

Abstract
Ultra-high-throughput sequencing (HTS) of fungal communities has been restricted by short read lengths and primer amplification bias, slowing the adoption of newer sequencing technologies to fungal community profiling. To address these issues, we evaluated the performance of several common internal transcribed spacer (ITS) primers and designed a novel primer set and work flow for simultaneous quantification and species-level interrogation of fungal consortia. Primer comparison and validation were predicted in silico and by sequencing a “mock community” of mixed yeast species to explore the challenges of amplicon length and amplification bias for reconstructing defined yeast community structures. The amplicon size and distribution of this primer set are smaller than for all preexisting ITS primer sets, maximizing sequencing coverage of hypervariable ITS domains by very-short-amplicon, high-throughput sequencing platforms. This feature also enables the optional integration of quantitative PCR (qPCR) directly into the HTS preparatory work flow by substituting qPCR with these primers for standard PCR, yielding quantification of individual community members. The complete work flow described here, utilizing any of the qualified primer sets evaluated, can rapidly profile mixed fungal communities and capably reconstructed well-characterized beer and wine fermentation fungal communities.
INTRODUCTION
Fungi comprise a critical component of most food and beverage fermentation systems, including beer (1), wine (2), and cheese (3). High-throughput, “next-generation” sequencing (HTS) tools have recently emerged, enabling sensitive profiling of microbial communities on an unprecedented scale by massively parallel sequencing of short (100- to 600-bp) DNA fragments amplified by PCR. The large number of sequences delivered by a single HTS run (104 to 108 reads) allows more sensitive description of diverse microbial communities and greater multiplexing, admitting greater per-run sequencing capacity. Pyrosequencing (4) has been used most frequently to study fungal communities of foods (5), the human gut (6), and soils (7), as 454 Life Sciences pyrosequencing platforms yield longer sequences (400 to 600 bp), which are presumably necessary for accurate identification of fungi. However, the greater sequencing coverage of Illumina sequencing platforms (∼108 reads/lane on the HiSeq2000) compared to pyrosequencing (∼106 reads/run) enables multiplexing of more samples per run and a magnitudes-lower cost per sample, popularizing these platforms for analysis of bacterial communities. No studies to date have utilized the superior coverage of Illumina sequencing platforms to analyze fungal communities, presumably because the shorter read length may compromise taxonomic resolution. However, the level of taxonomic discrimination of fungi achievable using such short amplicons must be established to confirm their utility for ultra-high-throughput diversity analysis of fungal communities.
Primer choice and taxonomic classification (8, 9) have been extensively described for HTS of bacterial communities, but these aspects have not been well established for fungi. Amplification bias exhibited by PCR primers toward different groups of fungi is a well-known phenomenon, including with short-amplicon sequences (10). The impact of different computational methods on taxonomic assignment of HTS reads has also been described previously (11). The influence of primer choice on accurate taxonomic assignment of very short HTS reads (e.g., those produced on Illumina platforms) has been tested for fungal large-subunit (LSU) ribosomal DNA (rDNA) (12) but has not been fully established for the internal transcribed spacer (ITS) region, the official fungal “barcode of life” (13). Optimal primer selection for short-amplicon HTS applications relies on several criteria: high coverage, taxonomic resolution, and accuracy and short amplicon length (14). The last concern is particularly important with very-short-amplicon HTS platforms (Illumina and Ion Torrent), to choose a DNA target yielding maximal taxonomic heterogeneity within a very short fragment (<250 bp). In response to this, Ihrmark and coworkers (15) designed and compared several new primer sets targeting the ITS2 locus (see Fig. S1 in the supplemental material) to the commonly used ITS1/ITS4 primer pair by pyrosequencing of artificial mixed fungal communities, demonstrating more reliable community reconstruction with the shorter ITS2 locus amplicons amplified by their primers. Toju et al. (16) also designed and evaluated the taxonomic coverage of novel ITS primers in silico. However, none of these authors evaluated the accurate taxonomic resolution of ITS primers for short-amplicon HTS.
To confirm the usability of very-short-amplicon HTS reads for ultra-high-throughput analysis of fungal communities, we evaluated the amplification frequency, amplicon length, taxonomic coverage, and taxonomic assignment accuracy of artificial sequence reads in silico and tested the effect of commonly used ITS primers on accurate taxonomic assignment of a defined yeast mock community. We developed a new ITS primer set delivering high coverage and accurate taxonomic assignment of very short (150- to 250-bp) fungal amplicons and a work flow integrating paired quantitative PCR (qPCR)-HTS, enabling simultaneous quantification and characterization of complex fungal communities. The results demonstrate the utility of several ITS primer sets for short-amplicon HTS—particularly those targeting the ITS1 locus—as many exhibit high coverage and taxonomic classification accuracy. However, all primers tested failed to accurately profile the mock community, underlining the dangers of amplification and sequencing bias faced with HTS of ITS amplicons.
MATERIALS AND METHODS
Reference sequence database.
