

Abstract
Polaromonas sp. strain JS666 grows on cis-1,2-dichoroethene (cDCE) as the sole carbon and energy source under aerobic conditions, but the degradation mechanism and the enzymes involved are unknown. In this study, we established the complete pathway for cDCE degradation through heterologous gene expression, inhibition studies, enzyme assays, and analysis of intermediates. Several lines of evidence indicate that a cytochrome P450 monooxygenase catalyzes the initial step of cDCE degradation. Both the transient accumulation of dichloroacetaldehyde in cDCE-degrading cultures and dichloroacetaldehyde dehydrogenase activities in cell extracts of JS666 support a pathway for degradation of cDCE through dichloroacetaldehyde. The mechanism minimizes the formation of cDCE epoxide. The molecular phylogeny of the cytochrome P450 gene and the organization of neighboring genes suggest that the cDCE degradation pathway recently evolved in a progenitor capable of degrading 1,2-dichloroethane either by the recruitment of the cytochrome P450 monooxygenase gene from an alkane catabolic pathway or by selection for variants of the P450 in a preexisting 1,2-dichloroethane catabolic pathway. The results presented here add yet another role to the broad array of productive reactions catalyzed by cytochrome P450 enzymes.
INTRODUCTION
Extensive use of perchloroethene (PCE) and trichloroethene (TCE) as industrial solvents has resulted in soil and groundwater contamination throughout the world (1). Anaerobic bacteria can detoxify PCE and TCE by converting the compounds to ethene (2). Incomplete reduction, however, can lead to accumulation of toxic (cis-1,2-dichoroethene [cDCE]) and carcinogenic (vinyl chloride [VC]) intermediates in the environment (3) when contaminant plumes migrate into aerobic zones and reductive dehalogenation ceases.
Aerobic bacteria that use VC as a growth substrate are readily isolated from chloroethene-contaminated sites (4). VC (5) and ethene (6) are both assimilated via epoxidation catalyzed by an alkene monooxygenase, followed by conversion of the (chloro)epoxide to a 2-hydroxyalkyl coenzyme M by epoxyalkane coenzyme M transferase. The degradation mechanisms, biochemistry, and genes that encode VC and ethene degradation are analogous for the two compounds (5, 7, 8).
In contrast, Polaromonas sp. strain JS666 is the only isolate able to grow with cDCE as the sole carbon and energy source (9). cDCE degradation is a rate-limiting step in natural attenuation processes for the chlorinated ethenes at many contaminated sites (10, 11). The pathway for cDCE biodegradation and the enzymes involved are unknown. The genome of Polaromonas sp. strain JS666 contains a number of genes related to metabolism of xenobiotic compounds, but there is no obvious cluster of genes that encode the cDCE degradation pathway (12). Whole-genome expression microarrays and protein expression analysis (13) revealed upregulation of genes for enzymes potentially involved in the initial reactions of cDCE degradation. They included genes for cyclohexanone monooxygenase, glutathione-S-transferase, and cytochrome P450. Genes for (di)chloroacetaldehyde, (di)chloroacetic acid, and (chloro)glycolate transformation were also upregulated in a pattern expected for growth on cDCE or 1,2-dichloroethane (DCA). The observations suggested that the cDCE and DCA pathways might converge.
Coleman et al. (9) suggested epoxidation as the first step in cDCE degradation based on epoxidation of ethene by JS666 after growth on cDCE. The epoxidation of chlorinated ethenes by oxygenases is a widespread reaction in bacteria (14–16), but the toxic and highly reactive epoxide intermediates are not assimilated except in the case of VC biodegradation (5). A series of Escherichia coli strains with an engineered metabolic pathway for cDCE degradation based on epoxide formation and degradation has been constructed. The strains carry combinations of engineered toluene monooxygenase (17) and epoxide hydrolase (18) to catalyze transformation of chloroethenes and up to 4 other engineered proteins to overcome epoxide toxicity (19). Although the strains mineralize cDCE, they do not grow on it, and the pathway must be induced with isopropyl-β-d-thiogalactopyranoside.
Many oxygenases, such as the soluble methane monooxygenase from Methylosinus trichosporium OB3b (15), produce mixtures of acetaldehydes and epoxides from halogenated ethenes via an intramolecular halogen migration during oxidation. Similarly, in mammalian systems, cytochrome P450 enzymes catalyze the initial attack on chlorinated ethenes to produce a mixture of products. Nonconcerted reactions of human P450 2E1 or rat P450 2B1 oxidize TCE to the corresponding epoxide and 1,1,1-trichloroacetaldehyde (chloral), but the mechanisms controlling the partitioning to the two different intermediates are not fully understood (20).
Here, we describe the initial steps of cDCE biodegradation and genes that encode the enzymes involved. The understanding of the cDCE degradation pathway will provide the basis to detect, predict, and enhance cDCE degradation at contaminated sites as well as increase our understanding of bacterial evolution of catabolic pathways for synthetic chemicals.
MATERIALS AND METHODS
Growth conditions.
A 5-liter culture of Polaromonas sp. JS666 was routinely grown with cDCE as the sole carbon and energy source in a 7-liter bioreactor at 22°C. Minimal salts broth (MSB) (21) was modified as follows: phosphate buffer was 10 mM, and all other nutrients were supplied at half-strength. Neat cDCE was continuously supplied via syringe pump (8 to 50 μl/h) to maintain cDCE in the culture at 50 to 500 μM. The pH was maintained at 7.0 by the addition of NaOH. Oxygen addition was controlled to maintain dissolved oxygen at 3.5 mg/liter (40% saturation). The reactor was stirred at 400 rpm. Portions of the culture (1 to 2 liters) were harvested by centrifugation and suspended in MSB for use in experiments. Trypticase soy at one-quarter strength solidified with 18 g agar/liter (1/4-TSA) was used to test for the presence of contaminating bacteria. Strain JS666 was also grown in 160-ml serum bottles on half-strength MSB (1/2-MSB) with cyclohexanone (5 mM), glycolate (10 mM), succinate (10 mM), or DCA (0.25 mM) as the carbon source. Inocula were prepared from single colonies grown on agar plates containing cyclohexanone (5 mM) or from cells actively growing on cDCE.
Substrate transformation by whole cells.
Cells were suspended to an appropriate density (optical densities at 600 nm [OD600], ≈1 for JS666 and 10 to 60 for clones) in 50 ml of 1/2-MSB in 160-ml serum bottles sealed with Teflon-coated butyl rubber stoppers. Reactions were initiated by the addition of substrates, and cultures were incubated at 25°C with shaking (150 rpm). Substrate disappearance and/or product formation was monitored by gas chromatography (GC).
Respirometry.
Cells grown on various substrates were harvested by centrifugation, washed with potassium phosphate buffer (pH 7.2; 20 mM), and suspended in the same buffer. Oxygen uptake was measured polarographically at 25°C with a Clark-type oxygen electrode connected to a YSI model 5300 biological oxygen monitor.
Cloning and expression of putative cytochrome P450 monooxygenase.
