

Abstract
Microbial physiology plays a crucial role in whole-cell biotransformation, especially for redox reactions that depend on carbon and energy metabolism. In this study, regio- and enantio-selective proline hydroxylation with recombinant Escherichia coli expressing proline-4-hydroxylase (P4H) was investigated with respect to its interconnectivity to microbial physiology and metabolism. P4H production was found to depend on extracellular proline availability and on codon usage. Medium supplementation with proline did not alter p4h mRNA levels, indicating that P4H production depends on the availability of charged prolyl-tRNAs. Increasing the intracellular levels of soluble P4H did not result in an increase in resting cell activities above a certain threshold (depending on growth and assay temperature). Activities up to 5-fold higher were reached with permeabilized cells, confirming that host physiology and not the intracellular level of active P4H determines the achievable whole-cell proline hydroxylation activity. Metabolic flux analysis revealed that tricarboxylic acid cycle fluxes in growing biocatalytically active cells were significantly higher than proline hydroxylation rates. Remarkably, a catalysis-induced reduction of substrate uptake was observed, which correlated with reduced transcription of putA and putP, encoding proline dehydrogenase and the major proline transporter, respectively. These results provide evidence for a strong interference of catalytic activity with the regulation of proline uptake and metabolism. In terms of whole-cell biocatalyst efficiency, proline uptake and competition of P4H with proline catabolism are considered the most critical factors.
INTRODUCTION
In redox biocatalysis, especially when activation of molecular oxygen is involved, the whole cell still remains the preferred catalyst. Besides the capability to continuously regenerate redox cofactors via central carbon metabolism, the whole-cell approach provides advantages with respect to operational catalyst stability, e.g., via continuous (multicomponent) enzyme synthesis, assembly, and stabilization, as well as degradation of reactive oxygen species (1–3).
Oxygenases are of special interest for industrial applications, since they catalyze the highly specific oxyfunctionalization of unactivated C-H bonds under mild conditions, with molecular oxygen as the oxygen donor (4). The efficiency of an oxygenase-containing whole-cell biocatalyst depends on both enzyme and host cell performance. Hence, biocatalyst engineering must encompass both factors and be integrated with biochemical process engineering (5, 6). The technological progress in enzyme engineering of the recent decades now allows fast generation of enzyme variants with improved properties (7). For in vivo applications, intracellular conditions have to be considered, and host cell engineering often is necessary to allow the exploitation of the catalytic capability of (engineered) enzymes. The benefits of metabolic engineering have especially become evident in fermentation processes, as exemplified by the development of strains for the biotechnological production of indigo (8) and propanediol (9). Metabolic engineering can rely on a set of powerful tools, such as flux balance analysis (10) and metabolic flux analysis (MFA) (11–13), used, inter alia, for the optimization of the production of various amino acids in Corynebacterium glutamicum (14, 15).
The efficiency of whole-cell redox biocatalysts often depends on microbial physiology (16, 17), for example, because the provision of (co)substrates/cofactors becomes limiting (18, 19), or because a toxic or inhibiting product has accumulated (20, 21). With respect to cell physiology, 2-oxoacid-dependent nonheme Fe(II)-dependent dioxygenases (22, 23), such as proline-4-hydroxylases (P4Hs), are highly interesting. P4Hs catalyze the hydroxylation of proline at the trans-4 position, with the cosubstrate α-ketoglutarate (αKG) undergoing oxidative decarboxylation to succinate (Fig. 1). As such, P4H catalysis is expected to strongly interact with metabolism, as it depends on a tricarboxylic acid (TCA) cycle intermediate as a cosubstrate. Furthermore, the substrate proline is involved in protein synthesis and osmoprotection, as well as catabolism and anabolism, serving as potential source of carbon, nitrogen, and energy.
Fig 1.

Scheme of proline hydroxylation catalyzed by recombinant E. coli containing proline-4-hydroxylase.
P4Hs were initially discovered in the biosynthetic pathway of the antibiotic etamycin (24). The product trans-4-hydroxyproline is a valuable chiral building block (or catalyst) for the synthesis of, for example, amino acid analogues, natural products, and benzodiazepins (25). The discovery of the activity in Dactylosporangium sp. strain RH1 was soon followed by gene identification and cloning (26) and the development of Escherichia coli-based systems for trans-4-hydroxyproline production from proline (27) or glucose (28). Despite successful application and except for the deletion of putA, responsible for proline degradation (29), the interactions of the target reaction with host physiology are poorly characterized for respective whole-cell biocatalysts.
The identification of the factors limiting whole-cell biocatalyst performance is paramount in the development of an effective bioprocess and can provide considerable insight into a whole-cell biocatalyst operation. In this study, we investigated the interrelationship between microbial physiology and P4H activity, focusing on factors that influence P4H levels (e.g., temperature, medium composition, codon optimization) and their correlation with in vivo and in vitro proline hydroxylation activities. Furthermore, the interplay between proline hydroxylation and metabolic network operation and regulation was investigated by metabolic flux analysis and real-time PCR (RT-PCR).
MATERIALS AND METHODS
Strains and constructs.
Recombinant strains used in this study were all derived from the commercially available Escherichia coli strain BL21(DE3) [F− ompT gal dcm lon hsdSB(rB− mB−) λ(DE3)] (30, 31). Utilized plasmids are listed in Table 1, and the relevant sequences are given in File S1 in the supplemental material. The wild-type P4H gene sequence is deposited in GenBank (accession number D78338). The p4h1or gene, optimized for E. coli by using the codon adaptation index (CAI) maximization approach (32) (see Fig. S1.1 in the supplemental material), was isolated from vector pGA4_p4h1or by digestion with NdeI and BamHI and ligated into pET-24a digested with the same enzymes to give pET_p4h1or. The p4h1of gene was designed with a codon optimization strategy mimicking the codon usage of the host microorganism while still avoiding rare codons (<10% frequency) (see Fig. S1.2). An EcoRI restriction site was included at the end of the gene. The gene was synthesized by Epoch Biolabs (Missoury City, TX) and delivered in the commercially available vector pBSK (Stratagene, Santa Clara, CA). The resulting pBSK_p4h1of construct and pET_p4h1or were digested with NdeI and EcoRI. The p4h1of fragment and the pET vector backbone were purified and ligated to give pET_p4h1of. The vector pET_p4h1in was isolated as part of a screening effort on a p4h1of mutant library created by SeSaM (33) in collaboration with U. Schwaneberg at RWTH Aachen University.
Table 1.
Plasmids used in this study
| Plasmid | Features | Reference or source |
|---|---|---|
| pLysS | Encodes T7 phage lysozyme that eliminates expression from T7 promoter-containing plasmids when not induced, Cmr | 31 |
| pGA4 | Cloning vector, Apr | Geneart |
| pGA4_p4h1or | pGA4 containing p4h1or, Apr | This study |
| pBSK | Cloning vector, Apr | Stratagene |
| pBSK_p4h1of | pBSK containing p4h1of, Apr | This study |
| pET-24a | High-copy-number vector, lacI/PT7lac, Kmr | Novagen |
| pET_p4h1or | pET-24a containing p4h1or, Kmr | This studya |
| pET_p4h1of | pET-24a containing p4h1of, Kmr | This study |
| pET_p4h1in | pET-24a encoding an inactive P4H mutant, Kmr | This study |
See also Fig. S1.1 in the supplemental material.
Chemicals.
Labeled glucose isotopes were purchased from Cambridge Isotope Laboratories (Andover, MA). Nymeen S-215 was kindly donated by Kyowa Hakko Bio Co., Ltd. (Tokyo, Japan). All other chemicals were purchased from Sigma-Aldrich (Munich, Germany) or Carl-Roth (Karlsruhe, Germany) and were of the highest purity available.
Cultivation of microorganisms and recombinant gene expression.