A modified reference sequence database was constructed from the public UNITE fungal ITS database (24 September 2012 release; http://unite.ut.ee/) (17, 18). Sequences were removed that contained the taxonomy terms “unidentified,” “compost,” “fungal sp.,” “endophyte,” “isolate,” “enrichment,” “fungus,” “soil,” “glacial,” “leaf,” “mycorrhizal,” “mycorrhizae,” “ectomycorrhiza,” “clone,” “culture,” “Antarctic,” “sterile,” “symbiont,” and “cf” or the incomplete taxonomic hierarchies “mycota sp.,” “mycote sp.,” “mycete sp.,” “mycetes sp.,” “ales sp.,” “aceae sp.,” “idea sp.,” and “mycotina sp.” This was performed to remove sequences with incomplete or absent taxonomies, as well as environmental samples, which can hinder useful taxonomic classification when used as reference sequences, particularly for primer evaluation. Sequences containing strings of ambiguous characters of ≥5 nucleotides (nt) were also removed. A taxonomy training file, exactly six levels deep (root, subphylum, order, family, genus, and species), was generated from the corresponding taxonomy strings (located in the sequence locus tags provided by UNITE) to be compatible with implementation in the HTS analysis pipeline QIIME (19). Both files were hand checked for accuracy and consistency. The database contains 158,874 sequences covering all fungal lineages, and taxonomic composition to the genus level is detailed in the supplemental material. Only publicly available sequences from the UNITE database were used in our reference database, and they can be replicated by following the filtering criteria defined above.
Primer design and analysis.
A list of primers used in this study is presented in Table 1. These primers were selected for analysis as the most commonly used ITS primer sets, as well as several recently published primers. In addition, they were chosen to represent primer pairs targeting the small-subunit (SSU) rDNA, 5.8S rDNA, and LSU rDNA flanks of the ITS, in order to evaluate primers across the entire ITS. Figure S1 in the supplemental material presents the orientation of these primer sets relative to the fungal ITS region. The BITS2F/B58S3 primer set was designed based on an alignment of representative ITS sequences from the UNITE ITS reference database clustered at 90% similarity, using the de novo primer design function of Primer Prospector (20) (http://pprospector.sourceforge.net/). Primer specificity, amplicon size distribution, and taxonomic coverage were predicted using the primer analysis modules of Primer Prospector (default settings), checking against a representative subset of the UNITE ITS reference database clustered at 97% identity. Taxonomic accuracy of in silico PCR hits from the UNITE database was evaluated by trimming each individual hit to 150 bp or 250 bp, assigning taxonomy using BLAST against the UNITE reference database, and then comparing the accuracy of each assignment lineage at each taxonomic level from class to species. Primer Prospector was also used to generate linker sequences without homology to fungal sequences at the 5′ terminus of the primer binding site (limiting primer bias), check for bar code and primer secondary structure (eliminating bar codes with a ΔG of <−5 kcal/mol), and optimize degenerate primer bases, using the appropriate program modules. The resulting primer set designed for this study is delineated in Table S1 in the supplemental material.
Table 1.
Mean frequency of primer homology to UNITE database ITS sequences
| rDNA | Primer | Sequence (5′→3′) | Referencea | Non-3′ mismatch | 3′ mismatch | 3′ gaps | Non-3′ gaps | Weighted scoreb |
|---|---|---|---|---|---|---|---|---|
| SSU | BITS | ACCTGCGGARGGATCA | 2.511 | 0.886 | 0.028 | 0.008 | 2.642 | |
| ITS1 | TCCGTAGGTGAACCTGCGG | 24 | 3.940 | 1.153 | 0.055 | 0.003 | 3.431 | |
| ITS5 | GGAAGTAAAAGTCGTAACAAGG | 24 | 6.458 | 1.038 | 0.053 | 0.000 | 4.173 | |
| ITS1-F_KYO2 | TAGAGGAAGTAAAAGTCGTAA | 16 | 6.111 | 1.448 | 0.027 | 0.002 | 4.238 | |
| ITS1-F_KYO1 | CTHGGTCATTTAGAGGAASTAA | 16 | 6.353 | 1.508 | 0.138 | 0.003 | 5.340 | |
| NSI1 | GATTGAATGGCTTAGTGAGG | 23 | 6.108 | 1.498 | 0.056 | 0.001 | 5.565 | |
| ITS1F | CTTGGTCATTTAGAGGAAGTAA | 22 | 6.858 | 1.598 | 0.108 | 0.005 | 5.910 | |
| NSA3 | AAACTCTGTCGTGCTGGGGATA | 23 | 7.225 | 1.519 | 0.111 | 0.005 | 6.077 | |
| 5.8S Reverse | ITS2_KYO1 | CTRYGTTCTTCATCGDT | 16 | 0.429 | 0.132 | 0.013 | 0.000 | 0.425 |
| ITS2_KYO2 | TTYRCTRCGTTCTTCATC | 16 | 0.438 | 0.146 | 0.010 | 0.001 | 0.492 | |
| B58S3 | GAGATCCRTTGYTRAAAGTT | 0.533 | 0.165 | 0.012 | 0.000 | 0.534 | ||
| 58A2 | CTGCGTTCTTCATCGAT | 23 | 0.495 | 0.187 | 0.011 | 0.001 | 0.545 | |
| ITS2 | GCTGCGTTCTTCATCGATGC | 24 | 0.644 | 0.195 | 0.027 | 0.001 | 0.704 | |
| 5.8S Forward | ITS3_KYO2 | GATGAAGAACGYAGYRAA | 16 | 0.502 | 0.109 | 0.011 | 0.000 | 0.399 |
| gITS7 | GTGAATCATCGARTCTTTG | 15 | 0.506 | 0.135 | 0.014 | 0.000 | 0.461 | |
| ITS3_KYO1 | AHCGATGAAGAACRYAG | 16 | 0.402 | 0.142 | 0.012 | 0.001 | 0.478 | |
| fITS7 | GAACACAGCGAAATGTGA | 15 | 0.575 | 0.135 | 0.016 | 0.000 | 0.497 | |
| 58A2 | ATCGATGAAGAACGCAG | 23 | 0.502 | 0.185 | 0.012 | 0.001 | 0.531 | |
| 58A1 | GCATCGATGAAGAACGC | 23 | 0.514 | 0.191 | 0.010 | 0.001 | 0.569 | |
| ITS3 | GCATCGATGAAGAACGCAGC | 24 | 0.667 | 0.198 | 0.027 | 0.001 | 0.640 | |
| fITS9 | GAACACAGCGAAATGTGA | 15 | 1.629 | 1.085 | 0.016 | 0.001 | 1.901 | |
| LSU | ITS4_KYO1 | TCCTCCGCTTWTTGWTWTGC | 16 | 4.274 | 1.107 | 0.039 | 0.002 | 3.846 |
| ITS4 | TCCTCCGCTTATTGATATGC | 24 | 4.489 | 1.480 | 0.057 | 0.004 | 4.070 | |
| ITS4_KYO2 | RBTTTCTTTTCCTCCGCT | 16 | 4.181 | 1.351 | 0.015 | 0.001 | 4.253 | |
| ITS4_KYO3 | CTBTTVCCKCTTCACTCG | 16 | 3.804 | 1.446 | 0.016 | 0.002 | 4.703 | |
| NLB4 | GGATTCTCACCCTCTATGAC | 23 | 6.410 | 0.819 | 0.025 | 0.002 | 5.228 | |
| ITS4B | CAGGAGACTTGTACACGGTCCAG | 22 | 7.528 | 1.694 | 0.127 | 0.005 | 6.198 |
In cases where no reference is listed, this study is meant.
