

Abstract
Lysostaphin is a peptidoglycan hydrolase secreted by Staphylococcus simulans. It can specifically lyse Staphylococcus aureus and is being tested as a novel antibacterial agent. The protein contains an N-terminal catalytic domain and a C-terminal cell wall targeting domain. Although the two domains from homologous enzymes were structurally determined, the structural organization of lysostaphin domains remains unknown. We used hydrogen/deuterium exchange mass spectrometry (H/DX-MS) and site-directed disulfide cross-linking to probe the interface between the lysostaphin catalytic and targeting domains. H/DX-MS-mediated comparison of peptides from full-length lysostaphin and the separated domains identified four peptides of lower solvent accessibility in the full-length protein. Cross-linking analysis using cysteine pair substitutions within those peptides showed that two pairs of cysteines can form disulfide bonds, supporting the domain association role of the targeted peptides. The cross-linked mutant exhibited a binding capacity to S. aureus that was similar to that of the wild-type protein but reduced bacteriolytic activity probably because of restraint in conformation. The diminished activity was further reduced with increasing NaCl concentrations that can cause contractions of bacterial peptidoglycan. The lytic activity, however, could be fully recovered by reducing the disulfide bonds. These results suggest that lysostaphin may require dynamic association of the two domains for coordinating substrate binding and target cleavage on the elastic peptidoglycan. Our study will help develop site-specific PEGylated lysostaphin to treat systemic S. aureus infections.
INTRODUCTION
Staphylococcus aureus, one of the most common human pathogens, causes a wide range of serious infectious diseases, including pneumonia, osteomyelitis, endocarditis, and sepsis (1, 2). The development of resistance to penicillin, methicillin, and vancomycin has dramatically limited the treatment options for S. aureus infections (3–5). There is an urgent need to develop novel antibacterial agents with completely different modes of action. Lysostaphin is a peptidoglycan hydrolase secreted by Staphylococcus simulans biovar staphylolyticus, as a defense mechanism against S. aureus (6). It is expressed as a 493-amino-acid (aa) preproenzyme (UniProt entry P10547) and secreted via the signal peptide (residues 1 to 23). Removal of the propeptide (residues 24 to 247) by an extra cysteine protease generates the 246-aa mature lysostaphin (Fig. 1A), which can specifically lyse S. aureus by cleaving glycylglycine bonds in the pentaglycine cross bridges of the peptidoglycan (7).
Fig 1.

(A) Schematic representation of the domain architecture of preprolysostaphin. Lysostaphin is expressed in Staphylococcus simulans as a 493-aa preproenzyme (UniProt entry P10547) and secreted via the signal peptide. Removal of the propeptide during secretion by an extra cysteine protease generates the 246-aa mature lysostaphin, which consists of an N-terminal catalytic domain (CAT), a C-terminal bacterial cell wall targeting domain (CWT), and a short linker in the middle. The CAT domain is a zinc-containing metalloprotease that cleaves glycylglycine bonds in the pentaglycine cross bridges of S. aureus. The CWT domain belongs to the SH3b family, which is necessary for accurate cell wall recognition. The CWT domain directs the CAT domain to its substrates on the surface of S. aureus. (B) Sequence alignment of lysostaphin to ALE-1 and LytM using BLAST. ALE-1 is a staphylolytic enzyme produced by S. capitis EPK1. ALE-1 (residues 119 to 362) and lysostaphin (residues 248 to 493) share 83% sequence identity and 88% similarity. LytM is an autolysin from S. aureus. The CAT domains of LytM (residues 182 to 316) and lysostaphin (residues 248 to 384) share 48% sequence identity and 63% similarity. Amino acid residues conserved in these proteins are indicated by asterisks; conserved substitutions are indicated by colons. Zinc binding sites and the active site of the CAT domain are indicated by dots and an inverted triangle, respectively.
Previous in vitro and in vivo studies have shown that lysostaphin is an effective therapeutic agent against antibiotic-resistant S. aureus (8–14). For example, lysostaphin, given as a single dose (100 mg/kg) or twice daily (30 mg/kg) for 3 days, has been shown to be more effective than vancomycin (30 mg/kg, twice daily for 3 days) for the treatment of experimental aortic valve endocarditis caused by vancomycin-intermediate S. aureus in rabbits (15). Recombinant lysostaphin has been used in clinical trials (phase III) to treat antibiotic-resistant S. aureus infections in burn wounds in China. The results of phase II clinical trials indicate that multiple doses of lysostaphin were effective in controlling S. aureus growth in burn wounds, reducing the risk of systemic infections (Y. J. Mo and Q. S. Huang, unpublished data).
The mature lysostaphin protein (27 kDa) contains two domains: an N-terminal catalytic domain (CAT) and a C-terminal bacterial cell wall targeting domain (CWT), which are connected by a short linker (Fig. 1A) (16). The CAT domain is a member of the M23 family of zinc-containing endopeptidases that cleave peptide bonds between the third and the fourth glycine residues of pentaglycine cross bridges (17), while the CWT domain specifically binds to pentaglycine cross bridges in cross-linked peptidoglycans of S. aureus (18). The CAT domain alone has lost its ability to specifically target S. aureus cells (16), indicating that CWT-mediated substrate binding is important for the specificity of lysostaphin.