Recombinant E. coli BL21(DE3) strains were grown either on lysogenic broth (LB) or M9 medium (34) or M9* medium (35), both supplemented with USFe trace element solution (36). If required, appropriate antibiotics were added (30 μg liter−1 chloramphenicol and/or 50 μg liter−1 kanamycin). M9 and M9* cultures were supplemented with 0.5% and 1% (wt/vol) glucose, respectively, unless otherwise stated. If indicated, l-proline was applied at a concentration of 20 mM, and gene expression from pET-24a-derived constructs was induced by addition of 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG). Cultures were grown in baffled Erlenmeyer flasks in rotary shakers (2.5-cm amplitude, 250 rpm). Cell densities were monitored by measuring the optical density at 450 or 600 nm (OD450 or OD600), using a Libra S11 spectrophotometer (Biochrom Ltd., Cambridge, United Kingdom). For induced cells of each strain, growing exponentially in M9 with and without proline, correlation factors between the OD and dry biomass concentration (see File S3 in the supplemental material) were determined as described before (37).
Recombinant protein production was analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and different activity assays as described below. SDS-PAGE was performed according to literature protocols (38). Digital images of the gels were taken with an AlphaImager HP instrument (Biozym; Hessisch Oldendorf, Germany). The included software was used to improve contrast and brightness and to estimate the relative intensities of selected bands.
Activity assays with resting and permeabilized cells.
Resting cell activity assays were performed as described before (39) in duplicates with 0.5 mg of cell dry weight (CDW) in 500 μl KPi buffer (50 mM [pH 7.4], giving 1 gCDW liter−1). The reaction was started by adding 5 μl of a 2 M aqueous proline stock solution. Samples (130 μl) were taken 0, 10, and 20 min after substrate addition, diluted in 130 μl of ice-cold isopropanol, and centrifuged (5 min, 17,000 × g, 4°C). The supernatant was transferred to a new tube and stored at −20°C. Activity is expressed in U gCDW−1, with 1 U equal to 1 μmol of product formed per minute.
Procedures for permeabilized cells were adapted from the literature (40). In short, aliquots containing 1.5 mgCDW were harvested, centrifuged (17,000 × g, 5 min, 4°C), washed with 1 ml of 150 mM 2-(N-morpholino)ethanesulfonic acid (MES) buffer (pH 6.0), and resuspended in 1.35 ml of the same buffer. The exact cell concentration was determined based on the OD450, and 450-μl aliquots were distributed to two screw-cap Pyrex tubes and kept on ice. Then, 20 mM αKG (from a 1 M aqueous stock), 5 mM ascorbic acid (from a 100 mM aqueous stock), 1 mM ferrous sulfate (from a freshly prepared 100 mM aqueous stock), and 1.4% (vol/vol) Nymeen solution (4 g Nymeen S-215 in 10 ml xylene) were added. The tubes were adapted for 3 min in a shaking water bath at 30°C and 300 rpm, and the reaction was started by adding 5 μl of a 2 M aqueous proline stock, giving a final volume of 500 μl. Samples were taken 0, 5, and 10 min after proline addition and treated as described above for resting cell assays.
Preparation of crude extracts and separation of inclusion bodies.
Culture volumes between 20 and 50 ml were harvested in the exponential growth phase into weighed Falcon tubes, centrifuged (15 min, 4,650 × g, 4°C), and washed with 50 mM N-Tris(hydroxymethyl)methyl-3-aminopropanesulfonic acid (TAPS) buffer (pH 9.0). The cell wet weight (CWW) was determined, and the cells were resuspended in the same buffer containing 200 μM EDTA, 2 mM dithiothreitol, 10% (vol/vol) glycerol, and 0.5 mg ml−1 of DNase I to a concentration of roughly 300 gCWW liter−1. Cells were lysed via French press using a Sim Aminco instrument (SLM Instruments Inc., Urbana, IL) equipped with a MINI cell and with a piston diameter of 3/8 in. and an internal pressure of ∼15,8000 lb/in2. The resulting crude extract was clarified of cell residues and inclusion bodies by centrifugation of 200-μl aliquots at 17,000 × g for 10 min at 4°C (41, 42). Supernatants were transferred to new tubes, and pellets were resuspended in 40 μl of water. The protein content of each fraction (crude extract, supernatant, and pellet) was determined via Bradford assay (43). The three fractions were kept on ice for further analysis and/or stored at −20°C.
Activity assays with cell extracts.
Assay mixtures contained 250 μg of total protein, 20 mM l-proline, 4 mM FeSO4, and 150 mM MES buffer (pH 6.5) in a final volume of 500 μl and were incubated in a Thermomixer Comfort (Eppendorf, Hamburg, Germany) at 37°C and a 1,200 rpm shaking speed. After 3 s, a 100-μl reference (zero) sample was taken, and the reaction was started by addition of 5 mM αKG. Further 100-μl samples were taken after 2, 4, and 6 min. Samples were transferred to tubes containing 10 μl of 1 M ZnSO4 on ice and centrifuged at 17,000 × g for 5 min at 4°C. Supernatants were stored at −20°C.
RNA isolation and quantification.
Upon reaching an OD600 of 2.5, induced cells were sampled (2 ml) from M9 cultures grown at 30°C in the absence or presence of 4 mM proline. The total RNA was isolated using a NucleSpin RNA II kit (Macherey-Nagel, Düren, Germany). The RNA was purified of contaminating genomic DNA by using an Ambion Turbo DNA-free kit (Life Technologies, Darmstadt, Germany). The concentration of the purified RNA was measured with a Nanodrop 2000 spectrophotometer (Peqlab, Erlangen, Germany). A 2.5-μg sample of RNA (in 20 μl) was retrotranscribed to cDNA using the Superscript III first-strand kit (Life Technologies). The cDNA concentration was assumed to be the same as that of the template RNA.
The levels of four selected genes (p4h1of, p4h1or, putP, and putA) and a reference gene (rrsA) were quantified in triplicate by RT-PCR with a StepOne Plus system (Life Technologies) with custom TaqMan gene expression assays (Life Technologies) using 50 ng of cDNA and following the protocol provided with the instrument. TaqMan probes and primers were designed by Life Technologies. Raw data were analyzed with the proprietary software, and processed data are reported as the −ΔΔCT normalized to the internal rrsA level and to the appropriate reference sample (see Table 2). The PCR efficiencies of the assays for p4h1of and p4h1or were equivalent.
Table 2.
Relative mRNA levels of relevant genes in recombinant E. coli BL21(DE3)(pLysS)
| Gene | Meana ± SEM value for strain with indicated plasmid in the presence (+) or absence (−) of proline |
|||||
|---|---|---|---|---|---|---|
| pET_24a | pET_p4h1or | pET_p4h1of | ||||
| − | + | − | + | − | + | |
| p4h1or/of | ND | ND | 0b | 0.3 ± 0.2 | 3.5 ± 0.2 | 3.6 ± 0.2 |
| putP | 0b | 2.9 ± 0.2 | −(1.4 ± 0.2) | 2.0 ± 0.2 | −1.0 ± 0.2 | −0.2 ± 0.2 |
| putA | 0b | 0.5 ± 0.2 | −(0.5 ± 0.2) | 0.3 ± 0.2 | −0.8 ± 0.2 | −1.3 ± 0.2 |
mRNA levels are reported as ΔΔCT values. Fold changes in expression levels can be calculated as 2−ΔΔCT. Hence, a value of 3.3 indicates an ∼10-fold difference. ND, not detected.
Reference sample for the respective target gene(s).
Microbial physiology and 13C-based metabolic flux analysis.