Genomic DNA was extracted with a genomic DNA purification system (Promega, Madison, WI). A 3.5-kb fragment (positions 52538 to 56017 on pPol338; GenBank accession number NC_007950) containing genes encoding the putative cytochrome P450, ferredoxin reductase, ferredoxin, and a 562-bp portion of the intergenic region between the P450 and its upstream neighbor (Bpro_5302; Fig. 1) was amplified by PCR using primers 017F (CACCACGCTGCACCGTGTTATGTTTCAC) and 017R (TCAGTGCTGGCCGAGCGGCG). The PCR mixture (20 μl) contained genomic DNA (50 ng), primers (0.25 μM each), deoxynucleoside triphosphates (0.4 mM each), and Promega GoTaq Hot Start polymerase (2 U) and 1× buffer (Madison, WI). Amplifications (30 cycles) were carried out as follows: 95°C for 1 min, 56°C for 30 s, and 72°C for 1 min, after initial denaturation at 95°C for 10 min. The PCR products were treated with the Klenow fragment of DNA polymerase I (New England BioLabs, MA) and then cloned into the pET101/D-TOPO vector (Novagen, Gibbstown, NJ). The resulting plasmid, designated pJS593, was transformed into E. coli BL21 Star(DE3) for expression to create strain JS593.
Fig 1.
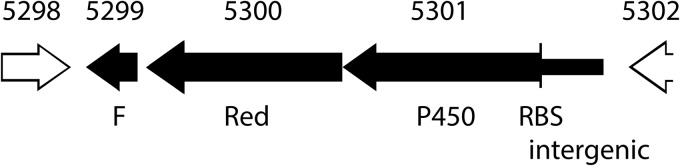
Map of region cloned into pET21a. Key: F, ferredoxin gene; Red, reductase gene; and P450, cytochrome P450 gene. Clones also contain the native ribosome binding site (RBS) and 562 bp of intergenic DNA. Gene loci are indicated above the genes.
JS593 was grown in 30 ml of terrific broth medium (TB) containing Overnight Express autoinduction system I (Novagen) at 37°C. When the OD600 reached 0.8 to 0.9, the temperature was reduced to 23°C and the cultures were incubated for an additional 14 to 20 h. Cells were harvested by centrifugation, washed twice with phosphate buffer (20 mM, pH 7.5), and stored on ice until used. Improved expression of the insert was obtained when pJS593 was moved into E. coli Rosetta 2; the resulting strain was designated JS595. γ-Aminolevulinic acid (0.5 mM) and FeSO4 (0.1 mM) were added at the start of the room temperature incubation (22).
Enzyme assays.
Cell extracts were prepared by passing cells twice through a French pressure cell at 20,000 lb/in2, followed by centrifugation at 160,000 × g. Cytochrome P450 monooxygenase activity was assayed by monitoring substrate disappearance (cDCE, DCA) and/or product formation by GC. Reaction mixtures (1 ml) consisted of cell extracts (1 to 4 mg protein) and substrates (80 to 160 nmol) in phosphate buffer (50 mM, pH 7.2) in 4-ml bottles sealed with Teflon-coated butyl rubber stoppers. Reactions were started by addition of NADH or NADPH (2.5 mM), and the bottles were incubated at 25°C. At appropriate intervals, the reactions were stopped by the addition of ZnSO4 (30 mM), and the mixtures were extracted with ethyl ether. Aldehyde dehydrogenase (23) was measured by following the reduction of NAD to NADH at 340 nm in the presence of (di)chloroacetaldehyde, cell extract (0.5 to 1 mg protein), and NAD (1 mM). Haloacid dehalogenase (24) was measured by chloride release in the presence of (di)chloroacetate and cell extract (0.5 to 1 mg protein).
Analytical methods.
cDCE and DCA in headspace samples were monitored with an Agilent 6890N GC equipped with a flame ionization detector (FID) and a Supelco 1% SP-1000 on 60/80 Carbopack B column, 6 ft by 1/8 in. (outer diameter) by 2.1 mm (inner diameter), as previously described (25), with the following modifications. The oven temperature was ramped at 20°C/min from 100°C to 150°C and then at 10°C/min to a maximum of 175°C and held until all compounds eluted. Some samples were spiked with TCE as an internal standard before extraction with ethyl ether. The ether extracts were analyzed by GC or GC-mass spectrometry (MS). Octane was measured in the headspace by GC-FID with a method similar to that for cDCE but with a Supelco 10% OV-101 on 80/100 Chromosorb W HP packed glass column, 6 ft by 1/4 in. (outer diameter) by 2 mm (inner diameter). cDCE epoxide, dichloroacetaldehyde, and chloroacetaldehyde in ether extracts were analyzed with an HP6890 GC equipped with either an electron-capture detector (ECD) or a 5973 mass detector. Compounds were separated on a DB-5MS column (30 m by 0.25 mm, 0.25-μm film thickness; J&W Scientific, CA). For GC-ECD, the oven temperature was held at 60°C with an injection temperature of 200°C and a detector temperature of 250°C. For GC-MS, the oven temperature was initially held at 35°C for 10 min, ramped at 20°C/min to 175°C, and held at the final temperature for 3 min. Absolute mass determination was performed by the Georgia Institute of Technology Bioanalytical Mass Spectrometry Facility.
Protein was measured with a Pierce bicinchoninic acid protein assay kit (Rockford, IL). Chloride was measured by the method of Bergmann and Sanik (26). Glyoxal was derivatized with 2,4-dinitro-phenylhydrazone (27) and then separated on a Phenomenex Synergi Polar-RP column (4 μm; 2.0 mm by 150 mm) with a Varian high-pressure liquid chromatography system equipped with a photodiode array detector. Solid-phase extraction (SPE) was performed with a MEPS C8 cartridge (SGE, Austin, TX).
Chemicals.
cDCE (97%, preservative free) was from Sigma-Aldrich. The major contaminant was trans-DCE, which was not metabolized by the cultures. All other chemicals were reagent grade. Dichloroacetaldehyde hydrate was converted to dichloroacetaldehyde by the method of Wakasugi (28). cDCE epoxide was synthesized chemically (29) and biologically (15) by transformation of cDCE by JS593. Products from both methods had the same GC retention times and parent masses [M+] at m/z 112, 114, and 116 which correspond to those of cDCE epoxide (30).
RESULTS
Hypothetical initial steps of cDCE degradation.
We investigated several possible reactions for the initial steps of cDCE biodegradation (Fig. 2). A reductase (Fig. 2, pathway I) or hydratase (pathway II) might channel cDCE into the DCA degradation pathway (24, 31). Glutathione-S-transferase might catalyze formation of glutathione conjugates during cDCE biodegradation (pathway III) (13). Monooxygenase enzymes could produce various amounts of an aldehyde (pathway IV) and cDCE epoxide (pathway V) (15, 32). Finally, although rare in aerobes, a reductive dehalogenase could catalyze removal of chlorine (pathway VI) (33).
Fig 2.

Potential initial steps for cDCE degradation in JS666 include reduction (I), hydration (II), glutathione addition (III), monooxygenation to an aldehyde (IV), monooxygenation to an epoxide (V), or reductive dehalogenation (VI). GSH, reduced glutathione; SG, glutathione conjugated to the organic compound, where the H (hydrogen) has been replaced by the organic compound.
Stimulation of oxygen uptake by potential intermediates of cDCE degradation.
Respirometry experiments do not provide rigorous conclusions about pathways but allow hypotheses to be made about enzyme induction and involvement of intermediates. The rates reflect not only preferences for the initial substrates but also stoichiometries and kinetics of subsequent reactions along with transport limitations. cDCE, DCA, and intermediates of the DCA degradation pathway stimulated oxygen uptake in cDCE- and DCA-grown cells of strain JS666 (Table 1). The results indicate (34) that cDCE is degraded via DCA or that the cDCE and DCA pathways converge. Alternatively, a monooxygenase induced by cDCE (13) could transform DCA into chloroacetaldehyde (31). Chloroacetaldehyde might then be degraded by an enzyme that has high constitutive activity but is also moderately upregulated during growth on cDCE, DCA, or cyclohexanone (Table 1). Cyclohexanone-grown cells did not oxidize cDCE, DCA, or intermediates of the DCA degradation pathway other than choroacetaldehyde, for which there was high constitutive activity in all cells.
