

Abstract
Toxoplasma gondii is an obligate intracellular parasite that permanently infects warm-blooded vertebrates through its ability to convert into a latent tissue cyst form. The latent form (bradyzoite) can reinitiate a life-threatening acute infection if host immunity wanes, most commonly in AIDS or organ transplant patients. We have previously shown that bradyzoite development is accompanied by phosphorylation of the parasite eukaryotic initiation factor 2 alpha subunit (eIF2α), which dampens global protein synthesis and reprograms gene expression. In this study, we analyzed the activities of two specific inhibitors of eIF2α dephosphorylation, salubrinal (SAL) and guanabenz (GA). We establish that these drugs are able to inhibit the dephosphorylation of Toxoplasma eIF2α. Our results show that SAL and GA reduce tachyzoite replication in vitro and in vivo. Furthermore, both drugs induce bradyzoite formation and inhibit the reactivation of latent bradyzoites in vitro. To address whether the antiparasitic activities of SAL and GA involve host eIF2α phosphorylation, we infected mutant mouse embryonic fibroblast (MEF) cells incapable of phosphorylating eIF2α, which had no impact on the efficacies of SAL and GA against Toxoplasma infection. Our findings suggest that SAL and GA may serve as potential new drugs for the treatment of acute and chronic toxoplasmosis.
INTRODUCTION
The protozoan parasite Toxoplasma gondii (phylum Apicomplexa) is an important life-threatening opportunistic pathogen in AIDS patients and other immunosuppressed individuals (1). During the course of infection, the host immune response induces this obligate intracellular parasite to convert from a proliferating stage (tachyzoite) into a latent tissue cyst stage (bradyzoite). Bradyzoite cysts can remain in host tissues for life, seemingly without overt health consequences. However, bradyzoites can reconvert to actively replicating tachyzoites when host immunity is attenuated, causing lifelong chronic infection. The recurrent reactivation of acute infection can lead to serious damage of the central nervous system (CNS) and heart, where bradyzoite cysts have a tendency to reside (1–3). While antifolates and other antibiotics are used to treat acute infection, the frontline drugs cause severe adverse effects and do not appear to exert activity against encysted parasites (1, 3, 4). Thus, there is an urgent need for new therapeutics to treat acute and latent toxoplasmosis.
The molecular mechanisms coordinating stage conversion are poorly understood. Tachyzoite-to-bradyzoite conversion can be induced in vitro by a range of different stresses, including pH extremes, heat shock, and exposure to sodium nitroprusside or mitochondrial inhibitors (3). Removal of the stress condition prompts the encysted bradyzoites to reconvert into proliferative tachyzoites. We previously showed that bradyzoite conversion triggered by exposure to different stress conditions is accompanied by phosphorylation of the alpha subunit of Toxoplasma eukaryotic initiation factor 2 (TgIF2α) (5, 6). The eukaryotic initiation factor 2 alpha subunit (eIF2α) is phosphorylated by a specialized group of protein kinases that possess a signature catalytic domain flanked by distinct regulatory domains, the latter of which allow their activation by disparate environmental stresses or signals (7). Phosphorylation of eIF2α results in a dramatic reduction in global protein synthesis, allowing cells time to organize a new pattern of gene expression designed to remedy the stress or undergo a developmental change (7). We identified four eIF2α kinases in Toxoplasma and confirmed that TgIF2α phosphorylation results in translational control during stress (5, 8–10). We also found that TgIF2α phosphorylation is maintained in mature bradyzoites, consistent with the dormant nature of this life cycle stage (6). These findings support the idea that TgIF2α phosphorylation could represent a potential new target for the treatment of Toxoplasma infection (8, 11).
During a screen for small molecules that protect rat PC12 cells from endoplasmic reticulum (ER) stress, salubrinal (SAL) was found to prevent host cell apoptosis by specifically preventing dephosphorylation of mammalian eIF2α (12). In a previous study, we found that treatment of Toxoplasma tachyzoites with SAL leads to TgIF2α phosphorylation and induction of the BAG1 and LDH2 bradyzoite-specific marker genes (6). More recently, guanabenz (GA), an α2-adrenergic receptor agonist used in the treatment of hypertension (13), has also been found to promote survival of human HeLa cells upon ER stress through inhibition of eIF2α dephosphorylation (14). In light of our previous work linking TgIF2α phosphorylation and microbial latency, we sought to determine the impact of eIF2α dephosphorylation inhibitors on Toxoplasma stage conversion in vitro. Results from this study suggest that the drugs SAL and GA inhibit Toxoplasma infection by a mechanism involving induced translational control that promotes and stabilizes the latent bradyzoite form. Consistent with the slowed growth observed in vitro, GA was also able to extend the life of mice infected with a hypervirulent strain of Toxoplasma.
MATERIALS AND METHODS
Chemicals.
Salubrinal (SAL) was purchased from Tocris Bioscience (no. 2347). Guanabenz acetate (GA) (no. G110) and clonidine (CLN) (no. C7897) were purchased from Sigma-Aldrich.
Parasite cultivation.
Toxoplasma tachyzoites were cultivated in human foreskin fibroblasts (HFFs) in Dulbecco's modification of Eagle's medium (DMEM) (Mediatech) containing 25 mM glucose and 4 mM glutamine and supplemented with 1% heat-inactivated fetal bovine serum (FBS) (Gibco/Invitrogen). Cultures were maintained in a humidified incubator at 37°C with 5% CO2. For the cultivation and infection of mouse embryonic fibroblast (MEF) cells, DMEM was supplemented with 10% heat-inactivated FBS,100 U/ml penicillin, and 100 μg/ml streptomycin.
