

Abstract
Serologic detection of Toxoplasma gondii IgG antibodies is widely accepted as a means to determine immune status and susceptibility to Toxoplasma infection during pregnancy. However, current commercial kits present some drawbacks, such as a requirement for whole-parasite antigen preparation or interassay variability. To address these problems, the purpose of this study was to produce a whole sequence of the recombinant T. gondii SAG1 antigen (rSAG1) to assess its diagnostic performance in Toxoplasma IgG screening and to explore a saliva-based method as a noninvasive alternative to serum-based testing. rSAG1 was expressed in recombinant bacteria as inclusion bodies, purified through one-step affinity chromatography, and refolded in native form by dialysis. A large amount was obtained, and the specific antigen immunoreactivity was confirmed by immunoblotting. Two rSAG1-based enzyme-linked immunosorbent assays (ELISAs) applied to paired serum and saliva samples were designed. The rSAG1-based ELISA evaluation consisted of testing intrinsic sensitivity and specificity of 49 serum samples from patients immune to toxoplasmosis and 42 serum samples from nonimmune controls identified by routinely used kits. To assess agreement between serum-based and saliva-based tests, the positive percent agreement (PPA) and negative percent agreement (NPA) between the 2 tests were estimated. The rSAG1 serum-based ELISA detected specific IgG with 100% sensitivity and specificity. The PPA and NPA between the serum-based and saliva-based tests varied according to the selected optical density threshold in saliva. Thus, for a selected cutoff of 0.14, the PPA was 100% and the NPA was 88.1%, whereas for a selected cutoff of 0.29, the PPA was 67.3% and the NPA was 100%.
INTRODUCTION
Toxoplasmosis is a common parasitic disease caused by the protozoan parasite Toxoplasma gondii. The infection is generally asymptomatic in immunocompetent adults, whereas intrauterine transmission of the parasite from the mother to the fetus during gestation can result in severe fetal and neonatal complications (1, 2). In Tunisia, North Africa, toxoplasmosis is a frequent event in childhood and is followed by a protective immunity against parasite reinfection. However, many women stay susceptible to toxoplasmosis at childbearing age (3, 4). Thus, most health care providers routinely screen for toxoplasmosis during the first prenatal visit in order to propose prophylactic recommendations (hygiene and diet guidelines) in nonimmune pregnant women (5) or to implement appropriate chemotherapy in case of recent primo-infection (6, 7).
Serologic confirmation of the T. gondii IgG antibody is indicative of exposure to the parasite and has become widely accepted as a means to determine the immune status and susceptibility to Toxoplasma infection. Among the various available serologic methods, the enzyme-linked immunosorbent assay (ELISA) for IgG detection is simple to perform and is commonly used. However, the type and purity of the antigen applied greatly affect its performance. Currently, many manual and automated systems are commercially available (8, 9). Most of them use whole-cell extracts of tachyzoites grown in mice or in tissue culture, which are often contaminated with extraparasitic material (10) or contain common protozoan antigens (11, 12), leading to interassay variability (13, 14, 15, 16). By the development of a second generation of more standardized diagnostic immunoassays based on specific immunodominant antigens, recombinant technology can contribute significantly to increase test performance (17). Among the several cloned genes encoding T. gondii antigens, the surface antigen 1 (SAG1) (also named P30) has proved to be a good candidate for serodiagnosis of toxoplasmosis (10, 18). In fact, it is a highly conserved antigen in most T. gondii strains examined (19, 20, 21), is extremely immunogenic, and is recognized during the acute and chronic phases of toxoplasmosis (22, 23, 24).
Nowadays, the detection of specific Toxoplasma antibodies relies on serum samples; nevertheless, blood collection remains an invasive procedure. Thus, the use of other biological fluids, such as saliva, would be more practical for Toxoplasma screening, especially under field conditions. This sampling method is safe, noninvasive, and easier and cheaper than blood sampling, and the compliance rate is high (25). In addition, specific antibodies in various infectious diseases have been sensitively and specifically detected in saliva samples collected with devices targeting the crevicular fluid, where IgG transudates are highly present (26). In regard to diagnosis of Toxoplasma infection, some researchers have suggested the usefulness of this practical sampling (27, 28). Recently, SAG1 was proposed as one of the major reactive antigens in a salivary immunoblotting test (29).
