

The function of the heart is to pump blood through the pulmonary and systemic vasculature. To do this, the atria and ventricles must contract in a precise sequence in response to a conducted electrical signal, the action potential (AP).
The cardiac AP originates in the sinoatrial node and is conducted to the atria and ventricles through electrical synapses: the gap junctions. The AP morphology varies with species, heart rate, location within the heart, developmental stage, and in response to neurohormones and drugs. Unlike the brief APs of skeletal muscle and neurons, which typically last ≈3-5 ms, the cardiac action potential is 100’s of milliseconds long and has five distinct phases (Figure 1). One of these components, sometimes considered one of little importance, the “rapid repolarization” period (also called “phase 1”) is the focus of a clever paper presented in the current issue of Journal of Molecular and Cellular Cardiology [1].
Figure 1.
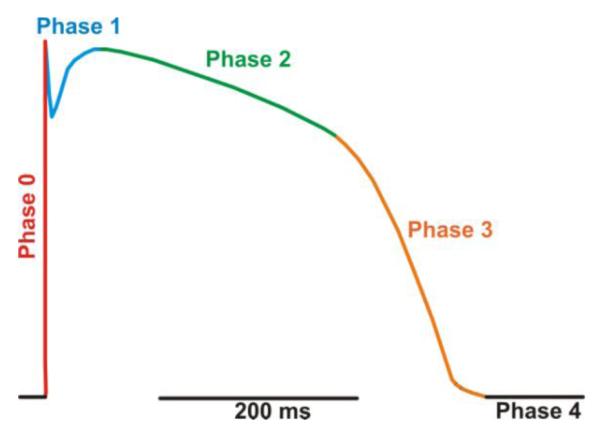
A typical action potential (AP) waveform of a healthy adult human ventricular myocyte (modified from Cooper et al. [1]).
In Fig. 1, we use a color-coded human AP to illustrate its five phases. Phase 0 (red) is the leading edge of the AP and corresponds to the period of rapid depolarization (depolarization rate ≈ 250 V/s) from the diastolic membrane potential of ≈ −90 mV to +50 mV. It is produced primarily by the activation of cardiac Na+ channels with a contribution from cardiac Ca2+ channels. Phase 1 (blue), also referred as the “notch”, is produced by termination of the depolarizing inward Na+ current due to inactivation and the concomitant activation of the transient outward K+ (Ito) and Na+/Ca2+ exchanger (INCX) currents. This results in a transient repolarization period (i.e. the notch) as the membrane potential goes from ≈ +50 to +30 mV (or more negative). Phase 2 (green) is the long (hundreds of milliseconds) plateau phase of the action potential during which the membrane potential changes little. This is produced by the balance of small, but non-inactivated Na+ and L-type Ca2+ current (ICa) components and hyperpolarizing K+ currents. In phase 3 (orange), reductions of Ca2+ and Na+ currents and increasing K+ current contribute to the repolarization of the myocyte. Towards the end of phase 3 the repolarizing actions of these K+ currents are opposed by an inward INCX produced by the Na+/Ca2+ exchanger operating in its Ca2+ extrusion mode. Phase 4 (black) is the diastolic membrane potential of quiescent ventricular myocytes of (≈ −90 mV). The detailed molecular identities of the membrane currents that contribute to each phase depend on the cardiac tissue type and anatomic location of the tissue as well as the specie and developmental age of the animal [2]. Similarly, the exact AP morphology depends on these same variables.
The coupling of cardiac membrane depolarization to Ca2+ release from the sarcoplasmic reticulum (SR) in myocytes (i.e. excitation-contraction (EC) coupling) takes place during the initial phases of the action potential when the L-type Ca2+ channels open. EC coupling is dependent on the positioning of the L-type Ca2+ channels within 15 nm of the junctional sarcoplasmic reticulum (jSR) to form a functional unit called a “couplon” [3]. The Ca2+ influx activates the nearby cluster of SR Ca2+ release channels in the couplon, the ryanodine receptors (RyRs) to produce Ca2+ sparks [4]. Synchronization of the Ca2+ sparks during the AP produces the cell-wide [Ca2+]i transient that activates contraction. Voltage and Ca2+-dependent inactivation rapidly terminate ICa. Because of the exquisite nano-anatomy of the couplon, the opening of single or small cluster of L-type Ca2+ channels could activate a Ca2+ spark [5-7]. This central component of EC coupling may thus link changes in AP morphology to EC coupling and the [Ca2+]i transient.
