

Abstract
Enterococcus faecalis encodes a biotin-dependent oxaloacetate decarboxylase (OAD), which is constituted by four subunits: E. faecalis carboxyltransferase subunit OadA (termed Ef-A), membrane pump Ef-B, biotin acceptor protein Ef-D, and the novel subunit Ef-H. Our results show that in E. faecalis, subunits Ef-A, Ef-D, and Ef-H form a cytoplasmic soluble complex (termed Ef-AHD) which is also associated with the membrane. In order to characterize the role of the novel Ef-H subunit, coexpression of oad genes was performed in Escherichia coli, showing that this subunit is vital for Ef-A and Ef-D interaction. Diminished growth of the oadA and oadD single deletion mutants in citrate-supplemented medium indicated that the activity of the complex is essential for citrate utilization. Remarkably, the oadB-deficient strain was still capable of growing to wild-type levels but with a delay during the citrate-consuming phase, suggesting that the soluble Ef-AHD complex is functional in E. faecalis. These results suggest that the Ef-AHD complex is active in its soluble form, and that it is capable of interacting in a dynamic way with the membrane-bound Ef-B subunit to achieve its maximal alkalinization capacity during citrate fermentation.
INTRODUCTION
The main activity of lactic acid bacteria (LAB) during fermentation is the catabolism of sugars present in food, producing lactic acid, but these microorganisms also have the capability to metabolize other substrates, such as citrate. Citrate fermentation by LAB leads to the production of aromatic compounds (diacetyl/acetoin) and CO2, which contributes to the formation of so-called eyes (or holes) in cheeses. Thus, the utilization of citrate in milk by LAB has a positive effect on the quality of the end products. Therefore, the interest of the dairy industry in understanding citrate utilization by LAB has promoted research of the proteins involved in this metabolic pathway (1).
Citrate fermentation in bacteria involves the intracellular conversion of citrate to oxaloacetate (OAA) and acetate by citrate lyase. OAA then is decarboxylated to pyruvate by oxaloacetate decarboxylase, which is then diverted into different metabolic pathways, depending on the metabolic needs. Oxaloacetate-decarboxylating enzymes of two different origins have been described (2). In Gram-negative pathogenic microorganisms, such as Klebsiella pneumoniae and Vibrio cholerae, for which the pathway has been extensively studied (3, 4), oxaloacetate decarboxylase is a multisubunit Na+ pumping membrane-bound complex (termed OAD), while in Gram-positive lactic acid bacteria, such as Lactococcus lactis, the reaction is catalyzed by a soluble, single-subunit enzyme, named CitM, which is a malic enzyme (ME) (5). In all microorganisms, the genes encoding these enzymes are clustered in the cit locus (Fig. 1A). In K. pneumoniae and V. cholerae, the expression of the OAD genes is induced together with other enzymes of the citrate fermentation pathway in the presence of citrate and anaerobiosis, conditions during which the tricarboxylic acid (TCA) cycle is not functional (3, 6), while in fermentative L. lactis, for instance, the cit cluster is induced by acidification of the medium (7). Genome sequences of selected Gram-positive bacteria of the class Lactobacillales have revealed the presence of genes encoding a membrane-bound complex homologous to the OAD complex of Gram-negative bacteria, but the function of these was never studied (8). In a recent publication, we have characterized the cit locus in Enterococcus faecalis in the same class (9). Surprisingly, the cit locus contained the coding sequences for both the OAD and ME types of oxaloacetate decarboxylases, which is a unique characteristic (2, 9) (Fig. 1A). Biochemical studies have confirmed that the enterococcal CitM (ME type) catalyzes oxaloacetate decarboxylation, but also that an E. faecalis citM mutant is still capable of using citrate as a carbon source, strongly suggesting that the OAD complex forms an active complex as well (2, 9).
Fig 1.
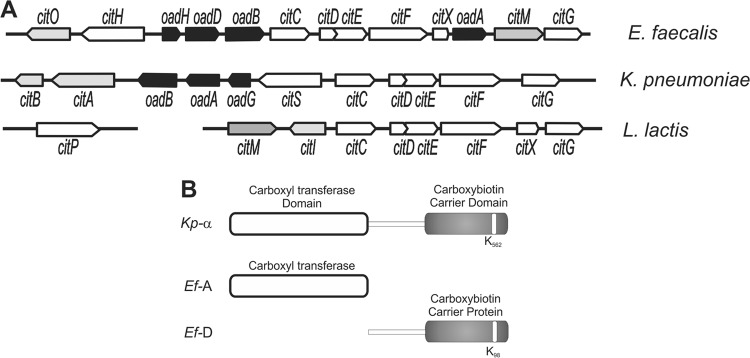
Genetic organization of the oxaloacetate decarboxylase involved in citrate utilization. (A) The cit locus is constituted by genes coding for citrate transporters (citH, citS, or the plasmid-encoded citP, depending on the family to which they belong), citrate lyase enzyme (citDEF), prosthetic group-binding citrate lyase (citC, citG, and citX), membrane bound-OAD complex (oadAHBD or oadGAB), cytoplasmic oxaloacetate decarboxylase (citM), and transcriptional regulators (citO, citAB, and citI). (B) Schematic representation of K. pneumoniae α subunit (Kp-α) and E. faecalis subunits Ef-A and Ef-D. The C-terminal part of Ef-D contains the AMKM conserved motif (22) around the biotin-accepting lysine residue, indicated in white.
The OAD complex of K. pneumoniae may be regarded as the prototype of the Na+-translocating decarboxylase (NaT-DC) family of enzymes. It consists of the two membrane-bound subunits β (Kp-β) and γ (Kp-γ) and the cytosolic α subunit (Kp-α), which is attached to the βγ complex via the γ subunit (4). The N-terminal domain of Kp-α catalyzes the transfer of the carboxyl group of oxaloacetate to the prosthetic biotin group of the C-terminal biotin acceptor domain (Fig. 1B). Decarboxylation of the carboxybiotin group is coupled to the translocation of Na+ ions out of the cells catalyzed by the Kp-β subunit. In E. faecalis, the E. faecalis OadA (Ef-A) and Ef-D subunits of the OAD complex are homologous to the N-terminal carboxyl transferase (49% identity) and the C-terminal biotin carrier protein (53% identity) domains of Kp-α, respectively (Fig. 1B). Ef-B shares 43% identity and 59% similarity to the paradigmatic Na+ pump from K. pneumoniae. Interestingly, we identified a coding sequence upstream of the oadD gene, renamed oadH, which encodes a putative polypeptide consisting of 120 amino acids (Fig. 1A).