Carbon balances were determined in M9 cultures grown in sealed flasks and sampled at regular intervals for quantification of glucose, acetate, proline, hydroxyproline, and biomass. CO2 and O2 levels were continuously monitored via gas sensors (Bluesens GmbH, Germany). For MFA experiments, precultures in M9 medium (the same conditions as the main culture, but with unlabeled glucose) were harvested in the exponential phase, washed once in M9 medium, and used to inoculate 20 ml of M9 medium containing 0.5% (wt/vol) of a 4:1 mixture of 1-13C-labeled and U-13C-labeled glucose in a 100-ml baffled Erlenmeyer flask. The initial OD600 was set to 0.01, and cultures were grown at 30°C and 250 rpm. For MFA, at least 3 samples (2 ml each) were taken when the OD600 reached 2. After centrifugation for 10 min at 17,000 × g and 4°C, the cells were washed once with sterile water, and the pellets were stored at −20°C. The pellets were treated, and proteogenic amino acids were analyzed as described in the literature (44). Hydrolyzed biomass was resuspended in 30 μl acetonitrile, to which 30 μl N-methyl-N-tert-butyldimethylsilyl-trifluoracetamide (MBDSTFA) was added. The raw data were analyzed with the software OpenFLUX (45). The detailed simulation protocol, relevant files, and data sources are included in File S3 of the supplemental material.
Analytics of extracellular metabolites.
A spectrophotometric method for hydroxyproline quantification in meat (46) was adapted to the microtiter plate format. The reagent solutions were prepared as described except that 100 mg liter−1 thimerosal was replaced with 200 mg liter−1 NaN3. To 100 μl of sample or standard (containing 1 to 50 μM hydroxyproline) in microtiter plates, 50 μl of oxidizing solution was added. After mixing and incubation at room temperature for 20 min, 50 μl of chromophore solution was added, followed by mixing, incubation for 22 min at 80°C, cooling on ice for 3 min, and incubation at room temperature for 2 min, after which the absorbance at 558 nm was measured with an Infinite M1000 microtiter plate reader (Tecan GmbH, Groedig, Austria). Calibration was performed together with sample analysis. Readings were stable for at least 40 min after derivatization. Quantification of proline and hydroxyproline and monitoring of other amino acids were performed by using high-performance liquid chromatography on an Elite LaChrom system (interface D-7000, pump L-2130, autosampler L-2200, column oven L-2300; Hitachi Ltd. Corp., Tokyo, Japan) equipped with a diode array detector (L-2450) in line with a Corona charged aerosol detector (ESA Biosciences, Chelmsford, MA). Analytes were separated using a Sielc Primesep100 column (4.6 by 250 mm, 100-Å pores, 5-μm particles; Sielc Technologies, Prospect Heights, IL) kept at 32°C. The mobile phase was composed of a gradient of 3 different eluents, described in File S3 of the supplemental material. Glucose and acetate were quantified via similar equipment, as described in the literature (44). Glucose measurements were routinely corroborated by spectrophotometric analysis with the Enzytec d-glucose kit (Scil Diagnostics GmbH, Martinsried, Germany).
RESULTS
Whole-cell proline-4-hydroxylase (P4H) activity can theoretically be limited by several factors, including P4H synthesis, hydroxyproline export, proline uptake, proline consumption by endogenous metabolism, and α-ketoglutarate (αKG) supply via central carbon metabolism.
Proline-4-hydroxylase production in E. coli.
With respect to P4H synthesis, the catalytic activity of P4H itself may lead to a depletion of the proline pool, which in turn may affect P4H production. Additionally, the comparison of the amino acid composition of P4H with that of the E. coli proteome revealed that P4H contains a high fraction of proline (8.5%, as opposed to an average of 4.3% for host proteins [47]; a relative abundance of 197%).
Initially, functional expression of a P4H gene (p4h1or) codon optimized for E. coli was evaluated by means of resting cell activity assays using different E. coli host strains and expression systems (results not shown), with E. coli BL21(DE3)(pLysS)(pET_p4h1or) showing the highest activity (the pET_p4h1or sequence is given in Fig. S1.1 in the supplemental material). Subsequently, the effects of amino acid availability on P4H production and activity were tested by cultivating E. coli BL21(DE3)(pLysS)(pET_p4h1or) in complex and glucose-containing minimal media at 30°C. After reaching full induction (3 to 4 h after IPTG addition), resting cell activities of cells grown in minimal media were constant and decreased in the stationary phase, while cells grown in LB displayed maximal activities upon entering the stationary phase. Maximal resting cell activities did not significantly differ for cells grown in M9 and M9* media (1.5 ± 0.6 U gCDW−1; mean ± standard deviation), but they were more than 4 times higher for cells grown in LB (6.5 ± 0.5 U gCDW−1). SDS-PAGE revealed that cells grown in minimal media produced only small amounts of P4H (see Fig. S2.1 in the supplemental material). Cultivation in complex medium led to high P4H levels (30 to 40% of total protein), but microscopic analysis and separation of soluble and insoluble protein fractions indicated extensive P4H-related inclusion body formation (>85% of P4H [see Fig. S2.2]). Whereas the soluble fraction retained the same activity as the crude cell extract in in vitro activity assays, the insoluble fraction was found to be inactive, as expected for enzyme molecules misfolded in inclusion bodies (48, 49).
Low cultivation temperatures have been reported to improve protein folding (50) and hence promote functional enzyme synthesis. Indeed, cultivation at 25°C instead of 30°C in LB medium almost doubled the resting cell activity (11.2 ± 0.9 U gCDW−1). P4H was produced at lower levels (3 to 5% of total protein) but with a good solubility of >90% (see Fig. S2.2 in the supplemental material). Growth at 37°C, as expected, caused the opposite effects: higher P4H levels, a larger insoluble P4H fraction, and lower activity (4.8 ± 0.4 U gCDW−1).
To improve P4H production in minimal medium (at 30°C), the addition of yeast extract (500 mg liter−1, estimated to contain 0.22 to 0.35 mM proline [51]) or proline (0.26 mM, as usually applied for auxotroph strains) was tested and was found to enhance resting cell activities (Fig. 2). However, activities were maximal 3 h after induction and decreased constantly afterwards (see Fig. S2.4 in the supplemental material), which may have been due to gradual proline depletion. In fact, higher proline levels (4 to 16 mM) allowed even higher resting cell activities throughout the exponential growth phase, equal to those achieved with cells grown in complex medium under otherwise-comparable conditions (Fig. 2; see also Fig. S2.4). The increase in P4H activity correlated with an increase in P4H levels (Fig. 2). Since p4h1or mRNA levels in the presence or absence of proline were identical (Table 2), we concluded that proline availability influenced functional P4H gene expression at the translational level. In comparison, addition of 20 mM proline to a BL21(DE3)(pLysS)(pET_p4h1in) culture (encoding an inactive P4H variant) did not increase the P4H level (see Fig S2.3), indicating that P4H activity plays a pivotal role in the observed proline dependency of P4H synthesis. The addition of proline to LB medium did not result in increased resting cell activities or P4H levels, indicating that LB contains enough proline (2.2 to 3.5 mM) to sustain efficient P4H synthesis.
Fig 2.
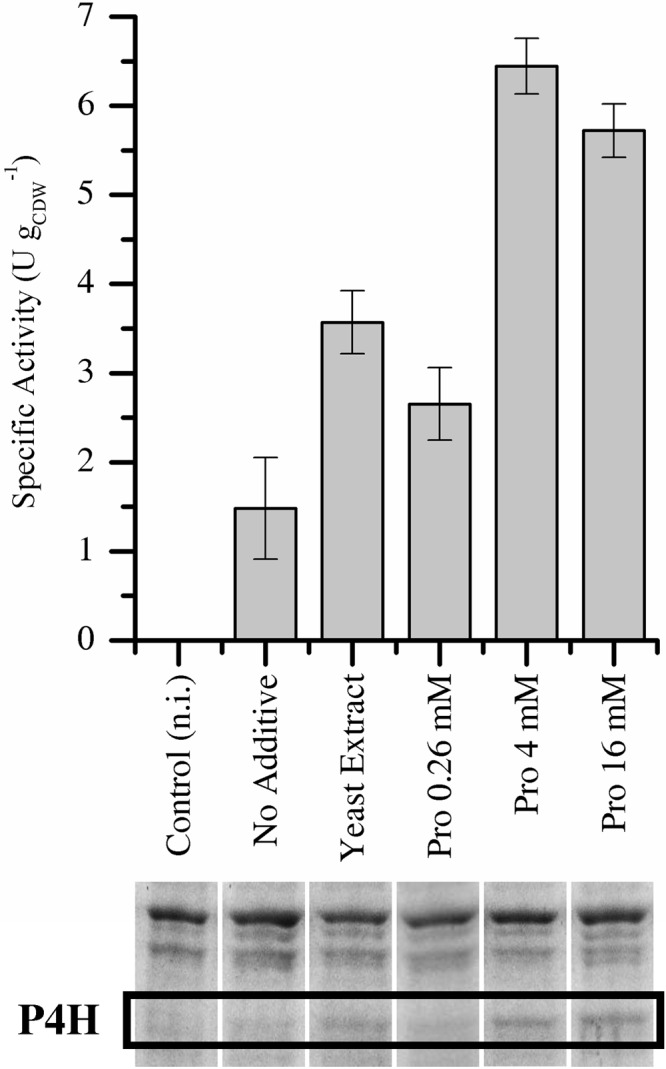
Specific resting cell activities and corresponding P4H levels of E. coli BL21(DE3)(pLysS)(pET_p4h1or) grown in minimal medium at 30°C in the presence of several additives. n.i., not induced.
