

Abstract
Pseudomonas aeruginosa produces three exopolysaccharides, Psl, Pel, and alginate, that play vital roles in biofilm formation. Pel is a glucose-rich, cellulose-like exopolysaccharide. The essential Pel biosynthesis proteins are encoded by seven genes, pelA to pelG. Bioinformatics analysis suggests that PelF is a cytosolic glycosyltransferase. Here, experimental evidence was provided to support this PelF function. A UDP-glucose dehydrogenase-based assay was developed to quantify UDP-glucose. UDP-glucose was proposed as the substrate for PelF. The isogenic pelF deletion mutant accumulated 1.8 times more UDP-glucose in its cytosol than the wild type. This suggested that PelF, which was found localized in the cystosol, uses UDP-glucose as substrate. Additionally, in vitro experiments confirmed that PelF uses UDP-glucose as substrate. To analyze the functional roles of conserved residues in PelF, site-directed mutagenesis was performed. The presence of the EX7E motif is characteristic for various glycosyltransferase families, and in PelF, E405/E413 are the conserved residues in this motif. Replacement of E405 with A resulted in a reduction of PelF activity to 30.35% ± 3.15% (mean ± standard deviation) of the wild-type level, whereas replacement of the second E, E413, with A did not produce a significant change in the activity of PelF. Moreover, replacement of both E residues did not result in a loss of PelF function, but replacement of the conserved R325 or K330 with A resulted in a complete loss of PelF activity. Overall, our data show that PelF is a soluble glycosyltransferase that uses UDP-glucose as the substrate for Pel synthesis and that conserved residues R325 and K330 are important for the activity of PelF.
INTRODUCTION
Pseudomonas aeruginosa is an opportunistic pathogen responsible for chronic pulmonary infections in cystic fibrosis patients. It causes persisting infections due to its ability to form biofilms (1). Inside the biofilm matrix, bacterial cells are protected from adverse effects of antibiotics and the host immune response (2). The biofilm matrix is mainly composed of extracellular DNA (eDNA), proteins, and exopolysaccharides (EPS). Three important exopolysaccharides that are synthesized and secreted by P. aeruginosa are alginate, Psl, and Pel (3).
Alginate is a polymer of manuronic acid and guluronic acids. Its biosynthesis pathway involves 13 genes (algC, algD, alg8, alg44, algK, algE, algG, algX, algL, algI, algJ, algF, and algA) (4). The exopolysaccharide, Psl, consists of a repeating pentasaccharide containing d-mannose, d-glucose, and l-rhamnose. It is synthesized and secreted by proteins encoded by the psl operon (pslA-pslO) (5).
The formation of a layer of polymer/cells at the air-liquid interface of a P. aeruginosa static culture is termed pellicle formation. It is controlled by the pel operon, which is composed of seven genes (pelA, pelB, pelC, pelD, pelE, pelF, and pelG), all of which are essential for pellicle formation (5, 6). Pellicle formation is attributed to the ability of P. aeruginosa to synthesize and secrete the Pel polysaccharide. Carbohydrate component analysis and cellulase treatment of exopolysaccharides produced by a Pel-deficient P. aeruginosa mutant and the wild type suggested that Pel is a glucose-rich cellulose-like polysaccharide (5, 7). In addition, another study showed that the pellicle is composed of lipopolysaccharide-like molecules (8).
The roles of the proteins encoded by the pel operon have only been determined for PelC and PelD. PelC is an outer membrane lipoprotein that is presumably involved in transportation of Pel to the bacterial cell surface (9), whereas PelD is a bis-(3′,5′)-cyclic dimeric GMP (c-di-GMP)-binding protein that is involved in posttranslational regulation of Pel production (10). The roles of PelA, PelB, PelE, PelF, and PelG have not yet been shown experimentally. However, on the basis of sequence homology, it has been predicted that PelG could be a member of the polysaccharide transporters (PST) family; PelD and PelE, the proposed inner membrane proteins, are presumably involved in the transfer of Pel across the cytoplasmic membrane; and PelA displays weak sequence homology with glycosylhydrolase enzymes (6). PelB has been proposed as a multidomain protein containing a periplasmic and an outer membrane domain. The C-terminal domain is proposed to contain a β-sheet structure and is suggested to be an outer membrane protein, like AlgE (11). This domain might function as a porin involved in polysaccharide secretion, whereas homology modeling has suggested that the N-terminal periplasmic domain shows similarity with anaphase-promoting complex/cyclosome subunit Cdc 16/Cut9 (PDB ID 2XPI). This domain might be involved in protein-protein interactions, due to the presence of tetratricopeptide-like repeats (TPR) (12). Bioinformatics analysis of PelF suggested that it is a glycosyltransferase (6).
Biosynthesis of polymers in bacteria is under extensive research. A comprehensive analysis of polymer biosynthesis in bacterial species has been previously published (13). Glycosyltransferases are required for initiation or elongation of carbohydrate chains during polysaccharide biosynthesis. These enzymes transfer an activated mono- or oligosaccharide residue to an existing acceptor molecule, forming a glycosidic bond. Glycosyltransferases use a nucleotide phospho-sugar (Leloir type) or an oligosaccharide (non-Leloir type) as the glycosyl donor, and monosaccharides, oligosaccharides, polypeptides, nucleic acids, and lipids act as acceptors to catalyze the formation of a glycosidic bond. The reaction can result in inversion or retention of the anomeric configuration of the donor sugar in the product and, as such, can be referred to as inverting or retaining glycosyltransferases, respectively (1). The Carbohydrate Active Enzyme (CAZy) database (www.cazy.org) has divided all glycosyltransferases into 94 families (as of July 2012) based on the classification described by Campbell et al. (14) and by Coutinho et al. (15). Despite great primary structure diversity, the tertiary structure of most glycosyltransferases is conserved. All structures of nucleotide-sugar-dependent glycosyltransferases solved to date have shown only two general folds, termed GT-A and GT-B (15–18). Bioinformatics analysis has suggested the presence of a third fold, GT-C (19). The GT-A and GT-B folds are Rossman-like folds that consist of two β/α/β domains. In the GT-A fold, both domains are closely associated, forming a compact globular structure that displays distinct nucleotide- and acceptor-binding sites (18). A DXD motif is commonly found in GT-A enzymes, in which the glutamic acid carboxylate groups coordinate a divalent cation (Mg2+ and Mn2+) and/or a ribose (14, 15). Although the DXD motif is considered to be a signature of GT-A glycosyltransferases, it has been found that this motif is not absolutely conserved (20). Alternatively, the DXD motif is present in many proteins that are not glycosyltransferases. The GT-B fold consists of two less-tightly associated domains facing each other in such a way that a cleft containing the active site is formed. In contrast to the GT-A fold, the GT-B fold lacks any DXD motif and, as such, works by a metal ion-independent mechanism (21). Most of the CAZy GT-4 family glycosyltransferases contain a conserved EX7E motif at their C terminus (15). These two conserved glutamic acid residues are suggested to be involved in catalytic activity, but their role has not yet been confirmed (15).
PelF belongs to glycosyltransferase family 4 (GT-4) in the CAZy database. Members of the GT-4 family are retaining glycosyltransferases that display a GT-B fold. In this study, the functional role of PelF was investigated. The in vivo and in vitro activities of PelF were studied with respect to its role in biosynthesis of Pel and the formation of the pellicle at the air-liquid interface. In this study, the key amino acid residues essential for the function of PelF were identified.
MATERIALS AND METHODS
Construction of the plasmid carrying a His10-tagged PelF gene.
To construct the His-tagged PelF-carrying plasmid, pelF was amplified by PCR with the primers PelF-His-Frw and PelF Rev (Table 1) by using plasmid pBBR1MCS-5::pelF (3) as the DNA template. The product was inserted into pGEMT-Easy (Promega, Sydney, Australia), and the resulting plasmid was propagated in Escherichia coli Top10 (Promega, Sydney, Australia) and isolated with a High Pure plasmid isolation kit according to the manufacturer's instructions (Roche). The DNA sequence was confirmed, and the plasmid was transformed into E. coli JM110 to avoid methylation of the XbaI site. pGEMT-easy-His10-PelF purified from E. coli JM110 was digested with NdeI and XbaI to obtain the fragment encoding His10-PelF. Plasmid pBBR1-MCS5::alg8 (22) was digested with NdeI and XbaI to remove the alg8 gene and ligated with the NdeI-XbaI fragment to obtain plasmid pBBR1-MCS5::His10-PelF, encoding His-tagged PelF. A ribosomal binding site was already available upstream of NdeI in the plasmid. All inserts cloned into the multiple-cloning site of the vector were under the control of a lac promoter.
Table 1.
