

Abstract
Poly(3-hydroxybutyrate) (PHB) granules are organelle-like multienzyme-polymer complexes (carbonosomes) and are widespread storage compounds in prokaryotes. The interaction of three PHB granule-bound proteins (PHB synthase PhaC1, phasin PhaP5, and PHB/DNA binding protein PhaM) was studied in vivo by bimolecular fluorescence complementation (BiFC) microscopy in Ralstonia eutropha. To this end, a mobilizable 2-plasmid system for arabinose-controlled expression of protein fusions with the N-terminal (YN) and C-terminal (YC) parts of the enhanced yellow fluorescent protein (eYfp) in Gram-negative bacteria was developed. Both plasmids were stably expressed in Escherichia coli and in transconjugants of R. eutropha. Homo-oligomerization of PhaC1, PhaP5, and PhaM and interactions between PhaC1 and PhaM and between PhaM and PhaP5 were detected in R. eutropha and colocalized with PHB granules under PHB-permissive conditions. PhaM-PhaC1 complexes were detected near the midcell/nucleoid region in the absence of PHB. Expression of BiFC complexes in R. eutropha with PhaM (PhaM homo-oligomers or PhaM-PhaC1 or PhaM-PhaP5 complexes) resulted in substantial cell elongation compared to wild-type cells and in BiFC signals that were generally located near the midcell/nucleoid region. Western blot analysis of wild-type cell extracts and proteome analysis of PHB granule-bound proteins revealed that PhaM and PhaP5 are expressed in R. eutropha and that PhaM is constitutively expressed independently of the presence or absence of PHB. Size exclusion chromatography analysis in combination with cross-linking experiments of purified PhaP5-His6 and PhaM-His6 showed that PhaP5 forms dimers and that PhaM is present in oligomeric (dodecamer) form. Implications of this finding for subcellular PHB localization and initiation of PHB granule formation in R. eutropha will be discussed.
INTRODUCTION
Ralstonia eutropha is the model organism for research on microbial synthesis of short-chain-length polyhydroxyalkanoates (PHASCL) such as poly(3-hydroxybutyrate) (PHB) or copolymers of 3-hydroxybutyrate and 3-hydroxyvalerate (PHB/HV). PHB and PHB/HV are produced on an industrial scale of 104 to 105 tons per annum worldwide. For overviews on polyhydroxyalkanoate (PHA) metabolism, see references 1 to 8. Despite many years of intensive research on the molecular biology of PHA accumulation in Eubacteria and Archaea (9, 10), several aspects of PHA granule formation and subcellular localization of PHB granules have not been solved. Two models of PHB granule initiation, (i) the budding model (PHB granules initiate in the cytoplasm membrane and the growing PHB granule blebs out from the membrane at late stages) and (ii) the micelle model (PHB synthase PhaC1 is soluble in the cytoplasm and forms micelles with other PhaC1 molecules and with the growing [hydrophobic] PHB chain) were discussed in the past (11). Recently, association of PHB granules with the cytoplasm membrane could be excluded in R. eutropha by electron cryotomography (12). In our lab, we identified two new proteins (PhaP5 and PhaM) by (bacterial) two-hybrid analysis that play important roles during formation and subcellular localization and for distribution of PHB granules between daughter cells after cell division (13, 14). PhaP5 represents the fifth phasin protein in R. eutropha. Phasins are PHB granule-associated proteins (PGAPs) with no (apparent) enzymatic function, and a pure structural function was assumed (15). PhaM represents a new type of PGAP with multiple functions, as (i) it interacts with PHB synthase PhaC1 and with PhaP5, (ii) represents a PGAP (phasin characteristic of PhaM), and (iii) is able to bind to the nucleoid (DNA binding characteristic), thereby controlling the distribution of PHB granules to daughter cells during replication of the nucleoid (16). PhaM has some features similar to those of PhaF in medium-chain-length PHA-accumulating pseudomonads (17). In total, up to 20 different polypeptides have been experimentally proven or are postulated to represent PGAPs in R. eutropha and to constitute the carbonosome organelle (18). Unfortunately, interactions between PhaM, DNA, PhaC1, and PhaP5 were demonstrated only in recombinant adenylate cyclase-deficient Escherichia coli reporter strains (E. coli does not synthesize PHB), and the interaction of PGAPs of R. eutropha in the presence of native PHB granules could not be studied in the native host until now.
The bimolecular fluorescence complementation (BiFC) technique is based on reconstitution of fluorescent complexes when two proteins fused to two nonfluorescent fragments of a fluorescent protein (in this case, enhanced yellow fluorescent protein [eYfp]) interact with each other (19–21). Several combinations of protein fragments (N-terminal part [YN] and C-terminal part [YC]) that can be used for bimolecular fluorescence complementation have been identified for eYfp (21). The YN and YC parts of eYfp can functionally reassociate if they come in close contact, e.g., by physical interaction of the respective fusion proteins, and up to about 10% of the fluorescence intensity of the intact holo-eYfp can be recovered (21). The BiFC approach is comparable to fluorescence resonance energy transfer (FRET) but is easier to perform and more suitable for the detection of weak interactions. To carry out BiFC in R. eutropha, two compatible broad-host-range plasmids were constructed that carry the genes for the N-terminal (YN) and C-terminal (YC) parts of eYfp together with a C-terminal multiple cloning site (MCS) for insertion of the gene of interest. We constructed fusions between three different PGAPs (PhaC1, PhaM, and PhaP5) and YN or YC in all permutations. The respective fusion genes were cloned under the control of an arabinose-dependent promoter (PBAD) in two mobilizable and R. eutropha-compatible broad-host-range plasmids (pBBR1MCS-2 [Kmr] and pCM62 [Tcr]) (22, 23). This BiFC approach enabled us to study protein-protein interaction and subcellular localization of PGAPs in R. eutropha under PHB-permissive (high C-to-N ratio) and nonpermissive conditions (C limitation).
MATERIALS AND METHODS
Bacterial strains, plasmids, and oligonucleotides.
Bacteria, plasmids, and primers used in this study are shown in Table 1. E. coli strain JM109 was used in cloning experiments and was grown on Luria broth (LB) supplemented with the appropriate antibiotics (depending on strains and plasmids; see BiFC conditions below). R. eutropha strains were routinely grown on nutrient broth (NB) medium at 30°C. Sodium gluconate (0.2% [wt/vol]) was added as indicated to promote PHB accumulation. Expression of fusion proteins for BiFC analysis was induced by the addition of 0.2% (wt/vol) arabinose.
Table 1.