Table 1.
Oxygen uptake by resting cells of JS666a
| Test substrate (concn [mM]) | Oxygen uptake (nmol min−1 mg−1 protein) after growth on: |
||||
|---|---|---|---|---|---|
| cDCE | DCA | Cyclohexanone | Glycolate | Succinate | |
| cDCE (1.0) | 13 ± 0 | 6 ± 2 | <1 | <1 | <1 |
| 1,2-DCA (1.0) | 11 ± 0 | 23 ± 6 | <1 | <1 | <1 |
| 2-Chloroethanol (1.0) | 7 ± 2 | 13 ± 0 | <1 | <1 | <1 |
| Chloroacetaldehyde hydrate (0.5) | 55 ± 1 | 47 ± 0 | 81 ± 6 | 39 ± 2 | 27 ± 5 |
| Dichloroacetaldehyde hydrate (0.5) | 18 ± 2 | 12 ± 0 | 6 ± 4 | 7 ± 1 | 3 ± 0 |
| Cyclohexanone (0.5) | 10 ± 1 | 1 ± 2 | 276 ± 36 | <1 | <1 |
| Chloroacetate (1.0) | 2 ± 0 | 26 ± 8 | <1 | 7 ± 2 | 2 ± 1 |
| Dichloroacetate (1.0) | ND | 10 ± 3 | |||
| Glycolate (1.0) | 7 ± 2 | 14 ± 2 | <1 | 95 ± 1 | 6 ± 2 |
| Succinate (1.0) | 7 ± 1 | 32 ± 1 | 41 ± 1 | ||
Oxygen uptake by resting cells of JS666 grown with various substrates. Reaction mixtures contained substrate, cells (0.29 to 0.31 mg of protein), and air-saturated phosphate buffer (20 mM, pH 7.2) to a final volume of 1.85 ml. cDCE, 1,2-DCA, and cyclohexanone were added from a 1.0 M stock in acetone, and the other substrates were dissolved in water. Acetone did not affect the rates of oxygen uptake. Data represent means and standard deviations from at least two measurements. ND, not determined; blank cell, not tested.
Involvement of oxygen in the initial steps of cDCE biodegradation.
cDCE-grown cells required oxygen to metabolize cDCE (Fig. 3) and DCA (Fig. 3B). Cell suspensions rapidly transformed cDCE and DCA in the presence of oxygen, but no transformation was detected in the absence of oxygen. The result is not consistent with reductase- or hydratase-catalyzed conversion of cDCE to DCA. cDCE degradation resumed immediately upon addition of air. The results suggest but do not prove that an oxygenase enzyme catalyzes the initial steps in cDCE and DCA degradation. The requirement for molecular oxygen also precludes involvement of a reductive dehalogenase.
Fig 3.

Dependence of cDCE and DCA degradation by JS666 on oxygen. (A) cDCE-grown cells were added to N2-sparged (□) and unsparged (•) serum bottles. Reactions were initiated by addition of cDCE to the bottles. Arrow, addition of 5 ml of air. (B) cDCE and DCA were added as neat solutions to N2-sparged serum bottles. Reactions were initiated by adding 1 ml of cDCE-grown sparged cells to bottles. Arrow, addition of 1 ml of O2. Error bars indicate 1 standard deviation.
Metyrapone and phenylhydrazine inhibition of cDCE biodegradation.
Many cytochrome P450 enzymes are inhibited by metyrapone and phenylhydrazine (35, 36). Both cDCE and DCA degradation were inhibited over 90% in intact cells of JS666 by 200 μM metyrapone or phenylhydrazine (Fig. 4A), as measured by either GC or stimulation of oxygen uptake. The oxygen uptake rates with chloroacetaldehyde and cyclohexanone were unchanged. The degree of inhibition of the initial rates of cDCE and DCA degradation was proportional to the concentration of metyrapone (Fig. 4B). The results indicate that a cytochrome P450 catalyzes early steps in both cDCE and DCA biodegradation in JS666.
Fig 4.

Effect of cytochrome P450 inhibitors on JS666. (A) Time course of cDCE degradation with no inhibitor (•), 200 μM metyrapone (○), or 200 μM phenylhydrazine (□). (B) Concentration-dependent effect of metyrapone on cDCE (•) and DCA (■) degradation by JS666. cDCE and DCA were provided at 100 μM.
cDCE degradation kinetics.
Strain JS666 grows on cDCE with stoichiometric accumulation of chloride (9). Chloride release was slightly delayed during the early stages of growth of JS666, which suggested a transient accumulation of degradation intermediates. During cDCE degradation by JS666 in the present study, small amounts of dichloroacetaldehyde transiently accumulated and then disappeared (data not shown), which suggested strongly that dichloroacetaldehyde is an intermediate of cDCE degradation. Trace amounts of cDCE epoxide also accumulated in the cDCE-degrading cultures. The detection of dichloroacetaldehyde and cDCE epoxide in cDCE-degrading cultures is consistent with the involvement of the cytochrome P450 monooxygenase, here called cDCE monooxygenase, in the initial steps of cDCE degradation in JS666.
Cloning and expression of cDCE monooxygenase.
Four genes are annotated as cytochrome P450 in the genome of JS666. Bpro_5301, which was upregulated 3.5-fold by growth on cDCE (13), is located on pPol338, the smaller of two megaplasmids in JS666. Bpro_5301 is adjacent to genes annotated as flavin adenine dinucleotide-dependent pyridine nucleotide disulfide oxidoreductase (Bpro_5300) and ferredoxin (Bpro_5299). The amino acid sequence of the cytochrome P450 is 76% identical to that of alkane hydroxylase (AphG) from Mycobacterium sp. strain HXN-1500, while the amino acid sequences of the oxidoreductase (AphH) and ferredoxin (AphI) are each 66% identical to the sequences from HXN-1500 (12, 37). AphH and AphI of HXN-1500 are the most closely related to the proteins of JS666 of any currently entered in GenBank. The three genes from JS666 were cloned together into E. coli (Fig. 1), and the expression of each component was confirmed by SDS-PAGE (Fig. 5). JS593 catalyzed the transformation of DCA and cDCE at 1.0 and 0.2 nmol/min/mg protein, respectively. No transformation of cDCE or DCA was observed in the absence of oxygen, but transformation was immediate upon the addition of oxygen. E. coli containing the pET vector alone did not catalyze transformation of any of the substrates.
Fig 5.
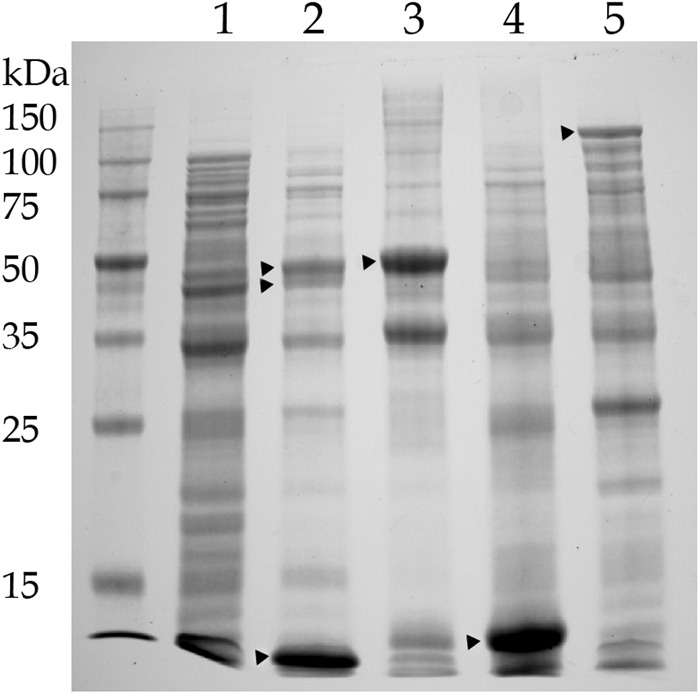
Expression of cytochrome P450. Lane 1, uninduced clone; lane 2, induced clone expressing Bpro_5301 (47.49 kDa), Bpro_5300 (43.18 kDa), and Bpro_5299 (11.29 kDa); lane 3, clone expressing Bpro_5301 with a 6× His tag (48.32 kDa); lane 4, clone expressing Bpro_5299 with a 6× His tag (12.12 kDa); lane 5, clone expressing LacZ (120 kDa).
