

Abstract
This study aimed to determine if biotic contaminants originating from pig production farms are disseminated into soil and groundwater microbial communities. A spatial and temporal sampling of soil and groundwater in proximity to pig production farms was conducted, and quantitative PCR (Q-PCR) was utilized to determine the abundances of tetracycline resistance genes (i.e., tetQ and tetZ) and integrase genes (i.e., intI1 and intI2). We observed that the abundances of tetZ, tetQ, intI1, and intI2 in the soils increased at least 6-fold after manure application, and their abundances remained elevated above the background for up to 16 months. Q-PCR further determined total abundances of up to 5.88 × 109 copies/ng DNA for tetZ, tetQ, intI1, and intI2 in some of the groundwater wells that were situated next to the manure lagoon and in the facility well used to supply water for one of the farms. We further utilized 16S rRNA-based pyrosequencing to assess the microbial communities, and our comparative analyses suggest that most of the soil samples collected before and after manure application did not change significantly, sharing a high Bray-Curtis similarity of 78.5%. In contrast, an increase in Bacteroidetes and sulfur-oxidizing bacterial populations was observed in the groundwaters collected from lagoon-associated groundwater wells. Genera associated with opportunistic human and animal pathogens, such as Acinetobacter, Arcobacter, Yersinia, and Coxiella, were detected in some of the manure-treated soils and affected groundwater wells. Feces-associated bacteria such as Streptococcus, Erysipelothrix, and Bacteroides were detected in the manure, soil, and groundwater ecosystems, suggesting a perturbation of the soil and groundwater environments by invader species from pig production activities.
INTRODUCTION
Over the last 25 years, pig production has largely shifted from small, integrated farming systems to concentrated animal feeding operations (CAFOs) that may house thousands of animals, with each pig typically producing about 1,500 kg of manure per production cycle. In September 2012 alone, the United States produced 67.5 million head of hogs and pigs, generating up to 1.0 × 1011 kg of fresh manure (1). The manure is generally stored in a waste lagoon prior to application to agricultural crop fields as fertilizer (2). During this stage, feces-associated contaminants can infiltrate soil beds to contaminate the groundwater supply and leach into the groundwater through compromised lagoon linings (3–5). Furthermore, manure generated from CAFOs contains the feces-associated microbiota, some of whose members may be opportunistic pathogens that impose health concerns when the public is exposed to these bacterial populations via the fecal-oral route. In addition, antibiotics are routinely used in the livestock industry for treatment and prevention of diseases and for growth promotion (6). Used in this manner, antibiotics may select for antibiotic-resistant bacteria in the gastrointestinal tract of pigs, and the resistance genes from such bacteria may be disseminated into the soil and groundwater in close proximity to pig production farms.
Further compounding the problem, the presence of mobile genetic elements, such as integrase genes, among the members of the fecal, soil, and groundwater microbiotas would facilitate horizontal transfer of genetic traits. A recent survey of over 600 fully or partially sequenced bacterial genomes revealed that about 10% of these contained known integrons (7). These integrons may contain gene cassettes (8) that encode traits such as antibiotic and heavy metal resistance and other virulence determinants (9, 10). Horizontal gene transfer is now recognized to play an important role in microbial evolution and adaptation, particularly in relation to traits that are related to human and animal health threats (11). Because horizontal gene transfer can effectively expand the gene pool available to resident soil and groundwater bacteria, it is important to understand whether biotic contamination from pig production activities can affect the abundance of mobile genetic elements and their persistence in the environment.
Past studies utilized culture-based and molecular biology-based approaches to reveal that antibiotic resistance genes and mobile genetic elements in the soil persist for up to 6 months when manure is applied to soil microcosms (12, 13). Furthermore, repeated manure application resulted in an accumulation of biotic contaminants (14). However, such studies have been restricted to microcosms conducted in the laboratory and do not reflect the extent of perturbation in uncontrolled environmental settings. For instance, the biotic contaminants may be exposed to UV irradiation or local weather conditions that would affect their actual persistence and eventual dissemination (15, 16). Although past studies attempted to address this limitation by measuring the abundance of antibiotic resistance genes in groundwater in close proximity to pig production farms (17–19), no further insights were provided on the extent of perturbation in the collective soil and groundwater microbiota due to contamination events originating from the production farms.
This study aimed to determine if biotic contaminants originating from pig production farms are disseminated into soil and groundwater microbial communities. The abundances of tetracycline resistance genes (i.e., tetQ and tetZ) and mobile genetic elements (i.e., intI1 and intI2) in the soils and groundwaters in close proximity to pig production farms were monitored. Tetracycline resistance genes were selected because tetracycline remains one of the most commonly used antibiotics on pig production farms (20, 21). The abundances of integrase genes provided insights into the likelihood of horizontal gene transfers among the microbial communities of these ecosystems. Based on the abundances of the tetracycline resistance genes and integrase genes, soil and groundwater samples that were relatively more affected by pig production activities were identified. 16S rRNA-based pyrosequencing was then performed to comparatively examine the microbial communities among the soil and groundwater samples and to determine the presence of genera associated with opportunistic pathogens.
MATERIALS AND METHODS
Study sites.
A total of three pig production sites, referred to here as sites A, C, and E, were identified for this study. The operation and site hydrogeology of sites A and C have been described previously (17, 18). Briefly, site A is a 4,000-pig finishing operation. This facility operates with a two-stage waste handling system in which a concrete settling basin collects most of the solid manure prior to the supernatant liquid passively moving into an unlined earthen lagoon. Site C is a farrowing and nursery operation that houses up to 2,500 sows. The facility in site C uses a single-stage unlined lagoon. Lagoon water is recycled to partially fill and flush the shallow pits below confinement buildings. Site E is a 2,300-hog finishing facility that uses a deep-pit lagoon (5). The described manure management strategies remained unchanged throughout the course of sampling, from 2005 to 2010. At all sites, manure was applied by first mixing the manure in the lagoon. The mixed manure was then pumped directly from the lagoon through a flexible pipe to a tractor, from which the manure was injected directly into the soil (I. Krapac, personal communication). For each sampling time series denoted in Fig. 1, manure was applied as a single dose, and no subsequent application was carried out at that particular field throughout the sampled duration. Chlortetracycline was used at all three sites for disease treatment, prophylaxis, and growth promotion. The exact amount of usage was not provided by the farmers. The geographic locations of the facilities cannot be disclosed due to a confidentiality agreement with the producers.