ALE-1 (UniProt entry O05156), a homologue of lysostaphin, is a staphylolytic enzyme produced by Staphylococcus capitis EPK1 (19). It is expressed as a 362-aa preprotein and secreted via a signal peptide (residues 1 to 35). ALE-1 (residues 119 to 362) and lysostaphin (residues 248 to 493) share 83% sequence identity and 89% similarity (Fig. 1B). Unlike lysostaphin, ALE-1 does not require proteolytic processing to remove its propeptide (residues 36 to 118). The N-terminal propeptide is not essential for bacteriolytic activity (19). Indeed, the prolysostaphin with N-terminal propeptide is poorly active with respect to the mature form (20). Currently, there are few reports about ALE-1 as an antibacterial agent. LytM (UniProt entry O33599) is an autolysin from S. aureus with roles in cell wall turnover and daughter cell separation (21, 22). It is expressed as a 316-aa preprotein and secreted via a signal peptide (residues 1 to 23). The CAT domains of LytM (residues 182 to 316) and lysostaphin (residues 248 to 384) share 48% sequence identity and 63% similarity (Fig. 1B). The truncated form of LytM (residues 185 to 316) corresponding to the CAT domain shows higher bacteriolytic activity than the full-length protein (residues 24 to 316) (23). Compared with lysostaphin and ALE-1, LytM does not contain the CWT domain (Fig. 1B). When used as an antibacterial agent in a mouse model of staphylococcal infection, the LytM CAT domain exhibits no beneficial effect and a high degree of environmental sensitivity, which markedly differs from the characteristics of lysostaphin (24).
Crystal structures of the individual domains are available for the CAT domain of LytM (PDB code 2B13) and the CWT domain of ALE-1 (PDB code 1R77). The presumable active site of the CAT domain consists of an extended and narrow groove (25). The CWT domain belongs to the bacterial SH3-like family (SH3b) and is necessary for accurate cell wall recognition. Its substrate binding groove has been mapped by mutagenesis analysis (26, 27). The structural organization of the two domains in lysostaphin or homologous proteins, however, is still unknown. In fact, it is difficult to obtain crystals for lysostaphin, either alone or in complex with peptidoglycan analogs (26, 27).
Despite extensive characterization of lysostaphin, it remains unclear how the two domains are coordinated to efficiently cleave bacterial peptidoglycan. In this study, we identified several peptides that may be involved in domain-domain interactions between the CAT and CWT domains by hydrogen/deuterium exchange mass spectrometry (H/DX-MS), which has been widely used to study macromolecular interaction interfaces (28–30). We introduced cysteine pair substitutions into the two domains and examined the formation of disulfide bonds between those residues that might be in close proximity. Finally, we investigated whether such introduction of conformational constraint by interdomain disulfide bonds inhibited the function of lysostaphin. The results from this study will improve our understanding of the bacteriolytic mechanism of lysostaphin.
MATERIALS AND METHODS
Protein expression and purification.
In order to express the mature lysostaphin and the separated domains in Escherichia coli, the synthesized gene encoding the mature lysostaphin protein (residues 248 to 493), the CAT domain (residues 248 to 387), or the CWT domain (residues 391 to 493) is flanked by 5′ NcoI containing the OmpA signal peptide-coding sequences and 3′ HindIII. Each DNA fragment was inserted into a pET28a vector and expressed in Escherichia coli BL21(DE3) (Novagen). Each recombinant protein was secreted via the OmpA signal peptide and sequentially purified by cation exchange (Capto MMC) and gel filtration (Sephadex G-50) chromatography. The purified proteins were stored at −20°C. Recombinant proteins were at least 95% pure, as judged by SDS-PAGE. Site-directed cysteine mutations were generated by PCR according to the standard procedure of the QuikChange lightning site-directed mutagenesis kit (Stratagene). All mutants were verified by DNA sequencing (Invitrogen).
Bacteriolytic activity assays.
The bacteriolytic activities of the proteins were determined using a modified turbidity assay as described previously (6, 31). S. aureus ATCC 6538 was grown to mid-log phase (optical density at 600 nm [OD600], about 0.5) and harvested by centrifugation (5,000 × g, 5 min). After washing twice with 10 mM phosphate-buffered saline (PBS) (pH 7.2), the cells were resuspended in PBS with a different concentration of NaCl to a final OD600 of 1.0. The turbidity assay was performed by adding 20 μl of the recombinant protein and 2 ml of the cell suspension to a 3-ml cuvette and then monitoring the decline in optical density at 600 nm at 1-min intervals (total of 15 min). Bacteriolytic activity assays were carried out at 37°C. All kinetic experiments were performed in triplicate. For testing the bacteriolytic activity of a double-cysteine mutant under reducing conditions, the sample was incubated with 20 mM dithiothreitol (DTT) for 5 min at room temperature, and 3 mM DTT was added to the assay buffer to prevent the formation of the interdomain disulfide bond.
Affinity binding assays.
Mid-log-phase S. aureus ATCC 6538 was collected and washed twice with 10 mM PBS (pH 7.2). The cells (1 mg [wet weight]) were resuspended in 90 μl of 10 mM PBS with 150 mM NaCl. Samples (10 μl) of the recombinant proteins (1.25 to 40 μM) were added to the cell suspension and incubated for 5 min with gentle shaking at 10°C. The binding experiments were carried out at a low temperature to reduce the bacterial lysis. The cells were pelleted and washed with 400 μl of 10 mM PBS with 150 mM NaCl. The supernatant containing unbound protein was collected and quantified using our previously described enzyme-linked immunosorbent assay (ELISA) method (32). Briefly, 96-well microtiter plates were coated overnight at 4°C with 4 ng/ml of rabbit anti-lysostaphin polyclonal antibody. After blocking with 1% bovine serum albumin (BSA), the plates were incubated with the recombinant proteins and the resulting supernatants. After washing, the plates were incubated for 1 h at 37°C with the horseradish peroxidase (HRP)-conjugated polyclonal antibody. HRP substrate (o-phenylenediamine dihydrochloride [OPD]) was added, and the mixture was incubated at room temperature until color developed (∼30 min). The standard curve was obtained by plotting the absorbance at A490 against each recombinant protein with a known concentration. The unknown samples were determined by the standard curve. The amount of bound protein was calculated as the total protein minus the unbound protein measured. These experiments were repeated three times.
Hydrogen/deuterium exchange mass spectrometry.