Products of transformation of cDCE by cDCE monooxygenase.
Resting cells of E. coli JS595 catalyzed the transformation of cDCE to 2,2-dichloroacetaldehyde (up to 88%; mean = 61% ± 12%, n = 12), accompanied by the appearance of an unidentified metabolite in the headspace. In preliminary experiments (Fig. 6), the recovery of 2,2-dichloroacetaldehyde was modest because the compound partitioned to the cells. In subsequent experiments that included multiple extractions with the cells included in the extraction, recovery of dichloroacetaldehyde improved. The rate of transformation of cDCE by JS593 or JS595 rapidly diminished so that it was only a fraction of the initial rate after 30 to 60 min. The production of dichloroacetaldehyde also diminished rapidly, while the unidentified metabolite continued to accumulate, which suggested that it resulted from abiotic transformation of 2,2-dichloroacetaldehyde. When JS666 was grown on cDCE, no dichloroacetaldehyde accumulated and only traces of the unknown metabolite were detected in the headspace (Fig. 6). The unidentified metabolite was never found in extracts of the aqueous phase, nor was it metabolized by JS666. The unidentified metabolite appeared as a major peak by GC-ECD but was undetectable by GC-FID and was barely detectable by GC-mass selection detector (MSD). The mass spectrum (Fig. 6B) did not match that of any of the known metabolites of chloroethenes/ethanes, although the exact mass was 112. Because the mass balance with JS595 cells and cell extracts of JS593 and JS595 indicated that cDCE is predominantly transformed into dichloroacetaldehyde and the wild type accumulates only traces of the unknown metabolite, attempts to identify the unknown were discontinued.
Fig 6.

(A) Degradation of cDCE by JS666 (solid lines and symbols; OD600 ≈ 1) and transformation by JS595 (dashed lines, open symbols; OD600 ≈ 40) and uninoculated control (open circle, dotted line). Circles, cDCE; squares, 2,2-dichloroacetaldehyde; diamonds, unknown metabolite. (B) Mass spectrum of unknown. (C) Mass spectrum of 2, 2-dichloroacetaldehyde.
Traces of cDCE epoxide occasionally accumulated in cDCE-degrading cultures, but lack of a published extinction coefficient prevented quantitation of the epoxide. The results indicate that cDCE monooxygenase specifically transforms cDCE mainly to dichloroacetaldehyde rather than to epoxide. They also indicate that other enzymes in JS666 but not in E. coli prevent the accumulation of dichloroacetaldehyde and the unknown metabolite.
Dichloroacetaldehyde production in cell extracts of JS593 and JS595.
Dichloroacetaldehyde was produced during cDCE transformation by extracts prepared from cells of JS593 or JS595 (Fig. 7). With dialyzed extracts, the reaction took place only when NADH was present in the reaction mixture. When DCA was the substrate, chloroacetaldehyde was produced. The enzyme activity was modest and unstable, but the results were reproducible. The results indicate that the cDCE monooxygenase catalyzes the transformation of both cDCE and DCA to chloroacetaldehydes that can be further degraded in JS666.
Fig 7.

Biotransformation of cDCE in cell extracts of JS595 expressing cDCE monooxygenase. Complete mixtures contained cDCE, phosphate buffer, cell extract, and NADH. Controls lacked either NADH or cell extract. Results are for cDCE in complete mixtures (•), cDCE in controls (■), final dichloroacetaldehyde in complete mixtures (○), and final dichloroacetaldehyde in controls (□). cDCE was allowed to equilibrate for 5 min, and then reactions were initiated by adding NADH.
Dichloroacetaldehyde transformation in cell extracts of JS666.
Dichloroacetaldehyde was degraded at a rate of 15.6 nmol min−1 mg−1 protein in extracts of cells grown on cDCE (Table 2), which is consistent with the rate of cDCE degradation by whole cells (17 nmol min−1 mg−1 protein). The apparent Km of the dichloroacetaldehyde dehydrogenase was 120 ± 12 μM. Genes that encode dichloroacetaldehyde dehydrogenase were not identified experimentally. There are several potential candidates for the genes that encode chloroacetaldehyde dehydrogenase and dichloroacetaldehyde dehydrogenase, but we do not have sufficient evidence from microarray data to establish which ones encode the activities that we measured in cells and cell extracts. The haloacid dehalogenase upregulated by cDCE catalyzes transformation of (di)chloroacetic acid (13, 64), and (di)chloroacetic acid was transformed in cell extracts of JS666 (Table 2). The results indicate that cDCE degradation via dichloroacetaldehyde is the productive pathway in JS666.
Table 2.
Enzyme activities measured in cell extract of Polaromonas sp. JS666
| Enzyme assayed | Substrate | Sp acta (nmol min−1 mg−1 protein) |
|---|---|---|
| Aldehyde dehydrogenase | Dichloroacetaldehyde | 15.6 ± 0.1 |
| Chloroacetaldehyde | 24.8 ± 5.2 | |
| Haloacid dehalogenase | Dichloroacetate | 15.0 ± 2.8 |
| Chloroacetate | 10.3 ± 1.6 |
Data represent means and standard deviations from at least two measurements.
Transformation of alkanes by cDCE monooxygenase.
Bpro_5301 is most closely related to a gene encoding a medium-chain alkane hydroxylase and was previously shown to enable the catalysis of n-alkanes to primary alcohols (38). Various alkanes were transformed by E. coli JS595 (Table 3). The rate of transformation of cDCE was an order of magnitude faster than the rates of transformation of the alkanes. The primary alcohols expected as the products of the alkane transformation (38, 39) were detected in SPE extracts of all cultures by GC-MS and were the only products of hexane and cyclohexane transformation. 2-Octanol and 2-octanone were detected in octane transformations along with the major 1-octanol peak. Perillyl alcohol was the major product of limonene transformation but was accompanied by two other unidentified peaks. The results indicate that although cDCE monooxygenase can still transform n-alkanes, it appears to be specialized for cDCE.
Table 3.