The present study focuses on the characterization of the OAD complex of E. faecalis (termed Ef-OAD). Results reported here show that the novel Ef-H subunit is involved in the cytoplasmic Ef-AHD complex formation. Also, we demonstrate that the presence of the membrane-bound Ef-B subunit is required for full alkalinization of the internal medium of E. faecalis cells during citrate fermentation.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Cultures of E. faecalis were grown at 37°C, without shaking, in Luria-Bertani medium (LB; Difco, NJ), initial pH 7.0, and supplemented with 33 mM trisodium citrate (LBC). Erythromycin was added, when appropriate, in a 5 μg ml−1 concentration. Growth was monitored by measuring the optical density at 600 nm (OD600) in a Beckman DU640 spectrophotometer. In order to compare the growth parameters (biomass and growth rate) of wild-type and oad mutant strains, E. faecalis was also cultivated in sterile 96-well microplates (Cellstar) in a total volume of 200 μl at 37°C. Exponentially growing cultures were diluted to an initial OD600 of 0.10 in LBC broth, and the OD600 was registered automatically every 1 h in a PowerWave XS microplate reader (BioTek Instrument Inc., VT). Data presented here correspond to the mean values from three independent experiments in which growth curves were assayed in duplicate.
E. coli DH5α and EC101 strains (Table 1) were used as cloning hosts, whereas E. coli BL21(DE3) was used for expression of recombinant proteins. E. coli strains were routinely grown aerobically at 37°C in LB medium and transformed as previously described (16). Aerobic growth was achieved by gyratory shaking at 250 rpm. The corresponding antibiotics (100 μg ampicillin ml−1, 150 μg erythromycin ml−1, or 50 μg kanamycin ml−1) were included in the medium in order to select cells harboring the different plasmids.
Table 1.
Strains, plasmids and primers used in this study
| Strain, plasmid, or primer | Description or sequencea (5′ to 3′) | Source, reference, or restriction enzyme |
|---|---|---|
| E. faecalis | ||
| JH2-2 | Fusr Rifr; plasmid-free wild-type strain | 13 |
| JHB1 | JH2-2 isogenic derivative citO mutant | 9 |
| JHA | JH2-2 isogenic derivative oadA mutant | This work |
| JHB | JH2-2 isogenic derivative oadB mutant | This work |
| JHD | JH2-2 isogenic derivative oadD mutant | This work |
| E. coli | ||
| DH5α | fhuA2 Δ(argF-lacZ)U169 phoA glnV44 Φ80dlacZΔM15 gyrA96 recA1 relA1 endA1 thi-1 hsdR17; used as an intermediate host for cloning | 12 |
| EC101 | supE thi Δ(lac-proAB) (F′ traD36 proAB lacIqZΔM15) repA; Kanr; used as the host for pBV-Gh constructs | 14 |
| BL21(DE3) | F− ompT hsdSB(rB− mB−) gal dcm (DE3) | 15 |
| Plasmids | ||
| pGEM-T Easy | Plasmid for intermediate cloning | Promega |
| pBV-Gh | Plasmid used for the construction of deletion mutants | 10 |
| pBV-GhA | pBV-Gh derivative carrying 500 bp upstream and downstream portions of oadA gene | This work |
| pBV-GhB | pBV-Gh derivative carrying 530 bp upstream and downstream portions of oadB gene | This work |
| pBV-GhD | pBV-Gh derivative carrying 520 bp upstream and downstream portions of oadD gene | This work |
| pET28a | His-tagged protein expression plasmid | Novagen |
| pET-A | pET28a derivative expressing His6-OadA | This work |
| pBAD24 | Arabinose-inducible plasmid | 11 |
| pBAD-D | pBAD24-His derivative expressing His6-OadD | This work |
| pMal-c2X | NEB | |
| pMal-H | pMal-c2X derivative expressing MBP-OadH | This work |
| pET-ADB | pET28a derivative expressing His6-OadA, OadD, and OadB | This work |
| pET-AHDB | pET28a derivative expressing OAD complex subunits | This work |
| Primers | ||
| OadAUp-For | CACACACCATGGGTTGTTAGAAAC | NcoI |
| OadAUp-Rev | CCCGTAAAGCTTTTCTGTAAAACG | HindIII |
| OadALo-For | AAAGATAAGCTTCGCGGAGAATATG | HindIII |
| OadALo-Rev | GCTAAACCATGGTGGTTTAACACCG | NcoI |
| OadBUp-For | AAACCATGGATGTTACCCAAGGCC | NcoI |
| OadBUp-Rev | CGGGGATCCTAACAATGGACCG | BamHI |
| OadBLo-For | CAAGGATCCGTGCTGCGGGTATTTC | BamHI |
| OadBLo-Rev | TTACCATGGCAGATTCTTTTCATTG | NcoI |
| OadDUp-For | CGGGGTACCAATGCTTATATACCAATG | KpnI |
| OadDUp-Rev | ACTGAATTCTCTTTCCCATCAATTG | EcoRI |
| OadDLo-For | CAGGAATTCATGTTACCCAAGGC | EcoRI |
| OadDLo-Rev | CCAAGGTACCCAATCGAAGCAGC | KpnI |
| OadA1 | GACACGAAAGGAGCAGCCATATGAG | NdeI |
| OadA2 | CGGAAAGCTTGCCCGTTCTATTCTG | HindIII |
| OadB1 | GTATTCGTTCTCGAGTCCGTCCTATG | XhoI |
| OadD1 | GCATCCATGGGATTACGCAAATTTAAAATTTC | NcoI |
| OadD2 | GCTAGGATCCCTAGTTAATTGTTATTAATGGTTCC | BamHI |
| OadD3 | GGGCTGTCAGAAGAAGCTTAGCTAGTTG | HindIII |
| OadH1 | AGAGAATTCATGGGCCTATTTAAGTC | EcoRI |
| OadH2 | CCCAAGCTTTTAAATTTGCGTAAC | HindIII |
| OadH3 | CTATAATGAAGCTTTACCAAGAATTAGTG | HindIII |
Fus, fusidic acid; Rif, rifampin; Tet, tetracycline; Em, erythromycin.