Recent studies suggest that modification of the ventricular action potential is a critical early step in the chain of events linked to the development hypertrophy and heart failure [8-12]. Indeed, changes in the AP waveform can be observed as early as 48 hours after myocardial insult, even in the absence of measurable cardiac hypertrophy. These changes include reductions in the rate of depolarization and peak depolarization during phase 0, a decrease in the repolarization during phase 1 (or even complete loss of the notch), and an increase in the duration of the action potential.
Decreased [Ca2+]i transients and contractility are also a hallmark of pathological hypertrophy and heart failure. The hypothesized mechanisms underlying these changes in function can be subdivided in two general categories. The first involves a decrease in the coupling strength between L-type Ca2+ channels and ryanodine receptors [13]. The second mechanism involves decreased Ca2+ storage in the sarcoplasmic reticulum. The later mechanism is likely due to decreased SERCA function and enhanced Na+/Ca2+ exchanger expression.
In the current issue of the Journal of Molecular and Cellular Cardiology, an article from Mark Cannell’s laboratory [1] revisited the following important question: what is the relationship between changes in action potential waveform and EC coupling during heart failure? In this study, they focused on the importance of the loss of the notch during phase 1 of the human cardiac AP during heart failure (HF) with respect to the magnitude and the kinetics of local and global [Ca2+]i transients. Although the functional importance of phase 1 of the action potential on EC coupling has been examined before under physiological [14] and pathological [15] conditions, the elegant study by Cooper et al. [1] provides novel mechanistic insights into the mechanisms underlying changes in EC coupling during HF.
By voltage clamping single cardiac myocytes from rat and rabbit using both normal (notched AP) and HF human AP (without notches), the authors show that EC coupling is significantly impaired during a failing AP. ICa during a failing AP shows a reduced peak amplitude and slower inactivation and the cell-wide (global) [Ca2+]i transient also shows reduced peak amplitude and slower rise-time. The authors show that the observed change in ICa and [Ca2+]i transient amplitude is not caused by action potential prolongation during a failing AP. By varying an artificially simulated notch voltage while fixing the action potential duration, the authors show that both, peak and integrated ICa, as well as [Ca2+]i transient amplitude decrease with decreased notch repolarization.
Using confocal microscopy, the authors further show how EC coupling is impaired when a failing AP causes spatial heterogeneity in Ca2+ transient and Ca2+ “spike” activation [16]. Due to impaired E-C coupling, the SR load is increased, which the authors demonstrate when a Ca2+ transient invoked by a normal AP is larger in amplitude when it is immediately preceded by an AP with HF morphology. Increased SR Ca2+ can lead to increased risk of arrhythmogenesis [17].
The recent work by Dong et al [18] is at odds with the studies by Cooper et al. [1], Sah et al. [14], and Harris et al. [15]. Dong et al. used the dynamic clamp to quantitatively examine the influence of Ito on [Ca2+]i and contraction of canine left ventricular endocardial and epicardial myocytes. In endocardial myocytes, where the native Ito is small, simulation of a large Ito increased phase 1 repolarization, but significantly decreased [Ca2+]i transient and cell shortening. Accordingly, subtraction of the native Ito via the dynamic clamp enhanced contractility in epicardial cells. On the basis of these data, Dong et al [18] concluded that there is an inverse correlation between Ito levels and myocyte contractility and [Ca2+]i transient amplitude in canine epicardial and endocardial myocytes. Further experiments may be needed to resolve the conflicts.