Transformation of alkanes by cytochrome P450a
| Substrate | Product(s) | Sp act (nmol min−1 mg−1 protein) |
|
|---|---|---|---|
| Replicate 1 | Replicate 2 | ||
| cDCE | Dichloroacetaldehyde | 0.39 | 0.26 |
| Hexane | 1-Hexanol | 0.005 | 0.01 |
| Cyclohexane | Cyclohexanol | 0.07 | 0.10 |
| Octane | 1-Octanol, 2-octanol, 2-octanone | 0.07 | 0.06 |
| Limonene | Perillyl alcohol, 2 unidentified peaks | 0.06 | 0.08 |
E. coli JS595 was provided with cDCE or alkane. Products were extracted and analyzed by GC-MS.
DISCUSSION
Several lines of evidence indicate that cDCE monooxygenase is responsible for the initial steps in cDCE biodegradation in JS666. cDCE was degraded only in the presence of oxygen, degradation was inhibited by cytochrome P450-specific inhibitors, heterologously expressed cytochrome P450 monooxygenase catalyzes the transformation of cDCE to dichloroacetaldehyde, and Bpro_5301 is upregulated by cDCE (13). The transient accumulation of dichloroacetaldehyde in cDCE-degrading cultures and the dichloroacetaldehyde dehydrogenase activities in cell extracts of JS666 support the proposed pathway (Fig. 8). Transformation of dichloroacetaldehyde and dichloroacetate in cell extracts of JS666 (Table 2) supports a downstream pathway of cDCE degradation similar to that of 1,2-dichloroethane (Fig. 8). Chloroglycolate would be expected to spontaneously dechlorinate to glyoxalate (40). The ability of JS666 to metabolize glyoxalate is inferred from the growth of JS666 on glycolate and the presence of putative glyoxalate pathway genes encoding isocitrate lyase (Bpro_2101) and malate synthase (Bpro_4517).
Fig 8.

Proposed cDCE and DCA degradation pathways. Solid arrows, major pathways supported by the results of this study; dashed arrows, potential minor pathways.
cDCE monooxygenase is also responsible for initiation of DCA degradation. Genes that encode the enzymes of the DCA degradation pathway are expressed in cells of JS666 grown on cDCE (13, 24), and clones that catalyzed the transformation of cDCE also catalyzed the transformation of DCA. The inhibition of DCA degradation by metyrapone in cells grown on DCA, the transformation of DCA to chloroacetaldehyde by cDCE monooxygenase, and the failure to transform DCA in the absence of oxygen support the hypothesis that strain JS666 degrades DCA by an oxidative pathway (Fig. 8).
JS666 could grow only on low concentrations of DCA (100 to 200 μM), which is consistent with previous studies indicating that JS666 could not grow on 200 μM to 10 mM DCA (9, 12). Although the same genes are responsible for cDCE and DCA degradation, growth on DCA is limited to concentrations an order of magnitude less than concentrations that support growth on cDCE. A contributing factor possibly lies in the greater solubility of DCA in the aqueous phase. It is also possible that chloroacetaldehyde is more toxic to JS666 than dichloroacetaldehyde.
The degradation of DCA has been most extensively studied in Xanthobacter autotrophicus GJ10 (24). The strain constitutively expresses two different dehalogenases; however, the hydrolytic pathway (Fig. 2, pathway II) in strain GJ10 is inconsistent with the results from JS666. Pseudomonas sp. strain DE2 (41) and Pseudomonas sp. strain DCA1 (31) degrade DCA by the oxidative pathway proposed for degradation of DCA by JS666 (Fig. 8). The monooxygenase from DE2 and DCA1 has not been identified. It seems likely that the reaction might be catalyzed by a P450 monooxygenase similar to the cDCE monooxygenase. Sequence information is not available for strain DE2 or DCA1, so the relatedness of the enzymes is unknown. It is known, however, that strain DCA1 transforms cDCE after growth on DCA (31).
Monooxygenation of cDCE and DCA by the same enzyme in JS666 thus accounts for the results of the simultaneous adaptation studies as well as for why cDCE degradation is induced in DCA-grown cells. The regulation of the metabolic pathway, however, is not understood. It seems likely that the regulation is in the early stages of evolution, as evidenced by long and unpredictable acclimation periods when cells are switched from more favored substrates to cDCE.
Cytochrome P450-catalyzed oxidation of chlorinated ethenes can yield multiple products. The formation of chloroacetaldehydes during the oxidation of 1,1-dichloroethene (42) and trichloroethene (32) by cytochromes P450 from rat liver microsomes has been reported. A transient chloroethene intermediate bound in the cytochrome P450 can be partitioned to epoxide, a chloroacetaldehyde via chloride migration, and a dead-end product due to an irreversible porphyrin N-alkylation (32, 43, 44). Theoretical studies on terminal olefin oxidation by cytochrome P450 suggest that different environments in the protein pocket of the enzyme may lead to different product ratios of epoxides, aldehydes, and suicide products (44). The mass balance derived from the studies with intact cells of JS595 suggests that cDCE monooxygenase in JS666 is specialized to favor dichloroacetaldehyde rather than epoxide production from cDCE.
Chloroacetaldehyde and dichloroacetaldehyde react with DNA (45), tRNA (46), and proteins (47), forming adducts of uncertain toxicity (48). Neither dichloroacetaldehyde nor epoxide was detected in JS666 cultures that were grown continuously on cDCE. Dichloroacetaldehyde was transiently detected only during the brief lag phase following transfer and dilution of cDCE-grown cultures into fresh medium. The simultaneous induction studies showed that dichloroacetaldehyde and/or chloroacetaldehyde dehydrogenase has high constitutive activity that is upregulated to higher activities by growth on cDCE and DCA (Table 1). High activity of the dehydrogenase would protect the cells from the effects of toxic intermediates (49) and channel the carbon into a productive pathway. To our knowledge, this is the first report of a productive pathway resulting from the P450-catalyzed conversion of an alkene to an aldehyde rather than an epoxide.
The rapid loss of activity in E. coli clones expressing the P450 complex is reminiscent of product toxicity to methane monooxygenase when oxygenated compounds are produced during transformation of TCE (epoxide) and chloroform (phosgene) (15, 50). For JS593 and JS595, the result is probably due to the toxicity of dichloroacetaldehyde. Chloroacetaldehyde (51) and dichloroacetaldehyde (52) react with free sulfhydryl groups, particularly with cysteine and glutathione. Cytochromes P450 have a critical cysteine ligand in the active site that binds and orients the heme moiety in the Cys pocket (53). Four cysteines act as the ligands that bind iron to ferredoxin/rubredoxin (54). When JS666 is grown on cDCE, a glutathione-S-transferase is upregulated over 100-fold (13). This, in addition to the high constitutive activity of chloroacetaldehyde/dichloroacetaldehyde dehydrogenase, would help to protect the cytochrome P450 complex from damage by dichloroacetaldehyde (55), but neither protection is available to the heterologously expressed proteins. Because dichloroacetaldehyde and chloroacetaldehyde are converted to hydrates in aqueous solutions (56), it was not possible to test the inactivation of the P450 enzyme with exogenous (di)chloroacetaldehyde. Addition of the hydrates had no effect on enzyme activity.
Conflicting carbon isotope fractionation results during cDCE degradation by JS666 suggested two different initial steps of cDCE degradation. Abe et al. (57) suggested the epoxidation of cDCE as the initial step of cDCE degradation by JS666 on the basis of the typical carbon isotope fractionation (−8.5‰) for epoxidation and a small chlorine isotope fractionation (−0.3‰). In contrast, the cleavage of a C—Cl bond was suggested by Jennings (58) to be the initial step of cDCE degradation by JS666 on the basis of the large carbon isotope fractionation (−17.4 to −22.4‰) during cDCE degradation. The cytochrome P450-catalyzed mechanism proposed here would be consistent with the latter results. Future clarification of the reaction mechanism and the factors controlling fractionation with purified enzymes will enable application of isotope fractionation to monitor aerobic cDCE degradation in the environment (59).