The p4h1or gene was designed using solely the codon with the highest abundance in E. coli. This method may lead to depletion of tRNAs charged with amino acids highly represented in the heterologous protein and cause translational errors (52). To confirm that P4H production was limited by the availability of charged tRNAs, a different codon optimization strategy mimicking the relative codon usage of the host was tested (e.g., for proline, the CCA, CCG, and CCT codons were used, rather than CCG only). Expression of the resulting gene, p4h1of (see Fig. S1.3 in the supplemental material for comparison of sequences), cloned into pET-24a to give pET_p4h1of, enabled a 2- to 4-fold increase in P4H levels under all conditions tested (Fig. 3) and caused an ∼20% decrease in growth rate upon induction (with and without proline). P4H was found to be the main component of the insoluble protein fraction, with 40 to 50% of P4H being soluble. Since p4h1of mRNA levels were ∼10-fold higher than with p4h1or (Table 2), the increase in P4H synthesis seems not only to be due to an increased availability of charged prolyl-tRNAs but also to other gene sequence-related factors, such as mRNA stability.
Fig 3.

SDS-PAGE results for the soluble fractions of cell extracts of E. coli BL21(DE3)(pLysS) carrying either pET_p4h1or (lanes 1 and 2) or pET_p4h1of (lanes 3 and 4), grown at 25°C or 30°C in M9 medium with 0.5% (wt/vol) glucose in the absence (lanes 1 and 3) or presence (lanes 2 and 4) of 20 mM proline. Lane M, protein size marker.
So far, three factors have been found to positively influence P4H production and activity in whole cells: cultivation temperature, proline availability, and codon usage. To investigate if these effects are additive, E. coli BL21(DE3)(pLysS) containing either pET_p4h1or or pET_p4h1of were grown in minimal medium at 25°C in the presence or absence of proline, giving four distinct levels of soluble P4H per gram of CDW (Fig. 3). Even in the absence of proline, BL21(DE3)(pLysS)(pET_p4h1of) produced high P4H levels, comparable to those of the strain carrying pET_p4h1or in the presence of proline, indicating a remarkable impact of codon usage on P4H synthesis. The beneficial effects of codon optimization and proline addition on P4H synthesis were additive, as exemplified by the highest P4H levels observed with BL21(DE3)(pLysS)(pET_p4h1of) grown on glucose and proline (Fig. 3). With this construct too the beneficial effect of proline on P4H levels was solely due to improved translation, since mRNA levels were unchanged (Table 2). However, the differences in soluble P4H levels were not reflected in resting cell activities. All strains/conditions tested, with the exception of BL21(DE3)(pLysS)(pET_p4h1or) grown in the absence of proline, led to similar resting cell activities, in the range of 10 to 12 U gCDW−1 (Fig. 4a).
Fig 4.

Comparison of specific activities of resting whole cells (a), permeabilized cells (b), and cell extracts (c) of E. coli BL21(DE3)(pLysS) carrying either pET_p4h1or or pET_p4h1of, grown in M9 medium at 25°C with 0.5% (wt/vol) glucose in the presence or absence of 20 mM proline. See Materials and Methods for experimental details.
When the same strains were grown in the presence of proline at 30°C instead of 25°C, soluble P4H levels were lower with pET_p4h1or, but almost identical with pET_p4h1of (Fig. 3). Cells cultivated at 30°C also reached a maximum resting cell activity (see Fig. S2.6a in the supplemental material), above which the activity did not correlate with soluble P4H levels. However, with 6 to 6.5 U gCDW−1, this activity was lower than for cells grown at 25°C. Thus, the maximum resting cell activity is a function of the cultivation temperature and obviously depends on temperature-related variations of host metabolism.
Soluble P4H levels correlate with the activity of permeabilized cells and cell extracts, but not resting cells.
In order to test if P4H was produced at a level at which the intracellularly available P4H activity was not fully exploited and was limited by host-related factors, P4H activity was tested independently of substrate uptake and cellular metabolism by using permeabilized cells and cell extracts (of cells grown at 25°C) provided with proline, αKG, and Fe(II).
Permeabilized cells and resting whole cells of BL21(DE3)(pLysS)(pET_p4h1or) grown in the absence of proline displayed similar activities, suggesting that, in this case, the resting cell activity was limited by the available P4H activity (Fig. 4). In all other cases, permeabilized cells showed higher activities than resting cells, correlating well with soluble P4H levels (Fig. 3 and 4). Because of the different assay conditions, the activity of crude extracts could not be directly compared to that of resting or permeabilized cells. However, their relative activity mirrored that of permeabilized cells, as did the amounts of soluble P4H, confirming that, in three of the four cases, the catalytic potential of P4H was not fully exploited in intact whole cells.
Results for cells grown at 30°C mirrored those at 25°C (see Fig. S2.6 in the supplemental material). In the same three out of four cases, activities of permeabilized cells and cell extracts did not correlate with those of resting cells. The proportionality between in vitro activities and enzyme levels was similar to that of cells grown at 25°C. The fact that pET_p4h1of-containing cells not only showed similar expression levels at 25°C and 30°C (Fig. 3), but also similar in vitro activities (Fig. 4; see also Fig. S2.6), whereas resting cell activities differed, gives further evidence for a dependency of the maximal resting cell activity on temperature-related variations in host physiology. The results obtained so far indicate clearly that, above a certain intracellular level of functional P4H, the available catalytic activity is not fully exploited and that resting cell activities are limited by host-related factors.
Metabolic flux redistribution in cells containing active proline-4-hydroxylase.
The investigation of intracellular rates by means of MFA can provide quantitative insight into the flux redistribution upon introduction of catalytic activity and into critical fluxes (e.g., proline uptake, proline catabolism, αKG supply) that may be responsible for the observed in vivo limitations. 13C-based MFA was performed during exponential growth on glucose or glucose and proline in M9 medium using E. coli BL21(DE3)(pLysS) containing either pET_p4h1or, pET_p4h1of, or pET-24a. A cultivation temperature of 30°C was chosen as a compromise between the optima found for functional gene expression (25°C) and P4H activity (37°C). The pseudo steady-state assumption was found to be valid for OD600 levels below 3 to 4. The carbon balance was closed (98 to 100%) for exponentially growing cells. The glyoxylate shunt was initially included in the metabolic model but was poorly resolved, showing a tendency toward low fluxes, and thus was considered not to be physiologically relevant under the examined conditions (see File S3 in the supplemental material for details). The respective flux was set to 0 in the reported simulations (Fig. 5). Resting cell activities of these cultivations were also measured and were comparable to those of cultivations with nonlabeled glucose (see above and also Fig. S2.6a in the supplemental material).
Fig 5.

