplementary Material
ACKNOWLEDGMENTS
We thank Elena Cattaneo (Milano University, Milan, Italy) for providing the striatal cell line (ST14A) and Jan-Jong Hung (National Cheng Kung University, Tainan City, Taiwan) for the cDNA constructs of Rpt6-myc and Rpt6S120A-myc. We are grateful to Ya-Yun Cheng for technical support with viral infection and Daniel P. Chamberlin for reading and editing the manuscript.
None of us has any conflict of interest to declare.
This work was supported by grants from the National Science Council (NSC96-2321-B-001-015, NSC97-2321-B-001-012, NSC98-2321-B-001-012, and NSC100-2320-B-001-0110MY3) and Academia Sinica (AS-94-TP-B17 and AS-97-TP-B02), Taiwan.
Footnotes
Published ahead of print 28 December 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.01434-12.
REFERENCES
- 1.The Huntington's Disease Collaborative Research Group 1993. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72:971–983 [DOI] [PubMed] [Google Scholar]
- 2.Martin JB, Gusella JF. 1986. Huntington's disease. Pathogenesis and management. N. Engl. J. Med. 315:1267–1276 [DOI] [PubMed] [Google Scholar]
- 3.Krobitsch S, Kazantsev AG. 2011. Huntington's disease: from molecular basis to therapeutic advances. Int. J. Biochem. Cell Biol. 43:20–24 [DOI] [PubMed] [Google Scholar]
- 4.Tai HC, Schuman EM. 2008. Ubiquitin, the proteasome and protein degradation in neuronal function and dysfunction. Nat. Rev. Neurosci. 9:826–838 [DOI] [PubMed] [Google Scholar]
- 5.Wang XH, Zhang L, Mitch WE, LeDoux JM, Hu J, Du J. 2010. Caspase-3 cleaves specific 19 S proteasome subunits in skeletal muscle stimulating proteasome activity. J. Biol. Chem. 285:21249–21257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang F, Su K, Yang X, Bowe DB, Paterson AJ, Kudlow JE. 2003. O-GlcNAc modification is an endogenous inhibitor of the proteasome. Cell 115:715–725 [DOI] [PubMed] [Google Scholar]
- 7.Bennett EJ, Shaler TA, Woodman B, Ryu KY, Zaitseva TS, Becker CH, Bates GP, Schulman H, Kopito RR. 2007. Global changes to the ubiquitin system in Huntington's disease. Nature 448:704–708 [DOI] [PubMed] [Google Scholar]
- 8.Wang J, Wang CE, Orr A, Tydlacka S, Li SH, Li XJ. 2008. Impaired ubiquitin-proteasome system activity in the synapses of Huntington's disease mice. J. Cell Biol. 180:1177–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hunter JM, Lesort M, Johnson GV. 2007. Ubiquitin-proteasome system alterations in a striatal cell model of Huntington's disease. J. Neurosci. Res. 85:1774–1788 [DOI] [PubMed] [Google Scholar]
- 10.Chiang MC, Lee YC, Huang CL, Chern Y. 2005. cAMP-response element-binding protein contributes to suppression of the A2A adenosine receptor promoter by mutant Huntingtin with expanded polyglutamine residues. J. Biol. Chem. 280:14331–14340 [DOI] [PubMed] [Google Scholar]
- 11.Giampa C, Laurenti D, Anzilotti S, Bernardi G, Menniti FS, Fusco FR. 2010. Inhibition of the striatal specific phosphodiesterase PDE10A ameliorates striatal and cortical pathology in R6/2 mouse model of Huntington's disease. PLoS One 5:e13417 doi:10.1371/journal.pone.0013417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hebb AL, Robertson HA, Denovan-Wright EM. 2004. Striatal phosphodiesterase mRNA and protein levels are reduced in Huntington's disease transgenic mice prior to the onset of motor symptoms. Neuroscience 123:967–981 [DOI] [PubMed] [Google Scholar]
- 13.Andremont A, Lancar R, An Le N, Hattchouel JM, Baron S, Tavakoli T, Daniel MF, Tancrede C, L MG. 1996. Secular trends in mortality associated with bloodstream infections in 4268 patients hospitalized in a cancer referral center between 1975 and 1989. Clin. Microbiol. Infect. 1:160–167 [DOI] [PubMed] [Google Scholar]