The presence of multiple pathways for cDCE degradation was suggested by two distinguishable modes of cDCE degradation by JS666 (13). One is faster and supports growth of JS666, whereas the other is slower, seems to be cometabolic, and probably involves some misrouting of intermediates. It seems likely that both pathways operate simultaneously in growing cells. Transcriptomics and proteomics indicate that cDCE causes upregulation of several genes that are candidates for encoding the competing initial reactions (13). Transcriptomics previously indicated that cyclohexanone monooxygenase is upregulated in JS666 grown on cDCE (13), and there is modest consumption of cyclohexanone by cDCE-grown cells (Table 1); however, the enzyme does not appear to be directly involved in cDCE degradation (65). Three lines of evidence support this conclusion. (i) There is no detectable oxygen uptake when DCA or cDCE is provided to cyclohexanone-grown cells, (ii) there is a 2- to 3-day lag period before resumption of growth upon switching between cDCE and cyclohexanone (in either direction) as growth substrates, and (iii) the simultaneous presence of cDCE and cyclohexanone in cultures of JS666 grown on cDCE reduces the rate of consumption of cDCE. cDCE degradation via dichloroacetaldehyde seems to be the major pathway, on the basis of the results presented here. The roles of cyclohexanone monooxygenase and other genes upregulated during growth on cDCE are under investigation.
The reductase and ferredoxin that constitute the electron transport chain for the cDCE monooxygenase are most closely related to the corresponding proteins involved in hexane and limonene monooxygenation in Mycobacterium sp. strain HXN-1500 (37). The cytochrome P450 from JS666 is slightly more closely related to a cytochrome P450 from Mycobacterium austroafricanum IFP 2173 that is upregulated in the presence of the branched alkane 2-ethyl hexyl nitrate. The HXN-1500 and IFP 2173 cytochrome P450 amino acid sequences are identical in 418/420 sites. The genes in JS666 are in the same orientation and order as those in HXN-1500. The context of the cytochrome P450 gene from M. austroafricanum is unknown. Cytochrome P450 alkane hydroxylases are among several alkane degradation systems (60) that catalyze the transformation of medium-chain-length alkanes (C6 to C10) to alkanols, which are further degraded to fatty acids by alcohol dehydrogenase (AlkJ), aldehyde dehydrogenase (AlkH), and acyl coenzyme A synthetase (AlkK). In JS666, putative alkKJH and pyrroloquinolinequinone (PQQ)-dependent dehydrogenase genes are located upstream of the cytochrome P450 monooxygenase genes, which is consistent with the growth of JS666 on heptane and octane (12). Mattes et al. suggest that the gene cluster was recently recruited from alkane-assimilating bacteria on the basis of the high G+C content, the association with several transposase genes, and the gene organization (12). Mattes et al. reported about 10-fold faster growth of JS666 on cDCE than on octane and JS595 transformed cDCE and DCA 4 to 6 times faster than octane (12). Purified enzymes catalyzed the transformation of octane to 1-octanol at a rate of 0.6 nmol min−1 mg−1 protein (38), only 10-fold faster than cells of JS595 (Table 3). The evidence suggests that although JS666 can still grow on n-alkanes, the enzymes and organism appear to be better adapted for growth on chlorinated alkenes/alkanes than on n-alkanes.
Instability and loss by JS666 of the ability to degrade cDCE under nonselective conditions (13), along with the lack of a clear organization of the genes involved, suggest that the system was recently assembled and sustained at the field site in response to contamination by cDCE. The most likely scenario for the assembly of the pathway would involve the recruitment and expression of the genes encoding the cytochrome P450 monooxygenase, which would allow the relatively common 1,2-dichloroethane-degrading bacteria (24, 31) to grow with cDCE as the sole source of carbon and energy. Genes of a 1,2-dichloroethane catabolic pathway might have been recruited for dichloroacetaldehyde degradation. Homologs of the cytochrome P450 monooxygenase seem to be readily available in soil, considering the widespread distribution of CYP153A alkane hydroxylases (61). Similar scenarios for assembly of chlorobenzene (62) and diphenylamine (63) degradation pathways by recruitment of genes from disparate pathways were strongly supported. Alternatively, a preexisting pathway for DCA degradation might employ a cytochrome P450 monooxygenase to catalyze the initial step. The presence of cDCE might then select for variants of the P450 that could catalyze the conversion of cDCE to dichloroacetaldehyde. Screening for cytochrome P450 monooxygenase genes in 1,2-dichloroethane-degrading bacteria will provide more insight about the evolution of the cDCE degradation pathway.
Uncertainty about the cDCE degradation pathway and intermediates has been a deterrent to the use of strain JS666 to remediate sites contaminated with cDCE and to the assessment of cDCE degradation in the environment. The determination of the catabolic pathway and genes that encode the enzymes involved provides the basis to detect, predict, and enhance cDCE degradation in the environment as well as increase our understanding of bacterial evolution of catabolic pathways for synthetic chemicals.
ACKNOWLEDGMENTS
This work was supported by the Strategic Environmental Research and Development Program (SERDP ER-1557), Geosyntec Consultants, and the DuPont Corporate Remediation Group.
We thank Evan Cox (Geoxyntec Consultants) for patient management and support of this project. We thank Sarah Craven for many helpful discussions and assistance with clone expression. We thank Barbara Sherwood Lollar and Scott Mundle (Department of Geology, University of Toronto), Charlene Bayer (Georgia Tech Research Institute), and F. P. Guengerich (Vanderbilt University) for helpful discussions.
Footnotes
Published ahead of print 25 January 2013
REFERENCES
- 1.Agency for Toxic Substances and Disease Registry 2011. ATSDR 2011 substance priority list. Agency for Toxic Substances and Disease Registry, U.S. Department of Health and Human Services, Atlanta, GA [Google Scholar]
- 2.Maymó-Gatell X, Chien Y, Gossett JM, Zinder SH. 1997. Isolation of a bacterium that reductively dechlorinates tetrachloroethene to ethene. Science 276:1568–1571 [DOI] [PubMed] [Google Scholar]