Construction of E. faecalis oad-defective strains.
The oad-deficient strains were constructed by introducing a deletion into the corresponding oad gene using the thermosensitive vector pBVGh (10). Oligonucleotides used for the amplification of upstream and downstream fragments of oadA, oadD, and oadH genes are indicated in Table 1. Fragments were purified, restricted, and ligated into the corresponding sites of the pBVGh vector, except for the fragment used for generating the oadD deletion, which was first cloned into the pGEM-T Easy vector (Promega), released by digestion with NotI, and finally ligated into the corresponding site of pBVGh. Cloned fragments were checked by sequencing at the University of Maine DNA sequencing facility. Finally, the protocol to generate the chromosomal deletion in E. faecalis was monitored as previously described by Blancato and Magni (10).
Cloning and expression.
The oadA gene from E. faecalis JH2-2 was amplified by PCR using the OadA1 forward primer and OadA2 reverse primer (Table 1). The amplified DNA fragment was cloned into the pGEMT-easy vector, digested with NdeI and BamHI, and finally ligated into the same sites of a predigested pET28a expression vector (Novagen, Darmstadt, Germany), yielding pET-A (Table 1). To obtain pBAD-D, the oadD gene was amplified by PCR using E. faecalis JH2-2 chromosomal DNA as the template and OadD1 and OadD2 forward and reverse primers (Table 1). The amplimer was cloned into the pGEM-T Easy vector, digested with NcoI and PstI, and finally cloned into the same site of a His tag carrying a derivative of the pBAD24 vector, thus yielding pBAD-D (Table 1). For the construction of a plasmid expressing the OadH subunit fused to maltose binding protein (MBP), oadH was amplified using primers OadH1, introducing an EcoRI restriction site, and OadH2, introducing a HindIII site. After digestion of the PCR product with the mentioned restriction enzymes, it was introduced into the predigested pMal-c2X vector to yield plasmid pMal-H (Table 1). For the construction of plasmids carrying oadADB, the region coding for oadD and oadB was amplified using E. faecalis JH2-2 chromosomal DNA as the template with primers OadD3 (HindIII) and OadB1 (XhoI), whereas for oadAHDB the region spanning oadH, oadD, and oadB was amplified with primers OadH3 and OadB1, introducing a HindIII and XhoI restriction site, respectively. Both amplimers were digested with the cited enzymes and cloned in the pET-A plasmid predigested with the same enzymes. The resulting plasmids were named pET-ADB and pET-AHDB, respectively (Table 1).
Purification of recombinant proteins.
To obtain high levels of soluble recombinant His-tagged OadA or OadD protein, 100-ml cultures of E. coli BL21(DE3) cells carrying pET-A or pBAD-D, respectively, were grown in LB at 37°C until an OD600 of 0.6. At this point, cells were induced by addition of 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG) or 0.5% arabinose, respectively, and incubated at 25°C for 20 h with slow shaking (100 rpm) for OadA or at 37°C for 3 h with vigorous shaking (250 rpm) for OadD. Cultures were then harvested by centrifugation and resuspended in ice-cold buffer (50 mM Tris-HCl, pH 8.0, 3 mM MgCl2, 1 mM EDTA, 1 mM phenylmethanesulfonyl fluoride [PMSF], and 10% glycerol). Cells were disrupted using a French press, and cell debris was removed by centrifugation as previously described (5). Both proteins then were purified from the soluble fraction by affinity chromatography using a Ni2+-nitrilotriacetic acid (NTA) column according to the protocol recommended by Novagen. The purified enzymes were then dialyzed against the resuspension buffer supplemented with 20% glycerol and finally stored at −80°C for further studies. Protein concentrations were determined by the Lowry method using bovine serum albumin as a standard.
Induction of E. coli strains carrying pET-ADB and pET-AHDB plasmids was performed as described above for pET-A, with the following modifications. The induction period was 4 h, and the culture was vigorously shaken at 250 rpm. Cells were then harvested by centrifugation, resuspended in lysis solution A (LSA; 50 mM K2HPO4, pH 8.0, 200 mM NaCl, 10% [vol/vol] glycerol) and finally disrupted as described above. Protein complexes were purified by affinity chromatography on monomeric avidin resin (Thermo Scientific) (see below).
For the purification of OadH fused to maltose binding protein (MBP), 100-ml cultures of E. coli BL21(DE3) cells carrying plasmid pMal-H (Table 1) were grown in 0.5% glucose-supplemented LB at 37°C until an OD600 of 0.6. At this point, cells were induced by addition of 0.5 mM IPTG and incubated at 37°C for 3 h at 250 rpm. Cells were then harvested by centrifugation and resuspended in ice-cold amylose buffer (AB; 20 mM Tris-HCl, pH 7.4, 100 mM NaCl, 1 mM PMSF, 1 mM dithiothreitol, and 10% glycerol). Cell lysate was prepared as explained above and mixed with 300 μl of amylose resin preequilibrated with AB. Protein binding to the resin was performed for 2 h at 4°C. The column was then washed with 5 ml of AB buffer, and pure protein was eluted with AB buffer containing 10 mM maltose.
Preparation of E. faecalis cell extracts.