Metabolic fluxes in recombinant E. coli BL21(DE3)(pLysS) strains during exponential growth at 30°C in M9 medium on 5 g liter−1 glucose (4:1 mixture of 1-13C-labeled and U-13C-labeled glucose) in the absence or presence of 4 mM proline. Rates were normalized to the glucose uptake rate. For external fluxes, the experimental error is reported. The 90% confidence intervals on intracellular fluxes were obtained via Monte Carlo analysis. The error on glutamate and proline demand was estimated from data in the literature on biomass precursor demand. *, the glyoxylate shunt flux was set to 0 in the reported simulations (see File S3 in the supplemental material for experimental details).
Expression of p4h1or did not significantly alter growth rates but slightly reduced the biomass yields on glucose (Table 3), suggesting that p4h1or expression did not hamper growth but still increased the energy demand, while expression of p4h1of reduced both biomass yields on glucose and growth rates.
Table 3.
Physiological data for recombinant E. coli BL21(DE3)(pLysS) strains grown at 30°C in M9 medium with 5 g/liter glucose in the absence or presence of 4 mM prolinea
| Parameter | Mean ± SEM value for strain with indicated plasmid and presence (+) or absence (−) of proline |
|||||
|---|---|---|---|---|---|---|
| pET_24a |
pET_p4h1or |
pET_p4h1of |
||||
| − | + | − | + | − | + | |
| μ (h−1) | 0.38 ± 0.01 | 0.43 ± 0.01 | 0.41 ± 0.01 | 0.46 ± 0.01 | 0.34 ± 0.01 | 0.34 ± 0.01 |
| rglc (mmol/g/h) | −(5.1 ± 0.1) | −(4.7 ± 0.1) | −(6.3 ± 0.2) | −(5.6 ± 0.1) | −(4.9 ± 0.1) | −(4.6 ± 0.1) |
| rpro (mmol/g/h) | NA | −(1.0 ± 0.1) | NA | −(1.1 ± 0.1) | NA | −(0.7 ± 0.1) |
| race (mmol/g/h) | 1.7 ± 0.1 | 2.2 ± 0.1 | 1.8 ± 0.1 | 2.4 ± 0.1 | 1.5 ± 0.1 | 2.1 ± 0.1 |
| rhyp (mmol/g/h) | ND | ND | 0.07 ± 0.01 | 0.19 ± 0.03 | 0.12 ± 0.02 | 0.34 ± 0.03 |
| rhyp (U g−1) | ND | ND | 1.2 ± 0.1 | 3.5 ± 0.7 | 2.0 ± 0.3 | 5.7 ± 0.2 |
| Yx/s (gCDW/gglc) | 0.42 ± 0.02 | 0.51 ± 0.02 | 0.36 ± 0.01 | 0.46 ± 0.01 | 0.38 ± 0.01 | 0.40 ± 0.01 |
| Yx/s (gCDW/molC) | 12.5 ± 0.3 | 12.8 ± 0.4 | 10.8 ± 0.2 | 10.1 ± 0.2 | 11.5 ± 0.2 | 10.8 ± 0.2 |
| Yace (mol/molglc) | 0.33 ± 0.02 | 0.47 ± 0.02 | 0.29 ± 0.02 | 0.44 ± 0.02 | 0.31 ± 0.02 | 0.45 ± 0.03 |
| Yhyp (mol/molpro) | NA | ND | NA | 0.17 ± 0.04 | NA | 0.51 ± 0.1 |
μ, specific growth rate; r, specific rate for substrate uptake or product formation (negative values indicate uptake); glc, glucose; pro, proline; ace, acetate; hyp, trans-4-hydroxyproline; Yx/s, yield coefficient for biomass on glucose; Yace and Yhyp, yield coefficients for products; ND, not detected; NA, not applicable.
Upon proline addition, growth was associated with higher acetate production rates in all cases. In the reference strain containing pET-24a, proline was cometabolized with glucose, leading to higher growth rates and yields on glucose. Proline uptake followed a counterintuitive trend: higher proline hydroxylation rates were associated with lower proline uptake rates, even when normalized to the glucose uptake rate (15% with pET_p4h1of, compared to 22% with pET-24a). Thus, the expression levels of the put operon, encoding the major proline transporter PutP (29, 53) and the proline dehydrogenase PutA, were monitored by RT-PCR (Table 2). For cells containing pET-24a, the putP mRNA levels showed an ∼8-fold increase upon induction by proline. In contrast, put operon transcription was clearly downregulated in the presence of P4H, indicating strong interference of P4H activity with the regulation of the put operon. As a consequence, proline addition to pET_p4h1of-containing E. coli did not enhance growth rate or yield on glucose (Table 3).
Specific hydroxyproline formation rates depended on the intracellular level of functional P4H, and the recombinant strains slowly produced hydroxyproline even in the absence of proline. In the presence of proline, specific hydroxyproline formation rates were considerably higher (3- to 4-fold) and differed for the pET_p4h1or and pET_p4h1of constructs (3.5 and 5.7 U gCDW−1, respectively). Thus, growing cells behaved differently than resting cells, which showed an activity of 6 to 6.5 U gCDW−1 with both constructs under comparable conditions (see above), suggesting that growing cells may encounter different limitations than resting cells.
When we examined the fluxomes (Fig. 5) during growth in the absence of proline, differences in the flux distribution of the three strains were related to the different biomass yields on glucose. Relative fluxes (normalized to the glucose uptake rate) were very similar close to the glucose entry point and gradually diverged along the glucose utilization pathway, differing most in the TCA cycle, where relative fluxes were up to one-third higher in the recombinant strains than in the reference strain. This reflects the increased energy demand upon P4H synthesis. The low proline hydroxylation rate during growth in the absence of proline significantly increased the proline synthesis rate but otherwise had only a minor impact on the global flux distribution.
In the presence of proline, the situation was more complex. For all strains, the flux distribution above the phosphoenolpyruvate (PEP) node was virtually unaffected by the presence of proline. This is in accordance with experiments with cells grown on 100% fully labeled glucose and unlabeled proline, which showed that proline carbon only flowed into amino acids synthesized from TCA cycle intermediates and not into gluconeogenesis (data not shown).
For BL21(DE3)(pLysS)(pET-24a), the presence of proline significantly changed the flux distribution below the PEP node compared to growth on glucose only. The flux of proline carbon into αKG was accompanied by 36 and 41% reductions of the relative and absolute TCA cycle fluxes toward αKG, respectively, whereas the flux from αKG to succinate was unchanged. The flux from PEP to oxaloacetate was strongly reduced (70% reduction of the relative flux), indicating that the lower fluxes in the first steps of the TCA cycle reduced the demand for anaplerosis. Concomitantly, a higher acetate production rate was observed, correlating with the lower PEP/pyruvate (PYR) demand for oxaloacetate and citrate synthesis.
The pET_p4h1of-containing strain combined the lowest proline uptake rate and the highest P4H activity and consequently showed the lowest impact of proline on metabolism (as reflected by the low mRNA-levels found for the put operon, Table 2). Proline was degraded to glutamate to a much lower extent than in the control strain, and the net flux between αKG and glutamate reverted toward glutamate synthesis, indicating that the glutamate obtained from proline was not sufficient to cover the glutamate demand for biomass synthesis. Hence, the effect of proline on the initial steps of the TCA cycle was less pronounced (24% decrease in relative flux compared to growth on glucose only). The flux between αKG and succinate was significantly lower, due to the withdrawal of αKG for proline hydroxylation. The net anaplerotic flux between PEP and oxaloacetate decreased to a lower but still considerable extent (41% reduction of relative flux).
The strain containing pET_p4h1or showed an intermediate behavior with respect to the pET_p4h1of-containing strain and the control. The lower P4H activity of the pET_p4h1or-containng strain was associated with intermediate expression of the put operon (Table 2) and a reduced yield of hydroxyproline on proline (Table 3), all evidence of less efficient competition of P4H activity with proline metabolism for proline utilization.