Fig 1.

Soil samples were collected in 2005 to 2007 from three agricultural crop fields that applied manure supplied by sites A, C, and E (pig production farms). The first sampling point of each time series is highlighted in bold, which also denotes that soil samples were collected before manure application. The subsequent sampling points and the spatial distance from the first sampling point denote the duration that passed after manure was applied. Manure was applied as a single dose, and no subsequent application was carried out at that particular field throughout the sampled time series.
Sample collection.
Each of the pig facilities applies manure to local farm fields, and time series studies were conducted in two fields from each facility (n = 6 fields) in order to quantify changes in antibiotic resistance genes, integrase genes, and microbial community composition in soil following manure application during 2005 to 2007. Each of the six fields was sampled as part of a time series that began just prior to manure application and then continued periodically for up to 2 to 16 months (Fig. 1). A 4 × 4 grid was constructed at each 0.16-km2 crop field to form 16 grid nodes per field. At each grid node, four soil samples that were 1 m from the grid node were collected at a depth of 0 to 20 cm by use of a hand corer. All samples collected from the grid nodes were mixed to form a composite sample that would represent a single sampling point at that localized site (Fig. 1). A total of 28 composite soil samples were collected throughout the course of 2005 to 2007. A previous geological-hydrological survey indicated that the local residents at site E utilized the groundwater as a water resource (5). Therefore, two sampling trips to site E were initiated, in June 2010 (trip a) and July 2010 (trip b), to collect groundwater samples. For each trip, 500 ml of manure slurry was sampled from the waste lagoon, and 1-liter groundwater samples were collected from the individual facility well and from monitoring wells E1, E4, E6, and E7 (Fig. 2). The facility well has a well depth of 36.6 m, and the monitoring wells have shallower well depths ranging from 5.3 to 7.1 m into the sandstone. A total of 10 groundwater and 2 manure samples were collected based on previously described procedures (17, 22), with the exception of groundwater from the facility well. Groundwater from the facility well was collected from a tap that is connected to the facility well and located inside the facility office. The tap was turned on to flush out the groundwater for about 3 min before the water samples were collected. Both facility well and monitoring well E1 are up-gradient of the waste lagoon and therefore served as negative controls in which no biotic contaminants originating from the waste lagoon were anticipated. In contrast, monitoring wells E4, E6, and E7 are directly adjacent to the waste lagoon and are anticipated to be susceptible to potential contamination events.
Fig 2.
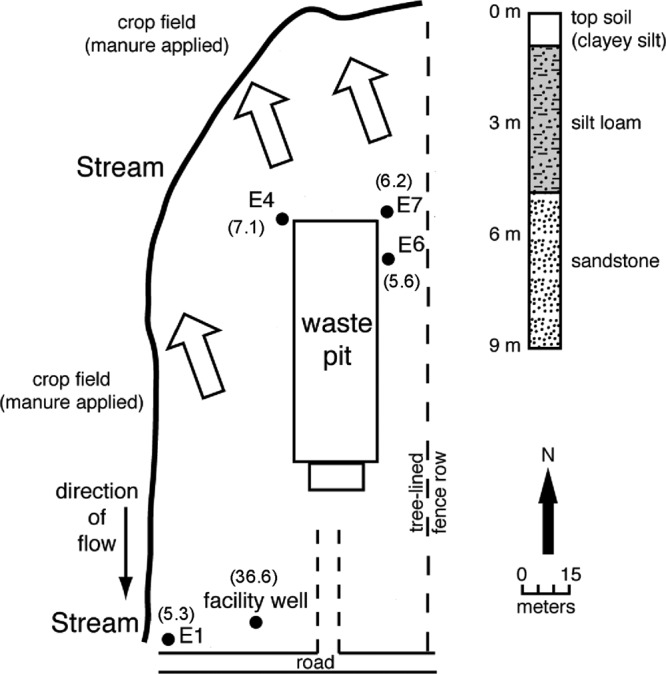
Map of groundwater wells at the site E pig production farm. The locations of groundwater wells are indicated by black circles, and the numbers in parentheses are well depths (m). An animal confinement building is situated above the waste lagoon. The direction of groundwater flow is indicated by large open arrows. Based on the direction of the groundwater flow and proximities to the waste lagoon, monitoring well E1 and the facility well served as background wells, where contamination was least anticipated. A corresponding stratigraphic column on the right indicates the characteristics of sand layers.
Genomic DNA extraction and bar-coded PCR.
Groundwater samples were filtered on a 0.22-μm polycarbonate membrane (Millipore, Billerica, MA), and genomic DNA was extracted using a modified protocol (23). Genomic DNA was extracted from soil samples by use of a FastDNA Spin kit for soil (MP Biomedicals, Solon, OH). Soil DNA was further purified of potential PCR contaminants by incubation at 65°C for 15 min in a solution of 1% cetyltrimethyl ammonium bromide and 0.7 M NaCl, followed by extraction with 24:1 chloroform-isoamyl alcohol, ethanol precipitation, and resuspension in Tris-EDTA (TE) buffer. To perform amplicon pyrosequencing, the V4-V5 region of the 16S rRNA gene was amplified from the genomic DNA by using the universal forward 519F (5′-fusion A-bar code-CAGCMGCCGCGGTAATWC-3′) and reverse 926R (5′-fusion B-bar code-CCGTCAATTCMTTTRAGTT-3′) primers. PCR mixtures comprised 10 ng of genomic DNA, 25 ml of premix F (Epicentre Biotechnologies, WI), 200 nM (each) forward and reverse primers, and 0.5 U of Ex Taq DNA polymerase (TaKaRa Bio, Japan), and the volume was brought up to 50 ml with molecular biology-grade water. PCR was performed with a 30-cycle thermal program (denaturation, 95°C for 30 s; annealing, 55°C for 45 s; and extension, 72°C for 60 s), and the amplicons were excised from the gel, concentrated, and purified with a Wizard DNA purification kit (Promega, Madison, WI).
Pyrosequencing and data analysis.