Strains, plasmids, and oligonucleotides used in this study
| Strain, plasmid, or oligonucleotide | Description or sequence (5′–3′) | Source or reference |
|---|---|---|
| Bacterial strains | ||
| E. coli TOP10 | E. coli cloning strain | Invitrogen |
| P. aeruginosa PAO1 ΔpslA ΔpelF | Markerless, isogenic pslA and pelF deletion double mutant derived from PAO1 | 3 |
| P. aeruginosa PAO1 ΔpslA | Markerless, isogenic pslA deletion double mutant derived from PAO1 | 3 |
| P. aeruginosa PAO1 ΔpslA Δalg8 | Markerless, isogenic pslA and alg8 deletion double mutant derived from PAO1 | 3 |
| Plasmids | ||
| pBBR1MCS-5 | Gmr, broad-host-range vector, Plac | 57 |
| pBBR1MCS-5::His10 -PelF | NdeI-XbaI fragment comprising His10 gene tagged at 5′ end of pelF inserted into vector pBBR1MCS-5 | This study |
| Oligonucelotidesa | ||
| PelF-His-Frw | AAACATATGCACCACCATCACCACCATCACCACCATCACACCGAACACACCGCTCCGACGGCGC | |
| PelF Rev | AAATCTAGATCATGCAATCTCCGTGGCTTCGCGG | |
| PelF(His)E405-AFrwL | CAGCGCCGCGCAGCCGCTGGTGATCCTCG | |
| PelF(His)E405-AFrwS | AGCCGCTGGTGATCCTCG | |
| PelF(His)E405-ARevL | GCGCGGCGCTGATCGAGGTGAGGACCATCAG | |
| PelF(His)E405-ARevS | ATCGAGGTGAGGACCATCAG | |
| PelF(His)E413-AFrwL | CCTCGCCGCCTGGGCTGCCGGCGCCCCGGTG | |
| PelF(His)E413-AFrwS | GGGCTGCCGGCGCCCCGGTG | |
| PelF(His)E413-ARevL | AGGCGGCGAGGATCACCAGCGGCTGCGCTTC | |
| PelF(His)E413-ARevS | ATCACCAGCGGCTGCGCTTC | |
| PelF(His)D303-AFrwL | CCTCGCCGCCTGGACCGGCGCCCTCGAACGG | |
| PelF(His)D303-AFrwS | GGACCGGCGCCCTCGAACGG | |
| PelF(His)D303-ARevL | AGGCGGCGAGGTCGATGCCGTTGGGGATCAC | |
| PelF(His)D303-ARevS | TCGATGCCGTTGGGGATCAC | |
| PelF(His)D362-AFrwL | TCCGGCCTATGCCAGCGAATGCCGCAGCCTG | |
| PelF(His)D362-AFrwS | CCAGCGAATGCCGCAGCCTG | |
| PelF(His)D362-ARevL | CATAGGCCGGATCTTCCTCCTCCGGACCGAC | |
| PelF(His)D362-ARevS | TCTTCCTCCTCCGGACCGAC |
Underlined nucleotides indicate changes to introduce site-directed mutations.
In vivo activity of PelF.
The plasmid pBBR1-MCS5::His10-PelF was used to transform ΔpelF mutants of P. aeruginosa for complementation experiments. The in vivo activity of PelF was assessed in pellicle formation assays and Congo red binding assays, as previously described (3).
Subcellular localization of PelF.
P. aeruginosa strains were grown for 12 to 14 in LB medium, diluted 1:50 in new LB medium, and then grown for 8 h to obtain an optical cell density at 600 nm of 1.5 to 1.6. The cells were collected by centrifugation at 5,000 × g for 10 min at 4°C. Cell sediments were washed twice with saline (150 mM NaCl, pH 7.2), and then the cells were resuspended in 50 mM HEPES buffer (pH 7.4). The cell suspension was sonicated at 30% intensity for 10 cycles of 15 s followed by 10 s of cooling. Cellular debris and unlysed cells were removed by centrifugation at 15,000 × g for 20 min at 4°C, and the supernatant was centrifuged at 100,000 × g for 2 h. The supernatant (soluble fraction) was transferred to a clean tube and used for sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotting. The sediments were resuspended in 800 μl of 50 mM HEPES buffer (pH 7.4) and washed by centrifugation at 100,000 × g for 2 h. The washing procedure was repeated twice. This washed pellet was used for SDS-PAGE and immunoblotting.
Analysis of proteins.
Protein extracts from P. aeruginosa containing plasmids pBBR1-MCS5 (negative control) and pBBR1-MCS5::His10-PelF (carrying His10-PelF) were separated by SDS-PAGE (23). Proteins were electroblotted onto a nitrocellulose membrane (Protran BA 83; Schleicher & Schuell) and then incubated with HisProbe-horseradish peroxidase conjugate (HisProbe-HRP; Pierce). Immunoblots were developed using a chemiluminescence protocol according to the manufacturer's manual (SuperSignal West HisProbe; Pierce).
Enrichment of PelF.
The His10-tagged PelF was enriched from protein extracts by using His-Spin protein miniprep (HSMP) columns (Zymo Research) in HSMP buffer (50 mM HEPES [pH 7.4], 150 mM NaCl, 10 mM β-mercaptoethanol, and 1× Roche EDTA-free Complete protease inhibitor). P. aeruginosa strains containing pBBR1-MCS5:His10-PelF were grown in 500 ml pseudomonas isolation (PI) medium for 14 h at 37°C with shaking. Cells were collected by centrifugation at 15,000 × g for 2 min, and the cell pellet was washed twice with saline (150 mM NaCl, pH 7.2) and resuspended in 5 ml of HSMP buffer containing 10 mM imidazole. The cell pellet was lysed by sonication as described above for subcellular fractionation. The cell lysate was centrifuged at 15,000 × g for 20 min to remove unlysed cells and cell debris, and 5 ml of supernatant was collected for further treatment. A volume of 250 μl of supernatant was incubated in a His-Spin protein miniprep column for 3 min and then centrifuged to remove unbound proteins. This was repeated until the whole 5 ml of supernatant had been processed through the column. Finally, the column was washed thrice with HSMP buffer containing 50 mM imidazole, and protein was eluted with 100 μl of HSMP buffer containing 300 mM imidazole. Pseudomonas aeruginosa PAO1 ΔpslA ΔpelF cells containing plasmid pBBR1-MCS5 were subjected to the same method to obtain the PelF-deficient elution fraction. Both the His10-PelF-containing and PelF-deficient elution fractions were analyzed by SDS-PAGE.
Quaternary structure analysis by gel filtration chromatography.
Elution fractions containing PelF were loaded onto a Superdex S-200 10/300 GL column (GE Healthcare, Piscataway, NJ) preequilibrated with 50 mM phosphate (pH 7.6), 150 mM NaCl, and 10 mM β-mercaptoethanol. A flow rate of 0.3 ml/min was used.
PelF and copurified protein identifications.
Proteins that were abundant and with an apparent molecular mass expected for PelF, as well as three other proteins with apparent molecular masses of 88 kDa, 35 kDa, and 19 kDa, were analyzed by tryptic peptide fingerprinting using matrix-assisted laser desorption ionization–time of flight–time of flight mass spectrometry (MALDI-TOF-TOF/MS).
Quantification of UDP-glucose in cell lysates.
Overnight cultures of Pel-producing and Pel-deficient mutants were diluted 1:50 in fresh PI medium and incubated at 37°C for 12 to 14 h until an optical cell density at 600 nm of 2.0 was obtained. Cultures were collected by centrifugation at 5,000 × g for 10 min at 4°C, and cells were washed twice with saline. Washed cells were diluted in HSMP buffer containing 10 mM imidazole. Cells were lysed by sonication as described above. The cell lysate was centrifuged at 15,000 × g for 20 min at 4°C to remove cellular debris and unlysed cells. Finally, the supernatant was centrifuged at 100,000 × g for 2 h to obtain the soluble fraction. To account for the amount of bacteria in the supernatant of each sample, quantification from the UDP-glucose assay was normalized to the amount of total protein in each sample. Total protein concentrations in all supernatants (subcellular fractions) were measured using the Bradford protein assay kit (Bio-Rad). A volume of 50 μl of each soluble fraction was added to the corresponding well of a 96-well flat-bottomed Greiner μClear plate, and the absorbance at 340 nm was recorded. Each well containing a soluble fraction was mixed with 0.01 units of UDP-glucose dehydrogenase (Calbiochem, La Jolla, CA) and 4 mM NAD+ (Sigma). The reaction mixtures were incubated at 30°C for 0, 10, 15, 20, 25, 30, 35, 40, and 45 min, and the absorbance at 340 nm was recorded after each incubation period. The quantity of NADH produced in the reaction at each time point was calculated using the absorbance at 340 nm (εNADH, 6,220 m−1 cm−1). The moles of NADH produced per ml of samples was calculated using Beer's law, and total UDP-glucose used in the reaction was calculated.
PelF activity.
Soluble subcellular fractions (30 μl) obtained from PelF-deficient mutants were added to 96-well flat-bottomed plates. Half of these wells were mixed with 20 μl of the PelF-enriched elution fraction containing 20 μg of total protein, and the remaining wells were mixed with 20 μl of the PelF-deficient elution fraction as a negative control. Mixtures were incubated at 37°C for 1 h. After this incubation period, each well was mixed with 0.01 units of UDP-glucose dehydrogenase and 4 mM NAD+, mixtures were incubated for 0 to 45 min at 30°C, and the absorbance at 340 nm was measured. The concentration of UDP-glucose was calculated using Beer's Law as described above.
Purification of Pel oligomers.