Strains, plasmids, and primers used in this study
| Strain, plasmid, or primer | Relevant characteristic(s) or feature(s) or primer sequencea (5′ to 3′) | Reference or source |
|---|---|---|
| Strains | ||
| E. coli | ||
| JM109 | Cloning strain | |
| S17-1 | Conjugation strain | 43 |
| R. eutropha | ||
| H16 | Wild-type strain | DSMZ 428 |
| H16 ΔphaC1 | Chromosomal deletion of phaC1 | 36 |
| H16 ΔphaP5 | Chromosomal deletion of phaP5 | 14 |
| H16 ΔphaM | Chromosomal deletion of phaM | 13 |
| Plasmids | ||
| pBBR1MCS-2 | Broad-host-range vector, Kmr | 22 |
| pCM62 | Broad-host-range vector, Tcr | 23 |
| pDMW7 | Source of araC-PBAD and rrnB | 44 |
| pEYFP-C1 | Source of eyfp-c1 | Clontech |
| pBBR1MCS-2-PphaC-phaM | Constitutive expression of PhaM (phaC promoter) | 16 |
| pBBR1MCS-2-PphaC-eyfp-phaM | Constitutive expression of eYfp-PhaM fusion (phaC promoter) | 13 |
| pBBR-EYFP-C1 | Plasmid coding for eYfp under the control of the PBAD promoter based on pBBR1MCS-2 | This study |
| pBBR-YN-C1 | Plasmid coding for the N-terminal part of eYfp (YN, amino acids 1 to 154) under the control of the PBAD promoter based on pBBR1MCS-2 | This study |
| pBBR-YC-C1 | Plasmid coding for the C-terminal part of eYfp (YC, amino acids 155 to 238) under the control of the PBAD promoter based on pBBR1MCS-2 | This study |
| pBBR-YN-phaC1 | phaC1 ligated into the XhoI and SpeI sites of pBBR-YN-C1 | This study |
| pBBR-YN-phaP5 | phaP5 ligated into the XhoI and KpnI sites of pBBR-YN-C1 | This study |
| pBBR-YN-phaM | phaM ligated into the KpnI and SpeI sites of pBBR-YN-C1 | This study |
| pCM-EYFP-C1 | Plasmid coding for eYfp under the control of the PBAD promoter based on pCM62 | This study |
| pCM-YC-C1 | Plasmid coding for the C-terminal part of eYfp (YC, amino acids 155 to 238) under the control of the PBAD promoter based on pCM62 | This study |
| pCM-YC-phaC1 | phaC1 ligated into the XhoI and SpeI sites of pCM-YC-C1 | This study |
| pCM-YC-phaP5 | phaP5 ligated into the XhoI and KpnI sites of pCM-YC-C1 | This study |
| pCM-YC-phaM | phaM ligated into the KpnI and SpeI sites of pCM-YC-C1 | This study |
| pJOE4036.1 | Rhamnose-inducible His tag expression vector, Ampr | J. Altenbuchner |
| pJOE4036.1-phaP5(His)6 | Expression vector for PhaP5 | This study |
| pJOE4036.1-phaM(His)6 | Expression vector for PhaM | 13 |
| Primers | ||
| rrnB_fwd_NsiI | CGGCATGCATTGAGTTTAAACGGTCTCCAGCTTGG | |
| rrnB_rev_NsiI | CGGCATGCATGTAGAAACGCAAAAAGGCCATCCG | |
| EYFP-C1_f_NdeI | GGGAATTCCATATGGTGAGCAAGGGCGAGG | |
| EYFP-C1_r_SpeI | GGACTAGTTGATCAGTTATCTAGATCCGGTGGATCC | |
| YC-C1_f_NdeI | GGGAATTCCATATGGCCGACAAGCAGAAGAACGGC | |
| YN-C1_r_XhoI | CCGCTCGAGATCTGAGTCCGGACATGATATAGACGTTGTGGCTGTTGTAG | |
| AdapA | CATGTGCATGCATATGGTTTAAACAGTACTAGTG | |
| AdapB | AATTCACTAGTACTGTTTAAACCATATGCATGCA | |
| PhaC1_C_f_XhoI | CCGCTCGAGGCATGGCGACCGGCAAAGGCGCGG | |
| PhaC1_C_r_SpeI | GGACTAGTTCATGCCTTGGCTTTGACGTATCGCCCAGGC | |
| PhaP5_C_f_XhoI | CCGCTCGAGGCATGGCCACGCCTCCC | |
| PhaP5_C_r_KpnI | GGGGTACCCTAGCCCTTGGATTTCGGCTTG | |
| PhaM_C_f_XhoI | CCGCTCGAGGCATGTTCGGACAGATTCCCG | |
| PhaM_C_r_KpnI | GGGGTACCTCAGGCTGCGCTGCTG | |
| PhaM_C_f_KpnI | GGGGTACCATGTTCGGACAGATTCCCGATTTCACC | |
| PhaM_C_r_SpeI | GGACTAGTTCAGGCTGCGCTGCTGCC |
Restriction sites are underlined. Start and stop codons are marked in bold type.
Construction of transferable BiFC expression plasmids.
For construction of BiFC expression plasmids, the two compatible and mobilizable broad-host-range vectors pBBR1MCS-2 (22) and pCM62 (23) were used. Fusions were expressed using the arabinose-inducible PBAD promoter (24). The genes coding for the N- and C-terminal parts of eYfp (YN, amino acids 1 to 154; YC, amino acids 155 to 238) were amplified via PCR from pEYFP-C1 (Clontech) using the primers EYFP-C1_f_NdeI and YN-C1_r_XhoI and primers YC-C1_f_NdeI and EYFP-C1_r_SpeI, respectively. For construction of fusions of PHB granule-associated proteins to the C terminus of the YN and YC fragments, the multiple cloning site (MCS; C1 fragment) of pEYFP-C1 was also included in the BiFC expression plasmids (Fig. 1).
Fig 1.

Plasmids used for BiFC analysis. A system based on two broad-host-range plasmids (pBBR1MCS-2 [22] and pCM62 [23]) was constructed to carry out BiFC in an R. eutropha background. N-terminal (YN) and C-terminal (YC) parts of eYfp together with a C-terminal MCS to clone the protein of interest were inserted in the two plasmids, respectively. The resulting plasmids, pBBR-YN-C1 and pCM-YC-C1, can be transferred to R. eutropha or other Gram-negative bacteria via conjugation and used to express BiFC fusions from the l-arabinose-inducible PBAD promoter.
Construction of the plasmids was carried out as follows: three transcriptional terminators (rrnB genes) of plasmid pDMW7 (25), a plasmid based on pBAD/Thio-TOPO (Invitrogen), were amplified using the primers rrnB_fwd_NsiI and rrnB_rev_NsiI and cloned into the NsiI site of pBBR1MCS-2-PBAD-phaM-eyfp (13), a plasmid based on pBBR1MCS-2 which harbors the PBAD promoter originally amplified from pDMW7, to minimize background expression of fluorescent fusion proteins during noninduced culture conditions from upstream-located promoters (e.g., kanamycin resistance gene). The orientation of the terminator fragments was checked by colony PCR and DNA sequencing; only constructs with desired orientations were used. The resulting plasmid pBBR1MCS-2-rrnB-PBAD-phaM-eyfp-c1 was cut with NdeI and SpeI to replace the phaM-eyfp gene by eyfp-c1 (amplified from pEYFP-C1 using primers EYFP-C1_f_NdeI and EYFP-C1_r_SpeI) and yc-c1, yielding the plasmids pBBR1MCS-2-rrnB-PBAD-eyfp-c1 (pBBR-EYFP-C1) and pBBR1MCS-2-rrnB-PBAD-yc-c1 (pBBR-YC-C1). To fuse the N-terminal eYfp fragment (YN) to the C1 fragment (MCS), pBBR-EYFP-C1 was cut with NdeI and XhoI and ligated with the yn fragment, yielding the plasmid pBBR1MCS-2-rrnB-PBAD-yn-c1 (pBBR-YN-C1). The rrnB-PBAD-eyfp-c1 and rrnB-PBAD-yc-c1 cassettes were amplified from pBBR-EYFP-C1 and pBBR-YC-C1 using the primers rrnB_fwd_NsiI and EYFP-C1_r_SpeI, cut with SphI and SpeI (the rrnB_fwd_NsiI primer also contained a SphI site), and subcloned into a derivative of pCM62 (see below) that had been cut with the same enzymes. The plasmid pLO11 (a derivative of pCM62 [26]) was cut with AflIII and EcoRI to obtain the pCM62 backbone and ligated with annealed oligonucleotides AdapA and AdapB, which contain internal SphI and SpeI sites for subcloning of fragments from pBBR1MCS-2-based plasmids in pCM62. The resulting plasmids were designated pCM-EYFP-C1 and pCM-YC-C1.