Is the decrease in ICa density associated with the absence of the repolarizing notch seen by Cooper et al. sufficient to explain the significant impairment in EC coupling observed during HF? The authors use a modified Monte Carlo stimulation of L-type Ca2+ channel and RyR gating, which takes into account Ca2+ dependent inactivation of L-type Ca2+ channels, to show that the simulated Ca2+ sparks agree well with the experimental data. The peak rate of Ca2+ spark production is halved, and the rate of initial spark triggering is slowed, while the appearance of late Ca2+ sparks due to stochastic L-type Ca2+ channel openings is increased. The authors report that the simulation shows that the change in EC coupling caused by the failing AP, with its absence of the repolarizing notch, can be entirely explained by the change in ICa flux.
In combination with the work by Harris et al. [15], the study by Cooper et al. [1] is likely to change all current models of EC coupling since there are so many influences in AP morphology. This is particularly relevant for our understanding of the development of EC coupling dysfunction during the onset of HF. Decreased Ito is a seemingly general response to myocardial insults, which has been detected as early as 48 hours following myocardial infarction [10]. Down-regulation of Ito precedes changes in SERCA, Na+/Ca2+ exchanger, and RyR proteins. The findings by Cooper et al. [1] would suggest that this, by itself, would directly translate into a change in EC coupling, which would later be exacerbated by functional decoupling of ICa and RyRs, down-regulation of SERCA, and/or up-regulation of Na+/Ca2+ exchanger.
Could up-regulation of Ito restore EC coupling during HF? A recent study by Jin et al. [19] suggest an answer to this important question. They showed that increasing Ito density by expressing the accessory subunit KChIP2 in failing ventricular myocytes attenuated HF and improved EC coupling. Thus, Ito could be a key, bi-directional regulator of EC coupling and a potential target for restoration of cardiac function during HF.
Although Cooper et al. [1]focused on HF, their findings also provide insights into the mechanisms underlying regional variations in EC coupling in the heart. Ito and hence phase 1 of the action potential varies throughout the heart. Ito is larger in epicardial than in endocardial myocytes of the left ventricular free wall. Yet, [Ca2+]i transients are larger in endocardial than in pericardial myocytes. In combination with the findings by Cooper et al. [1], this suggests that while the changes in the magnitude of phase 1 of the action potential could alter EC coupling, they are not sufficient to explain regional variations in [Ca2+]i transients in the heart. Under physiological conditions, regional and developmental variations in action potential waveform and the expression and function of Ca2+ handling proteins (e.g. Na+/Ca2+ exchanger, RyR2) are the likely culprits for heterogeneous EC coupling under physiological conditions [20].
The study by Cooper et al. [1] has broad implications on EC coupling. In ventricular myocytes, the probability of Ca2+ spark occurrence (PSpark) in a couplon during the action potential is proportional to the open probability (Po) of Cav 1.2 channels and the square of the Ca2+ current and local [Ca2+]i [7]. A recent study suggested that PSpark approaches unity in specific sites within a cell at the relatively positive membrane potentials (≈ +50 mV) attained during the plateau of the ventricular action potential [5]. This is surprising because, at +50 mV, Po is less than 1 (≈0.7) and the current through a single L-type Ca2+ channel is predicted to be very small (i.e. ≈ 10 fA) at physiological levels of extracellular Ca2+. However, as suggested by Cooper et al. [1], by depolarizing to +50 mV the myocyte promotes a high Po for L-type Ca2+ channels, which if followed by a pronounced notch to more negative potentials could enhance the driving force for Ca2+ influx and hence the unitary current of L-type Ca2+ channels. Indeed, according to the GHK equation repolarization from +50 to +30 during phase 1 would triple the Ca2+ influx (≈ 3.09-fold), which would increase PSpark by nine-fold. Although the opening of a single L-type Ca2+ channel can activate a couplon at less positive potentials, a larger number of L-type Ca2+ channels are likely to be needed at more positive potentials [6-Santana reference][5]. Indeed, experiments and modeling support the larger number of L-type Ca2+ channels activated to trigger a couplon at more positive potentials [5, 21][add Ramay & Sobie ?2009]. Phase 1 activation of Ca2+ sparks can thus be attributed to increased Po of the L-type Ca2+ channels and also the larger single channel current that would be observed without the notch. Coupled gating of L-type Ca2+ channels may also be a mechanisms for the concerted activation of multiple L-type Ca2+ channels and thereby contribute to SR Ca2+ release and a high PSpark [22]. A note of caution is in order, however: There are distinct cellular and molecular structural difference in human and rat ventricular myocytes (e.g. T-tubule organization, RyR2 number, relationship between L-type Ca2+ channels and RyR2 clusters, and more). To some extent excitation-contraction coupling in rat ventricular myocytes may not faithfully reproduce the local and cellular [Ca2+]i signals of human ventricular myocytes even when the AP is identical. Nevertheless, Cooper et al. [1] have provided important new evidence suggesting that the notch of the ventricular AP could be an important determinant of PSpark under diverse and varied physiological and pathological conditions.