Parasite proliferation assays.
Ten thousand tachyzoites were allowed to infect confluent HFF cells grown on coverslips in DMEM plus 1% FBS. At 4 h postinfection, infection medium was replaced, supplemented with the indicated concentration of SAL, GA, CLN, or vehicle (dimethyl sulfoxide [DMSO]). To determine the number of tachyzoites per vacuole, infected HFFs were fixed with 0.5 ml ice-cold methanol for 15 min at −20°C and nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) to facilitate counting of parasites by fluorescence microscopy in at least 50 random vacuoles.
For experiments using MEF cells, ∼50,000 MEF cells were seeded on glass coverslips in 12-well plates for approximately 16 to 20 h prior to infection with 25,000 ME49 tachyzoites in DMEM plus 10% FBS supplemented with 100 U/ml penicillin and 100 μg/ml streptomycin. At 4 h postinfection, infection medium was replaced with medium supplemented with the indicated concentrations up to 8 μM SAL, 15 μM GA, or vehicle (DMSO), as indicated. In mammalian cells, SAL and GA were reported to be nontoxic at concentrations of up to 100 μM and 50 μM, respectively (12, 14). In contrast to these reports, we observed toxic effects of GA on HFFs at concentrations higher than 25 μM, which could be due to the extended incubation time of up to 7 days.
At the indicated time points, cells were fixed in 3% paraformaldehyde for 30 min at room temperature. Following incubation in 1× phosphate-buffered saline (PBS) supplemented with 0.1 M glycine, samples were then permeabilized for 20 min with 1× PBS supplemented with 0.2% Triton X-100 and 3% bovine serum albumin (BSA) (fraction V; Omnipur) and subsequently blocked for 1 h with 3% BSA in 1× PBS. To visualize tachyzoites for counting, samples were incubated for 1 h with a monoclonal mouse anti-Sag1 antibody (1:5,000, C86319M; Meridian Life Science) in blocking solution, followed by incubation for 1 h with a goat anti-mouse antibody (1:2,000, A11029; Molecular Probes) conjugated with Alexa 488 (1:2,000, A11029; GE Healthcare). All incubation steps were carried out at room temperature.
To determine the number of parasites using the PCR-based B1 assay, MEF cells were seeded in 12-well plates and infected as described above. At 4 h and 24 h postinfection, infection medium was replaced, with supplementation with the indicated concentrations of SAL, GA, or vehicle (DMSO). At 48 h postinfection, genomic DNA was isolated from the infected cells, and parasite counts were determined using quantitative PCR with primers for the Toxoplasma B1 gene as described previously (15).
Monitoring tachyzoite-bradyzoite interconversion.
Five hundred ME49 strain tachyzoites were inoculated onto confluent HFF monolayers grown on glass coverslips. Four hours postinfection, culture medium was replaced with alkaline medium (pH 8.1), which was replaced daily. After 7 days in alkaline stress medium, infected monolayers were washed and incubated in normal medium (pH 7.2) plus SAL, GA, or vehicle (DMSO) to induce reconversion into tachyzoites. A parallel sample was maintained in alkaline medium to prevent reactivation. All media were replaced daily. When infection resumed in the vehicle control (as determined by host cell lysis observed by light microscopy, usually 3 to 4 days after switching from alkaline to normal medium), infected monolayers were fixed in ice-cold methanol, and bradyzoite cysts were visualized by indirect fluorescent-antibody assay (IFA) following staining with fluorescein isothiocyanate (FITC)-conjugated Dolichos biflorus lectin (1:200, FL-1031; Vector Laboratories) as described previously (6).
To determine the number of tachyzoites released into the medium upon reactivation of infection, confluent HFF monolayers grown in T-25 25-cm2 tissue culture flasks were infected with 106 ME49 tachyzoites and converted into bradyzoites using the alkaline pH method described above. At day 8, alkaline medium was washed out and replaced with normal medium supplemented with the appropriate concentrations of SAL, GA, or vehicle (DMSO), and replaced daily to induce reactivation of infection. After 3 to 4 days, the reactivated tachyzoites present in the culture supernatant were counted using a Neubauer hematocytometer.
To measure induced expression of bradyzoite marker genes upon treatment with GA, confluent HFF monolayers were grown in T-25 25-cm2 tissue culture flasks and infected with ∼106 ME49 tachyzoites. Four hours postinfection, the infection medium was replaced with medium supplemented with 15 μM GA or vehicle (DMSO) and replaced daily. After 5 days, intracellular parasites were released by syringe passage and washed twice with ice-cold PBS. Total RNA was isolated using an RNeasy Plus kit (Qiagen). cDNA was synthesized using an oligo(dT) primer (Qiagen) and the Omniscript kit (Qiagen). Quantitative PCR was carried out as described previously (6) using the following gene-specific oligonucleotides: β-tubulin (for normalization), sense (ATGTTCCGTGGTCGCATGT) and antisense (TGGGAATCCACTGAACGAAGT); LDH2, sense (TGAGGAACGTGTGGTCACAGA) and antisense (TTGGTAACCCCCGTGAAAAA); BAG1, sense (TGAGCGAGTGTCCGGTTATTT) and antisense (TAGAACGCCGTTGTCCATTG).
Recovery of bradyzoites from SAL and GA treatment.