In general, TCA cycle fluxes were influenced by both proline metabolism and proline hydroxylation, but it must be noted that, even at the highest P4H activity achieved with p4h1of, the flux toward αKG was still 7 times higher than the proline hydroxylation rate. Under the same conditions, the proline uptake rate was just twice as high as the proline hydroxylation rate.
DISCUSSION
Dependency of P4H synthesis on proline availability.
The addition of proline to a glucose-containing minimal medium had a positive effect on both P4H activity and level. This effect was proportional to the proline amount added, up to a certain threshold (between 0.26 and 4 mM), above which P4H levels and activity were constant. In combination with the effects of proline or yeast extract addition and codon usage (Fig. 2, 3, and 4a), this is strong evidence for regulation of enzyme synthesis by substrate availability. The MFA results (Fig. 5) indicated that P4H activity substantially contributes to intracellular proline consumption. Furthermore, resting cell activities decreased during growth with low proline concentrations (see Fig. S2.4 in the supplemental material), inferring that proline depletion entails a decreasing P4H synthesis rate. It therefore seems that P4H activity can reduce its own synthesis by reducing the availability of its reaction substrate (proline) in a sort of negative feedback mechanism. The unchanged p4h1or mRNA levels in the presence and absence of proline indicated a regulation at the translational level, likely mediated via the level of charged prolyl-tRNAs. Following this hypothesis, the strain containing pET_p4h1or and grown in the absence of proline encountered a limitation, where the only prolyl-tRNA utilized for the synthesis of the proline-rich P4H became depleted. Depending on the strain and on cultivation conditions, the P4H amount ranged between 5 and 20% of the total protein (Fig. 3; see also Fig. S2.1). Considering the 197% abundance of proline in P4H relative to the proteome, 10 to 40% of prolyl-tRNAs are used for P4H synthesis. Proline addition allows a faster reloading of the tRNA, while the codon usage in p4h1of distributes the proline demand across three different tRNAs, reducing the limitation on P4H synthesis (32). The increased P4H synthesis associated with the alternative codon usage cannot, however, be attributed only to improved P4H translation, since higher mRNA levels were reached with the p4h1of gene, likely due to increased mRNA stability. Such regulation of heterologous protein production by substrate availability may generally be shared by cells producing amino acid-transforming enzymes (e.g., transaminases, amino acid dehydrogenases, amino acid oxidases). Additional levels of regulation, such as induction of proline uptake by proline or increased uncoupling and inactivation of P4H in the absence of proline, may also contribute to this regulatory phenomenon.
Limitations in resting cells and effect of cultivation temperature.
The lowering of cultivation temperature directly correlated with higher levels of soluble P4H (fewer inclusion bodies [see Fig. S2.2 in the supplemental material), which in turn were associated with higher resting cell activities, similarly to what Klein and Huttel observed (49). Interestingly, the lower growth temperature was also responsible for a higher resting cell activity when the latter was not limited by functional P4H levels (Fig. 3 and 4; see also Fig. S2.6). The activities of permeabilized cells and cell extracts (Fig. 4; see also Fig. S2.6) and the correlating P4H levels (Fig. 3) indicated that the difference in resting cell activities after growth at 25°C or 30°C is not related to different functional P4H levels. Growth temperature was reported to influence not only recombinant gene expression and protein folding but also the expression of native genes involved in metabolism. Gadgil et al. (54), who investigated the transcriptome of E. coli grown at 37°C, 33°C, and 28°C, reported a negative correlation between growth temperature and expression of several TCA cycle genes and putP. If proline uptake limited the catalytic activity, the resting cell activities should reflect putP mRNA levels (Table 2). However, the strain carrying pET_p4h1or grown in the presence of proline showed a significantly higher putP mRNA level than the strain containing pET_p4h1of, but not a higher resting cell activity (Fig. 4a). Furthermore, while resting cells have a low proline demand for protein synthesis and catabolism, potentially leading to higher proline availability for biocatalysis, TCA cycle activity has been described to be low (20). Along this line of discussion, proline hydroxylation in resting cells appears to be limited by the cosubstrate supply. Alternatively, both resting and growing cell activities may be limited by export of hydroxyproline, which is not a natural metabolite of E. coli and for which the transport has not been characterized.
Metabolic flux redistribution and limitations in catalytically active growing cells.
During redox biocatalysis in living recombinant microorganisms, host metabolism may be influenced by several factors, including recombinant protein synthesis, the activity of the recombinant enzyme (e.g., cofactor/cosubstrate withdrawal, uncoupling), as well as the substrate supply and product formation (toxicity, cometabolism). Heyland et al. (44) studied the effect of various dmpA expression levels in recombinant E. coli. In contrast to P4H, the catalytic activity of the β-aminopeptidase DmpA is not directly connected to host metabolism. Whereas dmpA overexpression affected several physiological parameters (e.g., growth rate, acetate production rate, yield on glucose) proportionally to the expression level, it also influenced metabolic flux distribution, but independently of the expression level. In the present study, the flux distribution also was found to be influenced by mere overexpression (less efficient glucose utilization as emphasized by lower biomass yields on glucose and higher relative TCA activities). In this case, the variation of fluxes depended on both expression levels and proline hydroxylation rates, the latter dependency being a true consequence of the interaction between the catalytic reaction and cell metabolism.
In growing cells, proline hydroxylation appeared to be limited by different factors than in resting cells. Similarly to styrene epoxidation by styrene monooxygenase-containing E. coli (20), oxygenase activities in growing cells were lower than corresponding resting cell activities. In growing cells, the fluxes toward αKG were in all cases significantly higher than both proline hydroxylation and proline uptake rates, whereas the proline uptake rate of the more active strain containing pET_p4h1of exceeded the hydroxylation rate only by a factor of 2 and was 30% lower than that of the nonexpressing reference strain (Table 3). In contrast, the less active strain containing pET_p4h1or showed almost no reduction of the proline uptake rate. These uptake rates correlated well with the respective putP-mRNA levels (Table 2). The main regulator of putP expression is the protein PutA (55), which is also responsible for the first steps in proline degradation. In the absence of proline, the put operon is repressed by DNA-bound PutA (56), but once proline enters the cytoplasm and binds PutA, the protein translocates to the membrane, relieving the repression on both putA and putP expression and concomitantly initiating proline metabolism. The Km values of PutP, PutA, and P4H for proline are 3.3 μM (57), 58 mM (58), and 1 mM (unpublished data), respectively. Given an extracellular concentration of 4 mM, one can assume that PutP works at Vmax, while the reactions catalyzed by PutA and P4H may be proline limited. Given these assumptions, proline uptake is directly proportional to the amount of PutP, and a lower proline uptake rate implies a lower PutP level, as was confirmed via the determination of putP mRNA levels (Table 2). PutA and P4H activities are instead determined both by the respective enzyme levels and by the intracellular proline concentration. In the presence of proline and absence of P4H, no competition for proline exists and expression of putP and putA is maximal (Table 2). In the strain containing pET_p4h1of, a competition between P4H and PutA for proline exists, leading to lower intracellular proline levels, according to the lower Km of P4H. Thus, less proline is sensed by PutA, leading to downregulated putP and putA expression levels (Table 2) and consequently a lower proline uptake rate. This phenomenon is termed catalysis-induced reduction of substrate uptake. In cells containing pET_p4h1or, less P4H is available and the competition of P4H with PutA for proline has only minor impacts on putP and putA expression and on proline uptake (Tables 2 and 3). Following this line of argument, proline uptake is the main limitation of P4H activity in growing cells containing high P4H levels.
With variations in the molecular mechanism of regulation, substrate-induced substrate uptake is extremely common for all major carbon and energy sources (gluconate and related compounds [59], long-chain fatty acids [60], and several sugars, e.g., arabinose [61] and galactose [62]). Thus, catalysis-induced reduction of substrate uptake is likely to be observed in other processes involving recombinant whole-cell biocatalysis and native substrate uptake systems. In such cases, deregulation of substrate uptake may be necessary to increase catalytic activities. To further investigate this issue, future studies will focus on metabolically engineered strains with changed characteristics regarding proline uptake and degradation as well as αKG supply and consumption. Furthermore, uncoupling characteristics of P4H will be investigated.