- 14.Schilling G, Becher MW, Sharp AH, Jinnah HA, Duan K, Kotzuk JA, Slunt HH, Ratovitski T, Cooper JK, Jenkins NA, Copeland NG, Price DL, Ross CA, Borchelt DR. 1999. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum. Mol. Genet. 8:397–407 [DOI] [PubMed] [Google Scholar]
- 15.Chou SY, Lee YC, Chen HM, Chiang MC, Lai HL, Chang HH, Wu YC, Sun CN, Chien CL, Lin YS, Wang SC, Tung YY, Chang C, Chern Y. 2005. CGS21680 attenuates symptoms of Huntington's disease in a transgenic mouse model. J. Neurochem. 93:310–320 [DOI] [PubMed] [Google Scholar]
- 16.Ehrlich ME, Conti L, Toselli M, Taglietti L, Fiorillo E, Taglietti V, Ivkovic S, Guinea B, Tranberg A, Sipione S, Rigamonti D, Cattaneo E. 2001. ST14A cells have properties of a medium-size spiny neuron. Exp. Neurol. 167:215–226 [DOI] [PubMed] [Google Scholar]
- 17.Chou SY, Weng JY, Lai HL, Liao F, Sun SH, Tu PH, Dickson DW, Chern Y. 2008. Expanded-polyglutamine huntingtin protein suppresses the secretion and production of a chemokine (CCL5/RANTES) by astrocytes. J. Neurosci. 28:3277–3290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu FC, Wu GC, Hsieh ST, Lai HL, Wang HF, Wang TW, Chern Y. 1998. Expression of type VI adenylyl cyclase in the central nervous system: implication for a potential regulator of multiple signals in different neurotransmitter systems. FEBS Lett. 436:92–98 [DOI] [PubMed] [Google Scholar]
- 19.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685 [DOI] [PubMed] [Google Scholar]
- 20.Kim YC, Wu SY, Lim HS, Chiang CM, Kodadek T. 2009. Non-proteolytic regulation of p53-mediated transcription through destabilization of the activator.promoter complex by the proteasomal ATPases. J. Biol. Chem. 284:34522–34530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Phillips GR, Huang JK, Wang Y, Tanaka H, Shapiro L, Zhang W, Shan WS, Arndt K, Frank M, Gordon RE, Gawinowicz MA, Zhao Y, Colman DR. 2001. The presynaptic particle web: ultrastructure, composition, dissolution, and reconstitution. Neuron 32:63–77 [DOI] [PubMed] [Google Scholar]
- 22.Wang YT, Chuang JY, Shen MR, Yang WB, Chang WC, Hung JJ. 2008. Sumoylation of specificity protein 1 augments its degradation by changing the localization and increasing the specificity protein 1 proteolytic process. J. Mol. Biol. 380:869–885 [DOI] [PubMed] [Google Scholar]
- 23.Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. 1989. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 77:61–68 [DOI] [PubMed] [Google Scholar]
- 24.Chern Y, Lai HL, Fong JC, Liang Y. 1993. Multiple mechanisms for desensitization of A2a adenosine receptor-mediated cAMP elevation in rat pheochromocytoma PC12 cells. Mol. Pharmacol. 44:950–958 [PubMed] [Google Scholar]
- 25.Xiao X, McCown TJ, Li J, Breese GR, Morrow AL, Samulski RJ. 1997. Adeno-associated virus (AAV) vector antisense gene transfer in vivo decreases GABA(A) alpha1 containing receptors and increases inferior collicular seizure sensitivity. Brain Res. 756:76–83 [DOI] [PubMed] [Google Scholar]
- 26.Chen CC, Sun CP, Ma HI, Fang CC, Wu PY, Xiao X, Tao MH. 2009. Comparative study of anti-hepatitis B virus RNA interference by double-stranded adeno-associated virus serotypes 7, 8, and 9. Mol. Ther. 17:352–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martire A, Calamandrei G, Felici F, Scattoni ML, Lastoria G, Domenici MR, Tebano MT, Popoli P. 2007. Opposite effects of the A2A receptor agonist CGS21680 in the striatum of Huntington's disease versus wild-type mice. Neurosci. Lett. 417:78–83 [DOI] [PubMed] [Google Scholar]