Paired-end pyrosequencing was performed by the Roy J. Carver Biotechnology Center, University of Illinois. A total of 239,624 16S rRNA sequences (i.e., 16S pyrotags) were obtained (see Table S1 in the supplemental material). Raw sequence reads were checked for quality based on previously described procedures (24), and a pyrosequencing depth of approximately 5,990 sequences was obtained for each sample. RDP Classifier was used for taxonomic assignments of the 16S pyrotags at the 95% confidence level (25). Bray-Curtis similarity matrix analysis with square root transformation was conducted by Primer-E v5.2.4. Rarefaction curves (see Fig. S1A to E) were generated based on previously described procedures (24), and the number of operational taxonomic units (OTUs) identified at a depth of 2,300 pyrotags was noted for comparisons of microbial richness (see Table S2).
Q-PCR analysis of tetracycline resistance genes and integron genes.
Primers TetQ and TetZ target the tetracycline resistance genes, encoding a ribosomal protection protein and an efflux pump, respectively (see Table S3 in the supplemental material) (26, 27). Primers IntI1 and IntI2 target integron classes 1 and 2, respectively (see Table S3) (28). Quantitative PCR (Q-PCR) was performed on all soil, groundwater, and manure samples. Reaction mixtures were prepared based on previously described protocols (24). Amplification efficiencies obtained from the standard curves for the tetQ, tetZ, intI1, intI2, and 16S rRNA genes were 2.04, 2.06, 1.84, 1.83, and 1.93, respectively. The coefficients of determination for standard curves ranged from 0.937 to 0.999. The sequences for Q-PCR standards are listed in Table S4.
Statistical method.
The nonpairwise two-tailed-distribution t test was conducted to determine the statistical significance of differences between microbial richness values for soil and groundwater samples. The t test was conducted at the 95% confidence level, using Microsoft Excel, version 14.0.6123.5001.
Pyrosequence accession links.
The pyrotag sequences determined in this study have been deposited in the Short Read Archive (SRA) of the European Nucleotide Archive (ENA). The study accession number is ERP002328.
RESULTS
Abundances of biotic contaminants and 16S rRNA genes in surface soil.
Soil samples collected before manure application (premanure soil samples) during the first time series (i.e., S1A0, S1C0, and S1E0) had lower abundances of biotic contaminants than premanure soil samples collected during the second time series (i.e., S2A0, S2C0, and S2E0). For example, S1A0 and S1C0 had 8.6 × 101 and 3.7 × 101 copies/ng DNA of total biotic contaminants, respectively, while S1E0 had no detectable biotic contaminants. For the second time series, premanure soil samples had 2.2 × 102, 2.8 × 102, and 8.1 × 102 copies/ng DNA (S2A0, S2C0, and S2E0, respectively) (Fig. 3). The tetQ, tetZ, intI1, and intI2 genes that were present in premanure soils (i.e., S1A0, S2A0, S1C0, S2C0, S1E0, and S2E0) were compared with those in the first set of postmanure soils (soils collected after manure application) (i.e., S1A1, S2A1, S1C1, S2C1, S1E1, and S2E1) collected from each field. Total abundances of tetZ, tetQ, intI1, and intI2 in the soils increased from 2.4 × 102 to 1.9 × 104 copies/ng DNA after manure application. Among the biotic contaminants, there were 500-fold, 9-fold, 6-fold, and 123-fold increases in tetQ, tetZ, intI1, and intI2 levels, respectively (Fig. 3A to C). The fate of these biotic contaminants varied across the different fields. For example, the highest abundances of intI1 and intI2, namely, 3.4 × 103 and 3.3 × 104 copies/ng DNA, respectively, were observed in soil samples that were treated with manure from site C (Fig. 3B). The collective levels of integrase genes and tetracycline resistance genes in soil samples S1C1, S1C2, S1C3, and S1C4 remained elevated above the background (i.e., S1C0) for up to 16 months (Fig. 3B). The collective levels of biotic contaminants in postmanure soil samples remained approximately 2-fold higher than the amounts in premanure soils collected from fields associated with site A. This increase in abundance of biotic contaminants remained apparent 2 to 3 months after manure was first applied (Fig. 3A). In contrast, the abundance of biotic contaminants in postmanure soil samples retrieved from fields associated with site E decreased back to the premanure levels after 1 month (Fig. 3C). The soil samples had abundances of 2.4 × 104 to 9.3 × 107 copies of 16S rRNA genes per ng of DNA (Fig. 3A to C), with a microbial richness of 447 to 1,650 OTUs, based on a pyrosequencing depth of 2,300 pyrotags (see Fig. S2A in the supplemental material).
Fig 3.

Summed abundances of 16S rRNA genes, tetracycline resistance genes, and integron genes detected in soil samples from agricultural crop fields that applied manure from site A (A), soil samples from agricultural crop fields that applied manure from site C (B), soil samples from agricultural crop fields that applied manure from site E (C), and groundwater samples from site E (D). Soil samples collected before manure application are highlighted in bold. The duration that passed after manure was first applied is denoted after each sample name. Groundwater samples from each well were collected on June and July 2010 at site E and are labeled with the name of the well followed by “a” and “b,” respectively.
Abundances of biotic contaminants and 16S rRNA genes in groundwater.
Biotic contaminants (i.e., tetZ, tetQ, intI1, and intI2) were detected in most of the groundwater samples from site E, except for both samples collected from monitoring well E1 and those collected from wells E6 and E7 during the second sampling trip (Fig. 3D). Total abundances of 4.7 × 101 and 1.5 × 104 copies/ng DNA of biotic contaminants were detected in the groundwater samples retrieved from wells E6 and E7, respectively, during the first sampling trip (Fig. 3D). Both groundwater samples retrieved from the E4 monitoring well were positive for tetZ, tetQ, intI1, and intI2. There was a higher abundance of tetQ than tetZ detected in well E4 groundwater samples, and the abundances of tetQ detected were up to 1.5 × 103 copies/ng DNA. Similarly, there was a higher abundance of intI2 than intI1 in well E4 groundwater samples, and the abundances of intI2 were 9.6 × 103 and 1.5 × 102 copies/ng DNA for the first and second sampling trips, respectively (Fig. 3D). Biotic contaminants were also detected in the groundwater microbiota collected from the facility well, and the total abundance of these biotic contaminants was 200-fold higher for the second sampling trip (3.1 × 103 copies/ng DNA) than for the first one (1.5 × 101 copies/ng DNA). Compared with the manure, which had tetZ, tetQ, intI1, and intI2 present at high average abundances, i.e., 1.6 × 104, 8.5 × 105, 2.2 × 105, and 4.2 × 104 copies/ng DNA, respectively, the groundwater had consistently lower levels of these biotic contaminants (Fig. 3D). The groundwater samples also had a microbial richness of 258 to 938 OTUs, identified at a sequencing depth of 2,300 pyrotags, and this was 1.8-fold lower than that of the manure at the same sequencing depth (see Fig. S2B in the supplemental material).