Purified proteins (1 μl; 10 mg/ml) were diluted in D2O buffer (19 μl, 50 mM Tris, pD 7.0). The samples were incubated at 4°C for 30, 60, 300, 900, and 10,000 s and quenched with 20 μl of 0.45% trifluoroacetic acid (TFA) at 0°C before being flash frozen in liquid nitrogen. Deuterated proteins were thawed and digested for 5 min with agarose-immobilized pepsin slurry (Thermo Fisher Scientific) at 0°C. The digested samples (0.5 μl each) were mixed with a matrix solution [0.5 μl; 5 mg/ml of (R)-cyano-4-hydroxycinnamic acid in 1:1:1 acetonitrile–ethanol–0.1% TFA, pH 2.5] and 0.5 μl of the mixed samples was spotted on a matrix-assisted laser desorption ionization (MALDI) plate. After the spots were dried in a vacuum desiccator, the MALDI plate was immediately loaded into an UltrafleXtreme mass spectrometer working in linear mode. Mass spectra were acquired (three samples from each exchange time point) and analyzed with flexAnalysis 3.3 (Bruker Daltonics). The centroid value of the isotopic envelope of each peptide (intensity-weighted average of m/z values) was determined with MagTran 1.03 (33). All experiments were carried out under nearly identical conditions, and the data were not adjusted for back exchange.
Peptide identification.
Lysostaphin (1 mg/ml) was diluted in 0.2% TFA (1:1) and digested for 5 min with agarose-immobilized pepsin slurry (Thermo Fisher Scientific) at 0°C. The digested peptides were loaded onto an ultraperformance liquid chromatography (UPLC) system and separated through a C18 trap (2 mm by 1 cm; Thermo Fisher Scientific, San Jose, CA) and a C18 column (0.1 mm by 10 mm; Hypersil Gold, Thermo Electron) over a 15-min linear gradient of 5 to 50% CH3CN–0.3% formic acid. The separated peptides were electrosprayed directly into a high-resolution orbitrap mass spectrometer (LTQ-Orbitrap XL, Thermo Electron, San Jose, CA). The total ion intensities of the peptides were detected in the m/z range of 300 to 2,000. Mass scanning was followed by collision-induced dissociation to acquire a tandem mass spectrum (MS/MS). The MS/MS *.raw data files were converted into *.mgf files, which were submitted to Mascot (Matrix Science, London, United Kingdom) for peptide identification. The MS/MS MASCOT search was also performed against a decoy (reverse) sequence, and ambiguous identifications were ruled out. The MS/MS spectra of all of the peptide ions from the MASCOT search were further manually inspected, and only those that were verifiable were used in the coverage. Peptides used for H/DX analysis had high MASCOT scores (greater than 20) and signal-to-noise ratios in matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS).
Homology modeling of the CAT and CWT domains.
We employed homology modeling to predict the three-dimensional (3D) models of the lysostaphin CAT and CWT domains using the crystal structures of the LytM CAT domain (residues 185 to 315) (PDB code 2B13) and the ALE-1 CWT domain (residues 270 to 362) (PDB code 1R77) as templates. The two structures share about 63% and 89% amino acid sequence similarities, respectively, with the lysostaphin domains (see Fig. S1 in the supplemental material). Initial models of the CAT and CWT domains were built using MODELLER (34). All molecular graphics were created using PyMOL (35).
RESULTS
H/DX-MS analysis.
H/DX-MS has been broadly used to study molecular interactions (28–30, 36–38). To explore the potential lysostaphin CAT-CWT interface, we tried to use H/DX-MS to identify lysostaphin peptides of lower solvent accessibility in the full-length protein in comparison with those in the separated CAT and CWT domains. To perform the H/DX-MS experiment, we generated recombinant proteins for lysostaphin and the separated domains (Fig. 2) and tested the biochemical activities of the proteins to ensure that they are functional. The CAT domain was active by itself at the low salt concentrations, although the activity was about 3-fold lower than that of the full-length lysostaphin (Fig. 3A). Further analysis showed that the bacteriolytic activity of the CAT domain was dramatically reduced by the presence of NaCl, which had little effect on the full-length lysostaphin (Fig. 3B). In contrast, the CWT domain alone displayed similarly strong binding to S. aureus under 150 mM NaCl compared with the full-length lysostaphin protein in our affinity assays using live cells (Fig. 3C).
Fig 2.
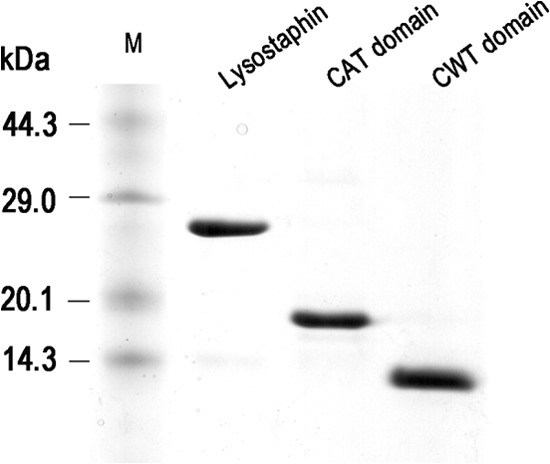
SDS-PAGE analysis of recombinant lysostaphin, CAT domain, and CWT domain purified by cation exchange (Capto MMC) and gel filtration (Sephadex G-50) chromatography. M, protein molecular mass marker (TaKaRa). The gel was stained with Coomassie brilliant blue.
Fig 3.

Functional analysis of the purified recombinant proteins. (A) The bacteriolytic activities of the recombinant CAT domain (triangles) and full-length lysostaphin (squares) were tested on mid-log-phase cultures of S. aureus ATCC 6538 suspended in 10 mM PBS (pH 7.2). The assays were carried out at 37°C. The decline in OD600 was monitored at 1-min intervals. (B) Effects of NaCl on the lytic activities of the recombinant CAT domain and full-length lysostaphin. (C) Binding affinities of the recombinant CWT domain and full-length lysostaphin. The recombinant proteins were added to S. aureus suspended in PBS with 150 mM NaCl. The binding experiments were carried out at a low temperature (10°C) to reduce the bacterial lysis. After centrifugation, the supernatants containing unbound proteins were collected and quantified using ELISA. The amount of bound protein was calculated as the total protein minus the unbound protein. These results, presented as mean ± standard error of the mean, are averages of results for three replicates.