E. faecalis strains were grown for 10 h in LBC in a final volume of 1 liter. Cells were then harvested by centrifugation and resuspended in lysis solution N (LSN; 100 mM Na2HPO4, pH 7.0, 10% [vol/vol] glycerol) for neutravidin (NeutrAvidin Ultralink Resin; Thermo Scientific) affinity purifications or LSA for monomeric avidin (Thermo Scientific) affinity purifications (see below). LS was supplemented with 1 mM PMSF and also with the lytic enzymes mutanolysin (50 U ml−1; Sigma) and lysozyme (2.5 mg ml−1; Sigma). Cell resuspensions were incubated for 1 h at 37°C. The cells were disrupted by passing them three times through a French pressure cell at 10,000 lb/in2. The suspension was centrifuged at 10,000 × g for 25 min to remove cell debris. The clear supernatant was separated, and soluble (S) and membrane (M) protein fractions were prepared from it. Thus, supernatant was ultracentrifuged for 45 min at 170,000 × g. The obtained supernatant (S fraction) was frozen at −20°C, whereas the pellet containing the membrane fraction was resuspended in 1 ml of a 50 mM K2HPO4, pH 7.0, 1 M NaCl solution. This fraction was then subjected to a new ultracentrifugation step, and the resulting pellet was homogenized with 200 μl 50 mM K2HPO4, pH 8.0, 400 mM NaCl, 20% (vol/vol) glycerol, and 2% Triton X-100. Finally, the sample was ultracentrifuged using the same conditions as those described above, and the obtained supernatant (M fraction) was kept at −20°C until use.
Immunoblot analysis.
For Western blot analysis, samples containing 30 μg of total protein were loaded onto a sodium dodecyl sulfate polyacrylamide electrophoresis (SDS-PAGE) gel (16). After this, samples were transferred to a nitrocellulose membrane. Rabbit polyclonal antiserum against E. faecalis OadA and OadH proteins was generated using the purified His-tagged OadA and MBP-fused OadH proteins as the antigens (Bioterio FCByF, UNR). Rabbits were immunized subcutaneously with 200 μg of purified protein. Polyclonal antibodies were purified (16) and used at a 1:1,000 dilution. The OadA- or OadH-antibody complexes were visualized using goat anti-rabbit IgG (H+L) alkaline phosphatase (AP)-conjugated antibodies (Bio-Rad). Biotinylated proteins were detected with alkaline phosphatase-conjugated streptavidin (Thermo Scientific). p-Nitroblue tetrazolium (ICN) and 5-bromo-4-chloro-3-indolylphosphate (Sigma) were used as alkaline phosphatase substrates.
OAD isolation and identification.
The protein complex was isolated from the soluble (S) and membrane (M) protein fractions by affinity chromatography on neutravidin or monomeric avidin-Sepharose (Thermo Scientific; see Results for details) by following the manufacturer's suggested protocol. Briefly, each of the fractions was applied to 50 to 100 μl of resin slurry, previously equilibrated in the corresponding LS, and incubated at 4°C for 1 h. The matrix was then washed four times with 1 ml LS (containing 0.1% Triton X-100 when added to the M fraction) to elute the proteins that were not retained. Finally, bound proteins were eluted from the neutravidin resin by boiling for 5 min after addition of 100 μl of Laemmli loading buffer. The elution from the avidin resin was performed after 30 min of incubation at room temperature with 100 μl of LSA supplemented with 5 mM biotin. Samples were supplemented with 20% glycerol and finally stored at −80°C for further studies.
For the identification by mass spectrometry of the proteins detected in SDS-PAGE gels, bands were excised and sent to the Unidad de Bioquímica y Proteómica Analíticas of Institute Pasteur of Montevideo for analysis. For the cross-linking assays, the elution fractions obtained after the avidin affinity chromatography were incubated for 20 min with 1 mM glutaraldehyde (Sigma) at room temperature.
OAD complex dissociation in response to pH.
E. faecalis JH2-2 cell extracts were prepared and subsequently applied to 100 μl of neutravidin resin as explained above. After binding, the resin was washed with 1 ml 150 mM citrate buffer, pH 7.0, and then sequentially eluted with 60-μl aliquots of citrate buffer adjusted to pH 3.0 to 6.0 for the acid range and with phosphate buffer adjusted to pH 7.0 to 11.0 for the alkaline range. After collecting the elution fractions, the resin was washed again with 1 ml citrate buffer, pH 6.0. Finally, bound proteins were eluted from the neutravidin resin by boiling for 5 min after addition of 100 μl of Laemmli loading buffer. All of the fractions were then analyzed by SDS-PAGE.
Loading of cells with the BCECF probe and internal pH measurements.
Loading of the cells with the fluorophore was performed as previously explained (2). Cells were loaded with the pH-sensitive fluorescent probe 2′,7′-bis-(2-carboxyethyl)-5(and -6)-carboxyfluorescein (BCECF) as previously described (17). The excitation wavelength was 503 nm, and fluorescent emission was recorded at 525 nm (slit widths were 4 and 16 nm, respectively). Cytoplasmic pH was determined from the fluorescence signal as previously described (17).
RESULTS
Heterologous expression of Ef-OAD subunits in E. coli.
The oadA and oadD genes, encoding Ef-A and Ef-D subunits of the E. faecalis OAD (Ef-OAD) complex, were cloned into pET28 and pBAD24-His plasmids (Table 1), respectively, and expressed in E. coli BL21(DE3). The His-tagged gene products were purified by Ni2+-NTA affinity chromatography and analyzed by SDS-PAGE (Fig. 2A and B). OadA eluted from the column at a concentration of 25 to 50 mM imidazole and showed an apparent molecular mass of 51 kDa, which was in line with the theoretical molecular mass of 52 kDa. The OadD protein required 150 mM imidazole for elution (Fig. 2B, lanes 6 and 7) and showed an apparent molecular mass of 24 kDa, which was considerably higher than the theoretical value of 14 kDa. The same procedure was not successful for heterologous expression of the membrane-bound Ef-B subunit in E. coli. In spite of the use of different cloning vectors producing the protein with different tags (His6 or biotin acceptor domain; Table 1) and expression hosts [E. coli BL21(DE3) and C43(DE3)], the Ef-B protein was never detected.
Fig 2.

Expression and purification of the different Ef-OAD subunits. (A) Ef-A affinity purification on Ni2+-NTA resin. BL21(DE3) cells carrying the pET28 derivative pET-A were induced by addition of IPTG (see Materials and Methods and Table 1). FT, flowthrough; W, wash; Im, imidazole. (B) Ef-D affinity purification on Ni2+-NTA resin. Cells carrying the pBAD24 derivative pBAD-D were induced as described previously. M, protein molecular markers. (C) Ef-H affinity purification on amylose resin. E. coli cells carrying the pMal-c2X derivative pMal-H were induced as described in Materials and Methods. Elutions were done with the addition of 10 mM maltose for all fractions. Recombinant proteins are indicated by the arrows.