Sources of Funding
Supported by NIH grants HL085686, P01 HL67849, HL081106 and Fondation Leducq and European Union Seventh Framework Program (FP7) “Identification and therapeutic targeting of common arrhythmia trigger mechanisms”. LFS is an Established Investigator of the AHA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Cooper PJ, Soeller C, Cannell MB. Excitation-contraction coupling in human heart failure examined by action potential clamp in rat cardiac myocytes. J Mol Cell Cardiol. 2010 Apr 26; doi: 10.1016/j.yjmcc.2010.04.012. [DOI] [PubMed] [Google Scholar]
- [2].Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiological reviews. 2005 Oct;85(4):1205–53. doi: 10.1152/physrev.00002.2005. [DOI] [PubMed] [Google Scholar]
- [3].Franzini-Armstrong C, Protasi F, Ramesh V. Shape, size, and distribution of Ca2+ release units and couplons in skeletal and cardiac muscles. Biophys J. 1999 Sep;77(3):1528–39. doi: 10.1016/S0006-3495(99)77000-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Cheng H, Lederer WJ, Cannell MB. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science. 1993;262(5134):740–4. doi: 10.1126/science.8235594. [DOI] [PubMed] [Google Scholar]
- [5].Inoue M, Bridge JH. Ca2+ sparks in rabbit ventricular myocytes evoked by action potentials: involvement of clusters of L-type Ca2+ channels. Circ Res. 2003 Mar 21;92(5):532–8. doi: 10.1161/01.RES.0000064175.70693.EC. [DOI] [PubMed] [Google Scholar]
- [6].Cannell MB, Cheng H, Lederer WJ. The control of calcium release in heart muscle. Science. 1995;268(5213):1045–9. doi: 10.1126/science.7754384. [DOI] [PubMed] [Google Scholar]
- [7].Santana LF, Cheng H, Gomez AM, Cannell MB, Lederer WJ. Relation between the sarcolemmal Ca2+ current and Ca2+ sparks and local control theories for cardiac excitation-contraction coupling. Circ Res. 1996;78(1):166–71. doi: 10.1161/01.res.78.1.166. [DOI] [PubMed] [Google Scholar]
- [8].Wang S, Ziman B, Bodi I, Rubio M, Zhou YY, D’Souza K, et al. Dilated cardiomyopathy with increased SR Ca2+ loading preceded by a hypercontractile state and diastolic failure in the alpha(1C)TG mouse. PLoS ONE. 2009;4(1):e4133. doi: 10.1371/journal.pone.0004133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Huang B, Qin D, El-Sherif N. Early down-regulation of K+ channel genes and currents in the postinfarction heart. Journal of cardiovascular electrophysiology. 2000 Nov;11(11):1252–61. doi: 10.1046/j.1540-8167.2000.01252.x. [DOI] [PubMed] [Google Scholar]
- [10].Rossow CF, Minami E, Chase EG, Murry CE, Santana LF. NFATc3-Induced Reductions in Voltage-Gated K+ Currents After Myocardial Infarction. Circ Res. 2004 May 28;94(10):1340–50. doi: 10.1161/01.RES.0000128406.08418.34. [DOI] [PubMed] [Google Scholar]
- [11].Rossow CF, Dilly KW, Yuan C, Nieves-Cintron M, Cabarrus JL, Santana LF. NFATc3-dependent loss of Ito gradient across the left ventricular wall during chronic beta adrenergic stimulation. J Mol Cell Cardiol. 2009 Feb;46(2):249–56. doi: 10.1016/j.yjmcc.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lue WM, Boyden PA. Abnormal electrical properties of myocytes from chronically infarcted canine heart. Alterations in Vmax and the transient outward current. Circulation. 1992 Mar;85(3):1175–88. doi: 10.1161/01.cir.85.3.1175. [DOI] [PubMed] [Google Scholar]