To identify possible peptide segments of lysostaphin produced in the H/DX-MS experiments, the protein was digested with agarose-immobilized pepsin, and the peptides were separated by UPLC and analyzed by electrospray ionization mass spectrometry. As shown in Fig. 4A, 14 peptides (including those with overlapping sequences) were analyzed. Those peptides cover 90% of the CAT domain and 58% of the CWT domain.
Fig 4.

H/DX-MS analysis of lysostaphin. (A) Amino acid sequence of lysostaphin. Secondary structures defined by the previous homologous CAT and CWT structures (PDB codes 2B13 and 1R77) were assigned above the lysostaphin sequence. The 14 peptides analyzed in H/DX-MS were noted below the sequence. These peptides cover 90% of the CAT domain and 58% of the CWT domain, respectively. (B) MALDI-TOF mass spectra of peptide 317–339 (2788.35 m/z) during the time courses (30, 60, 300, 900, and 10,000 s) of deuterium incorporation. The H/DX rate of this peptide from lysostaphin was significantly lower than that of the same peptide from the CAT domain. (C) Time courses (30, 60, 300, and 900 s) of deuterium incorporation for four peptides. The H/DX rates for the peptides from the full-length lysostaphin are lower than those for the peptides from the separated CAT or CWT domain. The data points, presented as mean ± standard error of the mean, are averages of results for three replicates. The number of deuterium incorporation is a relative value, because adjustment for the back exchange was not performed.
Four peptides (spanning amino acids 317 to 339, 248 to 260, 454 to 473, and 474 to 488 and here referred to in the form peptide XXX–XXX, where XXX–XXX is the range of amino acids) from full-length lysostaphin exhibited lower H/DX (hydrogen-deuterium exchange) rates than the same peptides from the separated domains (Fig. 4B and C; see Fig. S2 in the supplemental material). In these four peptides, peptides 317–339 and 454–473 were slightly less protected than the other two peptides (Fig. 4C). Among the remaining 10 peptides of no significant change in the H/DX rates (see Fig. S2), peptides 316–330 and 317–330 overlap peptide 317–339, allowing us to assign the residue 331 to 339 region responsible for the H/DX rate change. Collectively, the result indicated that at least four peptides were protected in the overall structure of lysostaphin.
Based on the crystal structures of the CAT domain of LytM (PDB code 2B13) and the CWT domain of ALE-1 (PDB code 1R77), the 3D models of the lysostaphin CAT (residues 248 to 384) and CWT (residues 402 to 493) domains were constructed using the homology modeling approach (Fig. 5; see Fig. S3 in the supplemental material). The flexible linker (residues 385 to 401) between the two domains was omitted because of the lack of corresponding template segments in the mentioned crystal structures. When superimposed, the predicted and experimentally determined structures are similar (root mean square deviation [RMSD], less than 0.5 Å) (see Fig. S3).
Fig 5.
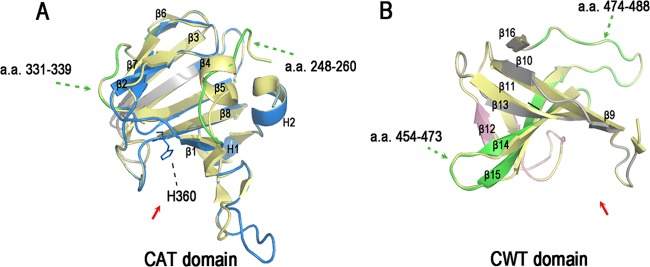
3D models of the lysostaphin CAT and CWT domains. (A) Homology model of the lysostaphin CAT domain (blue). The model was built using the crystal structure of the LytM catalytic domain (PDB code 2B13; yellow) as a template. H360 represents the active site of the CAT domain. (B) Homology model of the lysostaphin CWT domain (pink). The model was built using the crystal structure of the ALE-1 cell wall targeting domain (PDB code 1R77; yellow). The peptides with lower H/DX levels in lysostaphin (green) mapped onto the 3D models of the CAT domain and CWT domain, respectively. The peptides colored gray are not covered in the H/DX-MS analysis. The active grooves are indicated by red arrows. The secondary structures are labeled. These figures were created with PyMOL.
We then mapped the four peptides onto the 3D models of the CAT and CWT domains. Peptides 248–260 and 331–339 were mapped onto the CAT domain, while peptides 454–473 and 474–488 belong to the CWT domain (Fig. 5A and B). As lysostaphin exists as a monomer in solution based on gel filtration chromatography (Sephadex G-50), those peptides showing lower H/DX rates in full-length lysostaphin may be involved in interdomain interactions.
Site-directed disulfide cross-linking analysis within the low-H/DX peptides.
We used site-directed disulfide cross-linking to probe those peptides in close proximity. Lysostaphin does not contain cysteine. If two cysteines introduced in the two peptides, separately located in the CAT and CWT domains, were in close proximity because of domain association, they would have higher chances to form a disulfide bond. First, we carried out single-cysteine substitutions (Q256C, L258C, N260C, K332C, K336C, D339C, T464C, R470C, T477C, T482C, and V487C) within the low-H/DX peptides, which were surface exposed and not close to the catalytic and substrate-binding grooves of the CAT and CWT domains. To ensure that the introduced cysteines did not alter the protein fold, each of the single-cysteine mutants was tested for activity using a turbidity assay. All of the single-cysteine mutants exhibited bacteriolytic activity that was similar to that of the wild-type protein except for L258C, D339C, and R470C (Fig. 6A). The lack of activity in these three single-cysteine mutants could be due to large structural changes that would affect their catalytic or binding capacities.
Fig 6.