The plasmids pBBR-YN-C1 and pCM-YC-C1 were used for further construction of BiFC fusions. pBBR-EYFP-C1 and pCM-EYFP-C1 served as expression controls to study the strength of eYfp expression from the PBAD promoter system. The phaM, phaP5, and phaC1 genes were each amplified from R. eutropha genomic DNA (Table 1 shows the primers) and ligated into the XhoI and KpnI (phaM and phaP5) and XhoI and SpeI (phaC1) sites of pBBR-YN-C1 and pCM-YC-C1. The resulting constructs coded for C-terminal fusions of the proteins to the YN and YC fragments that were separated by the linker sequence SGLRSRG. Because of a low BiFC activity for PhaM constructs, additional plasmids using the longer and more flexible linker SGLRSRAQASNSAVDGT were constructed using the KpnI and SpeI sites. The elongation of the linker region resulted in a slight increase of the BiFC complementation efficiency. Therefore, only the results using the plasmids with the elongated linker region are shown for the PhaM fusions.
BiFC experimental conditions.
E. coli S17-1 was cotransformed with pBBR-YN and pCM-YC derivatives, and both plasmids were conjugatively transferred to R. eutropha H16. Transconjugants were selected on mineral salts medium supplemented with 0.5% (wt/vol) fructose, 15 μg ml−1 tetracycline, and 350 μg ml−1 kanamycin and purified by repeated transfer of single colonies to selection agar. Growth of recombinant R. eutropha strains for BiFC analysis in liquid culture was performed at 30°C and in the presence of 15 μg ml−1 tetracycline and 150 μg ml−1 kanamycin. A single colony of the strain of interest was used for inoculation of a 10-ml seed culture (100-ml Erlenmeyer flask filled with 10 ml of 0.8% [wt/vol] nutrient broth) and was incubated for 24 h with continuous shaking (150 to 180 rpm). One milliliter of this seed culture was transferred to 10 ml fresh NB medium supplemented with 0.2% l-arabinose and incubated for 24 h on a rotary shaker. Arabinose is not used as a carbon source by R. eutropha, but it induces transcription of the genes under the control of the PBAD promoter (24). The cells intermediately accumulated PHB granules during this time. After about 24 h (growth was slightly reduced compared to that of the wild type because of the presence of two antibiotics in the culture medium), the bacteria were in the stationary growth phase, as indicated by shortening of the cells and loss of Nile red-stainable PHB granules. For BiFC analysis and monitoring formation of PHB granules, 0.1 volume of the stationary-phase, PHB-free R. eutropha culture was used to inoculate 0.9 volume of fresh NB medium to which 0.2% sodium gluconate had been added to promote PHB accumulation (increase of C-to-N ratio). l-Arabinose (0.2% [wt/vol]) was also added to allow expression of BiFC fusions. Samples were taken at intervals as indicated and were immediately examined by fluorescence microscopy. Expression and size of BiFC fusions were studied by Western blot analysis of SDS-PAGE-separated whole-cell extracts using polyclonal anti-green fluorescent protein (anti-Gfp) antibodies from rabbit (Invitrogen).
Fluorescence microscopy.
Fluorescence microscopy was carried out as described previously (27, 28) on a Zeiss Axioplan fluorescence microscope using Metaview/Metamorph software (Visitron Systems). To ensure comparability between different experiments, fluorescence images were acquired immediately after the phase-contrast images using the same microscope settings (phase contrast, 400-ms exposure time; fluorescence, 4,000-ms exposure time). MBF ImageJ 1.43m (Wayne Rasband, National Institute of Mental Health, Bethesda, MD) was used for processing of images. All microscopic investigations were performed in triplicate. Typical images are shown.
Analytical size exclusion chromatography (gel filtration).
C-terminal His6-tagged PhaM and PhaP5 proteins were expressed in E. coli JM109 using the pJOE4036.1 expression vector and purified via standard nickel agarose (Amersham) affinity chromatography gravity flow columns. For analytical gel filtration analysis, the buffer was exchanged to 10 mM Tris-HCl, pH 8.0, 150 mM NaCl. Alternatively, 20 mM HEPES buffer (pH 7.5) was used for in vitro protein cross-linking experiments. Analytical gel filtration was carried out on an Äkta purifier system equipped with a Superdex-200 column (preparation-grade HR 10/30; geometric column volume [Vc] of 23.41 ml; equilibrated with 10 mM Tris-HCl, pH 8.0, 150 mM NaCl; operated at a flow rate of 0.5 ml/min). The column void volume (V0) was determined with Blue Dextran 2000 to 8.41 ml. Two hundred microliters of a 1.25-mg/ml purified PhaP5-His6 solution or 200 μl of a 0.5-mg/ml purified PhaM-His6 solution was loaded onto the column and eluted with equilibration buffer (detection at 280 nm). The apparent molecular weights of PhaP5-His6 and PhaM-His6 were determined after calibration of the column with standard proteins (Bio-Rad gel filtration standard, catalog no. 151-1901, and GE Healthcare low-molecular-weight gel filtration calibration kit, catalog no. 28-4038-41). To this end, the partition coefficient (Kav) for each protein was calculated using the elution volume (Ve) and the formula Kav = (Ve − V0)/(Vc − V0).
Cross-linking.
For in vitro cross-linking studies, 9.5 μl PhaM (50 μg/ml) or PhaP5 (150 μg/ml) in 20 mM HEPES buffer, pH 7.5, and 0.5 μl of a freshly prepared glutardialdehyde (1,5-pentanedial) solution (2.5%) were mixed and incubated at 37°C for 5 min. The reaction was stopped by addition of 1 μl of 1 M Tris-HCl, pH 8. As a negative control, 0.5 μl water was added instead of the glutardialdehyde solution. Additional controls were performed by repetition of the cross-linking experiments in the presence of 0.5% SDS. In some experiments, the proteins (PhaM and PhaP5) were heated to 95°C for 10 min prior to cross-linking. Samples were mixed with 5.5 μl 3× loading buffer, heated at 95°C for 5 min, and analyzed by SDS-PAGE and subsequent silver staining. Cross-linking experiments were repeated with variations in glutardialdehyde concentration and in cross-linking time. Similar results were obtained; a typical result is shown in Results.
Protein preparation for immunoblot expression analysis.
Expression of PhaM and PhaP5 was studied by Western blot (immunoblot) analysis after SDS-PAGE separation of soluble cell extracts of R. eutropha wild type and the PHB-negative ΔphaC1 deletion mutant. R. eutropha strains in which the phaP5 or phaM gene had been deleted (ΔphaP5 and ΔphaM mutants) and purified PhaM-His6 or PhaP5-His6 proteins served as negative and positive controls, respectively. Growth conditions were as described above for BiFC conditions except that no antibiotics and no inducer were present. Cells were harvested by centrifugation at time points as indicated, resuspended with phosphate-buffered saline to an optical density at 600 nm (OD600) of 100, mixed with 0.5 volume of 3× SDS-PAGE loading buffer, and heated to 95°C for 30 min with occasional vortexing to shear DNA and to solubilize proteins. The long heating time did not affect protein stability, as determined by use of purified PhaM and PhaP5 (not shown). PHB and cell debris were removed by centrifugation in Eppendorf microcentrifuge tubes for 1 min at 20,000 × g. Ten-microliter samples of the supernatant were loaded onto 12% acrylamide gels (cross-linking degree, acrylamide to bisacrylamide, 37.5:1) and separated by reducing (mercaptoethanol) SDS-PAGE. Immunoblot analysis was carried out using polyclonal mouse antibodies that had been raised against purified PhaM-His6 or PhaP5-His6. A commercial anti-mouse alkaline phosphatase-coupled antibody (Sigma) and the nitroblue tetrazolium–5-bromo-4-chloro-3-indolylphosphate (NBT-BCIP) system were used for signal detection.