Genera associated with opportunistic pathogens and fecal indicators.
Soil samples from fields associated with sites A, C, and E were further monitored for genera associated with opportunistic pathogens and fecal indicators (Table 1). A similar monitoring was performed on groundwater samples collected from site E (Table 2). The genus Acinetobacter was present in most of the groundwater and soil, at relative abundances of up to 0.69% of the total microbial community. Other genera, such as Treponema, Arcobacter, Yersinia, and Coxiella, were detected sporadically in both the soil and groundwater samples, at low relative abundances (<1%). In addition, genera associated with fecal bacterial indicators (e.g., Streptococcus, Staphylococcus, Erysipelothrix, Bacteroides, and Parabacteroides) were also detected ubiquitously in the soil and groundwater (Tables 1 and 2). To illustrate this point, the genus Streptococcus was consistently detected in most of the soils, albeit at low relative abundances (0.01 to 0.11%), while the genus Clostridium was detected in most of the soil and groundwater samples, at relative abundances ranging from 0.03 to 2.3% of the total microbial community. The genera Bacteroides and Parabacteroides were also present, particularly in groundwater retrieved from the E4 and E7 monitoring wells. Although not of fecal origin, the genus Legionella was detected ubiquitously in all soil and groundwater samples and was present at relative abundances ranging from 0.03 to 0.21% of the total microbial community.
Table 1.
Names and abundances of genera associated with pathogens that were detected in soil samples
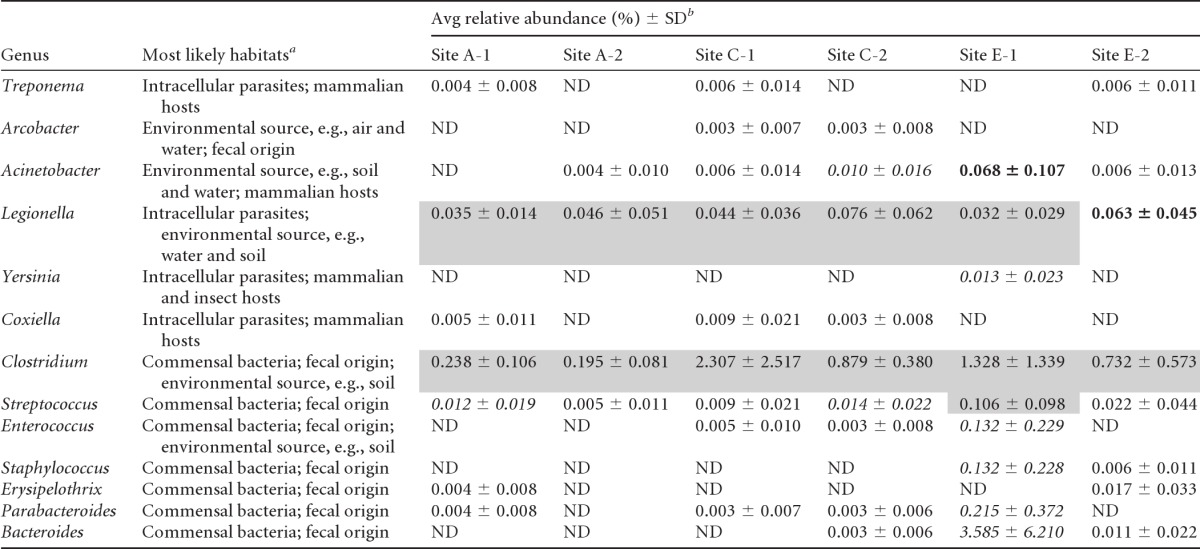
References related to the most likely habitats of each genus are found in the supplemental material.
Shading indicates a 76 to 100% frequency of occurrence among grab soil samples collected from the site, bold type indicates a 51 to 75% frequency, italics indicate a 26 to 50% frequency, and no formatting indicates a 0 to 25% frequency. ND, not detected at the current sequencing depth.
Table 2.
Names and abundances of genera associated with pathogens that were detected in groundwater and manure samples (n = 2 for each monitoring well)
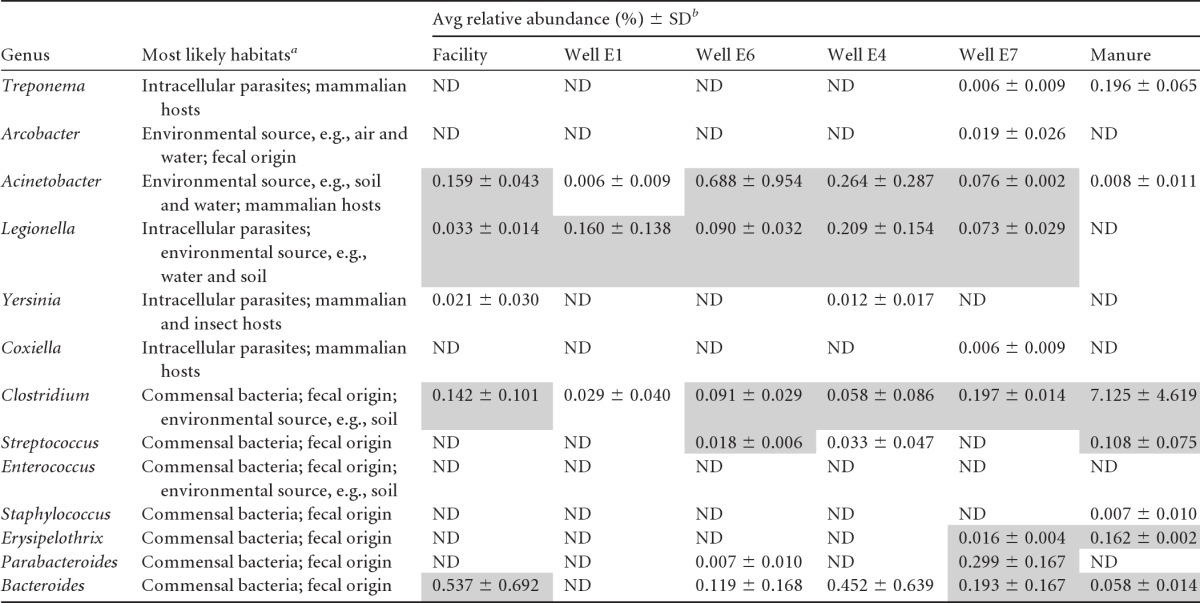
References related to the most likely habitats of each genus are found in the supplemental material.