- 3.Mohn WW, Tiedje JM. 1992. Microbial reductive dehalogenation. Microbiol. Rev. 56:482–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coleman NV, Mattes TE, Gossett JM, Spain JC. 2002. Phylogenetic and kinetic diversity of aerobic vinyl chloride-assimilating bacteria from contaminated sites. Appl. Environ. Microbiol. 68:6162–6171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coleman NV, Spain JC. 2003. Epoxyalkane:coenzyme M transferase in the ethene and vinyl chloride biodegradation pathways of Mycobacterium strain JS60. J. Bacteriol. 185:5536–5545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Allen JR, Clark DD, Krum JG, Ensign SA. 1999. A role for coenzyme M (2-mercaptoethanesulfonic acid) in a bacterial pathway of aliphatic epoxide carboxylation. Proc. Natl. Acad. Sci. U. S. A. 96:8432–8437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krum JG, Ensign SA. 2001. Evidence that a linear megaplasmid encodes enzymes of aliphatic alkene and epoxide metabolism and coenzyme M (2-mercaptoethanesulfonate) biosynthesis in Xanthobacter strain Py2. J. Bacteriol. 183:2172–2177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mattes TE, Coleman NV, Spain JC, Gossett JM. 2005. Physiological and molecular genetic analyses of vinyl chloride and ethene biodegradation in Nocardioides sp. strain JS614. Arch. Microbiol. 183:95–106 [DOI] [PubMed] [Google Scholar]
- 9.Coleman NV, Mattes TE, Gossett JM, Spain JC. 2002. Biodegradation of cis-dichloroethene as the sole carbon source by a β-proteobacterium. Appl. Environ. Microbiol. 68:2726–2730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fennell DE, Carroll AB, Gossett JM, Zinder SH. 2001. Assessment of indigenous reductive dechlorinating potential at a TCE-contaminated site using microcosms, polymerase chain reaction analysis, and site data. Environ. Sci. Technol. 35:1830–1839 [DOI] [PubMed] [Google Scholar]
- 11.Yang Y, McCarty PL. 2002. Comparison between donor substrates for biologically enhanced tetrachloroethene DNAPL dissolution. Environ. Sci. Technol. 36:3400–3404 [DOI] [PubMed] [Google Scholar]
- 12.Mattes TE, Alexander AK, Richardson PM, Munk AC, Han CS, Stothard P, Coleman NV. 2008. The genome of Polaromonas sp. strain JS666: insights into the evolution of a hydrocarbon- and xenobiotic-degrading bacterium, and features of relevance to biotechnology. Appl. Environ. Microbiol. 74:6405–6416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jennings LK, Chartrand MMG, Lacrampe-Couloume G, Lollar BS, Spain JC, Gossett JM. 2009. Proteomic and transcriptomic analyses reveal genes upregulated by cis-dichloroethene in Polaromonas sp. strain JS666. Appl. Environ. Microbiol. 75:3733–3744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ensign SA, Hyman MR, Arp DJ. 1992. Cometabolic degradation of chlorinated alkenes by alkene monooxygenase in a propylene-grown Xanthobacter strain. Appl. Environ. Microbiol. 58:3038–3046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fox BG, Borneman JG, Wackett LP, Lipscomb JD. 1990. Haloalkene oxidation by the soluble methane monooxygenase from Methylosinus trichosporium OB3b: mechanistic and environmental implications. Biochemistry 29:6419–6427 [DOI] [PubMed] [Google Scholar]
- 16.Koziollek P, Bryniok D, Knackmuss H-J. 1999. Ethene as an auxiliary substrate for the cooxidation of cis-1,2-ethene and vinyl chloride. Arch. Microbiol. 172:240–246 [DOI] [PubMed] [Google Scholar]
- 17.Canada KA, Iwashita S, Shim H, Wood TK. 2002. Directed evolution of toluene ortho-monooxygenase for enhanced 1-naphthol synthesis and chlorinated ethene degradation. J. Bacteriol. 184:344–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rui L, Cao L, Chen W, Reardon KF, Wood TK. 2004. Active site engineering of the epoxide hydrolase from Agrobacterium radiobacter AD1 to enhance aerobic mineralization of cis-1,2-dichloroethylene in cells expressing an evolved toluene ortho-monooxygenase. J. Biol. Chem. 279:46810–46817 [DOI] [PubMed] [Google Scholar]
- 19.Lee J, Cao L, Ow SY, Barrios-Llerena ME, Chen W, Wood TK, Wright PC. 2006. Proteome changes after metabolic engineering to enhance aerobic mineralization of cis-1,2-dichloroethylene. J. Proteome Res. 5:1388–1397 [DOI] [PubMed] [Google Scholar]
- 20.Guengerich FP. 2007. Mechanisms of cytochrome P450 substrate oxidation: MiniReview. J. Biochem. Mol. Toxicol. 21:163–168 [DOI] [PubMed] [Google Scholar]
- 21.Stanier RY, Palleroni NJ, Doudoroff M. 1966. The aerobic pseudomonads: a taxonomic study. J. Gen. Microbiol. 43:159–271 [DOI] [PubMed] [Google Scholar]
- 22.Gillam EMJ, Guo Z, Martin MV, Jenkins CM, Guengerich FP. 1995. Expression of cytochrome P450 2D6 in Escherichia coli, purification, and spectral and catalytic characterization. Arch. Biochem. Biophys. 319:540–550 [DOI] [PubMed] [Google Scholar]
- 23.van der Ploeg J, Smidt MP, Landa AS, Janssen DB. 1994. Identification of chloroacetaldehyde dehydrogenase involved in 1,2-dichloroethane degradation. Appl. Environ. Microbiol. 60:1599–1605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Janssen DB, Scheper A, Dijkhuizen L, Witholt B. 1985. Degradation of halogenated aliphatic compounds by Xanthobacter autotrophicus GJ10. Appl. Environ. Microbiol. 49:673–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carr CS, Hughes JB. 1998. Enrichment of high-rate PCE dechlorination and comparative study of lactate, methanol, and hydrogen as electron donors to sustain activity. Environ. Sci. Technol. 32:1817–1824 [Google Scholar]
- 26.Bergmann JG, Sanik J., Jr 1957. Determination of trace amounts of chlorine in naphtha. Anal. Chem. 29:241–242 [Google Scholar]
- 27.Nakajima K, Ohta K, Mostefaoui TA, Chai W, Utsukihara T, Horiuchi CA, Murakami M. 2007. Glyoxal sample preparation for high-performance liquid chromatographic detection of 2,4-dinitro-phenylhydrazone derivative: suppression of polymerization and mono-derivative formation by using methanol medium. J. Chromatogr. A 1161:338–341 [DOI] [PubMed] [Google Scholar]
- 28.Wakasugi T, Miyakawa T, Suzuki F, Itsuno S, Ito K. 1993. Preparation of dichloroacetaldehyde cyclic trimer and its depolymerization. Synth. Commun. 23:1289–1294 [Google Scholar]
- 29.Verce MF, Gunsch CK, Danko AS, Freedman DL. 2002. Cometabolism of cis-1,2-dichloroethene by aerobic cultures grown on vinyl chloride as the primary substrate. Environ. Sci. Technol. 36:2171–2177 [DOI] [PubMed] [Google Scholar]
- 30.Janssen DB, Grobben G, Hoekstra R, Oldenhuis R, Witholt B. 1988. Degradation of trans-1,2-dichloroethene by mixed and pure cultures of methanotrophic bacteria. Appl. Microbiol. Biotechnol. 29:392–399 [Google Scholar]
- 31.Hage JC, Hartmans S. 1999. Monooxygenase-mediated 1,2-dichloroethane degradation by Pseudomonas sp. strain DCA1. Appl. Environ. Microbiol. 65:2466–2470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miller RE, Guengerich FP. 1982. Oxidation of trichloroethylene by liver microsomal cytochrome P-450: evidence for chlorine migration in a transition state not involving trichloroethylene oxide. Biochemistry 21:1090–1097 [DOI] [PubMed] [Google Scholar]
- 33.Smidt H, de Vos WM. 2004. Anaerobic microbial dehalogenation. Annu. Rev. Microbiol. 58:43–73 [DOI] [PubMed] [Google Scholar]
- 34.Stanier R. 1947. Simultaneous adaptation: a new technique for the study of metabolic pathways. J. Bacteriol. 54:339–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bhushan B, Trott S, Spain JC, Halasz A, Paquet L, Hawari J. 2003. Biotransformation of hexahydro-1,3,5-trinitro-1,3,5-triazine (RDX) by a rabbit liver cytochrome P450: insight into the mechanism of RDX biodegradation by Rhodococcus sp. strain DN22. Appl. Environ. Microbiol. 69:1347–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raag R, Swanson BA, Poulos TL, Ortiz de Montellano PR. 1990. Formation, crystal structure, and rearrangement of a cytochrome P-450cam iron-phenyl complex. Biochemistry 29:8119–8126 [DOI] [PubMed] [Google Scholar]