In conclusion, the comprehensive physiological analysis of the P4H-based whole-cell biocatalyst allowed us to discover otherwise-nonobvious relationships between catalysis and physiology, such as the regulation of heterologous protein production by substrate availability and catalysis-induced reduction of substrate uptake. After optimization of P4H production by genetic and medium engineering, the limitations of proline hydroxylation were solely host related and not enzyme related. Furthermore, the findings emphasize the dependency of metabolism-coupled redox biocatalysis on the physiological state of the cells, e.g., they point to different limitations for growing and resting cells, and pave the way for metabolic engineering approaches that will help to further elucidate and resolve the physiological limitations.
ACKNOWLEDGMENTS
We gratefully acknowledge Jianan Fu, Patty Krabbe, and Katrin Rosenthal for their help with the experimental work, Ulrich Schwaneberg for collaboration on p4hof mutant library generation, and Lake-Ee Quek (University of Queensland, Brisbane, Australia) for help with the use of the OpenFLUX software.
This project was cofinanced by the EU-FP7 Oxygreen project (FP7 EU grant number 212281) and Evonik Industries AG.
Footnotes
Published ahead of print 1 March 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03640-12.
REFERENCES
- 1. Duetz WA, van Beilen JB, Witholt B. 2001. Using proteins in their natural environment: potential and limitations of microbial whole-cell hydroxylations in applied biocatalysis. Curr. Opin. Biotechnol. 12:419–425 [DOI] [PubMed] [Google Scholar]
- 2. Woodley JM. 2006. Microbial biocatalytic processes and their development, p 1–15 In Laskin A, Sariaslani S, Gadd G. (ed), Advances in applied microbiology, vol 60 Elsevier Academic Press Inc., San Diego, CA: [DOI] [PubMed] [Google Scholar]
- 3. Woodley JM. 2006. Choice of biocatalyst form for scalable processes. Biochem. Soc. Trans. 34:301–303 [DOI] [PubMed] [Google Scholar]
- 4. Blank LM, Ebert BE, Buehler K, Bühler B. 2010. Redox biocatalysis and metabolism: molecular mechanisms and metabolic network analysis. Antioxid. Redox Signal. 13:349–394 [DOI] [PubMed] [Google Scholar]
- 5. Bühler B, Schmid A. 2004. Process implementation aspects for biocatalytic hydrocarbon oxyfunctionalization. J. Biotechnol. 113:183–210 [DOI] [PubMed] [Google Scholar]
- 6. Kuhn D, Blank LM, Schmid A, Bühler B. 2010. Systems biotechnology: rational whole-cell biocatalyst and bioprocess design. Eng. Life Sci. 10:384–397 [Google Scholar]
- 7. Glieder A, Farinas ET, Arnold FH. 2002. Laboratory evolution of a soluble, self-sufficient, highly active alkane hydroxylase. Nat. Biotechnol. 20:1135–1139 [DOI] [PubMed] [Google Scholar]
- 8. Berry A, Dodge TC, Pepsin M, Weyler W. 2002. Application of metabolic engineering to improve both the production and use of biotech indigo. J. Ind. Microbiol. Biotechnol. 28:127–133 [DOI] [PubMed] [Google Scholar]
- 9. Nakamura CE, Whited GM. 2003. Metabolic engineering for the microbial production of 1,3-propanediol. Curr. Opin. Biotechnol. 14:454–459 [DOI] [PubMed] [Google Scholar]
- 10. Feist AM, Palsson BO. 2008. The growing scope of applications of genome-scale metabolic reconstructions using Escherichia coli. Nat. Biotechnol. 26:659–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wittmann C. 2007. Fluxome analysis using GC-MS. Microb. Cell Fact. 6:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sauer U. 2006. Metabolic networks in motion: 13C-based flux analysis. Mol. Syst. Biol. 2:62 doi:10.1038/msb4100109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Blank LM, Kuepfer L. 2010. Metabolic flux distributions: genetic information, computational predictions, and experimental validation. Appl. Microbiol. Biotechnol. 86:1243–1255 [DOI] [PubMed] [Google Scholar]
- 14. Becker J, Klopprogge C, Zelder O, Heinzle E, Wittmann C. 2005. Amplified expression of fructose 1,6-bisphosphatase in Corynebacterium glutamicum increases in vivo flux through the pentose phosphate pathway and lysine production on different carbon sources. Appl. Environ. Microbiol. 71:8587–8596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Asakura Y, Kimura E, Usuda Y, Kawahara Y, Matsui K, Osumi T, Nakamatsu T. 2007. Altered metabolic flux due to deletion of odhA causes l-glutamate overproduction in Corynebacterium glutamicum. Appl. Environ. Microbiol. 73:1308–1319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cornelissen S, Liu S, Deshmukh AT, Schmid A, Bühler B. 2011. Cell physiology rather than enzyme kinetics can determine the efficiency of cytochrome P450-catalyzed C-H-oxyfunctionalization. J. Ind. Microbiol. Biotechnol. 38:1359–1370 [DOI] [PubMed] [Google Scholar]
- 17. van Beilen JB, Duetz WA, Schmid A, Witholt B. 2003. Practical issues in the application of oxygenases. Trends Biotechnol. 21:170–177 [DOI] [PubMed] [Google Scholar]
- 18. Bühler B, Park JB, Blank LM, Schmid A. 2008. NADH availability limits asymmetric biocatalytic epoxidation in a growing recombinant Escherichia coli strain. Appl. Environ. Microbiol. 74:1436–1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chin JW, Khankal R, Monroe CA, Maranas CD, Cirino PC. 2009. Analysis of NADPH supply during xylitol production by engineered Escherichia coli. Biotechnol. Bioeng. 102:209–220 [DOI] [PubMed] [Google Scholar]
- 20. Julsing MK, Kuhn D, Schmid A, Bühler B. 2012. Resting cells of recombinant E. coli show high epoxidation yields on energy source and high sensitivity to product inhibition. Biotechnol. Bioeng. 109:1109–1119 [DOI] [PubMed] [Google Scholar]
- 21. Kuhn D, Fritzsch FSO, Zhang X, Wendisch VF, Blank LM, Bühler B, Schmid A. 2013. Subtoxic product levels limit the epoxidation capacity of recombinant E. coli by increasing microbial energy demands. J. Biotechnol. 163:194–203 [DOI] [PubMed] [Google Scholar]
- 22. Koehntop KD, Emerson JP, Que L. 2005. The 2-His-1-carboxylate facial triad: a versatile platform for dioxygen activation by mononuclear non-heme iron(II) enzymes. J. Biol. Inorg. Chem. 10:87–93 [DOI] [PubMed] [Google Scholar]
- 23. Hausinger RP. 2004. Fe(II)/alpha-ketoglutarate-dependent hydroxylases and related enzymes. Crit. Rev. Biochem. Mol. Biol. 39:21–68 [DOI] [PubMed] [Google Scholar]
- 24. Lawrence CC, Sobey WJ, Field RA, Baldwin JE, Schofield CJ. 1996. Purification and initial characterization of proline 4-hydroxylase from Streptomyces griseoviridus P8648: a 2-oxoacid, ferrous-dependent dioxygenase involved in etamycin biosynthesis. Biochem. J. 313:185–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Remuzon P. 1996. trans-4-Hydroxy-L-proline, a useful and versatile chiral starting block. Tetrahedron 52:13803–13835 [Google Scholar]
- 26. Shibasaki T, Mori H, Chiba S, Ozaki A. 1999. Microbial proline 4-hydroxylase screening and gene cloning. Appl. Environ. Microbiol. 65:4028–4031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shibasaki T, Mori H, Ozaki A. 2000. Enzymatic production of trans-4-hydroxy-L-proline by regio- and stereospecific hydroxylation of L-proline. Biosci. Biotechnol. Biochem. 64:746–750 [DOI] [PubMed] [Google Scholar]