Weighted score = (non-3′ mismatches × 0.4) + 3′ mismatches + non-3′ gaps + (3′ gaps × 3.0) + (final 3′ base mismatch × 3.0).
HTS work flow.
Samples of beer (1) and wine (2) were collected and prepared as described previously. Samples were amplified using the BITS/B58S3 primer set. Each forward primer contained a unique 8-nt bar code to enable sample multiplexing. A list of the bar codes used is presented in Table S1 in the supplemental material. PCR mixtures contained 5 to 100 ng of DNA template, 1× GoTaq Green master mix (Promega, Madison, WI), 1 mM MgCl2, and 5 pmol of each primer. Reaction conditions consisted of an initial 95°C for 2 min, followed by 35 cycles of 95°C for 30 s, 55°C for 30 s, and 72°C for 60 s, and a final extension of 72°C for 5 min. This PCR may be replaced with qPCR (see below) to achieve simultaneous community quantification. All samples were amplified in triplicate and combined prior to purification. Amplicons were mixed at roughly equivalent ratios based on electrophoretic band intensity and purified using a Qiaquick purification kit (Qiagen, Valencia, CA). Pooled samples were submitted to the University of California Berkeley Vincent Coates Genome Sequencing Laboratory for library preparation using the Illumina paired-end kit, cluster generation, and 150-bp paired-end sequencing on an Illumina HiSeq2000. Image analysis, base calling, and error estimation were performed using CASAVA 1.8.
In order to directly compare the short-sequence accuracies of the primer sets, a “mock community” was generated, consisting of DNA from eight yeast species deliberately combined at known ratios. These yeasts were grown and extracted as described in a previous report (2). rRNA operon copy number was enumerated by quantitative PCR as previously described (21) and DNA combined at known rRNA operon copy number ratios. Mock communities were amplified by five different 8-nt-bar-coded primer sets (BITS/B58S3, ITS1/ITS4, NSI1/58A2, ITS1F/ITS4, and 58A2/NLB4) using the thermal cycler programs previously described for each (22–24) and the reaction mixture listed above. Amplicons were prepared as described above and submitted to the University of California Davis Genome Center DNA technologies core for paired-end library preparation, cluster generation, and 250-bp paired-end sequencing on an Illumina MiSeq system. Image analysis, base calling, and error estimation were performed using CASAVA 1.8.
Data analysis.
Raw Illumina fastq files were demultiplexed, quality filtered, and analyzed using QIIME 1.5.0 (19). The 150-bp reads were truncated at any site of more than three sequential bases receiving a quality score of <Q20, and any read containing ambiguous base calls or bar code or primer errors were discarded, as were reads with <75% (of total read length) consecutive high-quality base calls (25). Primer sequences were trimmed from the ends of each sequence, and operational taxonomic units (OTUs) were assigned using the QIIME implementation of UCLUST (26), with a threshold of 97% pairwise identity. OTUs were classified taxonomically using a QIIME-based wrapper of BLAST (27) against the UNITE (17) database. Unassigned sequences (consisting of plant-associated sequences in the food samples) and any OTU comprising less than 0.001% of total sequences for each run were removed prior to further analysis, calibrating against a known mock community sample in each run (25). QIIME was used to calculate abundance-weighted Jaccard distance and binary Bray-Curtis dissimilarity between samples and to construct principal coordinate plots (PCoA).
Quantitative PCR.
To concurrently enumerate total fungal populations, qPCR may optionally replace classical PCR for amplicon generation in the work flow presented above. qPCR was performed in 20-μl reaction mixtures containing 2 μl of DNA template, 5 pmol of each respective primer, and 10 μl of TaKaRa SYBR 2× Perfect Real Time master mix (TaKaRa Bio Inc.). Reaction conditions involved an initial step at 95°C for 10 min, followed by 40 cycles of 30 s at 95°C, 30 s at 55°C, and 60 s at 72°C. Cell concentration was calculated by comparing sample threshold values (CT) to a standard curve of serially diluted genomic DNA extracted from a known concentration of Saccharomyces cerevisiae UCD522. All reactions were performed in triplicate in optical-grade 96-well plates on an ABI Prism 7500 Fast Real-Time PCR system (Applied Biosystems). The instrument automatically calculated cycle threshold (CT), efficiency (E), and confidence intervals. When integrated into the complete qPCR-HTS pipeline, amplicons generated by qPCR were pooled with other amplicons, purified, and processed as described above.