Analysis of interdomain disulfide bond formation. (A) The bacteriolytic activities of single-cysteine substitutions. (B) SDS-PAGE (with 8% Tricine) of purified double-cysteine substitutions with and without DTT. The interdomain disulfide bonds were found only in K332C/T464C and K336C/T464C, seen as bands of faster mobility. (C) Predicted structural organization of the lysostaphin CAT domain (blue) and CWT domain (pink). The peptides with lower H/DX levels in lysostaphin are colored green. Locations of the residues mutated to cysteines are colored red. The putative disulfide bonds and the interdomain linker are represented by black and yellow dashed lines, respectively.
We further engineered 16 cysteine pair substitutions into the two domains. The purified proteins were incubated with and without reducing agents and analyzed by 8% Tricine SDS-PAGE. Proteins with interdomain disulfide bonds would be presumably more compact in structure and move faster than the reduced forms. The K332C/T464C mutant indeed formed a faster-moving band in the absence of DTT (Fig. 6B). Meanwhile, a significant portion of the K336C/T464C mutant also formed an interdomain disulfide bond. In contrast, DTT did not alter the electrophoretic mobility of the other mutants. These suggest that the K332/T464 paired residues should have chances to be close to each other when lysostaphin adopts certain conformations (Fig. 6C). The result strongly supported the domain association role of the peptides 331–339 and 454–473.
Bacteriolytic activity of the conformationally restrained enzyme, the K332C/T464C mutant.
We next tested whether introduction of conformational constraint by an interdomain disulfide bond affected the bacteriolytic activity against S. aureus. As shown in Fig. 7A, the K332C/T464C mutant retained only ∼25% of the bacteriolytic activity of the wild-type protein (Fig. 3A). We further found that the bacteriolytic activity of the mutant was gradually reduced with increasing NaCl concentration but not fully abolished (Fig. 7B). The loss of activity was not caused by diminished substrate binding, as the mutant protein exhibited very similar binding capacity to S. aureus with the wild-type protein (Fig. 3C and 7C). To confirm that the loss of bacteriolytic activity was caused by disulfide linkage, we reduced the disulfide linkage with 20 mM DTT and added 3 mM DTT to the assay buffer to prevent the formation of the interdomain disulfide bond during turbidity assay. As expected, after DTT treatment, the bacteriolytic activity of the K332C/T464C mutant was fully recovered (Fig. 7D). In contrast, the wild-type protein without cysteine cross-link was almost not affected by DTT treatment (Fig. 7D). These results suggested that lysostaphin may require dynamic association of the two domains for the bacterial cell wall cleavage. The conformationally restrained enzyme likely could not efficiently access the cleavage sites, as it showed no change in binding to S. aureus (Fig. 7C).
Fig 7.

Functional analysis of the conformationally restrained enzyme, the K332C/T464C mutant. (A) Bacteriolytic activity of the K332C/T464C mutant (◆) and lysostaphin (■) against S. aureus suspended in 10 mM PBS. (B) Effect of NaCl on the lytic activity of the K332C/T464C mutant and lysostaphin. (C) Binding affinity of the K332C/T464C mutant (◆) and lysostaphin (■) to S. aureus cells suspended in PBS with 150 mM NaCl. (D) Bacteriolytic activities of the K332C/T464C mutant and lysostaphin with and without DTT. When treated, the proteins were mixed with 20 mM DTT for 5 min at room temperature. The assay buffer also contains 3 mM DTT to prevent the formation of the interdomain disulfide bonds. S. aureus cells were suspended in PBS with and without 150 mM NaCl. These results, presented as mean ± standard error of the mean, are averages of results for three replicates.
DISCUSSION
Lysostaphin is an effective anti-staphylococcal therapeutic agent that has been widely reported (13, 39). Our turbidity assay with live S. aureus cells showed that the lysostaphin CAT domain alone had strong lytic activity against S. aureus in low-ionic-strength buffer (Fig. 3A), whereas its bacteriolytic effect was dramatically reduced even in the presence of 50 mM NaCl (Fig. 3B). The CAT domain has an isoelectric point of 9.2 and can probably bind S. aureus via nonspecific electrostatic interactions leading to cell wall cleavage. It is possible that NaCl affected this nonspecific association, and therefore reduced cleavage, which is similarly found with the salt sensitivity of cationic antimicrobial peptide (40). In contrast, the efficacy of lysostaphin was less affected by the ionic strength (Fig. 3B). This result is in agreement with a recent report showing that lysostaphin was effective in the model of chronic S. aureus-infected eczema, while the catalytic domain of LytM was not (24). The most crucial factor is their different responses to environmental ionic strength. These results clearly show that CWT-mediated substrate binding is critical for the lytic activity of the CAT domain.
The CAT domain of LytM and CWT domain of ALE-1 were structurally determined (25, 26), but crystallization of the full-length lysostaphin or homologous enzymes has so far been unsuccessful. We think that the two domains of lysostaphin should adopt certain conformations for efficient substrate binding and target peptidoglycan cleavage. In order to probe the domain interface of the functionally important protein conformation, we performed H/DX-MS in combination with site-directed disulfide cross-linking analysis. H/DX-MS-mediated comparison of peptides from full-length lysostaphin and the separated domains identified four segments (248 to 260, 331 to 339, 454 to 473, and 474 to 488) of lower solvent accessibility in the full-length protein (Fig. 4), suggesting that the protein regions covered by those peptides might be involved in CAT-CWT domain associations. Two cysteine pairs engineered within the peptides 331-339 and 454-473 could form disulfide bonds (Fig. 6B), strongly supporting that they could be arranged in close proximity. The detected cysteine cross-links should not be caused by protein misfolding, as the bacteriolytic activities of those single-cysteine mutants were similar to that of the wild-type protein (Fig. 6A).
Our functional assay revealed that the conformationally restrained enzyme, the K332C/T464C mutant, showed binding capacity to S. aureus that was very similar to that of the wild-type protein (Fig. 7C) but significantly lower bacteriolytic activity (Fig. 7A), especially under high ionic strength (Fig. 7B). The lytic activity, however, could be recovered by adding DTT to disrupt the disulfide bond and remove the conformational restriction (Fig. 7D). These results suggested that lysostaphin may have a dynamic interface between the CAT and CWT domains for bacteriolytic activity. The elastic structures of bacterial cell wall peptidoglycan might require a conformationally dynamic enzyme for coordinated substrate binding and target cleavage.