As a measure of parasite infection of host cells following removal of alkaline stress, 500 ME49 strain tachyzoites were inoculated onto confluent HFF monolayers grown in 12-well plates and treated as described above. After 7 days, infected monolayers were washed and incubated in normal medium supplemented with the indicated concentration of SAL or GA and replaced daily. For controls, one set of samples was maintained in alkaline medium. After another 3 days (10 days postinfection), all infected monolayers were incubated in normal medium. One week later (17 days postinfection), host cells were fixed with methanol and stained with crystal violet. The degree of host cell lysis was determined with an Alpha Innotech imager (MultiImage II, AlphaView software, version 2.0.0.9).
Inhibition of TgIF2α dephosphorylation in the presence of SAL and GA.
ME49 tachyzoites were physically released from host cells via syringe passage. Filter-purified parasites were incubated in DMEM plus 1% FBS supplemented with 10 μM thapsigargin (T9033; Sigma) or vehicle (DMSO) for 1 h. Following a wash step, parasites were incubated for another hour in DMEM plus 1% FBS supplemented with the indicated concentration of SAL, GA, or vehicle (DMSO). All incubation steps were carried out at 37°C and 5% CO2. Western blot analysis of TgIF2α phosphorylation was carried out as previously described using polyclonal rabbit antibodies recognizing either total TgIF2α or TgIF2α phosphorylated at serine 71 (5, 6).
In vivo treatment of acute infection.
BALB/c mice were purchased from Harlan Laboratories (Indianapolis, IN) and infected via intraperitoneal (i.p.) injection with 100 tachyzoites of the RH strain, isolated via syringe passage from an HFF monolayer and resuspended in sterile 1× PBS. A total of 30 mice, which were separated into 5 groups consisting of 6 mice each (3 per cage), were used in the study. The tachyzoite suspension was kept at 37°C until injection (groups 1 to 4). Control mice were injected with a corresponding volume of 1× PBS (group 5). At 4 h postinfection, mice in group 1 were injected with vehicle, group 2 with 5 mg/kg of body weight GA, and groups 3 to 5 with 10 mg/kg GA. Group 1 received twice-daily injections of vehicle until the end of the study. For groups 2 and 3, this regimen was continued until mice were moribund (group 2, day 7/group 3, day 10). Groups 4 and 5 received injections of 10 mg/kg GA on day 2, and beginning on day 3, they were administered a total of 20 mg/kg GA in two injections of 10 mg/kg each (separated by 10 to 12 h), which was administered until the end of the study. GA was dissolved at a concentration of 2.5 mg/ml in physiological salt solution and sterile filtered prior to i.p. injection. Wet food was provided within the cage throughout the study. Mice were weighed before injection, and the injection volume was adjusted accordingly. All mice were checked twice daily for signs of infection and euthanized when moribund. Peritoneal fluid was analyzed microscopically for the presence of tachyzoites.
RESULTS
Salubrinal and guanabenz inhibit tachyzoite replication in vitro.
In a previous study, we showed that SAL can inhibit dephosphorylation of TgIF2α and induce expression of the BAG1 and LDH2 bradyzoite marker genes in type II ME49 tachyzoites (6). These findings support the idea that translational control mediated by TgIF2α phosphorylation is an important event in the formation of the latent stage (8). To further explore the role of translational control in Toxoplasma infection, we determined the effect of SAL on the replication of ME49 parasites cultured in HFF cells (Fig. 1A). Results show that SAL inhibits tachyzoite replication in a dose-dependent fashion with a 50% effective concentration (EC50) of ∼1.5 μM. Vehicle-treated samples contained 8 to 16 parasites per vacuole 32 h postinfection, but only 2 parasites per vacuole were present when 4 μM SAL was included in the medium (Fig. 1A).
Fig 1.
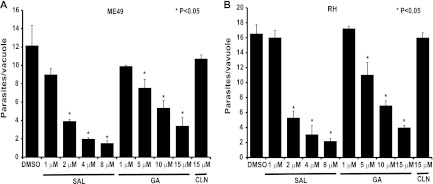
Salubrinal (SAL) and guanabenz (GA) reduce replication of Toxoplasma in vitro. HFF host cells infected with ME49 (A) or RH (B) strain parasites were cultured with the indicated concentration of SAL, GA, clonidine (CLN), or vehicle (DMSO). At 32 h postinfection, replication was assessed by counting tachyzoites in at least 50 random vacuoles. Histograms show averages for a representative experiment performed in triplicate. Error bars indicate standard deviations, and asterisks indicate a statistically significant difference relative to results for the vehicle control (P < 0.05, two-tailed unpaired t test).
We also measured the effects of GA on Toxoplasma replication in vitro since this drug was recently demonstrated to be an eIF2α dephosphorylation inhibitor (14). Like SAL, GA inhibited tachyzoite replication in a dose-dependent manner, with an EC50 of ∼6 μM (Fig. 1A). Since GA is also an α2-adrenergic receptor agonist (13), we examined the effect of the drug clonidine on parasite replication as a control. Like GA, clonidine is an α2-adrenergic receptor agonist used in the treatment of hypertension (16). Infected HFFs treated with15 μM clonidine showed no statistically significant difference in the average number of parasites per vacuole compared to results for the vehicle control (Fig. 1A), suggesting that the antiparasitic activities of GA do not involve α2-adrenergic receptor agonist activity. SAL and GA also had potent antiparasitic activity against the hypervirulent type I RH strain, with EC50s similar to those seen for the ME49 strain (Fig. 1B). As observed for the ME49 strain, we found that clonidine had no antiproliferative activity against RH strain tachyzoites.