Shaded cells denote that the particular genus was detected in both samples associated with the corresponding groundwater well. ND, not detected at the current sequencing depth.
Bacterial phyla in soil and groundwater microbial communities.
Figure 4A shows that all the soil samples were comprised predominantly of the phyla Actinobacteria (10.7%), Acidobacteria (12.1%), Proteobacteria (36.8%), and Bacteroidetes (6.2%), as well as unclassified bacteria (25.5%). With the exception of premanure soil samples S1C0 and S1E0, which contained unusually large amounts of Firmicutes and Bacteroidetes, respectively, there were no apparent changes in the soil microbial communities at the phylum level after manure application. Bray-Curtis similarity analyses revealed that regardless of the duration after manure application, the postmanure soil samples shared an average of 78.5% ± 3.0% similarity with the soil samples sampled before manure application (i.e., S1A0, S2A0, S2C0, and S2E0). The abundances of certain bacterial phyla were significantly different among the groundwater samples. For example, all the groundwater samples contained mainly 20.9 to 85.6% Proteobacteria and 3.6 to 48.3% unclassified bacteria (Fig. 4B). However, the phylum Acidobacteria was 6.6-fold more abundant in E1 and E6 than in E4 and E7 samples. In contrast, E4 and E7 groundwater had an about 11.6-fold higher abundance of the phylum Bacteroidetes. Bray-Curtis similarity analyses further revealed that the groundwater retrieved from the facility well clustered apart from the groundwater in the monitoring wells (i.e., E1, E4, E6, and E7) and shared an average similarity of 39.4% ± 1.1% with the other groundwater samples. The groundwater from monitoring wells was further clustered into two groups. To illustrate this, groundwater samples obtained from the E1 and E6 monitoring wells shared a similarity of 59.5% ± 0.2% with each other and a similarity of only 42.9% ± 1.8% with E4 and E7 groundwater. Similarly, groundwater samples obtained from the E4 and E7 monitoring wells shared a similarity of 53.7% ± 1.3% with each other.
Fig 4.

Percent abundances of bacterial phyla present in soil (A) and groundwater (B) samples. Soil samples collected before manure application are highlighted in bold. The duration that passed after manure was first applied is denoted after each sample name. Groundwater samples from each well were collected on June and July 2010 and are labeled with the name of the well followed by “a” and “b,” respectively.
Differences in bacterial genera among groundwater samples.
A further evaluation at the genus level revealed unique bacterial populations that were found in the facility waters as well as in the two clusters of groundwater sampled from monitoring wells (Fig. 5). Groundwater sampled from the facility well had relatively higher abundances of the genera Sulfuricurvum (1.6%), Sulfuricella (2.7%), and Lysobacter (7.0%) than the other groundwater samples. The high abundance of the phylum Acidobacteria in E1 and E6 groundwater was comprised of the uncultivated Gp1 (0.20 to 12.9%), Gp6 (0.76 to 11.4%), and Gp13 (4.5 to 14.3%) groups. Also, the genus Nitrospira accounted for 4.3% ± 0.1% of the total microbial community in E1 groundwater (Fig. 5). In contrast, the significantly higher abundance of the phylum Bacteroidetes in E4 and E7 groundwater was comprised of Flavobacterium (1.7%), Bacteroides (0.3%), and Parabacteroides (0.15%). The genus Sulfuritalea was also detected in both E4 and E7 groundwater samples, with average abundances of 24.8% and 6.5%, respectively, and these abundances were relatively higher than those in the remaining groundwater wells.
Fig 5.

Heat map of bacterial populations present in the groundwater at site E. Groundwater samples were clustered into three groups. Highly abundant bacterial populations associated with each group are boxed. Groundwater samples from each well were collected on June and July 2010 and are labeled with the name of the well followed by “a” and “b,” respectively.
DISCUSSION
This study aimed to monitor the presence of biotic contaminants in soil and groundwater ecosystems that are in close proximity to pig production farms and to investigate the extent of perturbation within the two indigenous microbial communities due to pig production activities. Q-PCR analysis detected spikes of at least 6-fold in the abundances of tetQ, tetZ, intI1, and intI2 in the soil environment after manure application. Similar to previous studies which demonstrated that antibiotic resistance genes can persist in the soil for up to 6 months (12, 13), the present studies showed that the abundances of tetracycline resistance and integrase genes in manure-treated soils can remain elevated above the initial abundances detected before manure application. The elevated abundance persisted for up to 16 months at certain agricultural crop sites, such as those that received manure from site C. Tetracycline resistance class tetQ genes are commonly associated with conjugative transposons, while tetZ genes have been reported to be associated with plasmids (29). Given that both class 1 and 2 integrase genes were detected and remained elevated in abundance for up to 16 months in the soils sampled from site C, the confluence of these factors has the potential to create a “microbial perfect storm” (30), defined as a phenomenon where novel microbial threats emerge with elevated frequency and that can create an environment that allows infectious diseases to emerge and become rooted in society (30).
Our findings further showed that soil samples that received manure from site C had the highest abundances of intI1 and intI2 and that the collective level of integrase genes and tetracycline resistance genes can remain elevated above the background for up to 16 months. Although the exact antibiotic usage was not disclosed, a nursery and farrowing operation such as site C is reported to typically feed sows an intake of 0.5 to 1 g of chlortetracycline per day, and in most cases, antibiotics are fed from about 1 week before breeding to approximately 2 to 3 weeks after breeding (31). Antibiotic usage and livestock management practices such as these may have possibly attributed to the persistent levels of biotic contaminants detected.