TRFLP.
As a comparison of HTS to a previously established method for profiling food fermentations, terminal restriction fragment length polymorphism (TRFLP) of beer (1) and wine (2) samples was performed as described for previous studies utilizing these samples. Briefly, PCR amplification was performed in triplicate with primers ITS1HEX (5′-[5HEX]TCCGTAGGTGAACCTGCGG-3′) and ITS4 (5′-TCCTCCGCTTATTGATATGC-3′) (24); purified using a QIAquick PCR purification kit (Qiagen, Valencia, CA); digested using the restriction enzymes HaeIII, DdeI, and HinfI; and submitted to the UC Davis Sequencing Core for ABI 3100 capillary electrophoresis fragment analysis. Taxonomic assignments of TRFLP peaks were made against an empirical ITS-TRFLP database (2). HaeIII profiles were used to construct plots and calculate distance matrices (as described for HTS data analysis above).
RESULTS
Primer analysis.
Amplification bias is a well-known phenomenon in fungal ITS primer sets (10), but only a few efforts (15, 16) have been made to design alternative primers with enhanced coverage, and no effort has been made to comprehensively evaluate the coverage, taxonomic resolution, and accuracy of existing ITS primers for very-short-amplicon HTS read lengths (<250 bp). We designed an alternative ITS primer set based on a multiple alignment of the UNITE database and computationally predicted its coverage of known fungal sequences in comparison with previously established ITS primers. To ascertain the usefulness of each primer for describing fungal communities, all primers were bioinformatically analyzed to assess overall taxonomic coverage of all fungal clades contained in the reference database. First, all primers listed in Table 1 were tested for overall database matches using the Primer Prospector “analyze primers” module (20). The weighted score calculated by this module is based on alignment against reference database sequences to predict overall coverage. Among ITS primers binding in the SSU rDNA, BITS exhibited the lowest weighted score, indicating increased coverage and diminished taxonomic bias through a lower mean count of gaps and mismatches across all species in the sequence database, but several other primers also scored very well, including ITS1 and ITS5 (Table 1). Among primers targeting the 5.8S rDNA, most primers had very low weighted scores, with ITS2_KYO1, ITS2_KYO2, and B58S3 as the lowest-scoring reverse primers. Among primers targeting the LSU rDNA, ITS4_KYO1 and ITS4 displayed the lowest weighted scores. Several primers exhibited very low database hits (as high weighted scores) and were eliminated from further testing.
Next, we used the taxonomic coverage module of Primer Prospector to predict the coverage of all fungal clades in both reference databases for each primer. Figure 1 presents the taxonomic coverage predicted for all fungal subphyla in the UNITE reference database, indicating very good coverage (≥60% relative coverage of most subphyla) by most primers matching the 5.8S rDNA (with the exception of fITS9). Agaricomycotina, Pezizomycotina, and Ustilaginomycotina exhibit the highest coverage (>80%) by the 5.8S primers (except fITS9 and ITS2), and primers B58S3, 58A2, ITS2_KYO1, and ITS3_KYO1 score highest among all 5.8S primers for all subphyla. In the SSU rDNA, BITS and ITS1 demonstrate highest relative coverage (19.1 to 63.3%) of all subphyla compared to other primers in the same binding region, but all primers display low relative coverage (2.9 to 19.1%) of Taphrinomycotina, suggesting potential amplification bias against this clade. In the LSU rDNA, ITS4 and ITS4_KYO1 showed the highest coverage (22 to 47%) for all subphyla, particularly the Pucciniomycotina (47% coverage). No hits were predicted for any primers for bacterial SSU/LSU sequences (data not shown), ensuring specificity for Eukarya. The complete results, presenting relative taxonomic coverage of all sequences at each hierarchical level from subphylum to genus, are provided in the supplemental material.
Fig 1.

Predicted subphylum coverage of UNITE database ITS sequences by primers located in terminal SSU (A), 5.8S (B), and terminal LSU (C) loci of the rRNA operon. Abbreviations for subphyla: Agar., Agaricomycotina; Ent., Entomophthoromycotina; In., incertae sedis; Kick., Kickxellomycotina; Muc., Mucoromycotina; Pezz., Pezizomycotina; Pucc., Pucciniomycotina; Sacc., Saccharomycotina; Taph., Taphrinomycotina; Ust., Ustilaginomycotina.