A single colony of P. aeruginosa Δalg8 ΔpslA (3), an alginate-negative and Psl-negative but Pel-producing mutant, was cultivated in 500 ml of PI medium as described above. The pellicle that formed at the air-liquid interface was mechanically removed and washed with MilliQ water. The pellicle was freeze-dried and weighed. The dried pellicle was suspended in 200 ml of 1% (wt/vol) NaOH and then boiled at 100°C for 30 min. After cooling, the digested pellicle material (Pel) was spun down by centrifugation at 7,000 × g for 20 min. After centrifugation, Pel was dissolved in Cross-Beaver reagent (200 ml of 12.1 M HCl mixed with 100 g of ZnCl2) and incubated for 15 h at room temperature. After incubation, the Pel in the Cross-Beaver reagent was slowly mixed with 4 volumes of 95% ethanol. The precipitate that formed after gentle shaking was centrifuged at 7,000 × g for 30 min. The supernatant was discarded, and the Pel sediment was dissolved in 5 ml phosphate buffer. Purified Pel along with other components was incubated with 15 μg/ml DNase I and 15 μg/ml RNase A at 37°C for 6 h. Then, pronase E was added to a final concentration of 20 μg/ml, and the solution was incubated at 37°C for a further 18 h. The Pel solutions were dialyzed against 5 liters of MilliQ water for 48 h and then freeze-dried. The dried Pel was subjected to acetic acid hydrolysis by heating it at 65°C for 2 h, and acetic acid was removed by evaporation. The dried material was scratched from the walls of the tube and weighed. Pel oligomers were dissolved at a concentration of 500 μg/ml in HSMP buffer and used for the in vitro UDP-glucose dehydrogenase-based assay.
In vitro UDP-glucose dehydrogenase-based assay for PelF activity.
Enzyme activity of UDP-glucose dehydrogenase was assessed as previously described (24). To assess the enzymatic activity of PelF, a reaction mixture containing 50 mM HEPES buffer (pH 7.4), 2 mM UDP-glucose, 5 μg Pel oligomers, and 20 μg PelF was made. According to the reaction requirements for known glycosyltransferase cofactors, MgCl2 or MnCl2 or CaCl2, each at a concentration of 5 mM, was added. To assess the roles that the contents of different subcellular fractions may play, 5-μl aliquots of soluble or inner membrane fractions containing 4 mg/ml protein were added to the reaction mixture. To study the roles of various-sized components in the soluble fractions, filtration of the soluble fraction was done through a filter with a 10-kDa cutoff. The retentate was dissolved in HSMP buffer containing 300 mM imidazole, and 5 μl retentate suspension containing 4 mg/ml of total protein was added to the reaction mixture. Similarly, filtrate containing 1 mg/ml of total protein was added to the reaction mixture. In all cases, the total volume was adjusted to 40 μl, and the mixtures were set in 96-well flat-bottomed Greiner μClear plates. Plates were incubated at 37°C for 0, 10, 15, 20, 25, 30, 35, 40, and 45 min. All wells were mixed postincubation with 0.01 units of UDP-glucose dehydrogenase (Calbiochem, La Jolla, CA) and 4 mM NAD+ (Sigma), and the total volume was adjusted to 50 μl. The reaction plates were incubated at 30°C for 1 to 10 min. The absorbance at 340 nm was measured. The moles of NADH produced per ml of sample was calculated by using Lambert-Beer's law, and total UDP-glucose used in the reaction was calculated.
UDP-glucose dehydrogenase-based assay using the heat inactivation and protease-soluble fraction.
Three 1.5-ml microcentrifuge tubes were marked A, B, and C. In each tube, 250 μl of soluble fraction containing 4 mg/ml total protein was poured. In tubes A and B, 100 μg/ml protease (Sigma-Aldrich, Auckland, New Zealand) was added, and the mixture was incubated for 3 h. Tube B was heated at 95°C for 20 min (as recommended by the manufacturer, to inactivate the protease). Tube C was also heated at 95°C for 20 min to inactivate total proteins. All tubes were placed in ice for subsequent use in assays. A volume of 5 μl from each tube was used to assess the activity of PelF in the presence of heat-treated and protease-treated soluble fractions. The assay was conducted as described above in the previous paragraph.
Site-directed mutagenesis of pelF.
The plasmid pBBR1MCS-5::His10-PelF, which encodes the His10-tagged protein PelF, was used as a template. Site-directed mutations in the coding sequence of PelF were created using the site-directed ligase-independent mutagenesis (SLIM) method (25–27). D303A, D362A, E405A, E413A, and E405A/E413A mutations were generated using four primers for each mutation. Instead of using all four primers in a single reaction, the primers were split into two pairs to produce two-tailed products, as described previously (27). Briefly, primers FrwL and RevS for respective mutations were used to produce product A, and primers RevL and FrwS were used to produce product B. Both products A and B were treated with DpnI to digest the template plasmids. The two PCR products were mixed in equimolar amounts and allowed to hybridize by incubation in H buffer (150 mM NaCl, 25 mM Tris, 20 mM EDTA; pH 8.0) at 99°C for 3 min, followed by three cycles of 65°C for 5 min and 30°C for 40 min. Competent E. coli TOP10 cells were transformed using the hybridized products. Plasmid-containing cells were selected on LB medium containing 10 μg/ml gentamicin. The plasmids were extracted and sequenced to confirm the mutations. For mutations D301A, D360A, R325A, K330A, and K333A, fragments PstI-D301A-SmaI and PstI-D360A-SmaI (Genscript, Piscataway, NJ) and PstI-R325A-SmaI, PstI-K330A-SmaI, and PstI-K333A-SmaI (Integrated DNA Technologies) (Table 2), harboring the respective mutations, were commercially synthesized by the indicated company through the company customscience (Auckland, New Zealand). The plasmid pBBR1MCS-5::His10-PelF was isolated and digested with enzymes PstI and SmaI. The 253-bp fragment was replaced with PstI-D301A-SmaI, PstI-D360A-SmaI, PstI-R325A-SmaI, PstI-K330A-SmaI, or PstI-K333A-SmaI fragments to generate D301A, D360A, R325A, K330A, and K333A mutations, respectively. Competent E. coli TOP10 cells were transformed using plasmids harboring the corresponding mutations. The mutations were confirmed by sequencing and/or by restriction digest analysis. Confirmed plasmids were transformed into P. aeruginosa PAO1 ΔpslA ΔpelF to observe pellicle formation. Congo red binding assays were performed as previously described, and the relative percentage of Congo red binding was determined (3). Congo red results are expressed as a percentage, with the percentages of Congo red bound to PelF-producing and PelF-deficient strains set as 100% and 0%, respectively.
Table 2.
Sequences of synthetic DNA fragments used in this study
| DNA fragment | Sequencea |
|---|---|
| D301A | CCCGGGTGATCCCCAACGGCATCGCCCTCGATGCCTGGACCGGCGCCCTCGAACGGCGGCCGCCGGGGATTCCGCCGGTGGTCGGGCTGGTCGGCCGGGTAGTGCCGATCAAGGACGTGAAGACCTTCATCCGCGCCATGCGCGGGGTGGTCAGCGCGATGCCGGAGGCGGAGGGCTGGATCGTCGGTCCGGAGGAGGAAGATCCGGACTATGCCAGCGAATGCCGCAGCCTGGTGGCCAGCCTCGGCCTGCAG |
| D360A | CCCGGGTGATCCCCAACGGCATCGACCTCGATGCCTGGACCGGCGCCCTCGAACGGCGGCCGCCGGGGATTCCGCCGGTGGTCGGGCTGGTCGGCCGGGTAGTGCCGATCAAGGACGTGAAGACCTTCATCCGCGCCATGCGCGGGGTGGTCAGCGCGATGCCGGAGGCGGAGGGCTGGATCGTCGGTCCGGAGGAGGAAGCTCCGGACTATGCCAGCGAATGCCGCAGCCTGGTGGCCAGCCTCGGCCTGCAG |
| R325A | CCCGGGTGATCCCCAACGGCATCGATCTCGACGCCTGGACCGGCGCCCTCGAACGGCGGCCGCCGGGGATTCCGCCGGTGGTCGGGCTGGTCGGCGCCGTAGTGCCGATCAAGGATGTGAAGACCTTCATCCGCGCCATGCGCGGTGTGGTCAGCGCGATGCCGGAGGCGGAAGGCTGGATCGTCGGTCCGGAGGAGGAAGACCCGGACTATGCCAGCGAATGCCGCAGCCTGGTGGCCAGCCTCGGCCTGCAG |
| K330A | CCCGGGTGATCCCCAACGGCATCGATCTCGACGCCTGGACCGGCGCCCTCGAACGGCGGCCGCCGGGGATTCCGCCGGTGGTCGGGCTGGTCGGCCGGGTAGTGCCGATCGCCGATGTGAAGACCTTCATCCGCGCCATGCGCGGTGTGGTCAGCGCGATGCCGGAGGCGGAAGGCTGGATCGTCGGTCCGGAGGAGGAAGACCCGGACTATGCCAGCGAATGCCGCAGCCTGGTGGCCAGCCTCGGCCTGCAG |
| K333A | CCCGGGTGATCCCCAACGGCATCGATCTCGACGCCTGGACCGGCGCCCTCGAACGGCGGCCGCCGGGGATTCCGCCGGTGGTCGGGCTGGTCGGCCGGGTAGTGCCGATCAAGGATGTGGCCACCTTCATCCGCGCCATGCGCGGTGTGGTCAGCGCGATGCCGGAGGCGGAAGGCTGGATCGTCGGTCCGGAGGAGGAAGACCCGGACTATGCCAGCGAATGCCGCAGCCTGGTGGCCAGCCTCGGCCTGCAG |
Underlined and italicized nucleotides indicate PstI and SmaI restriction sites; underlined nucleotides indicate changes to introduce site-directed mutations; italicized and boldface nucleotides indicate silent mutations introduced to adjust the CG content, which was problematic during DNA synthesis; boldface nucleotides indicate silent mutations introduced to add a new restriction site, i.e., ClaI.