- 28.Hegde AN, Goldberg AL, Schwartz JH. 1993. Regulatory subunits of cAMP-dependent protein kinases are degraded after conjugation to ubiquitin: a molecular mechanism underlying long-term synaptic plasticity. Proc. Natl. Acad. Sci. U. S. A. 90:7436–7440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang F, Hu Y, Huang P, Toleman CA, Paterson AJ, Kudlow JE. 2007. Proteasome function is regulated by cyclic AMP-dependent protein kinase through phosphorylation of Rpt6. J. Biol. Chem. 282:22460–22471 [DOI] [PubMed] [Google Scholar]
- 30.Oyler GA, Higgins GA, Hart RA, Battenberg E, Billingsley M, Bloom FE, Wilson MC. 1989. The identification of a novel synaptosomal-associated protein, SNAP-25, differentially expressed by neuronal subpopulations. J. Cell Biol. 109:3039–3052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rezvani K, Teng Y, Shim D, De Biasi M. 2007. Nicotine regulates multiple synaptic proteins by inhibiting proteasomal activity. J. Neurosci. 27:10508–10519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Benito E, Barco A. 2010. CREB's control of intrinsic and synaptic plasticity: implications for CREB-dependent memory models. Trends Neurosci. 33:230–240 [DOI] [PubMed] [Google Scholar]
- 33.Asai M, Tsukamoto O, Minamino T, Asanuma H, Fujita M, Asano Y, Takahama H, Sasaki H, Higo S, Asakura M, Takashima S, Hori M, Kitakaze M. 2009. PKA rapidly enhances proteasome assembly and activity in in vivo canine hearts. J. Mol. Cell. Cardiol. 46:452–462 [DOI] [PubMed] [Google Scholar]
- 34.Huang CL, Yang JM, Wang KC, Lee YC, Lin YL, Yang YC, Huang NK. 2011. Gastrodia elata prevents huntingtin aggregations through activation of the adenosine A(2A) receptor and ubiquitin proteasome system. J. Ethnopharmacol. 138:162–168 [DOI] [PubMed] [Google Scholar]
- 35.Giralt A, Saavedra A, Carreton O, Xifro X, Alberch J, Perez-Navarro E. 2011. Increased PKA signaling disrupts recognition memory and spatial memory: role in Huntington's disease. Hum. Mol. Genet. 20:4232–4247 [DOI] [PubMed] [Google Scholar]
- 36.Demartino GN, Gillette TG. 2007. Proteasomes: machines for all reasons. Cell 129:659–662 [DOI] [PubMed] [Google Scholar]
- 37.Li X, Wang CE, Huang S, Xu X, Li XJ, Li H, Li S. 2010. Inhibiting the ubiquitin-proteasome system leads to preferential accumulation of toxic N-terminal mutant huntingtin fragments. Hum. Mol. Genet. 19:2445–2455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bett JS, Cook C, Petrucelli L, Bates GP. 2009. The ubiquitin-proteasome reporter GFPu does not accumulate in neurons of the R6/2 transgenic mouse model of Huntington's disease. PLoS One 4:e5128 doi:10.1371/journal.pone.0005128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lindsten K, Menendez-Benito V, Masucci MG, Dantuma NP. 2003. A transgenic mouse model of the ubiquitin/proteasome system. Nat. Biotechnol. 21:897–902 [DOI] [PubMed] [Google Scholar]
- 40.Ortega Z, Diaz-Hernandez M, Maynard CJ, Hernandez F, Dantuma NP, Lucas JJ. 2010. Acute polyglutamine expression in inducible mouse model unravels ubiquitin/proteasome system impairment and permanent recovery attributable to aggregate formation. J. Neurosci. 30:3675–3688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bingol B, Wang CF, Arnott D, Cheng D, Peng J, Sheng M. 2010. Autophosphorylated CaMKIIalpha acts as a scaffold to recruit proteasomes to dendritic spines. Cell 140:567–578 [DOI] [PubMed] [Google Scholar]