SAL and GA induce tachyzoite conversion to latent bradyzoites in vitro.
Since SAL and GA treatment of tachyzoites induced expression of bradyzoite-specific marker genes (6) (see Fig. SA2 in the supplemental material), we next addressed if the antiproliferative effect of SAL and GA was due to conversion of tachyzoites to latent bradyzoites. Incubation of intracellular tachyzoites in alkaline media is a well-established method to induce formation of bradyzoite tissue cysts in vitro, which can be visualized by IFA using the Dolichos biflorus lectin that binds cyst wall protein (17). In dose-dependent fashion, both SAL and GA induced bradyzoite formation in vitro, with 4.0 and 10.0 μM concentrations yielding roughly the same number of cysts at day 7 as the alkaline-pH control, respectively (Fig. 2). Interestingly, tachyzoites exposed to 8.0 μM SAL for 7 days exhibited unusual morphology, with lectin-stained protein failing to reach the cyst wall (see Fig. SA1 in the supplemental material). These abnormal parasites also appeared to be limited to one per vacuole. These data suggest that SAL is lethal to parasites at doses exceeding 8.0 μM, but sublethal doses induce bradyzoite conversion, as found for GA. Higher doses of GA may also be lethal for Toxoplasma, but doses in excess of 25 μM begin to stress the host cell monolayer, leading to its detachment from the flask.
Fig 2.

SAL and GA induce tachyzoite conversion to latent bradyzoites in vitro. HFF cells infected with ME49 strain parasites were cultured with the indicated concentration of SAL, GA, or vehicle (DMSO) for 7 days. As a control, infected HFFs were grown in alkaline medium (pH 8.1) for 7 days. The frequency of cyst formation was assessed by counting Dolichos lectin-stained cysts in at least 25 random fields on day 7. The graph shows averages for an experiment performed in triplicate and normalized to the number of cysts present in the DMSO control sample. An independent experiment showed similar results. Error bars indicate standard deviations, and asterisks indicate a statistically significant difference relative to results for the vehicle control (P < 0.05, two-tailed unpaired t test).
SAL and GA inhibit reactivation of latent bradyzoites in vitro.
We previously reported that latent bradyzoites have sustained TgIF2α phosphorylation (6). If TgIF2α phosphorylation is critical to form and maintain the cyst form, one would predict that inhibitors of TgIF2α dephosphorylation would attenuate the reactivation of infection, i.e., block reconversion of bradyzoites into proliferative tachyzoites. The reactivation of infection can be examined in vitro since removal of the stress used to induce bradyzoite development allows rapid reconversion to tachyzoites. As described above, we induced formation of bradyzoite tissue cysts using alkaline medium for 7 days. The bradyzoite-containing monolayers were then washed and incubated in fresh medium of normal physiological pH (pH 7.2). As a control, a parallel sample of bradyzoites was retained in alkaline medium to prevent reactivation (Fig. 3A). As expected, the relative number of cysts was approximately 3.5-fold higher in the samples kept in alkaline medium than in the samples that had alkaline medium replaced with normal medium of pH 7.2 (Fig. 3B). However, in the presence of at least 4 μM SAL or 10 μM GA, the reactivation of latent bradyzoites was significantly inhibited, with the relative number of cysts being approximately 2.5- to 3.0-fold higher than that in the normal medium supplemented with DMSO (Fig. 3B). Samples with higher concentrations of SAL or GA contained as many bradyzoite cysts as the nonreactivated samples maintained in alkaline pH (Fig. 3B). We also determined the number of tachyzoites in the infected cell culture supernatant following removal of the alkaline medium as an independent measure of the reactivation of infection. Increasing concentrations of SAL or GA were found to reduce the number of tachyzoites in the reactivated cultures (Fig. 3C). These results strongly suggest that SAL and GA can inhibit the reactivation of latent bradyzoites upon stress removal in vitro.
Fig 3.

SAL and GA impede reactivation of latent bradyzoites in vitro. (A) The diagram outlines the experimental setup. ME49 tachyzoites (Tz) were differentiated into bradyzoite cysts (Bz) using alkaline (pH 8.1) medium for 7 days. The alkaline stress medium was then removed and replaced with normal medium (pH 7.2) containing indicated concentrations of SAL, GA, or vehicle (DMSO) for 3 days. Alkaline medium was retained in some samples as a positive control to prevent reactivation. Reactivation was assessed by counting Dolichos lectin-stained cysts in at least 25 random fields (B) or by counting reactivated tachyzoites present in culture medium (C). (B) The histogram shows the results from a representative experiment performed in triplicate and normalized to the number of cysts counted in the vehicle control. This experiment was repeated a total of three times, each with similar results. (C) The average for three independent experiments was normalized to the number of tachyzoites present in the vehicle control. In panels A and B, error bars indicate standard deviations and asterisks indicate a statistically significant difference relative to results for the vehicle control (P < 0.05, two-tailed unpaired t test).
To analyze whether SAL/GA-treated cysts suffer a permanent inability to reactivate, bradyzoite cysts subjected to SAL or GA for 3 days were allowed to recover in normal medium without the drug. The experimental setup is shown in Fig. 4A. After 7 days with no drugs or stress, the infected monolayers were fixed, and relative host cell lysis was determined as a measure of reactivated infection. As a standard, we measured the reactivation of bradyzoite cysts that were maintained on alkaline medium without drug for 3 days and then cultured in normal medium for 7 days. Reactivation of infection was detected in cultures exposed to 4 μM SAL or 10 μM GA (Fig. 4B). However, significantly less reactivation was detected in cultures treated with 8 μM SAL or 15 μM GA (Fig. 4B). These data further support the idea that SAL and GA prevent reconversion of bradyzoites into replicating tachyzoites and suggest that the failure to reactivate infection can persist for a time even after the drug is removed from culture. Furthermore, it is possible that the higher concentrations of SAL and GA are lethal to bradyzoites.