A previous study showed that the E. faecalis cit cluster did not contain a gene homologous to oadG of K. pneumoniae, nor was the gene found elsewhere on the E. faecalis genome (9). Closer inspection showed the presence of a coding sequence upstream of the oadD gene (Fig. 1A) which encodes a putative polypeptide consisting of 120 amino acids with a theoretical molecular mass of 14 kDa. The encoded protein, Ef-H, showed no sequence similarities with other proteins in the databases (RefSeq; searched in July 2012), but the oadH gene was conserved in the cit locus of all Gram-positive bacteria that contain the OAD complex. The Ef-H protein was successfully overexpressed in E. coli as an MBP fusion protein. The MBP-OadH fusion protein was purified by amylase affinity chromatography (Fig. 2C, lanes 5 to 8).
The His-OadA and MBP-OadH proteins were used for the polyclonal antibody preparations that were used in subsequent experiments.
Identification and cellular localization of the E. faecalis OAD complex.
E. faecalis wild-type strain JH2-2 and strain JHB1, a citO mutant unable to express citrate metabolism genes (9), were grown in LB broth supplemented with citrate, which is essential for the induction of cit cluster transcription. Western blotting of crude cell extracts using antibodies raised against the purified Ef-A and Ef-H proteins (see above) and alkaline phosphatase-conjugated streptavidin to detect biotinylated Ef-D clearly detected the subunits in strain JH2-2 but not in strain JHB1 (Fig. 3A, B, and C). Subunits Ef-A and Ef-D showed the same mobility as that seen after expression of the corresponding genes in E. coli, and the apparent molecular mass of Ef-H was 16 kDa, which correlates well with the theoretical size of 14 kDa. The results indicate that the E. faecalis OAD subunits Ef-A and Ef-D and the newly identified subunit Ef-H are expressed in medium containing citrate and are under the control of the transcriptional activator of the citrate metabolic pathway, CitO (9). Crude cell extracts of both E. faecalis strains were separated into soluble (S) and membrane (M) fractions. Biotinylated proteins and copurifying proteins were isolated by affinity chromatography using avidin Sepharose. SDS-PAGE analyses of the eluted proteins of the soluble fraction of strain JH2-2 revealed the presence of Ef-A and Ef-H (Fig. 3D). Remarkably, only a weak band was observed at the expected position of Ef-D, the biotinylated subunit. The identity of the bands was corroborated by mass spectroscopy analysis. A band running at approximately 120 kDa (band X) was identified by mass spectrometry as a high-molecular-weight complex formed mainly by the Ef-D subunit. As expected, in the elution fraction of the citO mutant strain JHB1, the Ef-OAD subunits were not detected (data not shown). Avidin affinity chromatography of the solubilized membrane fraction of the cells resulted in purification of the same Ef-OAD subunits as that observed in the soluble fraction (Fig. 3D). No bands were detected in the membrane fraction of the citO mutant (data not shown). These results show that the Ef-OAD subunits Ef-A, Ef-D, and Ef-H form a soluble complex in vivo, a fraction of which is bound to the membrane, presumably to the integral membrane subunit Ef-B.
Fig 3.
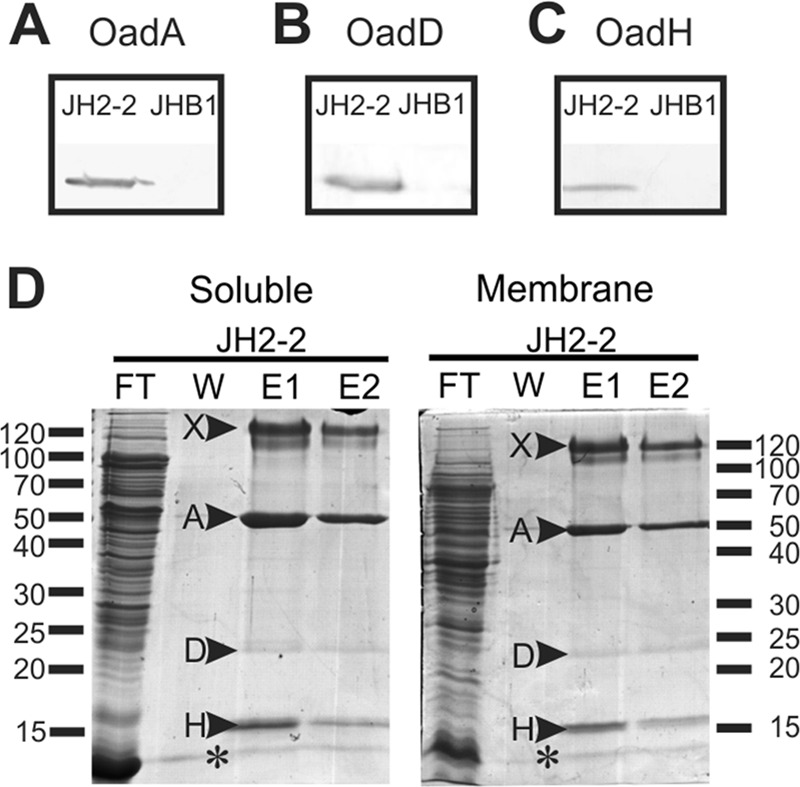
Isolation and identification of Ef-OAD subunits. Western blotting for the detection of Ef-A (A), Ef-D (B), and Ef-H (C) in crude extracts of wild-type (JH2-2) and citO mutant (JHB1) strains. To detect the presence of Ef-A and Ef-H subunits, polyclonal antibodies prepared in the laboratory were used (see Materials and Methods), whereas for the Ef-D protein, alkaline phosphatase-conjugated streptavidin, which permits the detection of biotinylated proteins, was utilized. (D) Soluble and membrane protein extracts corresponding to the wild type (JH2-2) were passed through an avidin resin. After washing (W), total biotinylated proteins were eluted with buffer containing 5 mM avidin. Two elution fractions (E1 and E2) were recovered, and all fractions were then run in an SDS-PAGE gel. Indicated bands (arrowheads) were excised from the gel and identified by mass spectrometry. Band A, Ef-A; band D, Ef-D; band H, Ef-H; band X, Ef-D protein aggregate. FT, flowthrough. An asterisk corresponds to avidin released from the column.