To assess the taxonomic usefulness of HTS reads from each primer, the classification accuracy of all primer hits predicted against the UNITE database was evaluated at each taxonomic level. Artificial 150-bp and 250-bp amplicons were generated from the primer hits predicted by Primer Prospector and taxonomically classified using BLAST against the UNITE reference database. The top BLAST hits were compared to the actual taxonomic lineage of each sequence at each taxonomic level. Net accuracy for the primers against all fungi is presented in Fig. 2, and that for all fungal classes is presented in Fig. S2 in the supplemental material. All primer pairs evaluated exhibited genus-level accuracies of >90% (>95% for ITS1 primers) for 150-bp reads and >95% for 250-bp reads. Primer pairs targeting the ITS1 locus exhibited species-level accuracy of >90% with both fragment sizes, but those targeting the ITS2 had much lower species-level accuracy (70 to 85%) with 150-bp reads, though this recovered to ∼90 to 95% with 250-bp reads (Fig. 2). The same trends could be observed when categorizing assignment accuracy by individual taxonomic classes (see Fig. S2 in the supplemental material). In general, ITS1 locus primer pairs displayed greater accuracy for most classes at the species and genus levels than did the equivalent read length of ITS2 amplicons, with the exception of NSI1/58A2. Species-level classification of Basidiomycota performed more reliably than those of Ascomycota and non-Dikarya, in general, by a narrow margin (see Fig. S2). The primer pairs 58A1/NLB4 and 58A2/NLB4 performed poorly, with <70% accurate classification of Dothidiomycetes, Sordariomycetes, and Glomeromycetes from 150-bp reads. These results demonstrate that ITS1 HTS sequences achieve a high species- and genus-level assignment success rate using BLAST for taxonomic classification of most fungal groups.
Fig 2.
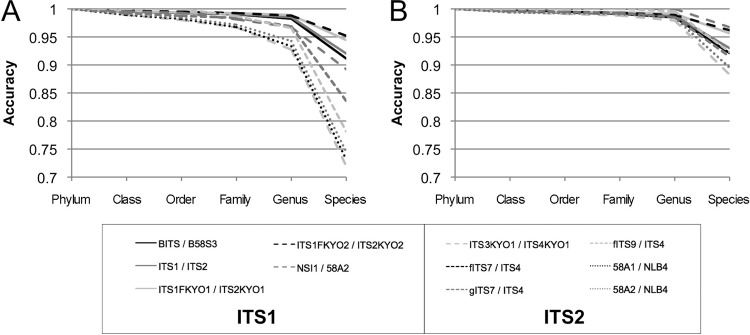
Average predicted taxonomic classification accuracy of selected primer sets against UNITE database sequences across all phyla. Predicted primer hits against database were trimmed to 150 bp (A) or 250 bp (B) and classified using BLAST. Accuracy represents the fraction of scores matching the correct taxon at each taxonomic level. Primer sets represent intended pairs targeting the ITS1 or ITS2.
Amplicon length unevenness can promote preferential amplification of shorter sequences (10), and shorter amplicons are preferential for maximizing read coverage of ultravariable DNA loci by the short read lengths employed by Illumina platforms. Short amplicon length is also crucial for efficient quantification with qPCR (28). Thus, amplicon lengths were predicted for all database matches for each primer to determine which primer sets would yield the shortest and most even amplicons. In general, whole-ITS and ITS1 subdomain amplicons displayed a greater distribution range of amplicon lengths than ITS2, but ITS1 amplicons were substantially shorter than ITS2 or whole-ITS amplicons (Table 2). This coincides with the findings of Bellemain and coworkers (10), who concluded that ITS1 was a preferential target for HTS efforts, as the shorter amplicon length and distribution would minimize preferential amplification of Ascomycota, which they found to contain systematically shorter ITS2 and whole-ITS regions than Basidiomycota. Amplicons were routinely longer for Basidiomycota than Ascomycota by 15 to 40 bp (ITS1 locus) and 30 to 50 bp (ITS2 locus). The shortest mean amplicon lengths were yielded by the BITS/B58S3 primer pair designed for this study, with a mean length of 183.6 (±46.8) bp, suggesting that most of these amplicons would be fully covered by current short sequencing reads. This primer pair is located at the 5′ terminus of the SSU rDNA, maximizing the amount of hypervariable ITS target sequence covered by a short sequencing read. Additionally, the amplicon is short enough to be efficiently amplified and quantified by qPCR, unlike the longer amplicons generated by other ITS primers, enabling qPCR quantification of the target.
Table 2.
Amplicon length distribution of primer hits categorized by ITS locus
| ITS locus | Forward primer | Reverse primer | Amplicon length distribution (mean ± SD) |
||
|---|---|---|---|---|---|
| Ascomycota | Basidiomycota | Non-Dikaryaa | |||
| ITS1 | BITS | B58S3 | 183.6 ± 46.8 | 219.8 ± 56.9 | 215.0 ± 95.9 |
| ITS1 | ITS2 | 218.1 ± 54.4 | 253.3 ± 60.0 | 201.0 ± 71.8 | |
| NSI1 | 58A2 | 357.2 ± 128.9 | 386.5 ± 105.7 | 269.1 ± 24.1 | |
| ITS1F_KYO1 | ITS2_KYO1 | 275.3 ± 103.2 | 285.3 ± 50.1 | 200.3 ± 54.2 | |
| ITS1F_KYO2 | ITS2_KYO2 | 270.6 ± 90.5 | 284.5 ± 42.1 | 216.4 ± 94.4 | |
| ITS2 | 58A1 | NLB4 | 478.8 ± 23.9 | 528.1 ± 30.3 | |
| 58A2 | NLB4 | 476.8 ± 23.9 | 525.9 ± 30.4 | ||
| ITS3F_KYO1 | ITS4_KYO1 | 310.5 ± 29.9 | 362.2 ± 35.3 | 376.0 ± 57.4 | |
| fITS7f | ITS4 | 258.5 ± 27.3 | 309.8 ± 35.6 | 312.7 ± 47.2 | |
| gITS7f | ITS4 | 259.9 ± 22.5 | 307.6 ± 34.7 | 312.9 ± 47.4 | |
| fITS9f | ITS4 | 324.4 ± 11.7 | 354.6 ± 32.9 | ||
| Whole ITS | BITS | ITS4 | 535.8 ± 81.8 | 618.0 ± 72.6 | 573.0 ± 132.4 |
| ITS1 | ITS4 | 547.2 ± 93.0 | 624.8 ± 79.7 | 582.4 ± 132.0 | |
| NSI1 | NLB4 | 1,066.6 ± 313.9 | 927.3 ± 182.6 | ||
| ITS1F_KYO1 | ITS4_KYO1 | 612.4 ± 131.1 | 664.9 ± 57.8 | 589.8 ± 155.6 | |
Missing values are due to noncoverage of non-Dikarya by that primer.