Identification of PHB granule-bound proteins via ESI-MS-MS.
For the identification of PHB granule-bound proteins, PHB granules were isolated by density gradient centrifugation as described earlier (29). Native PHB granules were washed three times with water to remove weakly bound proteins. After solubilization of granule-bound proteins by heating in SDS-PAGE loading buffer, samples were separated on a 10% acrylamide gel and stained with colloidal Coomassie brilliant blue. Identification of trypsin-digested proteins was carried out at the Life Science Centre, University Stuttgart-Hohenheim, using liquid chromatography-electrospray ionization-tandem mass spectrometry (LC-ESI-MS-MS).
Other techniques.
PHA content was determined by gas chromatography. For this, PHAs of vacuum-dried cells were subjected to acid methanolysis (30) and separated on an HP-5 column (Agilent). Protein determination was performed according to the method in reference 31. Conjugation experiments with E. coli S17-1 harboring the plasmid(s) of interest as donor and R. eutropha as recipient were performed by cross-streaking both strains directly on selective medium (mineral salts medium with 0.5% fructose and appropriate antibiotics). Transconjugants appeared after 2 to 3 days at 30°C and were purified and checked for the presence of plasmids.
RESULTS AND DISCUSSION
Construction of suitable vectors for controlled expression of YN and YC fusions with PGAPs in R. eutropha.
For the analysis of in vivo protein-protein interaction, a two-plasmid system for expression of the two proteins of interest was necessary. Hence, a BiFC expression system based on the two broad-host-range plasmids pBBR1MCS-2 (Kmr) and pCM62 (Tcr) was constructed as described in Materials and Methods. Both plasmids (pBBR-YN-C1 and pCM-YC-C1 [Fig. 1]) are equipped with the arabinose-dependent PBAD promoter, the araC regulator gene, and one of either the YN- or the YC-encoding DNA fragments followed by a multiple cloning site for in-frame fusion of test genes. Plasmids coding for fusions of YN and YC with PhaC1, PhaP5, or PhaM were constructed and verified by DNA sequencing of the cloned DNA fragments (Table 1). Plasmid pairs for expression of suitable combinations of fusion proteins (YN-PhaC1, YN-PhaP5, and YN-PhaM; YC-PhaC1, YC-PhaP5, and YC-PhaM) were cotransformed to E. coli S17-1 and subsequently transferred to R. eutropha H16 via conjugation. It was generally possible to transform both plasmids to E. coli S17-1 and to transfer both plasmids to R. eutropha in one step each using agar medium for selection that contained both antibiotics (kanamycin and tetracycline). All fusions were expressed both in E. coli and in recombinant R. eutropha. Expression of the respective YN- and/or YC-containing fusion proteins with expected sizes was confirmed by Western blot analysis of cell extracts using commercial polyclonal Gfp-specific antibodies (data not shown). Only the construct of the YC domain (without fusion protein) was poorly expressed in R. eutropha. The degree of expression of fusion proteins was dependent on the presence and concentration of arabinose in the range of 0 to 0.2% and is in agreement with the results of others (32). Control of the level of protein expression is important for BiFC analysis because a high level of overexpression can result in an increase of spontaneous formation of fluorescent aggregates in control experiments (21). The relative concentrations of all YN fusion proteins expressed in R. eutropha strains with 0.2% arabinose were almost the same as judged from the intensity of stained bands in Western blots (data not shown). In contrast, the concentrations of the YC fusions (derived from pCM-YC-C1 plasmids) after expression in recombinant E. coli were generally higher than those of pBBR1-YN-C1-derived YN fusion proteins. Presumably, pCM62-based plasmids are replicated with a higher copy number than that of pBBR1MCS-2.
BiFC analysis of PhaC1-PhaM and PhaM-PhaP5 interactions in R. eutropha.
PhaM had been previously identified to interact with the key enzyme of PHB synthesis, PHB synthase PhaC1, in a two-hybrid screening experiment in recombinant E. coli (13). The ability of PhaM to bind to PHB granules and to the nucleoid region in R. eutropha was also shown by expression of eYfp-PhaM and by transmission electron microscopy (TEM) analysis (13, 16). The interaction between PhaC1 and PhaM was recently independently confirmed by copurification of PhaM with streptavidin (Strep)-tagged PhaC1 (33). However, a proof for the direct in vivo interaction of PhaM with PHB synthase in R. eutropha was missing, and the subcellular localization of PhaM-PhaC1 complexes in R. eutropha was unknown. R. eutropha H16 harboring plasmids coding for YN-PhaM and YC-PhaC1 or for YN-PhaC1 and YC-PhaM was cultivated in NB medium with gluconate (PHB accumulation) and arabinose (expression of fusion proteins) as described in Materials and Methods. All images, including all controls, were acquired using the same settings to ensure comparability of the results. Generally, fluorescence from complemented eYfp was much weaker than that of comparable fusions with complete eYfp (not shown). Nevertheless, complemented fluorescence activity could be detected by increase of the exposure time to 4 s. The results of BiFC analysis of samples taken at different time points of growth are shown in Fig. 2. One of each fusion together with the empty N- or C-terminal domain of eYfp (YN-C1 or YC-C1) was used as a negative control. BiFC fluorescence was absent or hardly detectable in controls at any time point (representatively shown for the 3-h samples in Fig. 2B). In some cases, especially the combination of YN with a YC fusion, very weak fluorescence signals were detected. When BiFC activity of the YN-PhaM-plus-YC-PhaC1 combination was investigated, a fluorescence signal significantly above that of controls was detected (Fig. 2A) and thus revealed that PhaM and PhaC1 physically interacted in vivo. At zero time, when no PHB granules are present, the fluorescence was concentrated at midcell, very similar to experiments in which eYfp-PhaM fluorescence was expressed in the absence of PHB (see reference 13). It should be noted that the cell poles were free of fluorescence. We conclude that a YN-PhaM-YC-PhaC1 complex was formed and was concentrated in the nucleoid region of PHB-free cells. To our best knowledge, this is the first in vivo evidence that PhaC1 in the absence of PHB is not freely soluble as previously assumed (34) but is nonhomogeneously distributed in the cytoplasm in the form of a presumably nucleoid-associated PhaM-PhaC1 complex. Notably, investigation of PhaC1 localization by immunogold analysis using PhaC1-specific antibodies in TEM experiments that had been performed almost 20 years ago had revealed a nonrandom distribution of gold particles in PHB-poor cells: only about one-third of the number of gold particles (15 and 11 particles in the two cells of Fig. 2A of reference 35 in which the nucleoid region can be seen) was present in the cytoplasm, but the majority of gold particles (52 and 31, respectively; a few particles could not be assigned) were located in the nucleoid region. This is in contrast to experiments in which an overexpressed PhaC1-eYfp fusion was investigated and appeared to be soluble in the absence of PHB (36). This finding can be explained because the nucleoid association is masked by the constitutive overexpression of PhaC1-eYfp relative to PhaM in our previous publication. In conclusion, our BiFC data suggest that PhaM has the capacity to form a complex with PhaC1 and to direct PhaC1 to the nucleoid region in R. eutropha. At later time points, BiFC data for cells expressing YN-PhaM plus YC-PhaC1 showed that the fluorescence did not or only marginally increased and still was nonhomogeneously distributed in the cells, very similarly to eYfp-PhaM fluorescence (Fig. 2A). Since PhaM and PhaC1 are constitutively expressed in R. eutropha (see below and reference 37), we conclude that PhaC1-PhaM complexes are present and are bound to the nucleoid without PHB at zero time and with PHB granules at later time points. Staining of the cells with Nile red in combination with laser scanning microscopy and transmission electron microscopic analysis confirmed that (small) PHB granules had been formed and that the PHB granules colocalized with the dividing nucleoids near midcell if a PhaM fusion was present in any combination (Fig. 2C and D). We conclude that the YN-PhaM-YC-PhaC1 complex colocalized with PHB granules and that PHB granules are bound to the nucleoids. Practically the same results were obtained when the converse combination was examined (YN-PhaC1 plus YC-PhaM [Fig. 2A]).