Subunit interactions of the Ef-OAD complex.
The elution fractions obtained after avidin affinity chromatography of the soluble and membrane fractions of E. faecalis JH2-2 were treated with the unspecific cross-linker glutaraldehyde prior to SDS-PAGE analysis (Fig. 4A). The treatment resulted in the complete disappearance of the band corresponding to the Ef-H subunit, a significant decrease in intensity of the high-molecular-mass complex formed by the Ef-D subunit, and the appearance of a new band running at approximately 38 kDa (band B) with no significant difference between the soluble and membrane fractions. The new band, B, is likely to be a heterodimer formed by Ef-D and Ef-H (24 and 16 kDa, respectively), suggesting that Ef-H and Ef-D subunits are in close contact in the OAD complex.
Fig 4.

Interaction between Ef-OAD subunits. (A) Cross-linking assay. Soluble and membrane-eluted fractions from avidin affinity purification experiments were treated with a cross-linker agent (lanes 2 and 4) and analyzed in conjunction with nontreated samples (lanes 1 and 3) by SDS-PAGE. The appearance of a new band after the cross-linking is indicated by an arrowhead and the letter B (see the text for details). (B) Isolation of Ef-OAD subunits in different oad mutant strains. Soluble protein extracts corresponding to the wild type (JH2-2) and oadA- and oadB-deficient strains were passed through a neutravidin resin. Elution fractions were then run on an SDS-PAGE gel which was silver stained. M, molecular markers. (C, upper) Elution fractions from membrane protein extracts. (C, lower) Immunoblot analysis for the detection of Ef-A expression in the wild-type membrane fraction.
oadA, oadB, and oadD deletion mutants of E. faecalis were constructed to further study subunit interactions in the OAD complex. An oadH mutant strain could not be obtained in spite of using different genetic strategies. Subunit interactions in the soluble and membrane fractions of the wild-type and oad mutant strains were studied by pulldown experiments of the biotinylated Ef-D subunit. As observed before, the three subunits Ef-A, Ef-D, and Ef-H were detected in both fractions of the wild-type preparation (Fig. 4B and C), while the oadD deletion obviously results in the lack of detection of any of the OAD subunits (not shown). Deletion of oadA did not significantly affect the interaction between Ef-D and Ef-H in the soluble fraction, indicating a direct interaction between the latter two subunits. Notably, Ef-A was not detected in the membrane fraction of the oadB mutant, neither by silver staining of the gel nor by Western blotting using polyclonal antibodies against this subunit (Fig. 4C). This observation suggests that Ef-B is necessary for Ef-A to reach the membrane.
The OAD complex in crude cell extract of E. faecalis JH2-2 was immobilized on neutravidin resin to study the stability of the complex in both the acidic and alkaline range of pH. This resin interacts with the biotin-containing Ef-D subunit in a manner in which it can only be dissociated after harsh treatment, such as boiling. As shown in Fig. 5A, Ef-A and Ef-H were retained on the resin when pH diminished (lanes 5 to 8). As expected, final heat treatment of the resin released both of these subunits, as well as the Ef-D subunit. On the other hand, alkalinization induced the dissociation of the Ef-D and Ef-A subunits, whereas the Ef-H subunit was eluted solely after heat treatment. These experiments suggest an alkaline pH-dependent interaction between Ef-A and Ef-D, while Ef-H interacts with the Ef-D subunit independently of the external pH.
Fig 5.

In vitro analysis of Ef-OAD stability as a function of pH. E. faecalis cell extracts were first incubated in batch with a neutravidin resin. After the binding step, the resin was washed (W; lane 3) with 1 ml 150 mM citrate, pH 7.0, and eluted (lanes 4 to 8) with 60-μl aliquots of citrate (A) or phosphate buffers (B) according to the indicated pH range. As a final step, samples resuspended in Laemmli loading buffer were boiled (100°C). FT, flowthrough. M, molecular markers. An asterisk corresponds to avidin released from the column.
Expression plasmids containing Ef-A, Ef-D, and Ef-B subunits (pET-ADB; Table 1) and the whole complex (pET-AHDB; Table 1) were constructed for expression of the corresponding complexes in E. coli to further examine the role of subunit H. Pulldown experiments with Ef-D resulted in the copurification of the Ef-A and Ef-H subunits from extracts of cells carrying pET-AHDB, similar to that observed for E. faecalis JH2-2 cells (Fig. 6A). In the absence of the Ef-H subunit (plasmid pET-ADB), subunit Ef-A was not pulled down with Ef-D anymore (Fig. 6A), while immunoblots of the cell extract showed that Ef-A was synthesized from both plasmids (Fig. 6B). The results suggest that the Ef-H subunit is essential for the interaction between Ef-A and Ef-D.
Fig 6.

Ef-OAD complex expression in E. coli. (A) SDS-PAGE of first and second elution fractions (E1 and E2) of an avidin affinity chromatography purification of E. coli cell extracts from strains carrying different Ef-OAD subunits (pET-ADB and pET-AHDB). Relevant bands are indicated by arrowheads. W, third washing step. (B) Immunoblot detection of Ef-A expression from crude extracts of induced cells harboring pET-ADB and pET-AHDB plasmids.
Functionality of the OAD complex of E. faecalis.
Growth of the oadA, oadB, and oadD deletion strains of E. faecalis in the presence of citrate was monitored to analyze the involvement of the Ef-OAD complex in citrate metabolism (Fig. 7A). The parental strain reached a maximal OD of 1.2 at 10 h, whereas the oadA and oadD deletion mutant strains only grew to a maximal OD of 0.4, showing a profile similar to that of the wild-type strain when grown in the absence of citrate (data not shown). Thus, this experiment demonstrates that OadA and OadD are individually essential for citrate metabolism, and that the citM gene (encoding CitM, a decarboxylase of the malic enzyme type) is not capable of complementing the deficiency. Remarkably, the oadB mutant strain grew like the parental strain, with a delay in the start point of the second phase of growth (Fig. 7A) when the citrate pathway is transcriptionally induced (9). Apparently, the oadB gene product is not essential for citrate metabolism, and its mutant strain is capable of degrading oxaloacetate to pyruvate by the action of the cytoplasmic OadAHD complex.