RESULTS
Ability of N-terminally His10-tagged PelF to restore pellicle formation in pelF deletion mutants.
P. aeruginosa PAO1 ΔpelF ΔpslA (3) complemented with the plasmid pBBR1-MCS5::His10-PelF produced a pellicle at the air-liquid interface when grown in static culture, as shown by total Congo red staining of 72.5% ± 3.8% (mean ± standard deviation). In contrast, the pBBR1-MCS5-harboring mutant (negative control) was unable to form a pellicle at the air-liquid interface, and total Congo red staining of only 34% ± 4.1% was obtained.
Subcellular localization of PelF.
Envelope and cytosolic fractions of mutants PAO1 ΔpelF ΔpslA harboring pBBR1-MCS5::His10-PelF, PAO1 ΔpelF ΔpslA harboring pBBR1-MCS5, and PAO1ΔpslA were subjected to SDS-PAGE and immunoblotting with anti-His antibodies. Results showed that the His-tagged PelF was present only in the soluble fraction of PAO1 ΔpelF ΔpslA harboring pBBR1-MCS5::His10-PelF. The identified protein showed an apparent molecular mass of 58 kDa, which corresponds to the theoretical molecular mass of PelF (Fig. 1).
Fig 1.
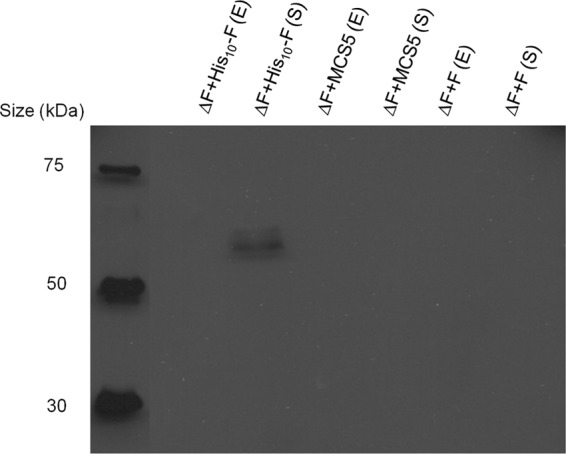
Immunoblot results using anti-His antibodies to determine the subcellular localization of PelF. Soluble (S) and envelope (E) fractions of P. aeruginosa PAO1 ΔpslA ΔpelF harboring pBBR1-MCS-5::His10-PelF, empty vector (pBBR1-MCS-5), and pBBR1-MCS-5::PelF (without His tag) were subjected to SDS-PAGE.
Purification and identification of PelF.
The soluble fraction of the His10-PelF-producing strain was subjected to affinity chromatography by using Ni-nitrilotriacetic acid–agarose in order to purify His-tagged PelF. His-tagged PelF was partially purified, as shown by the SDS-PAGE analysis. A distinct protein band exhibiting an apparent molecular mass of 58 kDa was obtained (Fig. 2). Tryptic peptide fingerprinting analysis in combination with MALDI-TOF/MS enabled identification of this 58-kDa protein as His10-PelF (Table 3). The native His10-PelF molecular mass was determined by gel filtration chromatography, and an apparent molecular mass of about 65 kDa was found. The ratio between estimated and actual molecular mass was 1.12, suggesting that the native PelF is present as a monomer. Three other proteins with apparent molecular masses of 88 kDa (band A), 34 kDa (band B), and 19 kDa (band C) (Fig. 2) coeluted with PelF. These proteins were absent in the elution fraction when we used the PelF-deficient mutant as the source. Tryptic peptide fingerprinting analysis in combination with MALDI-TOF/MS showed that the 88-kDa protein was an ATP-dependent protease (GI 15595976) and the 34-kDa protein was a hypothetical protein, PA4657 (GI 15599852) of P. aeruginosa. The third protein, with an apparent molecular mass of 19 kDa, showed the best match with a Gcn5-related N-acetyltransferase (GI 239721) of Serratia marcescens.
Fig 2.
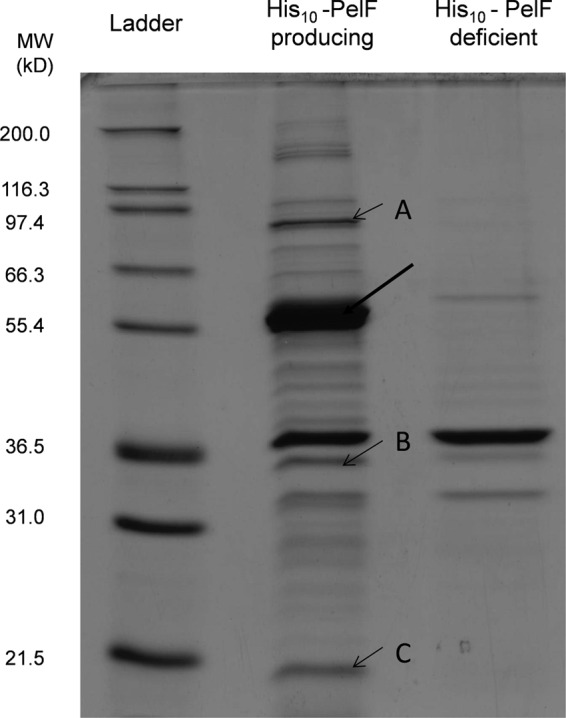
SDS-PAGE analysis of partially purified PelF. Soluble fractions of PelF-producing and PelF-deficient strains were subjected to a HSMP column, and elution fractions were analyzed by SDS-PAGE. An arrow (thick) indicates the presence of His10-PelF. Three proteins coeluting with PelF were identified: A, ATP-dependent protease (GI 15595976); B, PA4657 (GI 15599852) of P. aeruginosa; C, Gcn5-related N-acetyltransferase (GI 239721) (best match to Serratia marcescens).
Table 3.
PelF peptides identified by MALDI-TOF/MS
| Peptide no. |
Mr |
Missa | Scoreb | Expectedc | Peptided | ||
|---|---|---|---|---|---|---|---|
| Observed | Exptl | Calculated | |||||
| 2 | 1,201.5825 | 1,200.5752 | 1,200.6251 | 1 | 59 | 2.3e−007 | R.IGREDFLHSK.A |
| 3 | 1,216.5494 | 1,215.5421 | 1,215.5957 | 0 | 68 | 2.5e−008 | R.YYTEALMLGR.Y |
| 4 | 1,227.6030 | 1,226.5957 | 1,226.6520 | 0 | 68 | 2.4e−008 | R.WQAAQAVGLQR.V |
| 7 | 1,323.6367 | 1,322.6294 | 1,322.6870 | 0 | 79 | 1.5e−008 | R.YLLSEHGIYTK.E |
| 8 | 1,353.5862 | 1,352.5789 | 1,352.6360 | 0 | 88 | 7.3e−011 | K.ASWEAITAGYER.Y |
| 10 | 1,387.6714 | 1,386.6641 | 1,386.7255 | 0 | 105 | 1.6e−012 | R.AANPIVALYEGNR.Q |
| 12 | 1,413.8033 | 1,412.7960 | 1,412.8615 | 0 | 79 | 5.8e−010 | R.RPPGIPPVVGLVGR.V |
| 14 | 1,484.7013 | 1,483.6940 | 1,483.7630 | 0 | 94 | 2e−011 | R.AGEVVAIADPQATSR.A |
| 15 | 1,521.6864 | 1,520.6791 | 1,520.7479 | 0 | 117 | 2e−013 | R.SMQAPVFMLAEAAR.R |
| 16 | 1,537.6743 | 1,536.6670 | 1,536.7428 | 0 | (52) | 3.3e−007 | R.SMQAPVFMLAEAAR.R + oxidation (M) |
| 17 | 1,553.6725 | 1,552.6652 | 1,552.7377 | 0 | (29) | 5.7e−005 | R.SMQAPVFMLAEAAR.R + 2 oxidation (M) |
| 18 | 1,600.7485 | 1,599.7412 | 1,599.8078 | 1 | 29 | 0.00021 | R.VERYYTEALMLGR.Y |
| 19 | 1,614.7512 | 1,613.7439 | 1,613.8008 | 1 | 21 | 0.00039 | R.ELIEGADAEDRALGR.A |
| 20 | 1,839.8754 | 1,838.8681 | 1,838.9526 | 0 | 142 | 3.1e−016 | R.VIPNGIDLDAWTGALER.R |
| 23 | 1,968.8033 | 1,967.7960 | 1,967.8876 | 0 | 106 | 1.2e−012 | R.YCTDPSFVNYFWTLR.S + carbamidomethyl (C) |
| 25 | 2,090.0012 | 2,088.9939 | 2,089.0812 | 0 | 106 | 1.3e−012 | R.MLHSISTGYAGLLGCILQR.R + carbamidomethyl (C) |
| 28 | 2,733.1775 | 2,732.1702 | 2,732.2969 | 0 | 176 | 1.8e−018 | R.FFHYPETPDVEEGDALLDLLAEGR.I |
| 33 | 3,231.4546 | 3,230.4473 | 3,230.5731 | 0 | 127 | 9.8e−015 | K.IDLAQANWIAENPDEQLSTGLDAEVSYIR.R |
| 34 | 3,327.4819 | 3,326.4746 | 3,326.6108 | 0 | 127 | 9e−015 | R.HYPIPDNVLHIEEHFLETAWSSPNPQTR.Q |
| 35 | 3,359.5237 | 3,358.5164 | 3,358.6680 | 1 | 159 | 6.5e−018 | R.KIDLAQANWIAENPDEQLSTGLDAEVSYIR.R |
| 36 | 3,515.6355 | 3,514.6282 | 3,514.7691 | 2 | 61 | 1.5e−007 | R.KIDLAQANWIAENPDEQLSTGLDAEVSYIRR.L |
The number of missed cleavage sites.