The groundwater monitoring wells at site E that were situated adjacent to the manure lagoon (i.e., E4, E6, and E7) were positive for tetracycline resistance and integrase genes on at least one of the two sampling trips. In contrast, the background monitoring well E1, which was situated up-gradient from the waste lagoon, was negative for these biotic contaminants. These observations suggest that there was leaching of feces-associated contaminants from compromised lagoon linings, which in turn were horizontally disseminated to the groundwater wells, situated at shallow depths. Similar to well E1, the facility well was located up-gradient from the waste lagoon. However, we detected the presence of tetracycline resistance genes and integrase genes in the groundwater retrieved from the facility well. Groundwater from the facility well at site E is regularly utilized for cleaning purposes and for consumption. Further compounding the problem, private wells such as those located at site E are generally not subjected to U.S. EPA standards and may not be disinfected regularly prior to usage (32). The difference between the facility well and monitoring well E1 is the depth at which both wells are positioned in the aquifer (Fig. 2). The facility well was drilled 36.6 m into the aquifer, while well E1 was drilled to a shallower depth of 5.3 m. The detection of tetracycline resistance and integrase genes in the facility well, but not in well E1, suggested that there was a certain extent of vertical seepage of feces-associated contaminants that may have further affected the groundwater supplies. For example, leaching may have occurred from the bottom of the waste pit, which contained about 2 to 5 m of manure slurry. Depending on the local hydrogeology, the leachate can spread horizontally during the initial phase, and subsequently in a vertical manner, to reach the deep aquifer that is connected to the facility well. Alternatively, because the groundwater from the facility well was collected from a tap supply, the positive detection of biotic contaminants may have been due to compromises in the piping distribution network. Future studies would have to include hydrogeological and engineering surveys to ascertain these inferences. In addition, future studies should aim to isolate tetracycline-resistant bacteria from the facility well groundwater. Isolation of such bacterial species would provide insights into whether these resistance traits were indeed horizontally acquired from the pig fecal microbiota.
Based on the abundance of tetracycline resistance and integrase genes, it was inferred that the facility well and monitoring wells E4 and E7 were relatively more affected than wells E1 and E6. There was differentiation in the groundwater microbiotas that corresponded with the abundances of biotic contaminants. The groundwater microbiota in the facility well and monitoring wells E4 and E7 exhibited high abundances of the genera Sulfuritalea, Sulfuricurvum, and Sulfuricella (Fig. 5). These bacterial populations are able to oxidize the sulfur contained in pig manure and can also utilize nitrate as electron acceptors (33–35). A previous study detected about 140 mg/liter of SO42− in the facility well and 287 to 856 mg/liter of SO42− in the monitoring wells at site E (5), and these amounts were higher than that in manure (70.7 mg/liter), possibly indicating active sulfur-oxidizing activities in the groundwater by these bacterial genera. Feces-associated bacterial populations (e.g., Streptococcus and Bacteroides) were also detected in the groundwater (Table 2). Coupled with a past bacteriological survey at site E which revealed the presence of fecal coliforms and fecal streptococcus (5), these observations reiterated that fecal contamination was present in the groundwater at site E and that this contamination likely originated from the pig farms (36).
However, unlike the groundwater wells, which were in close spatial association with the waste lagoon and therefore exposed to a constant source of fecal contamination, the soil samples received only pulses of contamination that originated from the one-time manure application. These short pulses of contamination had no detectable impact on the microbial composition of the soil microbiota. It has been demonstrated that bacteria from manure are poorly adapted to the soil microcosm and are soon outnumbered by the soil microbiota (13). Similarly, a negative correlation between the diversity of the soil microbiota and the survival of pathogenic Escherichia coli populations has also been demonstrated (37). In agreement with these observations, the soil samples in this study exhibited a significantly (P value = 4.23 × 10−6) higher microbial richness than that of groundwater, and the higher soil microbial diversity possibly accounted for greater resilience toward perturbations by implanted bacterial populations in applied pig waste. The negative correlation is likely due to a decrease in the competitive ability of the invader pathogen to utilize the available resources that are present in the soils. Alternatively, the bacteria from manure may experience higher die-off rates than those in the groundwaters due to constant exposure to the full UV spectrum from sunlight when present in the soils.
At the current pyrosequencing depth of approximately 5,990 sequences per sample, it was revealed that the soil and groundwater samples contained a wide range of genera associated with opportunistic pathogens (Tables 1 and 2). Some of these genera are opportunistic human pathogens (e.g., Acinetobacter, Yersinia, and Enterococcus), while others are opportunistic human and animal pathogens (e.g., Treponema, Arcobacter, Coxiella, Staphylococcus, and Erysipelothrix). These genera associated with opportunistic pathogens can be classified further into three types: first, the genera Treponema, Legionella, Yersinia, and Coxiella, which contain species that are intracellular parasites and reside in living hosts; second, the genera Arcobacter, Acinetobacter, Clostridium, and Enterococcus, which originate from both environmental and fecal sources; and third, the genera Streptococcus, Staphylococcus, Erysipelothrix, Parabacteroides, and Bacteroides, which originate mainly from fecal sources. Among these, the genus Acinetobacter was present in the manure, soil, and groundwater ecosystems, with relative abundances of up to 0.69% of the total microbial community. Acinetobacter spp. are generally regarded as commensal, opportunist, relatively low-grade pathogens (38) but have now been identified as causative agents in cases of community-acquired infection and in 9% of nosocomial infections (39, 40). Besides Acinetobacter spp., feces-associated bacteria such as Streptococcus, Erysipelothrix, and Bacteroides were also detected in the manure, soil, and groundwater ecosystems, suggesting a perturbation of the soil and groundwater environments by invader species from pig production activities.
Lastly, intracellular parasites such as Yersinia and Coxiella were also detected in some of the manure-treated soils and in groundwaters sampled from monitoring well E4 or E7. The bacterial species Yersinia pestis and Coxiella burnetii are known to be etiologic agents of plague and Q fever, respectively. Dose-response experiments performed on mice have established that the median lethal doses (LD50s) are approximately 4.26 × 102 CFU (41) and 4.93 × 108 PFU (42) for Yersinia pestis and Coxiella burnetii, respectively. Given that only low relative abundances of the genera Yersinia and Coxiella were detected in the soil and groundwater, the potential health impact on farm workers and the public remains low. Furthermore, the short pyrosequencing length (∼370 nucleotides [nt]) in this study did not allow identification at the species level, and therefore we could not determine if Yersinia pestis and Coxiella burnetii were present.