- [13].Gomez AM, Valdivia HH, Cheng H, Lederer MR, Santana LF, Cannell MB, et al. Defective excitation-contraction coupling in experimental cardiac hypertrophy and heart failure. Science. 1997;276(5313):800–6. doi: 10.1126/science.276.5313.800. [DOI] [PubMed] [Google Scholar]
- [14].Sah R, Ramirez RJ, Backx PH. Modulation of Ca2+ release in cardiac myocytes by changes in repolarization rate: role of phase-1 action potential repolarization in excitation-contraction coupling. Circ Res. 2002 Feb 8;90(2):165–73. doi: 10.1161/hh0202.103315. [DOI] [PubMed] [Google Scholar]
- [15].Harris DM, Mills GD, Chen X, Kubo H, Berretta RM, Votaw VS, et al. Alterations in early action potential repolarization causes localized failure of sarcoplasmic reticulum Ca2+ release. Circ Res. 2005 Mar 18;96(5):543–50. doi: 10.1161/01.RES.0000158966.58380.37. [DOI] [PubMed] [Google Scholar]
- [16].Song LS, Sham JS, Stern MD, Lakatta EG, Cheng H. Direct measurement of SR release flux by tracking ‘Ca2+ spikes’ in rat cardiac myocytes. J Physiol. 1998 Nov 1;512:677–91. doi: 10.1111/j.1469-7793.1998.677bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Cheng H, Lederer WJ. Calcium sparks. Physiological reviews. 2008 Oct;88(4):1491–545. doi: 10.1152/physrev.00030.2007. [DOI] [PubMed] [Google Scholar]
- [18].Dong M, Yan S, Chen Y, Niklewski PJ, Sun X, Chenault K, et al. Role of the Transient Outward Current in Regulating Mechanical Properties of Canine Ventricular Myocytes. Journal of cardiovascular electrophysiology. 2010 Feb 1; doi: 10.1111/j.1540-8167.2009.01708.x. [DOI] [PubMed] [Google Scholar]
- [19].Jin H, Hadri L, Palomeque J, Morel C, Karakikes I, Kaprielian R, et al. KChIP2 attenuates cardiac hypertrophy through regulation of I(to) and intracellular calcium signaling. J Mol Cell Cardiol. 2010 Jan 4; doi: 10.1016/j.yjmcc.2009.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Dilly KW, Rossow CF, Votaw VS, Meabon JS, Cabarrus JL, Santana LF. Mechanisms underlying variations in excitation-contraction coupling across the mouse left ventricular free wall. J Physiol. 2006 Apr 1;572(Pt 1):227–41. doi: 10.1113/jphysiol.2005.102020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Polakova E, Zahradnikova A, Jr., Pavelkova J, Zahradnik I, Zahradnikova A. Local calcium release activation by DHPR calcium channel openings in rat cardiac myocytes. J Physiol. 2008 Aug 15;586(16):3839–54. doi: 10.1113/jphysiol.2007.149989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Navedo MF, Cheng EP, Yuan C, Votaw S, Molkentin JD, Scott JD, et al. Increased coupled gating of L-type Ca2+ channels during hypertension and Timothy syndrome. Circ Res. 2010 Mar 5;106(4):748–56. doi: 10.1161/CIRCRESAHA.109.213363. [DOI] [PMC free article] [PubMed] [Google Scholar]