The score is the −log10(P) value, where P is the probability that the observed match is a random event. Individual ion scores of >56 indicate identity or extensive homology (P ≤ 0.05).
Expected score based on BLAST search.
The sequence between the peptides was identified by MS. The amino acid before the period at the N terminal and that after the period at the C terminal indicate the cleavage sites.
Is UDP-glucose the substrate of PelF and a precursor for Pel synthesis?
In order to assess whether the presence or absence of PelF impacts the intracellular concentration of UDP-glucose, the proposed substrate for PelF, a UDP-glucose dehydrogenase-based assay was developed. The UDP-glucose dehydrogenase-based assay was performed as described in Materials and Methods. The UDP-glucose dehydrogenase displayed typical Michaelis-Menton kinetics under these conditions, yielding a Km of 77 μM and Vmax of 44 mol/minute, enabling sensitive detection of UDP-gluocse in the reaction mixture.
The UDP-glucose concentrations of the soluble fractions of each of the PelF-deficient and PelF-producing strains were analyzed using this new method and yielded 0.97 ± 0.06 and 0.533 ± 0.042 μmol/mg of protein, respectively.
When partially purified His10-PelF was added to the cell lysate of the PelF-deficient mutant, the concentration of UDP-glucose was reduced from 1.02 ± 0.06 to 0.66 ± 0.29 μmol/mg of total protein when incubated for 30 min. The same elution fraction obtained by purification of a strain lacking His10-PelF did not impact the total UDP-glucose concentration when added to cell lysate of the PelF-deficient mutant (Fig. 3).
Fig 3.

Concentration of UDP-glucose in the soluble fraction of various P. aeruginosa PAO1 mutants. PAO1ΔA (PAO1 ΔpslA), Psl-deficient/Pel-producing mutant; PAO1ΔFΔA (PAO1 ΔpelFΔpslA), Pel-deficient/Psl-deficent mutant; PAO1ΔFΔA (PAO1 ΔpelFΔpslA) + PelF, partially purified PelF added to soluble fraction of cell lysate from Pel-deficient/Psl-deficent mutant (*PelF was partially purified as described in Materials and Methods); PAO1ΔFΔA (PAO1 ΔpelFΔpslA) + No PelF, the elution fraction not containing PelF but containing coeluted proteins was added to the soluble fraction of the cell lysate from the Pel-deficient/Psl-deficient mutant. All experiments were conducted in triplicate, and mean values presented in the graphs. Standard deviations are shown as the error bars.
In vitro glycosyltransferase activity of PelF.
The UDP-glucose dehydrogenase-based assay was performed as described in Materials and Methods. Our data indicated that PelF did not show any activity when Pel oligomers were added to the reaction mixture. Similarly, no activity of PelF was observed when potential metal cofactors MnCl2, MgCl2, and CaCl2 were used in the assay (data not shown). To assess the role of undecaprenyl phosphate as an acceptor molecule, inner membrane (IM) fractions were added to the assay reaction mixture. UDP-glucose concentrations did not change in the presence or absence of IM, compared to the reaction mixture to which PelF was not added. Interestingly, when the soluble fraction of the ΔpelF mutant was added to the assay, PelF showed activity, reducing the UDP-glucose concentration from 53.9 ± 2.98 to 39.9 ± 2.84 nmol. When retentate fractions (>10 kDa) were added to the reaction mixture in the presence of PelF, the UDP-glucose concentration decreased from 57.65 ± 4.62 to 40.80 ± 2.27 nmol. UDP-glucose concentrations did not show any significant difference in the presence or absence of PelF when filtrate (<10 kDa) was added to the assay reaction mixture (Fig. 4).
Fig 4.

PelF activity as measured by UDP-glucose consumption. The residual UDP-glucose after reaction completion was determined. PelF+IM, PelF and 5 μl IM fraction (containing 4 mg/ml total protein); no PeLF+IM, partially purified suspension without PelF and 5 μl (containing 4 mg/ml total protein) inner membrane fraction; PelF+C, PelF and 5 μl soluble fraction (containing 4 mg/ml total protein); no PeLF+C, partially purified suspension without PelF and 5 μl soluble fraction (containing 4 mg/ml total protein); PelF+Ret, PelF and 5 μl retentate of soluble fraction (containing 4 mg/ml total protein) filtered; no PeLF+Ret, partially purified suspension without PelF and 5 μl retentate of soluble fraction (containing 4 mg/ml total protein); PelF+Filt, PelF and 5 μl filtrate of soluble fraction (containing 1 mg/ml total protein); no PeLF+Filt, partially purified suspension without PelF and 5 μl filtrate of soluble fraction (containing 1 mg/ml total protein). All experiments were conducted in triplicate, and mean values are presented. Standard deviations are shown as error bars.
Roles of other soluble proteins on PelF activity.
To understand the role of other soluble proteins (>10 kDa) in PelF activity, heat- and protease-treated soluble fractions were used in an assay. PelF showed no activity when proteins were inactivated by protease or/and heat. The UDP-glucose concentrations in assays were 57.60 ± 2.59, 60.55 ± 3.21, and 58.54 ± 3.57 nmol when heat-inactivated, protease-treated, or protease-treated/heat-treated soluble fractions were used, respectively (Fig. 5). In contrast, when PelF was used in the presence of the untreated soluble fraction, the UDP-glucose concentration was reduced to 41.2 ± 2.31 nmol.
Fig 5.

PelF activity measured by UDP-glucose consumption. The residual UDP-glucose was measured after completion of the assay. PelF+C, PelF and 5 μl untreated soluble fraction (containing 4 mg/ml total protein); PelF+C (HT), PelF and 5 μl heat-treated soluble fraction; PelF+C(PT), PelF and 5 μl protease-treated soluble fraction; PelF+C (HT/PT), PelF and 5 μl protease- and heat-treated soluble fraction.
Analysis of the catalytic mechanism of PelF by site-directed mutagenesis.
To identify amino acid residues involved in the catalytic mechanism of PelF, site-specific mutagenesis of conserved residues was performed. In PelF, E405 and E413 are proposed to be the two glutamic acids of the EX7E motif, based on sequence homology to previously characterized GT-4 glycosyltransferases. Replacement of neither E405 nor E413 abolished the activity of the enzyme. However, replacement of E405 with A did reduce the activity of the enzyme to 30.35% ± 3.15%, whereas replacement of E413 with A showed no significant effect on the activity of the enzyme, as assessed by pellicle formation in the Congo red binding assay (Fig. 6). The double mutant E405A/E413A showed an in vivo activity of 36.74% ± 3.24% of wild type. This was similar to the result with the single mutant E405A, suggesting that the second glutamic acid does not play a role in the enzymatic activity of PelF (Fig. 6). The roles of a conserved arginine and lysine had been shown previously in glycosyltransferases of the GT-4 family. To study the roles of the conserved lysine and arginine in PelF, R325, K330, and K333 were replaced by A. Interestingly, the mutants R325A or K330A were not able to form any pellicle at the air-liquid interface, as seen with the PelF-deficient strain. These data suggested that these amino acid residues play a critical role in PelF activity. The mutant K333A formed a pellicle similar to the wild type, with Congo red binding that was 96.12% ± 3.51% of that of the wild type.
Fig 6.

In vivo activities of PelF and its variants as determined by pellicle production at the air-liquid interface by various P. aeruginosa strains. Pellicle produced by P. aeruginosa strains harboring different variants of PelF after 96-h static cultures was quantified using the Congo red binding assay as described in Materials and Methods. (A) Congo red binding assay results. The percentages shown are the mean values of three independent assays. Standard deviations are presented as error bars. (B) Pellicle formation mediated by variants of PelF, compared with that of wild-type His10-PelF.