In summary, this study illustrates that both soil and groundwater are vulnerable to contamination originating from pig production farms. Collectively, manure-treated soils and feces-contaminated groundwater may be hot spots for horizontal gene transfer and may be critical zones for the exchange of genetic material between the species-rich environmental microbiota and antibiotic-resistant microorganisms. Future studies should aim to assess the direction of horizontal gene transfer by use of molecular and cultivation approaches and to determine if there is a possible exchange of mobile genetic elements between antimicrobial-resistant soil and groundwater bacteria and opportunistic pathogens. This study has provided a detailed monitoring of the biotic contaminants (i.e., tetracycline resistance genes, integrase genes, genera associated with opportunistic pathogens, and feces-associated bacteria) in both ecosystems. Coupled with our recent monitoring effort on the indoor bioaerosols on livestock production farms (24), our findings reiterate the need for better management practices and regulations to minimize dissemination of biotic contaminants originating from pig production farms into the environment.
ACKNOWLEDGMENTS
This work was funded by USDA grant 2009-351202-05021.
We thank Ivan Krapac for providing logistical support, the farm owners for granting access to the sampling sites, and Marie-Cécile Ploy and Olivier Barraud for the gift of Escherichia coli strains with intI1 and intI2 gene inserts. The technical assistance of Shannon Ortiz-Kofoed and Rushabh Shah is greatly appreciated.
Footnotes
Published ahead of print 8 February 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03760-12.
REFERENCES
- 1. USDA November 2012, posting date Quarterly hogs and pigs. USDA, Washington, DC: http://usda01.library.cornell.edu/usda/current/HogsPigs/HogsPigs-09-28-2012.pdf [Google Scholar]
- 2. Chee-Sanford JC, Mackie RI, Koike S, Krapac IG, Lin YF, Yannarell AC, Maxwell S, Aminov RI. 2009. Fate and transport of antibiotic residues and antibiotic resistance genes following land application of manure waste. J. Environ. Qual. 38:1086–1108 [DOI] [PubMed] [Google Scholar]
- 3. Arnon S, Dahan O, Elhanany S, Cohen K, Pankratov I, Gross A, Ronen Z, Baram S, Shore LS. 2008. Transport of testosterone and estrogen from dairy-farm waste lagoons to groundwater. Environ. Sci. Technol. 42:5521–5526 [DOI] [PubMed] [Google Scholar]
- 4. Sapkota AR, Curriero FC, Gibson KE, Schwab KJ. 2007. Antibiotic-resistant enterococci and fecal indicators in surface water and groundwater impacted by a concentrated swine feeding operation. Environ. Health Perspect. 115:1040–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Krapac IG, Dey WS, Roy WR, Smyth CA, Storment E, Sargent SL, Steele JD. 2002. Impacts of swine manure pits on groundwater quality. Environ. Pollut. 120:475–492 [DOI] [PubMed] [Google Scholar]
- 6. Cromwell GL. 2001. Antimicrobial and promicrobial agents, p 401–426 In Lewis A, Southern L. (ed), Swine nutrition, 2nd ed CRC Press, Boca Raton, FL [Google Scholar]
- 7. Boucher Y, Labbate M, Koenig JE, Stokes HW. 2007. Integrons: mobilizable platforms that promote genetic diversity in bacteria. Trends Microbiol. 15:301–309 [DOI] [PubMed] [Google Scholar]
- 8. Recchia GD, Hall RM. 1995. Gene cassettes: a new class of mobile element. Microbiology (Reading, England) 141:3015–3027 [DOI] [PubMed] [Google Scholar]
- 9. Wright MS, Baker-Austin C, Lindell AH, Stepanauskas R, Stokes HW, McArthur JV. 2008. Influence of industrial contamination on mobile genetic elements: class 1 integron abundance and gene cassette structure in aquatic bacterial communities. ISME J. 2:417–428 [DOI] [PubMed] [Google Scholar]
- 10. Leverstein-van Hall MA, Blok HE, Donders AR, Paauw A, Fluit AC, Verhoef J. 2003. Multidrug resistance among Enterobacteriaceae is strongly associated with the presence of integrons and is independent of species or isolate origin. J. Infect. Dis. 187:251–259 [DOI] [PubMed] [Google Scholar]
- 11. Ochman H, Lawrence JG, Groisman EA. 2000. Lateral gene transfer and the nature of bacterial innovation. Nature 405:299–304 [DOI] [PubMed] [Google Scholar]
- 12. Heuer H, Smalla K. 2007. Manure and sulfadiazine synergistically increased bacterial antibiotic resistance in soil over at least two months. Environ. Microbiol. 9:657–666 [DOI] [PubMed] [Google Scholar]
- 13. Heuer H, Focks A, Lamshöft M, Smalla K, Matthies M, Spiteller M. 2008. Fate of sulfadiazine administered to pigs and its quantitative effect on the dynamics of bacterial resistance genes in manure and manured soil. Soil Biol. Biochem. 40:1892–1900 [Google Scholar]
- 14. Heuer H, Solehati Q, Zimmerling U, Kleineidam K, Schloter M, Muller T, Focks A, Thiele-Bruhn S, Smalla K. 2011. Accumulation of sulfonamide resistance genes in arable soils due to repeated application of manure containing sulfadiazine. Appl. Environ. Microbiol. 77:2527–2530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McGrath BM, O'Halloran JA, Pembroke JT. 2005. Pre-exposure to UV irradiation increases the transfer frequency of the IncJ conjugative transposon-like elements R391, R392, R705, R706, R997 and pMERPH and is recA+ dependent. FEMS Microbiol. Lett. 243:461–465 [DOI] [PubMed] [Google Scholar]
- 16. Romero OC, Straub AP, Kohn T, Nguyen TH. 2011. Role of temperature and Suwannee River natural organic matter on inactivation kinetics of rotavirus and bacteriophage MS2 by solar irradiation. Environ. Sci. Technol. 45:10385–10393 [DOI] [PubMed] [Google Scholar]