Fig 2.

BiFC analysis between PhaC1 and PhaM and between PhaM and PhaP5 (hetero-oligomerization): BiFC assay of PhaM and PhaC1 and of PhaM and PhaP5 in R. eutropha H16 at different stages of PHB accumulation in NB-gluconate medium (for details, see Materials and Methods). (A) Interaction combination as indicated on the left. (B) Negative controls with strains carrying only one Pha fusion together with the corresponding empty eYfp fragment representatively shown for the 3-h samples. The upper left part of each image is shown in phase-contrast mode; the rest of the image is shown in fluorescence mode. All images were acquired and processed using the same settings. (C) Nile red stain of R. eutropha strains expressing different combinations of BiFC fusions. Images in panel C were taken from the 3-h and 5-h time points. (D) Effects of constitutive overexpression of PhaM (Nile red stain and TEM image; the bacterial nucleoid is marked with an arrowhead) and eYfp-PhaM (laser scanning microscopy with 4′,6-diamidino-2-phenylindole [DAPI] staining of the nucleoid and FM4-64 membrane stain) in R. eutropha. Note that PHB granules appear in close contact with the nucleoid region during overexpression of PhaM and of various BiFC or eYfp fusions of PhaM. Bars, 3 μm for all fluorescence microscopy images and 1 μm for the TEM picture.
Next, we studied interaction of PhaM with PhaP5 by BiFC analysis of expressed YN-PhaM plus YC-PhaP5 and the converse combination YN-PhaP5 plus YC-PhaM (Fig. 2A, bottom). Fluorescence complementation that was above the level of the controls was determined at all stages of PHB formation and clearly showed that PhaP5 and PhaM interacted in vivo in R. eutropha. BiFC activity appeared as “diffuse foci” independently of the presence or absence of PHB and colocalized with formed PHB granules and with the nucleoid region under PHB-permissive conditions. Staining of the bacteria with Nile red showed that only small PHB granules were formed, as shown earlier by TEM, but individual PHB granules could not be resolved by light microscopy (Fig. 2C and D). R. eutropha cells that expressed PhaM fusions tended to form longer cells than did the wild type, as had been found earlier for eYfp-PhaM- and PhaM-overexpressing cells (13, 16). BiFC activity was more concentrated in the center of cells and reminiscent of that of eYfp-PhaM-overexpressing cells (13). Since PhaP5, PhaM, and PhaC1 are all constitutively expressed, we assume that all three proteins form a complex and are attached to the nucleoid.
Figure 3 shows the results of experiments in which we tested homo-oligomerization of PhaP5, PhaM, and PhaC1 by BiFC. All BiFC signals were considerably stronger than controls and were stronger than those determined for interaction between different PGAPs (hetero-oligomerization). Cells expressing YN-PhaP5 plus YC-PhaP5 showed strong BiFC signals in the cytoplasm in the absence of PHB (zero time in Fig. 3A). Interestingly, the BiFC signal did not change in the first hour of growth under PHB-permissive conditions. At this time, most cells had already synthesized 2 ± 1 PHB granules as revealed by Nile red staining (Fig. 3A, bottom row). In conclusion, a fluorescent YN-PhaP5-YC-PhaP5 complex is formed but is not yet bound to PHB in the first hour under PHB-permissive conditions. We do not know whether this finding is a consequence of the overexpression of PhaP5 relative to other PGAPs. Interestingly, at later stages of PHB accumulation (3 and 5 h), the BiFC signal of the YN-PhaP5-YC-PhaP5 complex condensed to foci near the cell poles and colocalized with formed PHB granules. Similar results have been found for PhaP3-eYfp-, PhaP6-eYfp-, and PhaP7-eYfp-expressing cells (36). Strong BiFC signals were also confirmed for cells expressing YN-PhaM plus YC-PhaM or YN-PhaC1 plus YC-PhaC1 (Fig. 3B and C). In both cases, we found effects on PHB granule formation and localization similar to those for the fusions of eYfp-PhaM or the PhaC1-eYfp fusion, namely, cell elongation and diffuse fluorescence in the cell center (nucleoid region with many small PHB granules in the cell center) in the case of PhaM homo-oligomer fusion complexes and colocalization of normally formed PHB granules in the case of PhaC1 homo-oligomer complexes (Fig. 3B and C). The detection of a fluorescence signal for YN-PhaC1-YC-PhaC1 homo-oligomers at zero time indicated that PhaC1 is able to dimerize even in the absence of PHB. Our results also confirmed that the YN-PhaC1 and YC-PhaC1 fusions are biochemically active, as they both complement the PHB-minus phenotype of the ΔphaC1 strain. In the absence of PHB (zero time), the YC-PhaC1 and YN-PhaC1 complex was soluble probably because of overexpression relative to PhaM.
Fig 3.

BiFC analysis between PhaP5 and PhaP5, PhaM and PhaM, and PhaC1 and PhaC1 (homo-oligomerization). BiFC homo-oligomerization assay of PhaP5 (A), PhaM (B), and PhaC1 (C) in R. eutropha H16 (A and B) and H16 ΔphaC1 (C) at different stages of PHB accumulation (for details, see Materials and Methods). The respective negative controls are shown in Fig. 2. All images were acquired using the same settings as those in Fig. 2, except that less gamma and contrast correction was applied than that for the negative-control images, because signals obtained for the homo-oligomerization experiments were more intense than those for the images shown in Fig. 2. Nile red-stained cells for the PhaP5 homo-oligomerization experiment are shown in the bottom row of panel A. Note that the Nile red images are made from a separate sample and do not show the same cells as those in the two rows above. To determine the subcellular localization of BiFC complexes, overlay pictures of BiFC signals and phase-contrast images of single cells are shown in panel D. Note that PhaM BiFC complexes are localized at midcell at the nucleoid region (compare with Fig. 2D), whereas PhaP5 BiFC complexes localize near the cell poles. Bars, 3 μm.
Expression of PhaP5, PhaM, and other PGAPs in R. eutropha wild type.