Although PelF appears to belong to the GT4 family, which contains metal-independent retaining glycosyltransferases, it does appear to have two DXD motifs, which are known to be involved in the catalytic activity of metal-dependent glycosyltransferases. To exclude the possibility of these being involved in PelF's catalytic mechanism, all four aspartic acids were replaced by alanine (14, 15). The pellicle-forming abilities of the mutants D301A, D303A, D360A, and D362A were 95.86% ± 3.03%, 96.39% ± 2.47%, 95.21% ± 5.21%, and 97.51% ± 3.86%, respectively, compared to the wild type (Fig. 6).
DISCUSSION
Little is known about the structure and chemical nature of Pel. In this study, the production of PelF, a putative glycosyltransferase, was demonstrated, and its functional role in Pel formation was assigned. Here, experimental evidence for the subcellular localization of PelF and the use of UDP-glucose as the substrate for biosynthesis of the Pel polysaccharide was provided. Essential amino acid residues involved in the activity of PelF were identified. PelF, tagged with His10 at its N terminus, was able to restore pellicle formation when expressed in a PelF-deficient strain, suggesting that the addition of the extra amino acids at the N terminus of PelF did not interfere with the function of the protein. Bioinformatics analysis showed that PelF is a putative glycosyltransferase with no transmembrane helices. This suggested that PelF might be soluble and located in the cytosol. Subcellular fractionation confirmed that PelF is mainly found in the soluble cytosolic fractions of cells (Fig. 1).
Three proteins that coeluted with PelF had apparent molecular masses similar to PelA (104.5 kDa), PelE (36 kDa), and PelC (18.7 kDa). The absence of these proteins in the elution fraction derived from the pelF-deficient mutant suggested that these proteins potentially interact with PelF. However, these proteins were not found to be encoded by the pel operon. The hypothetical protein PA4657 (GI 15599852) in P. aeruginosa showed sequence similarity with FAD/NAD-dependent oxidoreductases. Previously, it was shown that FAD-dependent oxidoreductase is required to maintain disulfide bonds of bacterial proteins (28). PelF contains 6 cysteine residues that may be involved in disulfide bond formation. We speculate that PA4657 is required to maintain the disulfide bonds of PelF.
In a previous study, carbohydrate analysis of the total exopolysaccharides produced by Pel-deficient and Pel-producing strains showed that Pel might be composed of glucose (5). Hence, it was assumed that UDP-glucose could be a direct precursor of Pel synthesis and used as the substrate by PelF, the only putative glycosyltransferase encoded by the pel operon. Consequently, in an isogenic pelF knockout strain, i.e., in the absence of the putatively UDP-glucose-converting PelF, UDP-glucose might be present in the cytosol at elevated concentrations compared to the wild type. For this purpose, a UDP-glucose dehydrogenase-based assay was developed, and it confirmed that the UDP-glucose levels in the pelF knockout mutant were significantly elevated, suggesting that UDP-glucose is a substrate of PelF, a precursor of Pel, and that glucose is a constituent of the Pel polysaccharide (Fig. 3). UDP-glucose quantification has been done previously by using capillary zone electrophoresis (CE) (29) and high-performance liquid chromatography (30). MALDI-TOF/MS has also been used for detection of a range of metabolites in cells (31). Here, the use of UDP-glucose dehydrogenase to quantify the concentration of UDP-glucose in samples of cell extracts is reported. The low Km values of UDP-glucose dehydrogenase for UDP-glucose make this enzyme an ideal candidate for sensitive detection of UDP-glucose (24). The UDP-glucose dehydrogenase catalyzes a 2-fold oxidation of UDP-glucose (UDP-α-d-glucose) and reduces NAD(P)/NAD+ to NADH, the concentration of which can be easily monitored spectrophotometrically (32). Previously, it was reported that NADH does not inhibit UDP-glucose dehydrogenase by binding to its active site at higher concentrations (33, 34). In vitro PelF utilized UDP-glucose as a substrate in the presence of soluble components, with an apparent molecular mass of >10 kDa, indicating that PelF uses UDP-glucose as a donor substrate toward glycosylation of an unknown receptor molecule. In E. coli, during O-antigen biosynthesis, undecaprenyl phosphate acts as a receptor (35). That there was no PelF activity in the presence of the IM fraction suggested that PelF does not require undecaprenyl phosphate as an acceptor molecule for initiation of polysaccharide biosynthesis (Fig. 4). Interestingly, PelF only showed activity in the presence of a cytosolic soluble fraction containing macromolecules with a size of ≥10 kDa (Fig. 4). This suggested that the PelF activity requires the presence of another factor, i.e., an acceptor molecule and/or an activator protein. Heat and protease treatment of this soluble fraction abolished PelF activity, which suggested that another protein is required for PelF activity (Fig. 5). Previously, it was shown that the activation of some glycosyltransferases depends upon their interactions with auxiliary proteins. One glycosyltransferase, DesVII, which is involved in the biosynthesis of the macrolide antibiotics methymycin and pikromycin in Streptomyces venezuelae, was only activated if an auxiliary protein, DesVIII, was present (36–38). Similarly, other glycosyltransferases have been found to require another auxiliary protein for their activity (39–41). A recent study showed the role of two auxiliary proteins, Srm6 and Srm28, in the activation of two glycosyltransferases, Srm5 and Srm29, respectively, and both are required for the biosynthesis of spiramycin by Streptomyces ambofaciens (42). Activity of the glycosyltransferase EryCIII, which is required for biosynthesis of the antibiotic erythromycin D, is dependent upon an auxiliary protein, EryCII (43). Although the roles of these auxiliary proteins are not clear, it has been proposed that these auxiliary proteins are required to induce conformational changes of the glycosyltransferases, which consequently activate these enzymes (36, 42).
Based on amino acid sequence similarities, glycosyltransferases have been classified into 91 families in the CAZy database. Bioinformatics analysis suggested that PelF belongs to glycosyltransferase family 4 (CAZy) and the glycosyltransferase 1 family (Pfam), respectively. Members of this family are retaining glycosyltransferases. A conserved EX7E motif present in the C-terminal domains of the retaining glycosyltransferases has been previously reported as a characteristic of these glycosyltransferases (44). An alignment of the amino acid sequence of PelF with glycosyltransferases from the GT-4 family showed the presence of conserved amino acid residues and an EX7E motif in the C-terminal domain of the protein. Previous studies proposed that these conserved residues are involved in stabilizing the donor substrate and in glycosidic bond formation as a nucleophile (45–50). Here, it was shown that replacement of E405 in this motif with alanine caused a significant reduction in PelF activity, whereas mutation of the second glutamic acid, E413, had almost no impact on PelF activity, as shown by pellicle production at the air-liquid interface (Fig. 6), although according to the model proposed by Kapitonov and Robert (44) the second glutamic acid residue in the EX7E motif is anticipated to be the catalytic residue in retaining glycosyltransferases. However, it has been shown that this is not always the case. For example, for AceA, a mannosyltransferase from Acetobacter xylinum (45), human muscle glycogen synthase (46), and Alg11, an α1,2-mannosyltransferase from Saccharomyces cerevisiae (51), replacement of the first and not the second glutamic acid residue with alanine results in a significant reduction in enzyme activity. However, in Gpi3, which is involved in glycosylphosphatidylinositol biosynthesis in S. cerevisiae, the second glutamic acid residue has been shown to be important for the enzyme activity (48). For alg11, replacement of the first glutamic acid with alanine (E405A) significantly reduced activity, but complete loss of enzyme activity was not observed compared to the Δalg11 mutant. However, a double mutation (E405A/E413A) of both the first and second glutamic acid residues resulted in complete inhibition of Alg11 activity (51). Similarly, with PelF, the mutation of the first glutamic acid E405A did not abolish the activity completely (Fig. 6). Interestingly, a double mutation (E405A/E413A) in PelF also did not result in complete loss of enzyme activity. This indicated that the second glutamic acid is not essential for activity and that perturbation in the motif caused by replacing the second glutamic acid residue with alanine is tolerated by the enzyme, suggesting a distinct reaction mechanism that differs from other glycosyltransferases.
In some glycosyltransferases a DXD signature is present in which the carboxyl groups coordinate a divalent cation and/or a ribose (52, 53). Two conserved DXD motifs were found in PelF. Replacing all four aspartic acid residues with alanine showed no effect on enzyme activity, suggesting that these aspartic acid residues are not essential for the catalytic reaction mechanism, as had been described for other glycosyltransferases (Fig. 6).
In previous studies, a conserved arginine and/or lysine has also been shown to be required for enzyme activity of glycosyltransferases. In AceA (45) and Alg11 (51), replacement of the conserved K211 and K319 with A, respectively, significantly reduced the activity of these enzymes. Similarly, mutagenesis of conserved R604 in chitin synthase from S. cerevisiae results in a drastic decrease of the enzymatic activity (54). With PelF, when the respective K330 and R325 residues were replaced with A, the in vivo enzyme activities of each mutant, K330A or R325A, were abolished (Fig. 6). These data suggest that both K330 and R325 have an essential role in the enzymatic activity of PelF. Structural analysis has shown that R196 and K202 in the mannosyltransferase PimA from mycobacteria (49) and R300 and K305 in the glycogen synthase from E. coli form hydrogen bonds to the oxygens of the distal phosphate group of the donor nucleotide sugar (55). Using the Protein Homology Recognition Engine (PHYRE2 program) (56), a model structure of PelF based on the crystal structure of WaaG from E. coli (PDB ID 2IW1) was generated with 100% confidence (Fig. 7). Based on this structural model and the site-specific mutagenesis data, it is suggested that R325 and K330 in PelF are involved in forming a hydrogen bond with the phosphate oxygens of UDP-glucose. In the PelF structural model, E405, R325, and K330 form a UDP-glucose-binding pocket that could constitute the catalytic site of the enzyme (Fig. 7).