Peptidoglycan forms a complex three-dimensional architecture to maintain cell shape and protect cells against damages from outside, and the network is always made of glycan strands cross-linked by peptides (41–43). In S. aureus, short glycan strands, with a mean length of only 6 disaccharides (N-acetylmuramic acid-N-acetylglucosamine), are attached by peptide stems. The peptide stems are interconnected by pentaglycine cross bridges and form a zipper-like structure with a high degree of cross-linking (Fig. 8A). It is notable that the peptidoglycan is a flexible structure. The isolated staphylococcal cell walls can expand or contract 2-fold in response to changes in environmental ionic strength (44). As the contraction degree of live S. aureus cells increases, the peptidoglycan net becomes more relaxed. Then the narrow channels between two parallel glycan strands become winding; the relative distances between two pentaglycine cross bridges become variable. Lysostaphin, however, can easily adapt to the changing of the peptidoglycan net, as it possesses a dynamic interface and can explore multiple conformations by relative movements between the two domains (Fig. 8B). However, conformational constraints, such as cysteine cross-linking, can limit this capacity, resulting in significantly reduced performance (Fig. 7A and B). Similar structural dynamics have recently been described for the Atl amidase, an autolysin of S. epidermidis, which is composed of the catalytic domain N-acetylmuramyl-l-alanine amidase and the targeting domain repeats R1 and R2, specifically binding to the membrane-bound lipoteichoic acid (45). The interdomain flexibility of this autolysin allows its catalytic domain to access a large number of cleavage sites, thereby increasing the radius of action.
Fig 8.
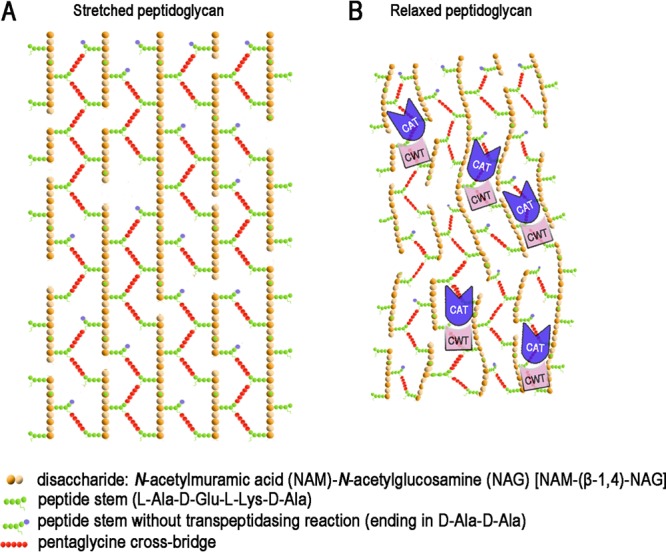
Schematic model for bacteriolytic mechanism of lysostaphin. Models illustrating the stretched (A) and relaxed (B) peptidoglycan of S. aureus. The proposed bacterial cell wall contains parallel glycan strands linked by peptide stems and pentaglycine cross bridges. The cell wall units and two lysostaphin domains are color coded. The dynamic interface enables lysostaphin to explore different conformational states on the elastic network of bacterial cell wall peptidoglycan, allowing the CAT domain to efficiently access and cleave the pentaglycine cross bridges.
Lysostaphin has been extensively studied as a therapeutic agent, with either topical or systemic administration, for the treatment of S. aureus infections (8–13). Now, recombinant lysostaphin is being effectively developed as a new topical antibacterial agent to control S. aureus infections in burn wounds in China (phase III clinical trials). However, a short half-life, immunogenicity, and enzymatic degradation in vivo have hampered the application of lysostaphin. Protein PEGylation, the covalent attachment of PEG chains to protein, is an effective method to reduce or overcome these problems (46, 47). It has been shown that PEGylation of lysostaphin, conjugating PEG to the ε-amino groups of lysine residues, improves in vivo half-life and reduces immunogenicity, while bacteriolytic activity of PEGylated lysostaphin is diminished (48). Nonspecific reaction of PEG with lysostaphin at different sites produces a number of isomers, most of which have decreased binding affinity to the big network of cell wall peptidoglycan (see Fig. S4A in the supplemental material). Site-specific PEGylation can minimize the loss of biological activity and avoid modification isomers (49). PEGylation at the thiol group of cysteine is the most specific method. As lysostaphin does not contain cysteine, site-specific PEGylation would require introducing this residue at surface-exposed positions that do not overlap the surfaces important for the enzymatic activity. Our study has revealed new structural elements that are involved in the bacterial cell wall cleavage and therefore will help in the design of PEGylated lysostaphin without losing activity (Fig. S4B).
In conclusion, lysostaphin CAT and CWT domains are dynamically associated with each other. The flexible interface likely enables lysostaphin to adapt the elastic network of cell wall peptidoglycan and therefore enhances the lytic activity against S. aureus. One important goal of our study in the future is to develop site-specific PEGylated lysostaphin as a new antibacterial agent. The results presented here will help select appropriate PEGylation sites without interfering with the activity of lysostaphin.
ACKNOWLEDGMENTS
We thank Guoquan Yan at Fudan University for technical assistance on mass spectrometry.
This work was supported by the major scientific and technological specialized project of China for Significant New Formulation of New Drugs (2008ZX09101-032) and the Jiangsu provincial science and technology achievement transformation project (BA2010089).
Footnotes
Published ahead of print 4 February 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02348-12.