Fig 7.

Physiological characterization of oad mutants. (A) Growth curves of wild-type E. faecalis (●) and oadA (■), oadB (▼), and oadD (⧫) derivative strains in LBC. (B) Cytoplasmic pH variation (pHint) of E. faecalis wild-type and mutant cells in response to citrate. Resting cells were loaded with the BCCEF fluorescent probe, and a pulse of 2 mM citrate was added at the time indicated by the arrow.
Resting cells of E. faecalis JH2-2 and the oadA, oadB, and oadD deletion mutants were loaded with the fluorescent dye BCECF, which allows monitoring of the alkalinization of the cytoplasm associated with citrate metabolism (18). Addition of 2 mM citrate to wild-type cells resuspended in a buffer of pH 5.8 resulted in immediate cytoplasmic alkalinization until a pH gradient of 1.0 U was reached after 2 min (Fig. 7B). In contrast, with the oadA and oadD mutants no changes in the cytoplasmic pH were observed upon addition of citrate, indicating the absence of decarboxylation activity. Interestingly, the oadB-deficient strain showed an intermediate increase in the internal pH (up to 0.6 U; Fig. 7B), in line with the results obtained for the growth curves. Addition of 3 mM glucose to wild-type and oadB-deficient strains showed the same levels of internal alkalinization, indicating the same physiological state of the cells (data not shown). These results indicate that the in vivo decarboxylation activity of Ef-AHD as well as Ef-AHDB complexes contributes to internal pH increase, and that the presence of the Ef-B subunit could be considered a surplus, from a physiological point of view, for E. faecalis.
DISCUSSION
The membrane oxaloacetate decarboxylase complex, which is associated with citrate metabolism, is found in phylogenetically distant bacteria. In pathogens belonging to the gammaproteobacterium subdivision, this complex is expressed in response to citrate under anaerobic conditions. Here, we investigated the presence of the OAD complex in Enterococcus faecalis, a disseminated microorganism associated with diverse human activities, from food fermentations to nosocomial infections. We have demonstrated that the OAD complex is essential for citrate metabolism in E. faecalis, whereas the activity of the soluble decarboxylase CitM seems irrelevant under these conditions (2). Moreover, the expression of OadA, OadD, and OadH subunits from OAD complex was detected for the first time in a Gram-positive bacterium. These proteins were isolated by affinity chromatography using an avidin resin, showing that Ef-D is biotinylated in vivo and constitutes a stable complex with the Ef-A and Ef-H subunits. The Ef-AHD complex was found in the soluble fraction as well as in the membrane. This suggests that these three subunits are tightly bound to the membrane but in equilibrium with a soluble subcomplex. Remarkably, after solubilization of the membrane, this tripartite complex is stable enough to be isolated by affinity chromatography.
Ef-A carboxyltransferase and Ef-D biotin-carrier subunits were shown to be vital for the conversion of OAA into pyruvate during citrate fermentation. These subunits are fused in K. pneumoniae and V. cholerae α subunits, constituting two different domains of the same protein. This might be a consequence of the evolutionary process experienced by these proteins, contributing to the association of different subunits into one polypeptide, as observed for other proteins carrying carboxyltransferase and biotin-carrier domains, such as pyruvate carboxylase (19). Interestingly, we have also identified a novel Ef-H subunit for which no homologues have been previously described. A clear homology relationship could be found between carboxyltransferase subunits in different OAD complexes present in LAB (between 73 and 82% identity). The same correlation is valid for biotin carriers (58 to 74%) and sodium pumps subunits (77 to 92%). OadH subunits instead show lower levels of identity to each other (33 to 55%). However, the oadH gene is conserved in the cit locus of all E. faecalis strains sequenced to date, in other enterococcal species (E. saccharolyticus, E. gallinarum, E. faecium, E. italicus, and E. casseliflavus), in other LAB, such as Lactobacillus (L. casei, L. sakei, and L. rhamnosus), and in opportunistic pathogens, such as Streptococcus (S. mutans and S. pyogenes). The diminished levels of identity have also been observed for γ subunits in other bacteria (20) and probably obey the rule that these adapter proteins evolve their sequences in such a way that they can interact with specific surface regions of the other subunits in order to keep complex integrity. Consistent with this hypothesis, specific interactions between Ef-H and Ef-D were observed (Fig. 4A). Additional information on the interaction between the complex subunits was obtained from OAD expression in E. coli and subsequent purification by avidin affinity chromatography, whereby it was seen that the absence of Ef-H impairs Ef-A isolation, suggesting a role of the former in the stabilization of the Ef-AD complex (Fig. 6A). Therefore, our genetic and biochemical results indicate that Ef-H fulfills the role of stabilizing the soluble Ef-AD complex and presumably the interaction of Ef-AHD with the membrane.
Another distinctive feature of the enterococcal OAD complex emerges from the stability analysis with regard to pH (Fig. 5). The dissociation between α and γ subunits of K. pneumoniae and V. cholerae complexes was shown previously to be induced by pHs lower than 6.5. This is presumably due to protonation of histidine residues (pKa of 6.5) involved in the assembly of these enzymes. Mutagenesis experiments demonstrated that His78 in K. pneumoniae (21) and His81 in V. cholerae (20), located in the C-terminal part of the γ subunit, are crucial for their interactions with the α subunit. In contrast, the treatment of Ef-OAD with buffers in the acidic range did not produce its dissociation; instead, pH values above 6.0 provoked the release of Ef-A from the complex (Fig. 5B). Amino acid residues that become deprotonated at pH >6.0 seem to be involved in the binding of Ef-A to the other subunits. Therefore, Ef-OAD integrity might also depend on the protonation state of histidine residues, since Ef-A dissociation is induced by pHs in the range of the histidine pKa. Remarkably, Ef-H interaction with Ef-D does not seem to depend on medium pH, since it is not dissociated from Ef-D at any of the pHs tested. Finally, this experiment suggests that the stability of Ef-OAD as a function of pH does not correlate with that corresponding to others OAD complexes so far studied, suggesting an evolutionary adaptation of this enzyme to the acidic environments in which LAB survive.