Proteome analysis of PHB granules isolated from R. eutropha wild type showed that PHB synthase (PhaC1), PhaM, phasins PhaP1 to PhaP5, PHB depolymerase PhaZa1, and regulator PhaR were expressed and were present in the granule fraction (data not shown). The BiFC studies shown above were performed with recombinant R. eutropha strains in which PGAPs such as PhaC1, PhaM, and PhaP5 were overexpressed from arabinose-controlled promoters of the respective plasmids. Therefore, the amount of PGAP produced is different from and probably artificially higher than that in the wild type. To find out whether and how much PhaP5 and PhaM protein is produced in the R. eutropha wild type, expression of PhaP5 and PhaM was studied by Western blot analysis of soluble cell extracts that were prepared from cells that had been grown in NB-gluconate medium. The constitutive expression of PhaC1 in R. eutropha had been repeatedly shown by others (37–39) and was not repeated in our study. The growth experiments were performed under the same condition (NB plus 0.2% gluconate) as that for the BiFC experiments, except that arabinose was not present in the growth medium (Fig. 4A). Samples were taken at zero time (no PHB) and after different periods of growth after transfer to fresh medium. Cells of all time points were adjusted to an equal turbidity (OD600 of 100), and Western blots of whole-cell extracts were prepared as described in Materials and Methods. The experiments were performed with R. eutropha H16 wild type and with a ΔphaC1 mutant to study whether the absence of PHB and PHB synthase protein could have an impact on expression of PhaP5 or PhaM. R. eutropha strains with deletion of the phaP5 or phaM gene (ΔphaP5 or ΔphaM strain) were used as negative controls to test the specificity of the antibodies.
Fig 4.

Expression of PhaM and PhaP5 and accumulation of PHB. R. eutropha H16 and the R. eutropha ΔphaC1 strain were grown on NB-gluconate medium, and optical densities at 600 nm and PHB contents of samples were recorded (A). CDW, dry weight of cells. Antisera against PhaM or PhaP5 reacted with respective proteins of the expected size in R. eutropha and with purified PhaM-His6 or PhaP5-His6, whereas no signals were observed in the respective deletion strains (H16 ΔphaM or ΔphaP5) (B). Expression of PhaM in H16 (C) and H16 ΔphaC1 (D) at different stages of growth was determined in whole-cell extracts by Western blotting (immunoblotting). Comparable loading of wells was performed using the same OD600 equivalents and checked by SDS-PAGE and Coomassie brilliant blue staining (not shown). Purified PhaM-His6 or PhaP5-His6 was used as a positive control, and whole-cell extracts of H16 ΔphaM and H16 ΔphaP5 corresponding to the 0-h time point were used as negative controls. One growth experiment with the respective Western blot out of two biological replicates is shown. WT, wild type.
As is evident from Fig. 4B, both PhaM and PhaP5 were expressed in R. eutropha H16. The bands in the Western blot for PhaM generally were more intense than the PhaP5 bands. PhaM proteins were present in the absence of PHB (0 h) and in the presence of PHB (1 h to 18 h) (Fig. 4C and D). The band intensities for PhaM of the 3-h and 5-h samples in Fig. 4C were slightly lower than those for the other time points. This apparent reduction in expression was caused by large amounts of PHB present in cells of these time points, resulting in increased turbidity. Since OD-normalized amounts of cells were used for cell extract preparation, the samples at 3 and 5 h contain slightly less protein than do samples of the other time points. Coomassie blue-stained loading controls and protein determination were in agreement with this explanation (data not shown). Similar results were obtained for determination of PhaP5 expression. However, the signals for PhaP5 were much weaker than those for PhaM and were near the detection limit (images not shown). In conclusion, the expression of PhaM did not significantly change during a batch culture on NB-gluconate medium and is in agreement with transcriptome studies of others (37).
The same principal results were obtained when the Western blot assays were performed with samples of the ΔphaC1 strain (Fig. 4D). We conclude that the presence of PHB or PHB synthase protein did not influence expression of PhaP5 and PhaM.
Determination of the quaternary structure of PhaP5 and PhaM.
Our two-hybrid and BiFC data indicated that several PGAPs in R. eutropha can interact in vivo, and some of the interacting proteins, such as PhaP5, interact with more than one other protein (PhaM and PhaC1). Moreover, at least two of the PGAPs (the transcriptional regulator PhaR and the recently discovered PhaM) are able to bind to DNA in addition to PHB. Depending on the number of interaction sites of a given interacting protein, the formation of a network in the cell or on the PHB granules is possible. Since the number of interaction sites of a protein depends also on the number of subunits, it was interesting to determine the quaternary structure of PhaP5 and PhaM. PhaC1 had been previously shown to be present as a dimer (11, 40). PhaP5-His6 and PhaM-His6 were expressed in recombinant E. coli and purified by nickel agarose affinity chromatography. PhaP5 and PhaM were almost homogeneous (purity, >95%) as revealed by SDS-PAGE analysis (Fig. 5); however, PhaM was rather unstable and disintegrated into several smaller fragments; the fragments still reacted with PhaM-specific antibodies (not shown); the presence of significant amounts of contaminating non-PhaM proteins could be excluded. The quaternary structures of PhaP5 and PhaM were determined by gel filtration on a Superdex-200 column that had been equilibrated with standard proteins of known molecular masses (Fig. 6). A Kav value of 0.505 was determined for purified PhaP5-His6, which corresponded to a molecular mass of the native protein in the range of approximately 42 kDa. With a theoretical molecular mass of 16.6 kDa, the quaternary structure of the native PhaP5-protein complex in solution was calculated to be a dimer or a trimer (2.5-mer). However, a trimer was rather unlikely from the in vitro cross-linking results (see below), and our results pointed toward a dimer.
Fig 5.
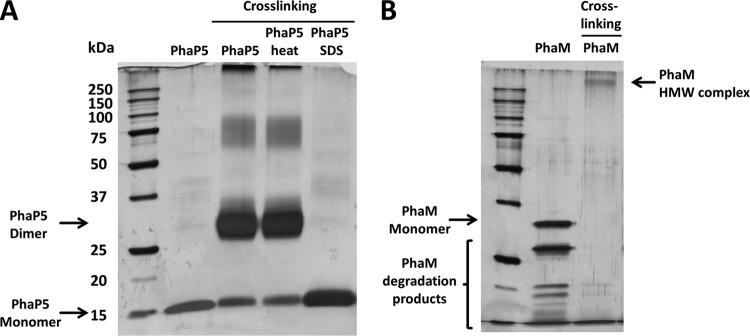
SDS-PAGE of PhaM and PhaP5 after chemical cross-linking. Oligomerization of PhaP5-His6 (A) and PhaM-His6 (B) was analyzed using glutardialdehyde cross-linking as described in Materials and Methods. Cross-linking of PhaP5 led to decrease of the monomer band and to the formation of a dominant ∼30- to 34-kDa product, which corresponds to a dimer and to minor products of higher apparent molecular masses corresponding to tetra- to hexamers. Formation of PhaP5 dimers was also observed after heating (95°C, 10 min) of PhaP5-His6 protein prior to cross-linking, but denaturation of PhaP5-His6 by preincubation with 0.5% SDS prevented the formation of oligomers. When samples with PhaM-His6 were subjected to cross-linking, the bands corresponding to monomers and to PhaM degradation products completely disappeared and high-molecular-weight products (HMW) with apparent molecular masses of >250 kDa were detected (B).
Fig 6.

Determination of quaternary structures of PhaP5 and PhaM. Analytical gel filtration was carried out on a Superdex-200 column (for details, see Materials and Methods). Calibration of the column (upper left) was performed using the following standard proteins: thyroglobulin (670 kDa), gamma globulin (158 kDa), conalbumin (75 kDa), ovalbumin (44 kDa), carbonic anhydrase (29 kDa), myoglobin (17 kDa), RNase (13.7 kDa), aprotinin (6.5 kDa), and vitamin B12 (1.35 kDa). Elution profiles of selected standard proteins (dashed line), purified PhaP5-His6 (gray line, Ve = 15.98 ml), and purified PhaM-His6 (black line, Ve = 11.21 ml) are shown. The void volume of the column (blue dextran) was determined as 8.41 ml.