Fig 7.
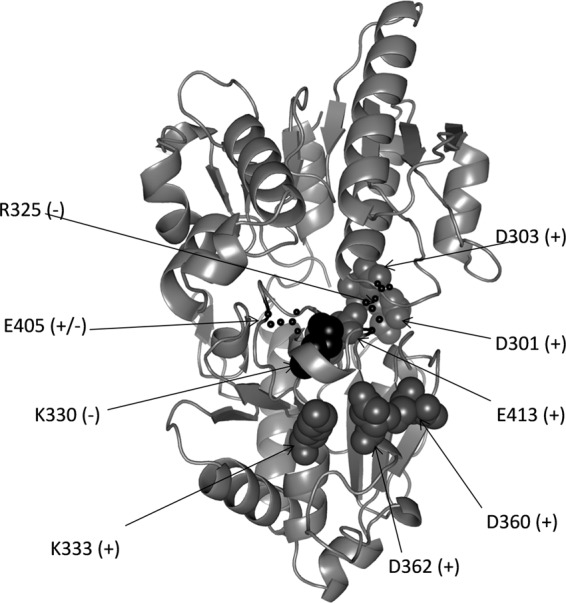
Structural model of PelF generated by PHYRE2, showing localization of the amino acid residues that were replaced with alanine. Selected amino acid residues were replaced with alanine by using site-directed mutagenesis as described in Materials and Methods. Residues targeted for replacement are shown as spheres. +, no impact on activity; +/−, partial loss of enzymatic activity; −, activity of the enzyme was lost.
Overall, our study suggested that PelF is a soluble glycosyltransferase that uses UDP-glucose as a donor substrate toward the biosynthesis of the Pel exopolysaccharide. Site-directed mutagenesis showed that E405, the first glutamic acid of the conserved EX7E motif, plays a significant but nonessential role in PelF activity and that R325 and K330 are essential for the activity of PelF.
ACKNOWLEDGMENTS
This study was supported by research grants to B.H.A.R. from Massey University. A.G. was supported by the Higher Education Commission (HEC) of Pakistan.
Footnotes
Published ahead of print 22 February 2013
REFERENCES
- 1. Singh PK, Schaefer AL, Parsek MR, Moninger TO, Welsh MJ, Greenberg EP. 2000. Quorum-sensing signals indicate that cystic fibrosis lungs are infected with bacterial biofilms. Nature 407: 762–764 [DOI] [PubMed] [Google Scholar]
- 2. Drenkard E. 2003. Antimicrobial resistance of Pseudomonas aeruginosa biofilms. Microbes Infect. 5: 1213–1219 [DOI] [PubMed] [Google Scholar]
- 3. Ghafoor A, Hay ID, Rehm BH. 2011. Role of exopolysaccharides in Pseudomonas aeruginosa biofilm formation and architecture. Appl. Environ. Microbiol. 77: 5238–5246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chitnis CE, Ohman DE. 1993. Genetic analysis of the alginate biosynthetic gene cluster of Pseudomonas aeruginosa shows evidence of an operonic structure. Mol. Microbiol. 8: 583–593 [DOI] [PubMed] [Google Scholar]
- 5. Friedman L, Kolter R. 2004. Genes involved in matrix formation in Pseudomonas aeruginosa PA14 biofilms. Mol. Microbiol. 51: 675–690 [DOI] [PubMed] [Google Scholar]
- 6. Vasseur P, Vallet-Gely I, Soscia C, Genin S, Filloux A. 2005. The pel genes of the Pseudomonas aeruginosa PAK strain are involved at early and late stages of biofilm formation. Microbiology 151: 985–997 [DOI] [PubMed] [Google Scholar]
- 7. Jackson KD, Starkey M, Kremer S, Parsek MR, Wozniak DJ. 2004. Identification of psl, a locus encoding a potential exopolysaccharide that is essential for Pseudomonas aeruginosa PAO1 biofilm formation. J. Bacteriol. 186: 4466–4475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Coulon C, Vinogradov E, Filloux A, Sadovskaya I. 2010. Chemical analysis of cellular and extracellular carbohydrates of a biofilm-forming strain Pseudomonas aeruginosa PA14. PLoS One 5: e14220 doi:10.1371/journal.pone.0014220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vasseur P, Soscia C, Voulhoux R, Filloux A. 2007. PelC is a Pseudomonas aeruginosa outer membrane lipoprotein of the OMA family of proteins involved in exopolysaccharide transport. Biochimie 89: 903–915 [DOI] [PubMed] [Google Scholar]
- 10. Lee VT, Matewish JM, Kessler JL, Hyodo M, Hayakawa Y, Lory S. 2007. A cyclic-di-GMP receptor required for bacterial exopolysaccharide production. Mol. Microbiol. 65: 1474–1484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Franklin MJ, Nivens DE, Weadge JT, Howell PL. 2011. Biosynthesis of the Pseudomonas aeruginosa extracellular polysaccharides, alginate, Pel, and Psl. Front. Microbiol. 2: 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Keiski CL, Harwich M, Jain S, Neculai AM, Yip P, Robinson H, Whitney JC, Riley L, Burrows LL, Ohman DE, Howell PL. 2010. AlgK is a TPR-containing protein and the periplasmic component of a novel exopolysaccharide secretin. Structure 18: 265–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rehm BHA. 2010. Bacterial polymers: biosynthesis, modifications and applications. Nat. Rev. Microbiol. 8: 578–592 [DOI] [PubMed] [Google Scholar]
- 14. Campbell JA, Davies GJ, Bulone V, Henrissat B. 1997. A classification of nucleotide-diphospho-sugar glycosyltransferases based on amino acid sequence similarities. Biochem. J. 326: 929–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Coutinho PM, Deleury E, Davies GJ, Henrissat B. 2003. An evolving hierarchical family classification for glycosyltransferases. J. Mol. Biol. 328: 307–317 [DOI] [PubMed] [Google Scholar]
- 16. Hu YN, Walker S. 2002. Remarkable structural similarities between diverse glycosyltransferases. Chem. Biol. 9: 1287–1296 [DOI] [PubMed] [Google Scholar]
- 17. Bourne Y, Henrissat B. 2001. Glycoside hydrolases and glycosyltransferases: families and functional modules. Curr. Opin. Struct. Biol. 11: 593–600 [DOI] [PubMed] [Google Scholar]
- 18. Unligil UM, Rini JM. 2000. Glycosyltransferase structure and mechanism. Curr. Opin. Struct. Biol. 10: 510–517 [DOI] [PubMed] [Google Scholar]
- 19. Liu J, Mushegian A. 2003. Three monophyletic superfamilies account for the majority of the known glycosyltransferases. Protein Sci. 12: 1418–1431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pak JE, Arnoux P, Zhou S, Sivarajah P, Satkunarajah M, Xing X, Rini JM. 2006. X-ray crystal structure of leukocyte type core 2 β1,6-N-acetylglucosaminyltransferase. Evidence for a convergence of metal ion-independent glycosyltransferase mechanism. J. Biol. Chem. 281: 26693–26701 [DOI] [PubMed] [Google Scholar]
- 21. Lairson LL, Henrissat B, Davies GJ, Withers SG. 2008. Glycosyltransferases: structures, functions, and mechanisms. Annu. Rev. Biochem. 77: 521–555 [DOI] [PubMed] [Google Scholar]
- 22. Remminghorst U, Rehm BH. 2006. In vitro alginate polymerization and the functional role of Alg8 in alginate production by Pseudomonas aeruginosa. Appl. Environ. Microbiol. 72: 298–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680–685 [DOI] [PubMed] [Google Scholar]
- 24. Stewart DC, Copeland L. 1998. Uridine 5′-diphosphate-glucose dehydrogenase from soybean nodules. Plant Physiol. 116: 349–355 [Google Scholar]
- 25. Hay ID, Rehman ZU, Rehm BH. 2010. Membrane topology of outer membrane protein AlgE, which is required for alginate production in Pseudomonas aeruginosa. Appl. Environ. Microbiol. 76: 1806–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chiu J, March PE, Lee R, Tillett D. 2004. Site-directed, ligase-independent mutagenesis (SLIM): a single-tube methodology approaching 100% efficiency in 4 h. Nucleic Acids Res. 32: e174 doi:10.1093/nar/gnh172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chiu J, Tillett D, Dawes IW, March PE. 2008. Site-directed, ligase-independent mutagenesis (SLIM) for highly efficient mutagenesis of plasmids greater than 8kb. J. Microbiol. Methods 73: 195–198 [DOI] [PubMed] [Google Scholar]
- 28. Wang C, Wesener SR, Zhang H, Cheng YQ. 2009. An FAD-dependent pyridine nucleotide-disulfide oxidoreductase is involved in disulfide bond formation in FK228 anticancer depsipeptide. Chem. Biol. 16: 585–593 [DOI] [PubMed] [Google Scholar]
- 29. Lehmann R, Huber M, Beck A, Schindera T, Rinkler T, Houdali B, Weigert C, Haring HU, Voelter W, Schleicher ED. 2000. Simultaneous, quantitative analysis of UDP-N-acetylglucosamine, UDP-N-acetylgalactosamine, UDP-glucose and UDP-galactose in human peripheral blood cells, muscle biopsies and cultured mesangial cells by capillary zone electrophoresis. Electrophoresis 21: 3010–3015 [DOI] [PubMed] [Google Scholar]