- 42.Djakovic SN, Marquez-Lona EM, Jakawich SK, Wright R, Chu C, Sutton MA, Patrick GN. 2012. Phosphorylation of Rpt6 regulates synaptic strength in hippocampal neurons. J. Neurosci. 32:5126–5131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hamilton AM, Oh WC, Vega-Ramirez H, Stein IS, Hell JW, Patrick GN, Zito K. 2012. Activity-dependent growth of new dendritic spines is regulated by the proteasome. Neuron 74:1023–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heck N, Betuing S, Vanhoutte P, Caboche J. 2012. A deconvolution method to improve automated 3D-analysis of dendritic spines: application to a mouse model of Huntington's disease. Brain Struct. Funct. 217:421–434 [DOI] [PubMed] [Google Scholar]
- 45.Klapstein GJ, Fisher RS, Zanjani H, Cepeda C, Jokel ES, Chesselet MF, Levine MS. 2001. Electrophysiological and morphological changes in striatal spiny neurons in R6/2 Huntington's disease transgenic mice. J. Neurophysiol. 86:2667–2677 [DOI] [PubMed] [Google Scholar]
- 46.Laforet GA, Sapp E, Chase K, McIntyre C, Boyce FM, Campbell M, Cadigan BA, Warzecki L, Tagle DA, Reddy PH, Cepeda C, Calvert CR, Jokel ES, Klapstein GJ, Ariano MA, Levine MS, DiFiglia M, Aronin N. 2001. Changes in cortical and striatal neurons predict behavioral and electrophysiological abnormalities in a transgenic murine model of Huntington's disease. J. Neurosci. 21:9112–9123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McNaught KS, Jnobaptiste R, Jackson T, Jengelley TA. 2010. The pattern of neuronal loss and survival may reflect differential expression of proteasome activators in Parkinson's disease. Synapse (New York) 64:241–250 [DOI] [PubMed] [Google Scholar]
- 48.Bingol B, Schuman EM. 2004. A proteasome-sensitive connection between PSD-95 and GluR1 endocytosis. Neuropharmacology 47:755–763 [DOI] [PubMed] [Google Scholar]
- 49.Jiang X, Litkowski PE, Taylor AA, Lin Y, Snider BJ, Moulder KL. 2010. A role for the ubiquitin-proteasome system in activity-dependent presynaptic silencing. J. Neurosci. 30:1798–1809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cepeda C, Cummings DM, Hickey MA, Kleiman-Weiner M, Chen JY, Watson JB, Levine MS. 2010. Rescuing the corticostriatal synaptic disconnection in the R6/2 mouse model of Huntington's disease: exercise, adenosine receptors and ampakines. PLoS Curr. 2010:pii=RRN1182 doi:10.1371/currents.RRN1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cepeda C, Wu N, Andre VM, Cummings DM, Levine MS. 2007. The corticostriatal pathway in Huntington's disease. Prog. Neurobiol. 81:253–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chiang MC, Chen HM, Lai HL, Chen HW, Chou SY, Chen CM, Tsai FJ, Chern Y. 2009. The A2A adenosine receptor rescues the urea cycle deficiency of Huntington's disease by enhancing the activity of the ubiquitin-proteasome system. Hum. Mol. Genet. 18:2929–2942 [DOI] [PubMed] [Google Scholar]
- 53.Huang NK, Lin JH, Lin JT, Lin CI, Liu EM, Lin CJ, Chen WP, Shen YC, Chen HM, Chen JB, Lai HL, Yang CW, Chiang MC, Wu YS, Chang C, Chen JF, Fang JM, Lin YL, Chern Y. 2011. A new drug design targeting the adenosinergic system for Huntington's disease. PLoS One 6:e20934 doi:10.1371/journal.pone.0020934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee YC, Yang YC, Huang CL, Kuo TY, Lin JH, Yang DM, Huang NK. 2012. When cytokinin, a plant hormone, meets the adenosine A2A receptor: a novel neuroprotectant and lead for treating neurodegenerative disorders? PLoS One 7:e38865 doi:10.1371/journal.pone.0038865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mievis S, Blum D, Ledent C. 2011. A(2A) receptor knockout worsens survival and motor behaviour in a transgenic mouse model of Huntington's disease. Neurobiol Dis. 41:570–576 [DOI] [PubMed] [Google Scholar]
- 56.Popoli P, Blum D, Domenici MR, Burnouf S, Chern Y. 2008. A critical evaluation of adenosine A2A receptors as potentially “druggable” targets in Huntington's disease. Curr. Pharm. Des. 14:1500–1511 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.