When purified PhaM-His6 (theoretical molecular mass, 27.6 kDa) was run on the Superdex-200 column, the protein eluted at a Kav value of 0.187 that corresponded to a molecular mass of ≈330 kDa and suggested that PhaM represents a dodecamer (theoretical molecular mass, 331.7 kDa). No significant differences in the values for the native molecular masses of PhaM and PhaP5 were found when the gel filtration experiment was repeated in buffers with different ionic strengths (not shown), and this indicated that neither PhaP5 nor PhaM interacted with the column material. We also mixed purified PhaP5 and PhaM prior to the gel filtration experiment to study if the two proteins interacted in vitro and formed complexes of the two proteins. However, gel filtration of the PhaP5 and PhaM mixture resulted in the same Kav values as those determined for runs with individual proteins, and no evidence for in vitro complex formation was found under the chosen experimental conditions.
Cross-linking experiments were performed to confirm the results on the quaternary structures of PhaP5 and PhaM; for this, the purified proteins were incubated in the presence of glutardialdehyde as described in Materials and Methods. After stopping the cross-linking reaction, the proteins were analyzed by SDS-PAGE. Cross-linked PhaP5 showed a strong band in the range of the dimer (≈33 kDa) and a very weak (broad) band at higher apparent molecular masses (tetra- to hexamer) (Fig. 5A). The band at the position of the monomer (17 kDa) was reduced in intensity, and no band was visible at the position at which a trimer of PhaP5 would have been expected (≈50 kDa). The same principal result was obtained when the experiment was repeated with a PhaP5 preparation that had been heated to 95°C for 10 min prior to the cross-linking condition. Apparently, PhaP5 is highly stable and cannot be denatured by heat treatment. Heat-stable proteins with phasin-like properties have been previously described for Rhodospirillum rubrum (41, 42). When cross-linked PhaP5 was run on the gel filtration column, the PhaP5 complex coeluted with native (not cross-linked) PhaP5, and this confirmed that PhaP5 apparently is a dimer.
PhaM showed a strong self-aggregation ability: all PhaM material was present in the high-molecular-mass region above 250 kDa after exposure to the cross-linking condition. The exact value of the apparent molecular mass could not be resolved by gel electrophoresis (Fig. 5B). Interestingly, no material of the monomer was visible anymore in the cross-linked sample. Even the degradation products of PhaM (see above) had “disappeared,” and this indicated that they also were cross-linked. Heating of PhaM to 95°C did not prevent cross-linking, but if the protein was incubated in the presence of 0.5% SDS prior to addition of cross-linking reagent, no cross-linking was observed (not shown). When PhaP5 and PhaM were mixed and cross-linked prior to SDS-PAGE, the same products appeared as those for the cross-linking of individual PhaP5 and individual PhaM. Apparently, PhaP5 and PhaM do not react with each other under these in vitro conditions. We cannot exclude the possibility that other factors or conditions are necessary for interaction of PhaP5 with PhaM.
ACKNOWLEDGMENTS
This work was supported by the Deutsche Forschungsgemeinschaft.
We thank Oliver Lenz for providing plasmid pLO11 and the Institute of Cell Biology (University Stuttgart) for anti-PhaM and anti-PhaP5 antisera. The support of University Stuttgart-Hohenheim (A. Huber, J. Pfannstiel, and O. Simon) in proteome analysis of nPHB granules is greatly acknowledged.
Footnotes
Published ahead of print 22 February 2013
REFERENCES
- 1. Anderson AJ, Dawes EA. 1990. Occurrence, metabolism, metabolic role, and industrial uses of bacterial polyhydroxyalkanoates. Microbiol. Rev. 54: 450–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Madison LL, Huisman GW. 1999. Metabolic engineering of poly(3-hydroxyalkanoates): from DNA to plastic. Microbiol. Mol. Biol. Rev. 63: 21–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pötter M, Steinbüchel A. 2006. Biogenesis and structure of polyhydroxyalkanoate granules. Microbiol. Monogr. 1: 110–136 [Google Scholar]
- 4. Grage K, Jahns AC, Parlane N, Palanisamy R, Rasiah IA, Atwood JA, Rehm BHA. 2009. Bacterial polyhydroxyalkanoate granules: biogenesis, structure, and potential use as nano-/micro-beads in biotechnological and biomedical applications. Biomacromolecules 10: 660–669 [DOI] [PubMed] [Google Scholar]
- 5. Sudesh K, Abe H, Doi Y. 2000. Synthesis, structure and properties of polyhydroxyalkanoates: biological polymers. Prog. Polym. Sci. 25: 1503–1555 [Google Scholar]
- 6. Pederson EN, McChalicher CWJ, Srienc F. 2006. Bacterial synthesis of PHA block copolymers. Biomacromolecules 7: 1904–1911 [DOI] [PubMed] [Google Scholar]
- 7. Gao X, Chen JC, Wu Q, Chen GQ. 2011. Polyhydroxyalkanoates as a source of chemicals, polymers, and biofuels. Curr. Opin. Biotechnol. 22: 768–774 [DOI] [PubMed] [Google Scholar]
- 8. Escapa IF, García JL, Bühler B, Blank LM, Prieto MA. 2012. The polyhydroxyalkanoate metabolism controls carbon and energy spillage in Pseudomonas putida. Environ. Microbiol. 14: 1049–1063 [DOI] [PubMed] [Google Scholar]
- 9. Han J, Hou J, Liu H, Cai S, Feng B, Zhou J, Xiang H. 2010. Wide distribution among halophilic archaea of a novel polyhydroxyalkanoate synthase subtype with homology to bacterial type III synthases. Appl. Environ. Microbiol. 76: 7811–7819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cai S, Cai L, Liu H, Liu X, Han J, Zhou J, Xiang H. 2012. Identification of the haloarchaeal phasin (PhaP) that functions in polyhydroxyalkanoate accumulation and granule formation in Haloferax mediterranei. Appl. Environ. Microbiol. 78: 1946–1952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stubbe J, Tian J. 2003. Polyhydroxyalkanoate (PHA) hemeostasis: the role of PHA synthase. Nat. Prod. Rep. 20: 445–457 [DOI] [PubMed] [Google Scholar]
- 12. Beeby M, Cho M, Stubbe J, Jensen GJ. 2012. Growth and localization of polyhydroxybutyrate granules in Ralstonia eutropha. J. Bacteriol. 194: 1092–1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pfeiffer D, Wahl A, Jendrossek D. 2011. Identification of a multifunctional protein, PhaM, that determines number, surface to volume ratio, subcellular localization and distribution to daughter cells of poly(3-hydroxybutyrate), PHB, granules in Ralstonia eutropha H16. Mol. Microbiol. 82: 936–951 [DOI] [PubMed] [Google Scholar]
- 14. Pfeiffer D, Jendrossek D. 2011. Interaction between poly(3-hydroxybutyrate) granule-associated proteins as revealed by two-hybrid analysis and identification of a new phasin in Ralstonia eutropha H16. Microbiology 157: 2795–2807 [DOI] [PubMed] [Google Scholar]
- 15. Steinbüchel A, Aerts K, Babel W, Follner C, Liebergesell M, Madkour MH, Mayer F, Pieper-Furst U, Pries A, Valentin HE. 1995. Considerations on the structure and biochemistry of bacterial polyhydroxyalkanoic acid inclusions. Can. J. Microbiol. 41(Suppl 1): 94–105 [DOI] [PubMed] [Google Scholar]