- 30. Dorion S, Rivoal J. 2003. Quantification of uridine 5′-diphosphate (UDP)-glucose by high-performance liquid chromatography and its application to a nonradioactive assay for nucleoside diphosphate kinase using UDP-glucose pyrophosphorylase as a coupling enzyme. Anal. Biochem. 323: 188–196 [DOI] [PubMed] [Google Scholar]
- 31. Edwards JL, Kennedy RT. 2005. Metabolomic analysis of eukaryotic tissue and prokaryotes using negative mode MALDI time-of-flight mass spectrometry. Anal. Chem. 77: 2201–2209 [DOI] [PubMed] [Google Scholar]
- 32. Ridley WP, Kirkwood S. 1973. The stereospecificity of hydrogen abstraction by uridine diphosphoglucose dehydrogenase. Biochem. Biophys. Res. Commun. 54: 955–960 [DOI] [PubMed] [Google Scholar]
- 33. Hinterberg B, Klos C, Tenhaken R. 2002. Recombinant UDP-glucose dehydrogenase from soybean. Plant Physiol. Biochem. 40: 1011–1017 [Google Scholar]
- 34. Stewart DC, Copeland L. 1999. Kinetic properties of UDP-glucose dehydrogenase from soybean nodules. Plant Sci. 147: 119–125 [Google Scholar]
- 35. Marolda CL, Vicarioli J, Valvano MA. 2004. Wzx proteins involved in biosynthesis of O antigen function in association with the first sugar of the O-specific lipopolysaccharide subunit. Microbiology 150: 4095–4105 [DOI] [PubMed] [Google Scholar]
- 36. Borisova SA, Zhang C, Takahashi H, Zhang H, Wong AW, Thorson JS, Liu HW. 2006. Substrate specificity of the macrolide-glycosylating enzyme pair DesVII/DesVIII: opportunities, limitations, and mechanistic hypotheses. Angew Chem. Int. Ed. Engl. 45: 2748–2753 [DOI] [PubMed] [Google Scholar]
- 37. Borisova SA, Zhao L, Melancon IC, Kao CL, Liu HW. 2004. Characterization of the glycosyltransferase activity of DesVII: analysis of and implications for the biosynthesis of macrolide antibiotics. J. Am. Chem. Soc. 126: 6534–6535 [DOI] [PubMed] [Google Scholar]
- 38. Hong JS, Park SH, Choi CY, Sohng JK, Yoon YJ. 2004. New olivosyl derivatives of methymycin/pikromycin from an engineered strain of Streptomyces venezuelae. FEMS Microbiol. Lett. 238: 391–399 [DOI] [PubMed] [Google Scholar]
- 39. Hong JS, Park SJ, Parajuli N, Park SR, Koh HS, Jung WS, Choi CY, Yoon YJ. 2007. Functional analysis of desVIII homologues involved in glycosylation of macrolide antibiotics by interspecies complementation. Gene 386: 123–130 [DOI] [PubMed] [Google Scholar]
- 40. Lu W, Leimkuhler C, Gatto GJ, Jr, Kruger RG, Oberthur M, Kahne D, Walsh CT. 2005. AknT is an activating protein for the glycosyltransferase AknS in L-aminodeoxysugar transfer to the aglycone of aclacinomycin A. Chem. Biol. 12: 527–534 [DOI] [PubMed] [Google Scholar]
- 41. Melancon CE, III, Takahashi H, Liu HW. 2004. Characterization of tylM3/tylM2 and mydC/mycB pairs required for efficient glycosyltransfer in macrolide antibiotic biosynthesis. J. Am. Chem. Soc. 126: 16726–16727 [DOI] [PubMed] [Google Scholar]
- 42. Nguyen HC, Karray F, Lautru S, Gagnat J, Lebrihi A, Huynh TD, Pernodet JL. 2010. Glycosylation steps during spiramycin biosynthesis in Streptomyces ambofaciens: involvement of three glycosyltransferases and their interplay with two auxiliary proteins. Antimicrob. Agents Chemother. 54: 2830–2839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Moncrieffe MC, Fernandez MJ, Spiteller D, Matsumura H, Gay NJ, Luisi BF, Leadlay PF. 2012. Structure of the glycosyltransferase EryCIII in complex with its activating P450 homologue EryCII. J. Mol. Biol. 415: 92–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kapitonov D, Yu RK. 1999. Conserved domains of glycosyltransferases. Glycobiology 9: 961–978 [DOI] [PubMed] [Google Scholar]
- 45. Abdian PL, Lellouch AC, Gautier C, Ielpi L, Geremia RA. 2000. Identification of essential amino acids in the bacterial alpha-mannosyltransferase aceA. J. Biol. Chem. 275: 40568–40575 [DOI] [PubMed] [Google Scholar]
- 46. Cid E, Gomis RR, Geremia RA, Guinovart JJ, Ferrer JC. 2000. Identification of two essential glutamic acid residues in glycogen synthase. J. Biol. Chem. 275: 33614–33621 [DOI] [PubMed] [Google Scholar]
- 47. Troutman JM, Imperiali B. 2009. Campylobacter jejuni PglH is a single active site processive polymerase that utilizes product inhibition to limit sequential glycosyl transfer reactions. Biochemistry 48: 2807–2816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kostova Z, Yan BC, Vainauskas S, Schwartz R, Menon AK, Orlean P. 2003. Comparative importance in vivo of conserved glutamate residues in the EX7E motif retaining glycosyltransferase Gpi3p, the UDP-GlcNAc-binding subunit of the first enzyme in glycosylphosphatidylinositol assembly. Eur. J. Biochem. 270: 4507–4514 [DOI] [PubMed] [Google Scholar]
- 49. Guerin ME, Kordulakova J, Schaeffer F, Svetlikova Z, Buschiazzo A, Giganti D, Gicquel B, Mikusova K, Jackson M, Alzari PM. 2007. Molecular recognition and interfacial catalysis by the essential phosphatidylinositol mannosyltransferase PimA from mycobacteria. J. Biol. Chem. 282: 20705–20714 [DOI] [PubMed] [Google Scholar]
- 50. Muniz JR, Alves CA, de Pieri C, Beltramini LM, Selistre-de-Araujo HS, Vettore AL, da Silva FR, Arruda P, Garratt RC, Oliva G, Souza DH. 2004. Overexpression, purification, biochemical characterization, and molecular modeling of recombinant GDP-mannosyltransferase (GumH) from Xylella fastidiosa. Biochem. Biophys. Res. Commun. 315: 485–492 [DOI] [PubMed] [Google Scholar]
- 51. Absmanner B, Schmeiser V, Kampf M, Lehle L. 2010. Biochemical characterization, membrane association and identification of amino acids essential for the function of Alg11 from Saccharomyces cerevisiae, an α1,2-mannosyltransferase catalysing two sequential glycosylation steps in the formation of the lipid-linked core oligosaccharide. Biochem. J. 426: 205–217 [DOI] [PubMed] [Google Scholar]
- 52. Wiggins CA, Munro S. 1998. Activity of the yeast MNN1 alpha-1,3-mannosyltransferase requires a motif conserved in many other families of glycosyltransferases. Proc. Natl. Acad. Sci. U. S. A. 95: 7945–7950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Breton C, Bettler E, Joziasse DH, Geremia RA, Imberty A. 1998. Sequence-function relationships of prokaryotic and eukaryotic galactosyltransferases. J. Biochem. 123: 1000–1009 [DOI] [PubMed] [Google Scholar]
- 54. Nagahashi S, Sudoh M, Ono N, Sawada R, Yamaguchi E, Uchida Y, Mio T, Takagi M, Arisawa M, Yamada-Okabe H. 1995. Characterization of chitin synthase 2 of Saccharomyces cerevisiae. Implication of two highly conserved domains as possible catalytic sites. J. Biol. Chem. 270: 13961–13967 [DOI] [PubMed] [Google Scholar]
- 55. Sheng F, Jia X, Yep A, Preiss J, Geiger JH. 2009. The crystal structures of the open and catalytically competent closed conformation of Escherichia coli glycogen synthase. J. Biol. Chem. 284: 17796–17807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kelley LA, Sternberg MJ. 2009. Protein structure prediction on the Web: a case study using the PHYRE server. Nat. Protoc. 4: 363–371 [DOI] [PubMed] [Google Scholar]
- 57. Kovach ME, Elzer PH, Hill DS, Robertson GT, Farris MA, Roop RM, 2nd, Peterson KM. 1995. Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166: 175–176 [DOI] [PubMed] [Google Scholar]