- 17. Chee-Sanford JC, Aminov RI, Krapac IJ, Garrigues-Jeanjean N, Mackie RI. 2001. Occurrence and diversity of tetracycline resistance genes in lagoons and groundwater underlying two swine production facilities. Appl. Environ. Microbiol. 67:1494–1502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Koike S, Krapac IG, Oliver HD, Yannarell AC, Chee-Sanford JC, Aminov RI, Mackie RI. 2007. Monitoring and source tracking of tetracycline resistance genes in lagoons and groundwater adjacent to swine production facilities over a 3-year period. Appl. Environ. Microbiol. 73:4813–4823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Koike S, Aminov RI, Yannarell AC, Gans HD, Krapac IG, Chee-Sanford JC, Mackie RI. 2010. Molecular ecology of macrolide-lincosamide-streptogramin B methylases in waste lagoons and subsurface waters associated with swine production. Microb. Ecol. 59:487–498 [DOI] [PubMed] [Google Scholar]
- 20. Callens B, Persoons D, Maes D, Laanen M, Postma M, Boyen F, Haesebrouck F, Butaye P, Catry B, Dewulf J. 2012. Prophylactic and metaphylactic antimicrobial use in Belgian fattening pig herds. Prev. Vet. Med. 106:53–62 [DOI] [PubMed] [Google Scholar]
- 21. Jensen VF, Emborg HD, Aarestrup FM. 2012. Indications and patterns of therapeutic use of antimicrobial agents in the Danish pig production from 2002 to 2008. J. Vet. Pharmacol. Ther. 35:33–46 [DOI] [PubMed] [Google Scholar]
- 22. Mackie RI, Koike S, Krapac I, Chee-Sanford J, Maxwell S, Aminov RI. 2006. Tetracycline residues and tetracycline resistance genes in groundwater impacted by swine production facilities. Anim. Biotechnol. 17:157–176 [DOI] [PubMed] [Google Scholar]
- 23. Hong PY, Wheeler E, Cann IK, Mackie RI. 2011. Phylogenetic analysis of the fecal microbial community in herbivorous land and marine iguanas of the Galapagos Islands using 16S rRNA-based pyrosequencing. ISME J. 5:1461–1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hong PY, Li X, Yang X, Shinkai T, Zhang Y, Wang X, Mackie RI. 2012. Monitoring airborne biotic contaminants in the indoor environment of pig and poultry confinement buildings. Environ. Microbiol. 14:1420–1431 [DOI] [PubMed] [Google Scholar]
- 25. Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM. 2009. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 37:D141–D145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Aminov RI, Chee-Sanford JC, Garrigues N, Teferedegne B, Krapac IJ, White BA, Mackie RI. 2002. Development, validation, and application of PCR primers for detection of tetracycline efflux genes of gram-negative bacteria. Appl. Environ. Microbiol. 68:1786–1793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Aminov RI, Garrigues-Jeanjean N, Mackie RI. 2001. Molecular ecology of tetracycline resistance: development and validation of primers for detection of tetracycline resistance genes encoding ribosomal protection proteins. Appl. Environ. Microbiol. 67:22–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Barraud O, Baclet MC, Denis F, Ploy MC. 2010. Quantitative multiplex real-time PCR for detecting class 1, 2 and 3 integrons. J. Antimicrob. Chemother. 65:1642–1645 [DOI] [PubMed] [Google Scholar]
- 29. Speer BS, Shoemaker NB, Salyers AA. 1992. Bacterial resistance to tetracycline: mechanisms, transfer, and clinical significance. Clin. Microbiol. Rev. 5:387–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Smolinski MS, Hamburg MA, Lederberg J. 2003. Microbial threats to health: emergence, detection, and response. National Academies Press, Washington, DC: [PubMed] [Google Scholar]
- 31. Cromwell GL. 2002. Why and how antibiotics are used in swine production. Anim. Biotechnol. 13:7–27 [DOI] [PubMed] [Google Scholar]
- 32. Macler BA, Merkle JC. 2000. Current knowledge on groundwater microbial pathogens and their control. Hydrogeol. J. 8:29–40 [Google Scholar]
- 33. Kodama Y, Watanabe K. 2004. Sulfuricurvum kujiense gen. nov., sp. nov., a facultatively anaerobic, chemolithoautotrophic, sulfur-oxidizing bacterium isolated from an underground crude-oil storage cavity. Int. J. Syst. Evol. Microbiol. 54:2297–2300 [DOI] [PubMed] [Google Scholar]
- 34. Kojima H, Fukui M. 2010. Sulfuricella denitrificans gen. nov., sp. nov., a sulfur-oxidizing autotroph isolated from a freshwater lake. Int. J. Syst. Evol. Microbiol. 60:2862–2866 [DOI] [PubMed] [Google Scholar]
- 35. Kojima H, Fukui M. 2011. Sulfuritalea hydrogenivorans gen. nov., sp. nov., a facultative autotroph isolated from a freshwater lake. Int. J. Syst. Evol. Microbiol. 61:1651–1655 [DOI] [PubMed] [Google Scholar]
- 36. Lamendella R, Santo Domingo JW, Yannarell AC, Ghosh S, Di Giovanni G, Mackie RI, Oerther DB. 2009. Evaluation of swine-specific PCR assays used for fecal source tracking and analysis of molecular diversity of swine-specific “Bacteroidales” populations. Appl. Environ. Microbiol. 75:5787–5796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. van Elsas JD, Chiurazzi M, Mallon CA, Elhottova D, Kristufek V, Salles JF. 2012. Microbial diversity determines the invasion of soil by a bacterial pathogen. Proc. Natl. Acad. Sci. U. S. A. 109:1159–1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Joly-Guillou ML. 2005. Clinical impact and pathogenicity of Acinetobacter. Clin. Microbiol. Infect. 11:868–873 [DOI] [PubMed] [Google Scholar]
- 39. Vincent JL, Bihari DJ, Suter PM, Bruining HA, White J, Nicolas-Chanoin MH, Wolff M, Spencer RC, Hemmer M. 1995. The prevalence of nosocomial infection in intensive care units in Europe. Results of the European Prevalence of Infection in Intensive Care (EPIC) study. JAMA 274:639–644 [PubMed] [Google Scholar]
- 40. Vaque J, Rossello J, Arribas JL. 1999. Prevalence of nosocomial infections in Spain: EPINE study 1990–1997. J. Hosp. Infect. 43(Suppl):S105–S111 [DOI] [PubMed] [Google Scholar]
- 41. Lathem WW, Crosby SD, Miller VL, Goldman WE. 2005. Progression of primary pneumonic plague: a mouse model of infection, pathology, and bacterial transcriptional activity. Proc. Natl. Acad. Sci. U. S. A. 102:17786–17791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Williams JC, Cantrell JL. 1982. Biological and immunological properties of Coxiella burnetii vaccines in C57BL/10ScN endotoxin-nonresponder mice. Infect. Immun. 35:1091–1102 [DOI] [PMC free article] [PubMed] [Google Scholar]