- 16. Wahl A, Schuth N, Pfeiffer D, Nussberger S, Jendrossek D. 2012. PHB granules are attached to the nucleoid via PhaM in Ralstonia eutropha. BMC Microbiol. 12: 262 doi:10.1186/1471-2180-12-262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Galán B, Dinjaski N, Maestro B, de Eugenio LI, Escapa IF, Sanz JM, García JL, Prieto MA. 2011. Nucleoid-associated PhaF phasin drives intracellular location and segregation of polyhydroxyalkanoate granules in Pseudomonas putida KT2442. Mol. Microbiol. 79: 402–418 [DOI] [PubMed] [Google Scholar]
- 18. Jendrossek D. 2009. Polyhydroxyalkanoate granules are complex subcellular organelles (carbonosomes). J. Bacteriol. 191: 3195–3202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Southward CM, Surette MG. 2002. The dynamic microbe: green fluorescent protein brings bacteria to light. Mol. Microbiol. 45: 1191–1196 [DOI] [PubMed] [Google Scholar]
- 20. Valdivia RH, Cormack BP, Falkow S. 2006. The uses of green fluorescent protein in prokaryotes. Methods Biochem. Anal. 47: 163–178 [PubMed] [Google Scholar]
- 21. Kerppola TK. 2008. Bimolecular fluorescence complementation (BiFC) analysis as a probe of protein interactions in living cells. Annu. Rev. Biophys. 37: 465–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kovach ME, Elzer PH, Hill DS, Robertson GT, Farris MA, Roop RM, Peterson KM. 1995. Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166: 175–176 [DOI] [PubMed] [Google Scholar]
- 23. Marx CJ, Lidstrom ME. 2001. Development of improved versatile broad-host-range vectors for use in methylotrophs and other Gram-negative bacteria. Microbiology 147: 2065–2075 [DOI] [PubMed] [Google Scholar]
- 24. Fukui T, Ohsawa K, Mifune J, Orita I, Nakamura S. 2011. Evaluation of promoters for gene expression in polyhydroxyalkanoate-producing Cupriavidus necator H16. Appl. Microbiol. Biotechnol. 89: 1527–1536 [DOI] [PubMed] [Google Scholar]
- 25. Heiss G, Trachtmann N, Abe Y, Takeo M, Knackmuss H-J. 2003. Homologous npdGI genes in 2,4-dinitrophenol- and 4-nitrophenol-degrading Rhodococcus spp. Appl. Environ. Microbiol. 69: 2748–2754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schwarze A, Kopczak MJ, Rögner M, Lenz O. 2010. Requirements for construction of a functional hybrid complex of photosystem I and [NiFe]-hydrogenase. Appl. Environ. Microbiol. 76: 2641–2651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hermawan S, Jendrossek D. 2007. Microscopical investigation of poly(3-hydroxybutyrate) granule formation in Azotobacter vinelandii. FEMS Microbiol. Lett. 266: 60–64 [DOI] [PubMed] [Google Scholar]
- 28. Jendrossek D, Selchow O, Hoppert M. 2007. Poly(3-hydroxybutyrate) granules at the early stages of formation are localized close to the cytoplasmic membrane in Caryophanon latum. Appl. Environ. Microbiol. 73: 586–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Handrick R, Reinhardt S, Focarete ML, Scandola M, Adamus G, Kowalczuk M, Jendrossek D. 2001. A new type of thermoalkalophilic hydrolase of Paucimonas lemoignei with high specificity for amorphous polyesters of short chain-length hydroxyalkanoic acids. J. Biol. Chem. 276: 36215–36224 [DOI] [PubMed] [Google Scholar]
- 30. Brandl H, Gross RA, Lenz RW, Fuller RC. 1988. Pseudomonas oleovorans as a source of poly(beta-hydroxyalkanoates) for potential applications as biodegradable polyesters. Appl. Environ. Microbiol. 54: 1977–1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72: 248–254 [DOI] [PubMed] [Google Scholar]
- 32. Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177: 4121–4130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cho M, Brigham CJ, Sinskey AJ, Stubbe J. 2012. Purification of a polyhydroxybutyrate synthase from its native organism, Ralstonia eutropha: implications in the initiation and elongation of polymer formation in vivo. Biochemistry 51: 2276–2288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Haywood GW, Anderson AJ, Dawes EA. 1989. The importance of PHB-synthase substrate specificity in polyhydroxyalkanoate synthesis by Alcaligenes eutrophus. FEMS Microbiol. Lett. 57: 1–6 [Google Scholar]
- 35. Gerngross TU, Reilly P, Stubbe J, Sinskey AJ, Peoples OP. 1993. Immunocytochemical analysis of poly-beta-hydroxybutyrate (PHB) synthase in Alcaligenes eutrophus H16: localization of the synthase enzyme at the surface of PHB granules. J. Bacteriol. 175: 5289–5293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pfeiffer D, Jendrossek D. 2012. Localization of poly(3-hydroxybutyrate) (PHB) granule-associated proteins during PHB granule formation and identification of two new phasins, PhaP6 and PhaP7, in Ralstonia eutropha H16. J. Bacteriol. 194: 5909–5921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Brigham CJ, Speth DR, Rha C, Sinskey AJ. 2012. Whole-genome microarray and gene deletion studies reveal regulation of the polyhydroxyalkanoate production cycle by the stringent response in Ralstonia eutropha H16. Appl. Environ. Microbiol. 78: 8033–8044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lawrence AG, Schoenheit J, He A, Tian J, Liu P, Stubbe J, Sinskey AJ. 2005. Transcriptional analysis of Ralstonia eutropha genes related to poly-(R)-3-hydroxybutyrate homeostasis during batch fermentation. Appl. Microbiol. Biotechnol. 68: 663–672 [DOI] [PubMed] [Google Scholar]
- 39. Peplinski K, Ehrenreich A, Döring C, Bömeke M, Reinecke F, Hutmacher C, Steinbüchel A. 2010. Genome-wide transcriptome analyses of the “Knallgas” bacterium Ralstonia eutropha H16 with regard to polyhydroxyalkanoate metabolism. Microbiology 156: 2136–2152 [DOI] [PubMed] [Google Scholar]
- 40. Stubbe J, Tian J, He A, Sinskey AJ, Lawrence AG, Liu P. 2005. Nontemplate-dependent polymerization processes: polyhydroxyalkanoate synthases as a paradigm. Annu. Rev. Biochem. 74: 433–480 [DOI] [PubMed] [Google Scholar]
- 41. Handrick R, Technow U, Reichart T, Reinhardt S, Sander T, Jendrossek D. 2004. The activator of the Rhodospirillum rubrum PHB depolymerase is a polypeptide that is extremely resistant to high temperature (121 degrees C) and other physical or chemical stresses. FEMS Microbiol. Lett. 230: 265–274 [DOI] [PubMed] [Google Scholar]
- 42. Handrick R, Reinhardt S, Schultheiss D, Reichart T, Schüler D, Jendrossek V, Jendrossek D. 2004. Unraveling the function of the Rhodospirillum rubrum activator of polyhydroxybutyrate (PHB) degradation: the activator is a PHB-granule-bound protein (phasin). J. Bacteriol. 186: 2466–2475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Simon R, Priefer U, Pühler A. 1983. A broad-host-range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram-negative bacteria. Nat. Biotechnol. 1: 784–791 [Google Scholar]
- 44. Heiss G, Hofmann KW, Trachtmann N, Walters DM, Rouvière P, Knackmuss H-J. 2002. npd gene functions of Rhodococcus (opacus) erythropolis HL PM-1 in the initial steps of 2,4,6-trinitrophenol degradation. Microbiology 148: 799–806 [DOI] [PubMed] [Google Scholar]