Mock community testing.
Computational analysis of primer coverage and specificity cannot adequately predict behavior under mixed biological conditions, so we tested the accuracy of these primers profiling a defined yeast community in a single 250-bp MiSeq run. Several highly scoring primer pairs were selected (representing ITS1, ITS2, and whole ITS) to directly compare their efficacies at reconstructing the taxonomic distribution of a known mock community comprising eight yeast strains mixed at known rRNA operon copy number ratios. No primer pair could reconstruct the known taxonomic distribution of this mock community with perfect accuracy (Fig. 3; see also Table S2 in the supplemental material), whether due to primer bias/mismatch, amplicon length bias, or computational bias (e.g., clustering of similar species as a single OTU), but most adequately profiled the dominant community members. To evaluate the accuracy of dominant-member mock community reconstruction by each primer pair, abundance-weighted Jaccard distance was calculated between each sample and used to construct principal coordinate plots (PCoA) to visualize the relationship to the expected community structure (see Fig. S3A in the supplemental material). 58A2/NLB4, BITS/B58S3, and ITS1F/ITS4 clustered close together with the expected community, indicating that these primer pairs are most effective at detecting the most dominant community members. To evaluate the accuracy of unweighted community profiling, a binary Bray-Curtis dissimilarity PCoA was also calculated between each sample and the expected community structure (see Fig. S3A). Primer pairs clustered separately with this statistic, though ITS1/ITS4, 58A2/NLB4, and BITS/B58S3 all clustered closer to the expected community, compared to ITS1F/ITS4 and NSI1/58A2. These data suggest that in practice, these primer pairs deviate from the expected mock community through the generation (or nonamplification) of low-abundance OTUs. This is a significant criterion for evaluating primer efficacy, as false-positive or -negative OTUs will distort community diversity and richness estimates that are important for ecological comparisons. Relative OTU abundance may be expected to shift with any PCR-based method for mixed-culture profiling, as a function of amplification bias. Given these results, mock community sequencing should be performed routinely for evaluation of new HTS primers to assess amplification bias issues under actual working conditions.
Fig 3.
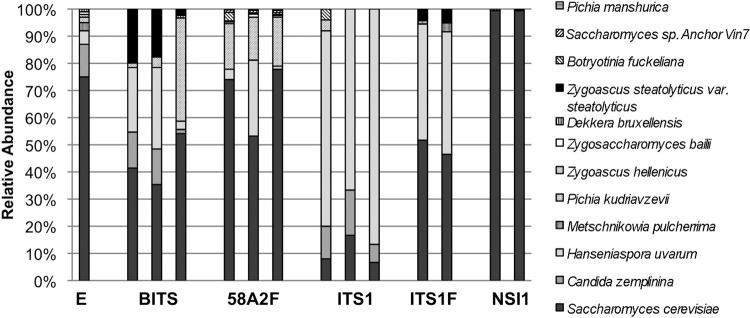
Taxonomic distribution of OTUs detected in a single yeast mock community reveals differential bias of ITS primer sets. Shown is the HTS community structure of a defined yeast mock community amplified with five different paired ITS primers. E, expected.
qPCR-HTS work flow.
One disadvantage of standard HTS protocols is that only relative OTU abundance is reported. To obtain absolute quantification, a second method must be employed; qPCR is one of the only techniques that can match the throughput of current HTS systems. We propose incorporating qPCR directly into the HTS library preparation work flow, optionally replacing standard PCR to streamline and concurrently perform community profiling and quantification, an option made possible by the short amplicon length of the BITS-B58S3 primer pair. To demonstrate the qPCR-HTS work flow using these primers, in which qPCR replaces standard PCR for amplicon generation, we performed qPCR of our yeast mock community and constituent yeast isolates. Standard curves of multiple yeasts were constructed to calculate accuracy and efficiency on multiple species. This reaction consistently achieved accurate standard curves of cultures as low as 103 cells/ml (R2 > 0.98) and reaction efficiency as high as 86.2% (see Fig. S4 in the supplemental material), supporting its use for quantification of mixed fungal samples.
Food fungal community profiling.
To assess the usability for HTS to describe food fungal communities, this method was used to profile beer (1) and wine fermentations that were previously analyzed with ITS-TRFLP (2). Results indicate some qualitative similarity between HTS with the BITS/B58S3 primer set and ITS-TRFLP using the ITS1/ITS4 primer set. In American coolship ale, the same dominant community members are observed, dominated by Saccharomyces cerevisiae, Brettanomyces bruxellensis, and several oxidative yeasts, principally Pichia kudriavzevii (see Fig. S5 in the supplemental material). Correspondence is weaker for the wine samples, as filamentous fungi were not represented in the empirical TRFLP database developed previously (2), and thus several fungal species detected by sequencing are either undetected or misidentified by TRFLP (Fig. 4). Sample similarity was further evaluated via abundance-weighted Jaccard distance PCoA (see Fig. S6 in the supplemental material). No distinct clusters could be established between methods for beer samples. However, wine samples cluster in a method-dependent fashion, indicating that OTU assignment differed between the two methods (see Fig. S6B). TRFLP infers OTU identity from restriction fragment size comparison to an empirical database, as opposed to sequence information, possibly leading to this disparity and highlighting a potential disadvantage of TRFLP. However, as the true composition of these samples is unknown, it is impossible to assess whether these differences are due to greater accuracy or greater noise with either method, and further evaluation must be performed to fully compare these techniques.