Fig 4.

SAL- and GA-treated tissue cysts are defective for reactivation. After cysts were generated for 7 days, the alkaline stress medium was removed and replaced with normal medium containing the indicated concentrations of SAL or GA for an additional 3 days. Alkaline medium (pH 8.1) was retained in some samples as a positive control to prevent reactivation. Following the incubation in normal medium without SAL or GA for another 7 days, the extent of reactivation of tachyzoite infection was assessed by determining the degree of host cell lysis relative to that for the alkaline control using an Alpha Innotech imager. (A) The diagram shows a time line of the experimental setup. (B) The histogram shows the results from two experiments, each performed in triplicate. Error bars indicate standard deviations, and asterisks indicate a statistically significant difference relative to results for the alkaline control (P < 0.05, two-tailed unpaired t test).
SAL and GA inhibit dephosphorylation of TgIF2α.
SAL and GA are recognized inhibitors of eIF2α dephosphorylation in mammalian cells (12, 14). An assay that takes advantage of the fact that TgIF2α becomes phosphorylated in purified tachyzoites exposed to pharmacological agents inducing endoplasmic reticulum (ER) stress has been established (6). Removal of the stress treatment causes rapid dephosphorylation of TgIF2α, but the inclusion of SAL can prevent dephosphorylation of TgIF2α. We tested if SAL and GA could inhibit dephosphorylation of TgIF2α in response to another ER stress agent, thapsigargin. Purified tachyzoites treated with thapsigargin for 60 min exhibited a concentration-dependent induction of TgIF2α phosphorylation (Fig. 5A and B). One hour after removing the stress, we observed nearly complete dephosphorylation of TgIF2α (Fig. 5C and D). However, SAL and GA inhibited this dephosphorylation of TgIF2α in a dose-dependent manner (Fig. 5C and D). These results suggest that both SAL and GA inhibit TgIF2α dephosphorylation in stressed tachyzoites.
Fig 5.

SAL and GA inhibit TgIF2α dephosphorylation. (A) Schematic diagram of experimental design. (B) Purified ME49 tachyzoites were treated with the indicated concentrations of thapsigargin (TG) or vehicle (DMSO) for 60 min. Following exposure to 10 μM TG for 60 min, tachyzoites were allowed to recover in the presence of vehicle (DMSO) or the indicated concentrations of SAL (C) or GA (D). Equal amounts of protein lysate were separated by SDS-PAGE, followed by immunoblotting using antibody that recognizes total TgIF2α or phosphorylated TgIF2α (TgIF2α∼P).
Antiparasitic activities of SAL and GA do not involve host eIF2α.
While the previous experiment indicates that SAL and GA are capable of adversely affecting the dephosphorylation of TgIF2α, the antiparasitic action of the drugs may also involve inhibition of host eIF2α dephosphorylation. We addressed the possibility that SAL or GA adversely affects the parasites by acting through the host cell in two different ways. First, we reasoned that if SAL and GA function through the host cells, treatment with either drug prior to infection may affect parasite replication. Such effects of drug preconditioning were reported by Radke et al., who demonstrated that pretreatment of HFF host cells with compound 1 (a trisubstituted pyrrole and bradyzoite inducer) prior to infection with tachyzoites reduced parasite proliferation by promoting bradyzoite formation (18). We therefore pretreated HFF monolayers with increasing concentrations of SAL, GA, or vehicle for 24 h prior to infection and then counted the number of parasites per vacuole 24 h postinfection. Preconditioning host cells with either SAL or GA had no significant effect on parasite replication (Fig. 6), suggesting that these drugs do not modify host cells in a way that makes them less hospitable for infection.
Fig 6.

Pretreatment of HFF cells with SAL or GA does not affect tachyzoite infection. Confluent HFF monolayers were treated with the indicated concentrations of SAL, GA, or vehicle (DMSO) for 24 h (“pre-treated host cells”). Following two washing steps in medium, the pretreated HFF cells were infected with ME49 tachyzoites. At 24 h postinfection, the number of parasites per vacuole was determined for at least 50 random vacuoles. As a control, replication in the presence of the indicated concentration of SAL, GA, or vehicle (DMSO) was also assessed (“no pre-treatment”). Histograms show averages from a triplicate experiment. Error bars indicate standard deviations, and asterisks indicate a statistically significant difference from results for the vehicle control (P < 0.01, two-tailed unpaired t test).
As an alternative way to test if the antiparasitic activities of SAL and GA involved effects on host eIF2α, we infected a mouse embryonic fibroblast (MEF) cell line engineered to express a nonphosphorylatable version of eIF2α (eIF2α-S51A), which would negate the effects of either drug on host translational control (19). Wild-type (S/S) and mutant (A/A) MEF cells were infected with ME49 tachyzoites in the presence or absence of SAL or GA, and the number of parasites per vacuole was determined 24 h postinfection. The antiparasitic effects of SAL and GA were identical in both S/S and A/A MEF cells, suggesting that host cell eIF2α is not a contributor (Fig. 7A). Due to difficulties in visualizing parasites within MEF cells at 48 h, we used quantitative PCR and primers specific for the Toxoplasma B1 gene to monitor infection levels. Once again, tachyzoite replication was sharply reduced in the presence of SAL or GA in both S/S and A/A cells (Fig. 7B). These results suggest that the mechanism of action of SAL and GA against Toxoplasma infection does not involve host cell eIF2α phosphorylation.