A previous model for OAD in Streptococcus mutans by Sobczak and Lolkema (8) proposed a shuttling of the carboxyl-carrier protein between the carboxyltransferase in the cytoplasm and the sodium pump in the membrane. According to our current results, a new model is presented in which Ef-D shuttles from the cytoplasm to the membrane, and Ef-A would be aided by the novel Ef-H subunit, which might be responsible for Ef-AHD interaction with Ef-B. This soluble subcomplex is capable of decarboxylating OAA in the cytoplasm but, when coupled to Ef-B, confers an additional energetic advantage to E. faecalis during growth with citrate. Further studies are under way to uncover the basis for such a difference.
ACKNOWLEDGMENTS
This work was supported by grants from the Agencia Nacional de Promoción Científica y Tecnológica of Argentina (ANPCyT; PICT2010-1828 and PICT 2008-1562), a European Union grant (BIAMFood; contract KBBE-211441), and a short-term fellowship from Boehringer Ingelheim Fonds Stiftung für Medizinische Grundlagenforschung Euro-Tango. G.D.R. and P.M. are fellows of CONICET, and V.S.B. and C.M. are Career Investigators of the same institution.
Footnotes
Published ahead of print 22 February 2013
REFERENCES
- 1. García-Quintans N, Blancato VS, Repizo GD, Magni C, López P. 2008. Citrate metabolism and aroma compound production in lactic acid bacteria, p 65–88 In Mayo B, López P, Pérez-Martínez G. (ed), Molecular aspects of lactic acid bacteria for traditional and new applications. Research Signpost, Kerala, India [Google Scholar]
- 2. Espariz M, Repizo G, Blancato V, Mortera P, Alarcon S, Magni C. 2011. Identification of malic and soluble oxaloacetate decarboxylase enzymes in Enterococcus faecalis. FEBS J. 278:2140–2151 [DOI] [PubMed] [Google Scholar]
- 3. Dahinden P, Auchli Y, Granjon T, Taralczak M, Wild M, Dimroth P. 2005. Oxaloacetate decarboxylase of Vibrio cholerae: purification, characterization, and expression of the genes in Escherichia coli. Arch. Microbiol. 183:121–129 [DOI] [PubMed] [Google Scholar]
- 4. Dimroth P, Jockel P, Schmid M. 2001. Coupling mechanism of the oxaloacetate decarboxylase Na(+) pump. Biochim. Biophys. Acta 1505:1–14 [DOI] [PubMed] [Google Scholar]
- 5. Sender PD, Martin MG, Peiru S, Magni C. 2004. Characterization of an oxaloacetate decarboxylase that belongs to the malic enzyme family. FEBS Lett. 570:217–222 [DOI] [PubMed] [Google Scholar]
- 6. Bott M, Meyer M, Dimroth P. 1995. Regulation of anaerobic citrate metabolism in Klebsiella pneumoniae. Mol. Microbiol. 18:533–546 [DOI] [PubMed] [Google Scholar]
- 7. Martín MG, Sender PD, Peiru S, de Mendoza D, Magni C. 2004. Acid-inducible transcription of the operon encoding the citrate lyase complex of Lactococcus lactis biovar diacetylactis CRL264. J. Bacteriol. 186:5649–5660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sobczak I, Lolkema JS. 2005. The 2-hydroxycarboxylate transporter family: physiology, structure, and mechanism. Microbiol. Mol. Biol. Rev. 69:665–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Blancato VS, Repizo GD, Suárez CA, Magni C. 2008. Transcriptional regulation of the citrate gene cluster of Enterococcus faecalis involves the GntR family transcriptional activator CitO. J. Bacteriol. 190:7419–7430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Blancato VS, Magni C. 2010. A chimeric vector for efficient chromosomal modification in Enterococcus faecalis and other lactic acid bacteria. Lett. Appl. Microbiol. 50:542–546 [DOI] [PubMed] [Google Scholar]
- 11. Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177:4121–4130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hanahan D. 1983. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166:557–580 [DOI] [PubMed] [Google Scholar]
- 13. Jacob AE, Hobbs SJ. 1974. Conjugal transfer of plasmid-borne multiple antibiotic resistance in Streptococcus faecalis var. zymogenes. J. Bacteriol. 117:360–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Law J, Buist G, Haandrikman A, Kok J, Venema G, Leenhouts K. 1995. A system to generate chromosomal mutations in Lactococcus lactis which allows fast analysis of targeted genes. J. Bacteriol. 177:7011–7018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Studier FW, Moffatt BA. 1986. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 189:113–130 [DOI] [PubMed] [Google Scholar]
- 16. Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 17. Molenaar D, Abee T, Konings WN. 1991. Continuous measurement of the cytoplasmic pH in Lactococcus lactis with a fluorescent pH indicator. Biochim. Biophys. Acta 1115:75–83 [DOI] [PubMed] [Google Scholar]
- 18. Magni C, de Mendoza D, Konings WN, Lolkema JS. 1999. Mechanism of citrate metabolism in Lactococcus lactis: resistance against lactate toxicity at low pH. J. Bacteriol. 181:1451–1457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Balsera M, Buey RM, Li XD. 2011. Quaternary structure of the oxaloacetate decarboxylase membrane complex and mechanistic relationships to pyruvate carboxylases. J. Biol. Chem. 286:9457–9467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dahinden P, Pos KM, Dimroth P. 2005. Identification of a domain in the alpha-subunit of the oxaloacetate decarboxylase Na+ pump that accomplishes complex formation with the gamma-subunit. FEBS J. 272:846–855 [DOI] [PubMed] [Google Scholar]
- 21. Schmid M, Wild MR, Dahinden P, Dimroth P. 2002. Subunit gamma of the oxaloacetate decarboxylase Na(+) pump: interaction with other subunits/domains of the complex and binding site for the Zn(2+) metal ion. Biochemistry 41:1285–1292 [DOI] [PubMed] [Google Scholar]
- 22. Samols D, Thornton CG, Murtif VL, Kumar GK, Haase FC, Wood HG. 1988. Evolutionary conservation among biotin enzymes. J. Biol. Chem. 263:6461–6464 [PubMed] [Google Scholar]