Fig 4.

Comparison of ITS sequencing to ITS-TRFLP of wine fungal communities. Botrytized wine fermentations were analyzed with BITS-B58S3 HTS (A) and ITS1-ITS4 TRFLP (B). Panel B was adapted from reference 2 with permission.
DISCUSSION
HTS has revolutionized the study of microbial ecology across a range of environments—including food fermentations—propelled by declining sequencing costs concurrent with increasing sequence coverage (14). Surprisingly, the most economical technologies currently available (namely, Illumina and Ion Torrent sequencing platforms) have yet to be applied to the study of fungal ecosystems via HTS—to our knowledge—presumably due to the short sequence length afforded by these systems and possible doubts about the taxonomic reliability of such short fragments. Several criteria define optimal primer characteristics for short-amplicon HTS, including target coverage, taxonomic resolution and accuracy, and short amplicon length (14). We have evaluated several ITS primer sets targeting different loci of the fungal ITS for each of these criteria, verifying many as valid choices for short-amplicon HTS applications. Primer pairs targeting the ITS1 locus generally appear to have the shortest mean amplicon lengths for all phyla, the smallest difference between Ascomycota and Basidiomycota amplicon lengths, and the highest species- and genus-level classification accuracy for short amplicon reads, arguing for the primacy of this locus for HTS, compared to ITS2 or whole ITS. The shortest amplicon length is displayed by the primer set designed for this study, BITS/B58S3, maximizing sequencing coverage of hypervariable ITS domains by very-short-amplicon HTS platforms and theoretically minimizing amplification bias (10). In addition, the amplicon size is adequately small for efficient use with qPCR quantification of fungal communities. The paired qPCR-HTS protocol presented here enables enumeration of total fungi and OTU profiling in a streamlined, two-step work flow. As the qPCR step quantifies the very same amplicon pool as serves as the template for the sequencing reaction, this paired work flow effectively permits absolute quantification of each individual OTU in a sample.
The complete HTS work flow described here, utilizing any of the qualified primer sets evaluated in this study, can rapidly profile complex fungal communities on ultra-high-throughput Illumina platforms, facilitating future exploration of fungal biomes with unprecedented coverage and sequencing depth. However, caution must be observed, as none of the primer sets could reliably reconstruct the mock yeast community sequenced in this study, indicating that the threat of amplification and sequencing bias remains prevalent even after judicious primer selection, likely due to the high amplicon length variability for all ITS loci.
ACKNOWLEDGMENTS
N.A.B. was supported by a Wine Spectator scholarship, the ASBC Foundation Brian Williams Scholarship (American Society of Brewing Chemists), and the 2012-2013 Dannon Probiotics Fellow Program (The Dannon Company, Inc.) during the completion of this work.
D.A.M. acknowledges Jim Orvis and John Thorngate from Constellation Brands Inc. for their helpful discussions and support.
Footnotes
Published ahead of print 1 February 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03870-12.
REFERENCES
- 1. Bokulich NA, Bamforth CW, Mills DA. 2012. Brewhouse-resident microbiota are responsible for multi-stage fermentation of American coolship ale. PLoS One 7(4):e35507 doi:10.1371/journal.pone.0035507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bokulich NA, Hwang CF, Liu S, Boundy-Mills K, Mills DA. 2012. Profiling the yeast communities of wine using terminal restriction fragment length polymorphism. Am. J. Enol. Vitic. 63:185–194 [Google Scholar]
- 3. Alegría A, Szczesny P, Mayo B, Bardowski J, Kowalczyk M. 2012. Biodiversity in Oscypek, a traditional Polish cheese, determined by culture-dependent and -independent approaches. Appl. Environ. Microbiol. 78:1890–1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Irzyk GP, Jando SC, Alenquer ML, Jarvie TP, Jirage KB, Kim JB, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J, Lohman KL, Lu H, Makhijani VB, McDade KE, McKenna MP, Myers EW, Nickerson E, Nobile JR, Plant R, Puc BP, Ronan MT, Roth GT, Sarkis GJ, Simons JF, Simpson JW, Srinivasan M, Tartaro KR, Tomasz A, Vogt KA, Volkmer GA, Wang SH, Wang Y, Weiner MP, Yu P, Begley RF, Rothberg JM. 2005. Genome sequencing in microfabricated high-density picolitre reactors. Nature 437:376–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jung MJ, Nam YD, Roh SW, Bae JW. 2012. Unexpected convergence of fungal and bacterial communities during fermentation of traditional Korean alcoholic beverages inoculated with various natural starters. Food Microbiol. 30:112–123 [DOI] [PubMed] [Google Scholar]
- 6. LaTuga MS, Ellis JC, Cotton CM, Goldberg RN, Wynn JL, Jackson RB, Seed PC. 2011. Beyond bacteria: a study of the enteric microbial consortium in extremely low birth weight infants. PLoS One 6(12):e27858 doi:10.1371/journal.pone.0027858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tedersoo L, Nilsson RH, Abarenkov K, Jairus T, Sadam A, Saar I, Bahram M, Bechem E, Chuyong G, Koljalg U. 2010. 454 pyrosequencing and Sanger sequencing of tropical mycorrhizal fungi provide similar results but reveal substantial methodological biases. New Phytol. 188:291–301 [DOI] [PubMed] [Google Scholar]