Fig 7.

SAL and GA reduce Toxoplasma replication regardless of host cell eIF2α phosphorylation. Tachyzoites were allowed to infect MEFs expressing wild-type (S/S) or nonphosphorylatable (A/A) eIF2α in culture medium supplemented with the indicated concentration of SAL, GA, or vehicle (DMSO). (A) At 24 h postinfection, parasite replication was assessed by counting tachyzoites visualized by IFA through labeling with an Alexa 488-conjugated anti-SAG1 antibody in at least 50 random vacuoles. The graph shows the averages for an experiment performed in triplicate. Error bars indicate standard deviations, and asterisks indicate a statistically significant difference relative to the vehicle control (P < 0.05, two-tailed unpaired t test). (B) At 48 h postinfection, parasite replication was assessed using the PCR-based B1 assay. The histograms show the averages for a representative experiment performed in triplicate. The experiment was repeated three independent times with similar results. Error bars indicate standard deviations, and asterisks indicate a statistically significant difference relative to results for the vehicle control (P < 0.01, two-tailed unpaired t test).
GA extends survival of mice acutely infected with Toxoplasma.
The data presented suggest that GA and SAL adversely affect TgIF2α dephosphorylation, reduce Toxoplasma replication, and promote bradyzoite formation and stability in vitro. To address whether GA affects Toxoplasma infection in vivo, BALB/c mice were infected with a lethal dose of 100 RH strain tachyzoites and treated with GA or vehicle every other day throughout the course of infection. In this model of acute infection, the hypervirulent RH parasites cause a moribund state 6 to 7 days postinfection (10). As expected, all infected mice treated with vehicle alone were moribund by day 7. In contrast, infected mice treated with GA did not reach a moribund state until up to 11 days. Mice treated with a total dosage of 5 mg/kg GA every other day (group 2) survived 3 days longer, and mice treated with a total dosage of 10 or 20 mg/kg every other day (groups 2 and 4, respectively) survived 4 additional days compared to the infected mice receiving vehicle (Fig. 8). The course of infection and the onset were similar for all mice within each group. At day 11, all uninfected mice treated with the highest dose of GA appeared normal, suggesting that this dosing regimen does not affect the viability of the mice. Tachyzoites were found in the peritoneal fluid of all infected mice, suggesting that mice succumbed to infection rather than to adverse effects of drug treatment.
Fig 8.

GA extends the survival of mice acutely infected with Toxoplasma. Mice in groups 1 to 4 were infected with a lethal dose of 100 Toxoplasma tachyzoites (RH strain) and treated with vehicle (group 1) or the indicated dose of GA (groups 2 to 4). To exclude adverse effects of GA on survival, a noninfected group receiving the same dose of GA as group 4 was included (group 5). Survival of each treatment group is presented for the time course of the experiment.
DISCUSSION
During the course of Toxoplasma infection, proliferating tachyzoites evade the immune system by converting into latent bradyzoite cysts, which can persist in host tissues for life. If the immune system of the host becomes compromised, bradyzoites can reconvert into destructive tachyzoites, leading to dangerous reactivation of parasite infection (3). Current treatments for toxoplasmosis produce serious adverse effects and are thought to primarily target only tachyzoites, underscoring the need for safer treatment and prophylactic options (1).
This study showed that the drugs SAL and GA significantly slow the replication of Toxoplasma tachyzoites in cultured HFF host cells, with EC50s of 1.5 and 6 μM, respectively (Fig. 1). Furthermore, since slowed parasite proliferation is a precursor for development into the latent bradyzoite cyst stage (20), we determined if SAL- or GA-treated tachyzoites were expressing cyst wall proteins, using FITC-conjugated Dolichos lectin staining. Our findings indicate that SAL and GA are potent promoters of tachyzoite-to-bradyzoite conversion (Fig. 2). Bradyzoite cysts treated with SAL or GA failed to reactivate into tachyzoites upon removal of the induction stress (Fig. 3B and C). Drug-treated cysts appeared to sustain an irreversible defect that rendered them incapable of reconverting to tachyzoites even when drug was removed (Fig. 4). While this could be due to residual drug retained in the cells, it opens the exciting possibility that high levels of TgIF2α phosphorylation in response to SAL and GA may be lethal to bradyzoites.
GA offers several significant advantages over SAL in terms of in vivo usage. First, GA is a safe, FDA-approved drug already in clinical use for hypertension. Second, GA crosses the blood-brain barrier, which is necessary to target latent bradyzoites (or reactivated tachyzoites) in the brain. These features, considered with the ability of GA to slow parasite replication in vitro, prompted our examination of GA in the mouse model of acute toxoplasmosis infection. Mice infected with the RH strain reached the moribund state after 6 days, as expected, but mice treated with GA survived at least 3 days longer (Fig. 8). Prolonged survival of mice infected with the hypervirulent RH strain, which has a 50% lethal dose (LD50) of ∼1 and does not readily form latent cysts, is promising. It is also noteworthy that the GA-treated mice showed little sign of toxicity at the dosages used here. Optimization of the dose of GA or combination with another antiparasitic may increase survival time. These encouraging results highlight the importance of characterizing the target of GA to develop more potent inhibitors. It is noted that GA may exert greater activity against a cyst-generating strain or be effective at blocking reactivation of infection in vivo, and therefore, future studies are needed to examine the efficacy of GA against chronic toxoplasmosis in the mouse model. High-throughput drug screening has found that GA exerts antiproliferative activity against various Plasmodium falciparum strains at concentrations between 4 and 11 μM (PubChem BioAssay; http://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?cid=5702062; see also reference 21), suggesting that GA may have activity against a number of parasitic protozoa.