- 37.van Beilen JB, Holtackers R, Luscher D, Bauer U, Witholt B, Duetz WA. 2005. Biocatalytic production of perillyl alcohol from limonene by using a novel Mycobacterium sp. cytochrome P450 alkane hydroxylase expressed in Pseudomonas putida. Appl. Environ. Microbiol. 71:1737–1744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scheps D, Honda Malca S, Hoffmann H, Nestl BM, Hauer B. 2011. Regioselective ω-hydroxylation of medium-chain n-alkanes and primary alcohols by CYP153 enzymes from Mycobacterium marinum and Polaromonas sp. strain JS666. Org. Biomol. Chem. 9:6727–6733 [DOI] [PubMed] [Google Scholar]
- 39.van Beilen JB, Funhoff EG, van Loon A, Just A, Kaysser L, Bouza M, Holtackers R, Rothlisberger M, Li Z, Witholt B. 2006. Cytochrome P450 alkane hydroxylases of the CYP153 family are common in alkane-degrading eubacteria lacking integral membrane alkane hydroxylases. Appl. Environ. Microbiol. 72:59–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reineke W. 2001. Aerobic and anaerobic biodegradation potentials of microorganisms, p 1–161 Beek B. (ed), The handbook of environmental chemistry: biodegradation and persistence, vol 2, part K Springer-Verlag, Berlin, Germany [Google Scholar]
- 41.Stucki G, Krebser U, Leisinger T. 1983. Bacterial growth on 1,2-dichloroethane. Experientia 39:1271–1273 [DOI] [PubMed] [Google Scholar]
- 42.Meunier B, de Visser SP, Shaik S. 2004. Mechanism of oxidation reactions catalyzed by cytochrome P450 enzymes. Chem. Rev. 104:3947–3980 [DOI] [PubMed] [Google Scholar]
- 43.Liebler DC, Guengerich FP. 1983. Olefin oxidation by cytochrome P-450: evidence for group migration in catalytic intermediates formed with vinylidene chloride and trans-1-phenyl-1-butene. Biochemistry 22:5482–5489 [DOI] [PubMed] [Google Scholar]
- 44.de Visser SP, Kumar D, Shaik S. 2004. How do aldehyde side products occur during alkene epoxidation by cytochrome P450? Theory reveals a state-specific multi-state scenario where the high-spin component leads to all side products. J. Inorg. Biochem. 98:1183–1193 [DOI] [PubMed] [Google Scholar]
- 45.Kohwi Y, Kohwi-Shigematsu T. 1988. Magnesium ion-dependent triple-helix structure formed by homopurine-homopyrimidine sequences in supercoiled plasmid DNA. Proc. Natl. Acad. Sci. U. S. A. 85:3781–3785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Biemat J, Ciesiołka J, Gornicki P, Adamiak RW, Krzyzosiak W, Wiewiórowski M. 1978. New observations concerning the chloroacetaldehyde reaction with some tRNA constituents. Stable intermediates, kinetics and selectivity of the reaction. Nucleic Acids Res. 5:789–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bond T, Henriet O, Goslan EH, Parsons SA, Jefferson B. 2009. Disinfection byproduct formation and fractionation behavior of natural organic matter surrogates. Environ. Sci. Technol. 43:5982–5989 [DOI] [PubMed] [Google Scholar]
- 48.European Food Safety Authority 2006. Opinion of the Scientific Panel on Plant Health, Plant Protection Products and Their Residues on a request from EFSA related to the evaluation of dichlorvos in the context of Council Directive 91/414/EEC. EFSA J. 343:1–45 [Google Scholar]
- 49.Reineke W, Knackmuss H-J. 1984. Microbial metabolism of haloaromatics: isolation and properties of a chlorobenzene-degrading bacterium. Appl. Environ. Microbiol. 47:395–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alvarez-Cohen L, McCarty PL. 1991. Product toxicity and cometabolic competitive inhibition modeling of chloroform and trichloroethylene transformation by methanotrophic resting cells. Appl. Environ. Microbiol. 57:1031–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schwerdt G, Kirchhoff A, Freudinger R, Wollny B, Benesic A, Gekle M. 2007. Mesna or cysteine prevents chloroacetaldehyde-induced cell death of human proximal tubule cells. Pediatr. Nephrol. 22:798–803 [DOI] [PubMed] [Google Scholar]
- 52.Liebler DC, Meredith MJ, Guengerich FP. 1985. Formation of glutathione conjugates by reactive metabolites of vinylidene chloride in microsomes and isolated hepatocytes. Cancer Res. 45:186–193 [PubMed] [Google Scholar]
- 53.Hasemann CA, Kurumbail RG, Boddupalli SS, Peterson JA, Deisenhofer J. 1995. Structure and function of cytochromes P450: a comparative analysis of three crystal structures. Structure 3:41–62 [DOI] [PubMed] [Google Scholar]
- 54.van Beilen JB, Neuenschwander M, Smits THM, Roth C, Balada SB, Witholt B. 2002. Rubredoxins involved in alkane oxidation. J. Bacteriol. 184:1722–1732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dowsley TF, Forkert P-G, Benesch LA, Bolton JL. 1995. Reaction of glutathione with the electrophilic metabolites of 1,1-dichloroethylene. Chem. Biol. Interact. 95:227–244 [DOI] [PubMed] [Google Scholar]
- 56.Elmore JD, Wong JL, Laumbach AD, Streips UN. 1976. Vinyl chloride mutagenicity via the metabolites chlorooxirane and chloroacetaldehyde monomer hydrate. Biochim. Biophys. Acta 442:405–419 [DOI] [PubMed] [Google Scholar]
- 57.Abe Y, Aravena R, Zopfi J, Shouakar-Stash O, Cox E, Roberts JD, Hunkeler D. 2009. Carbon and chlorine isotope fractionation during aerobic oxidation and reductive dechlorination of vinyl chloride and cis-1,2-dichloroethene. Environ. Sci. Technol. 43:101–107. [DOI] [PubMed] [Google Scholar]
- 58.Jennings LK. 2009. Proteomic and transcriptomic analyses reveal genes upregulated by cis-dichloroethylene in Polaromonas JS666. Cornell University, Ithaca, NY: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schmidt KR, Augenstein T, Heidinger M, Ertl S, Tiehm A. 2010. Aerobic biodegradation of cis-1,2-dichloroethene as sole carbon source: stable carbon isotope fractionation and growth characteristics. Chemosphere 78:527–532 [DOI] [PubMed] [Google Scholar]
- 60.Van Beilen JB, Li Z, Duetz WA, Smits THM, Witholt B. 2003. Diversity of alkane hydroxylase systems in the environment. Oil Gas Sci. Technol. 58:427–440 [Google Scholar]
- 61.Wang L, Wang W, Lai Q, Shao Z. 2010. Gene diversity of CYP153A and AlkB alkane hydroxylases in oil-degrading bacteria isolated from the Atlantic Ocean. Environ. Microbiol. 12:1230–1242 [DOI] [PubMed] [Google Scholar]
- 62.Muller TA, Werlen C, Spain J, van der Meer JR. 2003. Evolution of a chlorobenzene degradative pathway among bacteria in a contaminated groundwater mediated by a genomic island in Ralstonia. Environ. Microbiol. 5:163–173 [DOI] [PubMed] [Google Scholar]
- 63.Shin KA, Spain JC. 2009. Pathway and evolutionary implications of diphenylamine biodegradation by Burkholderia sp. strain JS667. Appl. Environ. Microbiol. 75:2694–2704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chan WY, Wong M, Guthrie J, Savchenko AV, Yakunin AF, Pai EF, Edwards EA. 2010. Sequence- and activity-based screening of microbial genomes for novel dehalogenases. Microb. Biotechnol. 3:107–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Alexander AK, Biedermann D, Fink MJ, Mihovilovic MD, Mattes TE. 2012. Enantioselective oxidation by a cyclohexanone monooxygenase from the xenobiotic-degrading Polaromonas sp. strain JS666. J. Mol. Catal. B Enzym. 78:105–110 [Google Scholar]