- 8. Soergel DAW, Dey N, Knight R, Brenner SE. 2012. Selection of primers for optimal taxonomic classification of environmental 16S rRNA gene sequences. ISME J. 6:1440–1444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu Z, Lozupone CA, Hamady M, Bushman FD, Knight R. 2007. Short pyrosequencing reads suffice for accurate microbial community analysis. Nucleic Acids Res. 35:e120 doi:10.1093/nar/gkm541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bellemain E, Carlsen T, Brochmann C, Coissac E, Taberlet P, Kauserud H. 2010. ITS as an environmental DNA barcode for fungi: an in silico approach reveals potential PCR biases. BMC Microbiol. 10:189 doi:10.1186/1471-2180-10-189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Porter TM, Golding GB. 2011. Are similarity- or phylogeny-based methods more appropriate for classifying internal transcribed spacer (ITS) metagenomic amplicons? New Phytol. 192:775–782 [DOI] [PubMed] [Google Scholar]
- 12. Porter TM, Golding GB. 2012. Factors that affect large subunit ribosomal DNA amplicon sequencing studies of fungal communities: classificiation method, primer choice, and error. PLoS One 7(4):e35749 doi:10.1371/journal.pone.0035749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schoch CL, Seifert KA, Huhndorf S, Robert V, Spouge JL, Levesque CA, Chen W. 2012. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. U. S. A. 109:6241–6246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bokulich NA, Mills DA. 2012. Next-generation approaches to the microbial ecology of food fermentations. BMB Rep. 45:377–389 [DOI] [PubMed] [Google Scholar]
- 15. Ihrmark K, Bodeker ITM, Cruz-Martinez K, Friberg H, Kubartova A, Schenck J, Strid Y, Stenlid J, Brandstrom-Durling M, Clemmensen KE, Lindahl BD. 2012. New primers to amplify the fungal ITS2 region—evaluation by 454-sequencing of artificial and natural communities. FEMS Microbiol. Ecol. 82:666–677 [DOI] [PubMed] [Google Scholar]
- 16. Toju H, Tanabe AS, Yamamoto S, Sato H. 2012. High-coverage ITS primers for the DNA-based identification of Ascomycetes and Basidiomycetes in environmental samples. PLoS One 7(7):e40863 doi:10.1371/journal.pone.0040863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kõljalg U, Larsson KH, Abarenkov K, Nilsson RH, Alexander IJ, Eberhardt U, Erland S, Hoiland K, Kjoller R, Larsson E, Pennanen T, Sen R, Taylor AF, Tedersoo L, Vralstad T, Ursing BM. 2005. UNITE: a database providing web-based methods for the molecular identification of ectomycorrhizal fungi. New Phytol. 166:1063–1068 [DOI] [PubMed] [Google Scholar]
- 18. Abarenkov K, Nilsson RH, Larsson K-H, Alexander IJ, Eberhardt U, Erland S, Høiland K, Kjøller R, Larsson E, Pennanen T, Sen R, Taylor AFS, Tedersoo L, Ursing BM, Vrålstad T, Liimatainen K, Peintner U, Kõljalg U. 2010. The UNITE database for molecular identification of fungi—recent updates and future perspectives. New Phytol. 186:1447–1452 [DOI] [PubMed] [Google Scholar]
- 19. Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Gonzalez Pena A, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. Qiime allows analysis of high-throughput community sequence data. Nat. Methods 7:335–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Walters WA, Caporaso JG, Lauber CL, Berg-Lyons D, Fierer N, Knight JR. 2011. PrimerProspector: de novo design and taxonomic analysis of barcoded polymerase chain reaction primers. Bioinformatics 27:1159–1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hierro N, Esteve-Zarzoso B, Gonzalez A, Mas A, Guillamon JM. 2006. Real-time quantitative PCR (QPCR) and reverse transcription-QPCR for detection and enumeration of total yeasts in wine. Appl. Environ. Microbiol. 72:7148–7155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gardes M, Bruns T. 1993. ITS primers with enhanced specificity for basidiomycetes—application to the identification of mycorrhizae and rusts. Mol. Ecol. 2:113–118 [DOI] [PubMed] [Google Scholar]
- 23. Martin KJ, Rygiewicz PT. 2005. Fungal-specific PCR primers developed for analysis of the ITS region of environmental DNA extracts. BMC Microbiol. 5:28 doi:10.1186/1471-2180-5-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. White T, Burns T, Lee S, Taylor J. 1990. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics, p 315–322 In Innis MA, Gelfand DH, Sninsky JJ, White TJ. (ed), PCR protocols: a guide to methods and applications. Academic Press, San Diego, CA [Google Scholar]
- 25. Bokulich NA, Subramanian S, Faith JJ, Gevers D, Gordon JI, Knight R, Mills DA, Caporaso JG. 2013. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 10:57–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461 [DOI] [PubMed] [Google Scholar]
- 27. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403–410 [DOI] [PubMed] [Google Scholar]
- 28. D'haene B, Vandesompele J, Hellemans J. 2010. Accurate and objective copy number profiling using real-time quantitative PCR. Methods 50:262–270 [DOI] [PubMed] [Google Scholar]