GA was found to bind to GADD34 (PPP1R15A), a regulatory protein of the PP1 dephosphorylation complex, thereby enhancing phosphorylated eIF2α (eIF2α∼P) levels and providing cytoprotection during cellular stress (14). It is noted that GA binds and interferes only with GADD34-mediated dephosphorylation of eIF2α, whereas SAL also associates with and blocks the function of CReP (PPP1R15B), which facilitates basal eIF2α dephosphorylation (12). Therefore, GA is thought to attenuate recovery of translation following stress, while SAL can trigger enhanced eIF2α∼P and repressed protein synthesis in the absence of additional stress. These mechanisms of action may explain why SAL is more potent against Toxoplasma than GA, with concentrations of 8.0 μM or more appearing to be lethal.
Given that Toxoplasma is an obligate intracellular parasite, we considered that the antiparasitic effects of SAL and GA might occur indirectly through interference with host cell eIF2α dephosphorylation. We addressed this issue with a number of independent approaches. First, as we have shown previously, SAL has a direct effect on TgIF2α phosphorylation independent of host cells (6). SAL has also been shown to increase RNA granule formation in extracellular Toxoplasma, which occurs during eIF2α phosphorylation, and SAL can enhance the levels of Plasmodium eIF2α phosphorylation (22, 23). Here we have further demonstrated this interaction by using extracellular tachyzoites treated with thapsigargin, which potently induces phosphorylation of eIF2α. Upon removal of the thapsigargin, TgIF2α is rapidly dephosphorylated, but inclusion of SAL or GA directly prevents the dephosphorylation of TgIF2α (Fig. 5). Second, we preconditioned the HFF host cells with SAL or GA prior to infection with Toxoplasma, but the drugs did not alter the host cell in a way that was detrimental to parasite invasion or replication (Fig. 6). Finally, SAL and GA reduced Toxoplasma infection even when we used host cells that were genetically engineered to be incapable of phosphorylating eIF2α (Fig. 7). Considered together, the data suggest that the mechanisms of action of SAL and GA do not involve host cell eIF2α phosphorylation. However, it is possible that SAL or GA can also exert their effects by one or more mechanisms that are independent of eIF2α phosphorylation. These independent mechanisms may involve either host or parasite stress response pathways.
Despite clear evidence that SAL and GA can directly prevent TgIF2α dephosphorylation, how this may occur in the parasite requires further investigation. Bioinformatics analysis of the ToxoDB showed the presence of an unequivocal PP1 homologue (TGME49_110700) but did not identify clear orthologues of PP1 regulatory proteins GADD34 or CReP. This could simply be a matter of primary sequence divergence in Toxoplasma; i.e., functional equivalents of these PP1 regulatory proteins may exist and serve as the target of SAL and/or GA. However, we cannot exclude the potential roles of additional targets in Toxoplasma at this time.
Recent studies in other parasites, such as Plasmodium, Leishmania, and Trypanosoma, have supported our earlier findings that eIF2α phosphorylation and translational control play a critical role in parasite development (11, 22, 24–26). The findings presented here further underscore the significance of translational control during parasite proliferation and development into latent stages. Given its proven safety record, our discovery of the antiparasitic effects of GA in particular could rapidly lead to a new prophylactic treatment to prevent reactivated toxoplasmosis and perhaps other protozoal infections as well.
ACKNOWLEDGMENTS
Research was supported by grants from NIH (AI084031 to W.J.S. and R.C.W.) and AHA (11POST7730010 to C.K.).
We thank Brad Joyce for helpful discussions and Kati McNamee, Sean Cooney, and Tamila Garbuz for technical assistance.
Footnotes
Published ahead of print 4 February 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01899-12.