REFERENCES
- 1. Lowy FD. 1998. Medical progress—Staphylococcus aureus infections. N. Engl. J. Med. 339:520–532 [DOI] [PubMed] [Google Scholar]
- 2. Whitby M, McLaws ML, Berry G. 2001. Risk of death from methicillin-resistant Staphylococcus aureus bacteraemia: a meta-analysis. Med. J. Aust. 175:264–267 [DOI] [PubMed] [Google Scholar]
- 3. Dombrowski JC, Winston LG. 2008. Clinical failures of appropriately-treated methicillin-resistant Staphylococcus aureus infections. J. Infect. 57:110–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lodise TP, Graves J, Evans A, Graffunder E, Helmecke M, Lomaestro BM, Stellrecht K. 2008. Relationship between vancomycin MIC and failure among patients with methicillin-resistant Staphylococcus aureus bacteremia treated with vancomycin. Antimicrob. Agents Chemother. 52:3315–3320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chambers HF, Deleo FR. 2009. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat. Rev. Microbiol. 7:629–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schindler CA, Schuhardt VT. 1964. Lysostaphin: a new bacteriolytic agent for the staphylococcus. Proc. Natl. Acad. Sci. U. S. A. 51:414–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Browder HP, Zygmunt WA, Young JR, Tavormina PA. 1965. Lysostaphin: enzymatic mode of action. Biochem. Biophys. Res. Commun. 19:383–389 [DOI] [PubMed] [Google Scholar]
- 8. Satishkumar R, Sankar S, Yurko Y, Lincourt A, Shipp J, Heniford BT, Vertegel A. 2011. Evaluation of the antimicrobial activity of lysostaphin-coated hernia repair meshes. Antimicrob. Agents Chemother. 55:4379–4385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Belyansky I, Tsirline VB, Martin TR, Klima DA, Heath J, Lincourt AE, Satishkumar R, Vertegel A, Heniford BT. 2011. The addition of lysostaphin dramatically improves survival, protects porcine biomesh from infection, and improves graft tensile shear strength. J. Surg. Res. 171:409–415 [DOI] [PubMed] [Google Scholar]
- 10. Miao J, Pangule RC, Paskaleva EE, Hwang EE, Kane RS, Linhardt RJ, Dordick JS. 2011. Lysostaphin-functionalized cellulose fibers with antistaphylococcal activity for wound healing applications. Biomaterials 32:9557–9567 [DOI] [PubMed] [Google Scholar]
- 11. Cui FY, Li GD, Huang JJ, Zhang JE, Lu M, Lu WY, Huan JN, Huang QS. 2011. Development of chitosan-collagen hydrogel incorporated with lysostaphin (CCHL) burn dressing with anti-methicillin-resistant Staphylococcus aureus and promotion wound healing properties. Drug Deliv. 18:173–180 [DOI] [PubMed] [Google Scholar]
- 12. Placencia FX, Kong L, Weisman LE. 2009. Treatment of methicillin-resistant Staphylococcus aureus in neonatal mice: lysostaphin versus vancomycin. Pediatr. Res. 65:420–424 [DOI] [PubMed] [Google Scholar]
- 13. Kumar JK. 2008. Lysostaphin: an antistaphylococcal agent. Appl. Microbiol. Biotechnol. 80:555–561 [DOI] [PubMed] [Google Scholar]
- 14. Yang XY, Li CR, Lou RH, Wang YM, Zhang WX, Chen HZ, Huang QS, Han YX, Jiang JD, You XF. 2007. In vitro activity of recombinant lysostaphin against Staphylococcus aureus isolates from hospitals in Beijing, China. J. Med. Microbiol. 56:71–76 [DOI] [PubMed] [Google Scholar]
- 15. Patron RL, Climo MW, Goldstein BP, Archer GL. 1999. Lysostaphin treatment of experimental aortic valve endocarditis caused by a Staphylococcus aureus isolate with reduced susceptibility to vancomycin. Antimicrob. Agents Chemother. 43:1754–1755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Baba T, Schneewind O. 1996. Target cell specificity of a bacteriocin molecule: a C-terminal signal directs lysostaphin to the cell wall of Staphylococcus aureus. EMBO J. 15:4789–4797 [PMC free article] [PubMed] [Google Scholar]
- 17. Schneewind O, Fowler A, Faull KF. 1995. Structure of the cell wall anchor of surface proteins in Staphylococcus aureus. Science 268:103–106 [DOI] [PubMed] [Google Scholar]
- 18. Grundling A, Schneewind O. 2006. Cross-linked peptidoglycan mediates lysostaphin binding to the cell wall envelope of Staphylococcus aureus. J. Bacteriol. 188:2463–2472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sugai M, Fujiwara T, Akiyama T, Ohara M, Komatsuzawa H, Inoue S, Suginaka H. 1997. Purification and molecular characterization of glycylglycine endopeptidase produced by Staphylococcus capitis EPK1. J. Bacteriol. 179:1193–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thumm G, Gotz F. 1997. Studies on prolysostaphin processing and characterization of the lysostaphin immunity factor (Lif) of Staphylococcus simulans biovar staphylolyticus. Mol. Microbiol. 23:1251–1265 [DOI] [PubMed] [Google Scholar]
- 21. Ramadurai L, Jayaswal RK. 1997. Molecular cloning, sequencing, and expression of lytM, a unique autolytic gene of Staphylococcus aureus. J. Bacteriol. 179:3625–3631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ramadurai L, Lockwood KJ, Nadakavukaren MJ, Jayaswal RK. 1999. Characterization of a chromosomally encoded glycylglycine endopeptidase of Staphylococcus aureus. Microbiology 145:801–808 [DOI] [PubMed] [Google Scholar]
- 23. Odintsov SG, Sabala I, Marcyjaniak M, Bochtler M. 2004. Latent LytM at 1.3 Å resolution. J. Mol. Biol. 335:775–785 [DOI] [PubMed] [Google Scholar]
- 24. Sabala I, Jonsson IM, Tarkowski A, Bochtler M. 2012. Anti-staphylococcal activities of lysostaphin and LytM catalytic domain. BMC Microbiol. 12:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Firczuk M, Mucha A, Bochtler M. 2005. Crystal structures of active LytM. J. Mol. Biol. 354:578–590 [DOI] [PubMed] [Google Scholar]