- 28. Shibasaki T, Hashimoto S, Mori H, Ozaki A. 2000. Construction of a novel hydroxyproline-producing recombinant Escherichia coli by introducing a proline 4-hydroxylase gene. J. Biosci. Bioeng. 90:522–525 [DOI] [PubMed] [Google Scholar]
- 29. Muro-Pastor AM, Maloy S. 1995. Proline dehydrogenase activity of the transcriptional repressor PutA is required for induction of the Put operon by proline. J. Biol. Chem. 270:9819–9827 [DOI] [PubMed] [Google Scholar]
- 30. Studier FW, Moffatt BA. 1986. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 189:113–130 [DOI] [PubMed] [Google Scholar]
- 31. Moffatt BA, Studier FW. 1987. T7 lysozyme inhibits transcription by T7 RNA-polymerase. Cell 49:221–227 [DOI] [PubMed] [Google Scholar]
- 32. Welch M, Villalobos A, Gustafsson C, Minshull J. 2009. You're one in a googol: optimizing genes for protein expression. J. R. Soc. Interface 6:S467–S476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wong TS, Tee KL, Hauer B, Schwaneberg U. 2004. Sequence saturation mutagenesis (SeSaM): a novel method for directed evolution. Nucleic Acids Res. 32:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, New York, NY [Google Scholar]
- 35. Panke S, Meyer A, Huber CM, Witholt B, Wubbolts MG. 1999. An alkane-responsive expression system for the production of fine chemicals. Appl. Environ. Microbiol. 65:2324–2332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bühler B, Bollhalder I, Hauer B, Witholt B, Schmid A. 2003. Use of the two-liquid phase concept to exploit kinetically controlled multistep biocatalysis. Biotechnol. Bioeng. 81:683–694 [DOI] [PubMed] [Google Scholar]
- 37. Blank LM, Ebert BE, Bühler B, Schmid A. 2008. Metabolic capacity estimation of Escherichia coli as a platform for redox biocatalysis: constraint-based modeling and experimental verification. Biotechnol. Bioeng. 100:1050–1065 [DOI] [PubMed] [Google Scholar]
- 38. Laemmli UK. 1970. Cleavage of structural proteins during assembly of head of bacteriophage T4. Nature 227:680–685 [DOI] [PubMed] [Google Scholar]
- 39. Schrewe M, Magnusson AO, Willrodt C, Bühler B, Schmid A. 2011. Kinetic analysis of terminal and unactivated C-H bond oxyfunctionalization in fatty acid methyl esters by monooxygenase-based whole-cell biocatalysis. Adv. Synth. Catal. 353:3485–3495 [Google Scholar]
- 40. Mori H, Shibasaki T, Uozaki Y, Ochiai K, Ozaki A. 1996. Detection of novel proline 3-hydroxylase activities in Streptomyces and Bacillus spp. by regio- and stereospecific hydroxylation of l-proline. Appl. Environ. Microbiol. 62:1903–1907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Batas B, Schiraldi C, Chaudhuri JB. 1999. Inclusion body purification and protein refolding using microfiltration and size exclusion chromatography. J. Biotechnol. 68:149–158 [DOI] [PubMed] [Google Scholar]
- 42. Anderson M, Blowers D, Hewitt N, Hedge P, Breeze A, Hampton I, Taylor L. 1999. Refolding, purification, and characterization of a loop deletion mutant of human Bcl2 from bacterial inclusion bodies. Protein Expr. Purif. 15:162–170 [DOI] [PubMed] [Google Scholar]
- 43. Bradford MM. 1976. Rapid and sensitive method for quantitation of microgram quantities of protein utilizing principle of protein-dye binding. Anal. Biochem. 72:248–254 [DOI] [PubMed] [Google Scholar]
- 44. Heyland J, Blank LM, Schmid A. 2011. Quantification of metabolic limitations during recombinant protein production in Escherichia coli. J. Biotechnol. 155:178–184 [DOI] [PubMed] [Google Scholar]
- 45. Quek LE, Wittmann C, Nielsen LK, Kromer JO. 2009. OpenFLUX: efficient modelling software for 13C-based metabolic flux analysis. Microb. Cell Fact. 8:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Matisek R, Steiner G. 2006. Lebensmittelanalytik Grundzüge Methoden Anwendungen. Springer-Lehrbuch, Heidelberg, Germany: (In German.) [Google Scholar]
- 47. Kane JF. 1995. Effects of rare codon lusters on high-level expression of heterologous proteins in Escherichia coli. Curr. Opin. Biotechnol. 6:494–500 [DOI] [PubMed] [Google Scholar]
- 48. Przybycien TM, Dunn JP, Valax P, Georgiou G. 1994. Secondary structure characterization of beta-lactamase inclusion-bodies. Protein Eng. 7:131–136 [DOI] [PubMed] [Google Scholar]
- 49. Klein C, Huttel W. 2011. A simple procedure for selective hydroxylation of L-proline and L-pipecolic acid with recombinantly expressed proline hydroxylases. Adv. Synth. Catal. 353:1375–1383 [Google Scholar]
- 50. Sorensen HP, Mortensen KK. 2005. Soluble expression of recombinant proteins in the cytoplasm of Escherichia coli. Microb. Cell Fact. 4:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sezonov G, Joseleau-Petit D, D'Ari R. 2007. Escherichia coli physiology in Luria-Bertani broth. J. Bacteriol. 189:8746–8749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kurland C, Gallant J. 1996. Errors of heterologous protein expression. Curr. Opin. Biotechnol. 7:489–493 [DOI] [PubMed] [Google Scholar]
- 53. Zhou A, Wozniak A, Meyer-Lipp K, Nietschke M, Jung H, Fendler K. 2004. Charge translocation during cosubstrate binding in the Na+/proline transporter of E. coli. J. Mol. Biol. 343:931–942 [DOI] [PubMed] [Google Scholar]
- 54. Gadgil M, Kapur V, Hu WS. 2005. Transcriptional response of Escherichia coli to temperature shift. Biotechnol. Prog. 21:689–699 [DOI] [PubMed] [Google Scholar]
- 55. Maloy SR, Roth JR. 1983. Regulation of proline utilization in Salmonella typhimurium: characterization of put::Mu d(Ap, lac) operon fusions. J. Bacteriol. 154:561–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ostrovsky De Spicer P, Maloy S. 1993. PutA protein, a membrane-associated flavin dehydrogenase, acts as a redox-dependent transcriptional regulator. Proc. Nat. Acad. Sci. U. S. A. 90:4295–4298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Amin A, Ando T, Saijo S, Yamato I. 2011. Role of Asp187 and Gln190 in the Na+/proline symporter (PutP) of Escherichia coli. J. Biochem. 150:395–402 [DOI] [PubMed] [Google Scholar]
- 58. Ostrander EL, Larson JD, Schuermann JP, Tanner JJ. 2009. A conserved active site tyrosine residue of proline dehydrogenase helps enforce the preference for proline over hydroxyproline as the substrate. Biochemistry 48:951–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Peekhaus N, Conway T. 1998. What's for dinner? Entner-Doudoroff metabolism in Escherichia coli. J. Bacteriol. 180:3495–3502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Black PN, Dirusso CC. 1994. Molecular and biochemical analyses of fatty acid transport, metabolism, and gene regulation in Escherichia coli. Biochim. Biophys. Acta 1210:123–145 [DOI] [PubMed] [Google Scholar]
- 61. Kolodrubetz D, Schleif R. 1981. Regulation of the L-arabinose transport operons in Escherichia coli. J. Mol. Biol. 151:215–227 [DOI] [PubMed] [Google Scholar]
- 62. Wilson DB. 1974. Regulation and properties of galactose transport-system in Escherichia coli K-12. J. Biol. Chem. 249:553–558 [PubMed] [Google Scholar]