REFERENCES
- 1. Montoya JG, Liesenfeld O. 2004. Toxoplasmosis. Lancet 363:1965–1976 [DOI] [PubMed] [Google Scholar]
- 2. Skariah S, McIntyre MK, Mordue DG. 2010. Toxoplasma gondii: determinants of tachyzoite to bradyzoite conversion. Parasitol. Res. 107:253–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sullivan WJ, Jr, Jeffers V. 2012. Mechanisms of Toxoplasma gondii persistence and latency. FEMS Microbiol. Rev. 36:717–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Katlama C, De Wit S, O'Doherty E, Van Glabeke M, Clumeck N. 1996. Pyrimethamine-clindamycin vs. pyrimethamine-sulfadiazine as acute and long-term therapy for toxoplasmic encephalitis in patients with AIDS. Clin. Infect. Dis. 22:268–275 [DOI] [PubMed] [Google Scholar]
- 5. Sullivan WJ, Jr, Narasimhan J, Bhatti MM, Wek RC. 2004. Parasite-specific eukaryotic initiation factor-2 (eIF2) kinase required for stress-induced translation control. Biochem. J. 380:523–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Narasimhan J, Joyce BR, Naguleswaran A, Smith AT, Livingston MR, Dixon SE, Coppens I, Wek RC, Sullivan WJ., Jr 2008. Translation regulation by eukaryotic initiation factor-2 kinases in the development of latent cysts in Toxoplasma gondii. J. Biol. Chem. 283:16591–16601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wek RC, Jiang HY, Anthony TG. 2006. Coping with stress: eIF2 kinases and translational control. Biochem. Soc. Trans. 34:7–11 [DOI] [PubMed] [Google Scholar]
- 8. Joyce BR, Konrad C, Wek RC, Sullivan WJ., Jr 2011. Translation control is critical during acute and chronic stages of toxoplasmosis infection. Expert Rev. Anti Infect. Ther. 9:1–3 [DOI] [PubMed] [Google Scholar]
- 9. Konrad C, Wek RC, Sullivan WJ., Jr 2011. A GCN2-like eukaryotic initiation factor-2 kinase increases the viability of extracellular Toxoplasma gondii parasites. Eukaryot. Cell 10:1403–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Joyce BR, Queener SF, Wek RC, Sullivan WJ., Jr 2010. Phosphorylation of eukaryotic initiation factor-2α promotes the extracellular survival of obligate intracellular parasite Toxoplasma gondii. Proc. Natl. Acad. Sci. U. S. A. 107:17200–17205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang M, Joyce BR, Sullivan WJ, Jr, Nussenzweig V. 14 December 2012. Translational control in Plasmodium and Toxoplasma parasites. Eukaryot. Cell. doi:10.1128/EC.00296-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, Kaufman RJ, Ma D, Coen DM, Ron D, Yuan J. 2005. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science 307:935–939 [DOI] [PubMed] [Google Scholar]
- 13. Holmes B, Brogden RN, Heel RC, Speight TM, Avery GS. 1983. Guanabenz. A review of its pharmacodynamic properties and therapeutic efficacy in hypertension. Drugs 26:212–229 [DOI] [PubMed] [Google Scholar]
- 14. Tsaytler P, Harding HP, Ron D, Bertolotti A. 2011. Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science 332:91–94 [DOI] [PubMed] [Google Scholar]
- 15. Costa JM, Pautas C, Ernault P, Foulet F, Cordonnier C, Bretagne S. 2000. Real-time PCR for diagnosis and follow-up of Toxoplasma reactivation after allogeneic stem cell transplantation using fluorescence resonance energy transfer hybridization probes. J. Clin. Microbiol. 38:2929–2932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Neil MJ. 2011. Clonidine: clinical pharmacology and therapeutic use in pain management. Curr. Clin. Pharmacol. 6:280–287 [DOI] [PubMed] [Google Scholar]
- 17. Soete M, Camus D, Dubremetz JF. 1994. Experimental induction of bradyzoite-specific antigen expression and cyst formation by the RH strain of Toxoplasma gondii in vitro. Exp. Parasitol. 78:361–370 [DOI] [PubMed] [Google Scholar]
- 18. Radke JR, Donald RG, Eibs A, Jerome ME, Behnke MS, Liberator P, White MW. 2006. Changes in the expression of human cell division autoantigen-1 influence Toxoplasma gondii growth and development. PLoS Pathog. 2:e105 doi:10.1371/journal.ppat.0020105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, Saunders T, Bonner-Weir S, Kaufman RJ. 2001. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol. Cell 7:1165–1176 [DOI] [PubMed] [Google Scholar]
- 20. Bohne W, Heesemann J, Gross U. 1994. Reduced replication of Toxoplasma gondii is necessary for induction of bradyzoite-specific antigens: a possible role for nitric oxide in triggering stage conversion. Infect. Immun. 62:1761–1767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yuan J, Johnson RL, Huang R, Wichterman J, Jiang H, Hayton K, Fidock DA, Wellems TE, Inglese J, Austin CP, Su XZ. 2009. Genetic mapping of targets mediating differential chemical phenotypes in Plasmodium falciparum. Nat. Chem. Biol. 5:765–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang M, Mishra S, Sakthivel R, Rojas M, Ranjan R, Sullivan WJ, Jr, Fontoura BM, Menard R, Dever TE, Nussenzweig V. 2012. PK4, a eukaryotic initiation factor 2alpha (eIF2alpha) kinase, is essential for the development of the erythrocytic cycle of Plasmodium. Proc. Natl. Acad. Sci. U. S. A. 109:3956–3961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lirussi D, Matrajt M. 2011. RNA granules present only in extracellular Toxoplasma gondii increase parasite viability. Int. J. Biol. Sci. 7:960–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang M, Fennell C, Ranford-Cartwright L, Sakthivel R, Gueirard P, Meister S, Caspi A, Doerig C, Nussenzweig RS, Tuteja R, Sullivan WJ, Jr, Roos DS, Fontoura BM, Menard R, Winzeler EA, Nussenzweig V. 2010. The Plasmodium eukaryotic initiation factor-2alpha kinase IK2 controls the latency of sporozoites in the mosquito salivary glands. J. Exp. Med. 207:1465–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chow C, Cloutier S, Dumas C, Chou MN, Papadopoulou B. 2011. Promastigote to amastigote differentiation of Leishmania is markedly delayed in the absence of PERK eIF2alpha kinase-dependent eIF2alpha phosphorylation. Cell. Microbiol. 13:1059–1077 [DOI] [PubMed] [Google Scholar]
- 26. Tonelli RR, Augusto Lda S, Castilho BA, Schenkman S. 2011. Protein synthesis attenuation by phosphorylation of eIF2alpha is required for the differentiation of Trypanosoma cruzi into infective forms. PLoS One 6:e27904 doi:10.1371/journal.pone.0027904 [DOI] [PMC free article] [PubMed] [Google Scholar]