- 26. Lu J, Fujiwara T, Komatsuzawa H, Sugai M, Sakon J. 2006. Cell wall-targeting domain of glycylglycine endopeptidase distinguishes among peptidoglycan cross-bridges. J. Biol. Chem. 281:549–558 [DOI] [PubMed] [Google Scholar]
- 27. Hirakawa H, Akita H, Fujiwara T, Sugai M, Kuhara S. 2009. Structural insight into the binding mode between the targeting domain of ALE-1 (92AA) and pentaglycine of peptidoglycan. Protein Eng. Des. Sel. 22:385–391 [DOI] [PubMed] [Google Scholar]
- 28. Ling J, Shima CH, Schriemer DC, Schryvers AB. 2010. Delineating the regions of human transferrin involved in interactions with transferrin binding protein B from Neisseria meningitidis. Mol. Microbiol. 77:1301–1314 [DOI] [PubMed] [Google Scholar]
- 29. Low D, Frecer V, Le Saux A, Srinivasan GA, Ho B, Chen JZ, Ding JL. 2010. The molecular interfaces of the galactose-binding protein tectonin domains in host-pathogen interaction. J. Biol. Chem. 285:9898–9907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Roberts VA, Pique ME, Hsu S, Li S, Slupphaug G, Rambo RP, Jamison JW, Liu T, Lee JH, Tainer JA, Ten EL, Woods VJ. 2012. Combining H/D exchange mass spectroscopy and computational docking reveals extended DNA-binding surface on uracil-DNA glycosylase. Nucleic Acids Res. 40:6070–6081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kusuma CM, Kokai-Kun JF. 2005. Comparison of four methods for determining lysostaphin susceptibility of various strains of Staphylococcus aureus. Antimicrob. Agents Chemother. 49:3256–3263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Huang QS, Zhang JE, Wu HY, Mo YJ. 2007. Detection of recombinant lysostaphin using antibody sandwich enzyme-linked immunoadsorbent assay. Sheng Wu Gong Cheng Xue Bao 23:117–121 (In Chinese.) [DOI] [PubMed] [Google Scholar]
- 33. Zhang Z, Marshall AG. 1998. A universal algorithm for fast and automated charge state deconvolution of electrospray mass-to-charge ratio spectra. J. Am. Soc. Mass Spectrom. 9:225–233 [DOI] [PubMed] [Google Scholar]
- 34. Sali A, Potterton L, Yuan F, van Vlijmen H, Karplus M. 1995. Evaluation of comparative protein modeling by MODELLER. Proteins 23:318–326 [DOI] [PubMed] [Google Scholar]
- 35. DeLano WL. 2002. The PyMOL Molecular Graphics System. DeLano Scientific, Palo Alto, CA: http://www.pymol.org/ [Google Scholar]
- 36. Anand GS, Law D, Mandell JG, Snead AN, Tsigelny I, Taylor SS, Ten EL, Komives EA. 2003. Identification of the protein kinase A regulatory RIα-catalytic subunit interface by amide H/2H exchange and protein docking. Proc. Natl. Acad. Sci. U. S. A. 100:13264–13269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhou B, Zhang JL, Liu SJ, Reddy S, Wang F, Zhang ZY. 2006. Mapping ERK2-MKP3 binding interfaces by hydrogen/deuterium exchange mass spectrometry. J. Biol. Chem. 281:38834–38844 [DOI] [PubMed] [Google Scholar]
- 38. Cao J, Hsu YH, Li S, Woods VJ, Dennis EA. 2013. Structural basis of specific interactions of Lp-PLA2 with HDL revealed by hydrogen deuterium exchange mass spectrometry. J. Lipid Res. 54:127–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Szweda P, Schielmann M, Kotlowski R, Gorczyca G, Zalewska M, Milewski S. 2012. Peptidoglycan hydrolases—potential weapons against Staphylococcus aureus. Appl. Microbiol. Biotechnol. 96:1157–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yu HY, Tu CH, Yip BS, Chen HL, Cheng HT, Huang KC, Lo HJ, Cheng JW. 2011. Easy strategy to increase salt resistance of antimicrobial peptides. Antimicrob. Agents Chemother. 55:4918–4921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dmitriev BA, Toukach FV, Holst O, Rietschel ET, Ehlers S. 2004. Tertiary structure of Staphylococcus aureus cell wall murein. J. Bacteriol. 186:7141–7148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vollmer W, Blanot D, de Pedro MA. 2008. Peptidoglycan structure and architecture. FEMS Microbiol. Rev. 32:149–167 [DOI] [PubMed] [Google Scholar]
- 43. Vollmer W, Seligman SJ. 2010. Architecture of peptidoglycan: more data and more models. Trends Microbiol. 18:59–66 [DOI] [PubMed] [Google Scholar]
- 44. Ou LT, Marquis RE. 1970. Electromechanical interactions in cell walls of gram-positive cocci. J. Bacteriol. 101:92–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zoll S, Schlag M, Shkumatov AV, Rautenberg M, Svergun DI, Gotz F, Stehle T. 2012. Ligand-binding properties and conformational dynamics of autolysin repeat domains in staphylococcal cell wall recognition. J. Bacteriol. 194:3789–3802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Harris JM, Chess RB. 2003. Effect of PEGylation on pharmaceuticals. Nat. Rev. Drug Discov. 2:214–221 [DOI] [PubMed] [Google Scholar]
- 47. Pasut G, Veronese FM. 2012. State of the art in PEGylation: the great versatility achieved after forty years of research. J. Control. Release 161:461–472 [DOI] [PubMed] [Google Scholar]
- 48. Walsh S, Shah A, Mond J. 2003. Improved pharmacokinetics and reduced antibody reactivity of lysostaphin conjugated to polyethylene glycol. Antimicrob. Agents Chemother. 47:554–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang C, Yang XL, Yuan YH, Pu J, Liao F. 2012. Site-specific PEGylation of therapeutic proteins via optimization of both accessible reactive amino acid residues and PEG derivatives. BioDrugs 26:209–215 [DOI] [PubMed] [Google Scholar]