The purpose of this study was to produce a recombinant SAG1 (rSAG1) antigen using the Escherichia coli expression system, evaluate its immunoreactivity after the purification and refolding steps, and then assess the diagnostic performance of the rSAG1 ELISA for detecting specific anti-T. gondii IgG in pregnant women. The percent agreement between saliva-based and serum-based ELISAs was also estimated.
MATERIALS AND METHODS
Preparation of recombinant SAG1.
Total RNA was isolated from about 107 freshly extracted T. gondii tachyzoites (RH strain maintained in Swiss mice by intraperitoneal inoculations) in a single-step procedure using the SV total isolation system kit (Promega, Madison, WI). The first-strand cDNA was synthesized from total RNA using avian myeloblastosis virus (AMV) reverse transcriptase and oligo(dT) primer (Promega, France) according to the manufacturer's protocol. The nucleotide sequence encoding amino acids 47 to 336 of SAG1 was amplified from the cDNA, under standard conditions, using Taq DNA polymerase (Amersham Biosciences, France). According to the sag1 sequence (GenBank accession no. X14080), sense (5′-GGATCCGAATTCGGATCCCCCTCTTGTTG-3′) and antisense (5′-CACCACTCGAGCGCCACACAAGCTGCCG-3′) primers were designed with the inclusion of BamHI and XhoI restriction sites (underlined), respectively. Thirty cycles of PCR were performed as follows: denaturation at 95°C for 1 min (10 min in cycle 1), annealing at 60°C for 1 min, and polymerization at 72°C for 2 min (5 min in cycle 30). The amplified product was purified, cleaved successively with BamHI and XhoI, and inserted into the expression vector plasmid pET22b(+) (Novagen, Madison, WI), linearized previously with the same enzymes. The sag1 fragment was ligated between the pelB leader sequence and the hexahistidine tail sequence in the C terminus to facilitate purification and immunoquantification. The plasmid containing the correct sequence was introduced into E. coli Origami(DE3) cells (Novagen).
The bacteria were grown at 37°C in Luria-Bertani (LB) medium supplemented with 100 μg/ml of ampicillin, 12.5 μg/ml of tetracycline, and 15 μg/ml of kanamycin. When the optical density (OD) at 600 nm reached 0.6, the expression of a hexahistidine-tagged rSAG1 was induced with 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG) (Sigma-Aldrich, Inc., Germany), and the temperature was shifted to 28°C for 20 h. Then, the cell pellet was suspended in lysis buffer (50 mM NaH2PO4-300 mM NaCl [pH 8], 1 mg/ml lysozyme, and protease inhibitor cocktail [Roche Applied Science]), and the mixture was lysed by sonication in an ice-water mixture bath for 5 min. The soluble fraction was collected by centrifugation at 7,000 × g for 30 min at 4°C. The washed pellet containing the insoluble inclusion body protein was extracted with 8 M guanidine-HCl in extraction buffer (50 mM NaH2PO4-300 mM NaCl, pH 8) supplemented with protease inhibitor cocktail (30, 31).
The solubilized protein was purified by affinity chromatography using a nickel-nitrilotriacetic acid (Ni-NTA) agarose column (Qiagen, GmbH, Germany) under denaturing conditions according to the manufacturer's instructions. Briefly, after being washed with 5 bed volumes of extraction buffer (pH 6.3) containing 6 M urea, the protein was eluted in extraction buffer containing 6 M urea using step-by-step decreasing pHs (pH 5.9 and 4.5). Fractions containing the purified recombinant SAG1 were stored at −20°C until use.
The purified denatured SAG1 was refolded through urea gradient dialysis. The protein was loaded into a dialysis bag with a membrane molecular mass cutoff of 13,000 Da and was dialyzed against 100 volumes of buffer (50 mM NaH2PO4-300 mM NaCl [pH 8]) at 4°C over 20 h. Denaturant was slowly removed by serial equilibration buffers with decreasing urea concentrations (30, 31). The urea concentration was reduced from 6 M to 3 M, then to 1.5 M, and finally to 0 M. The protein concentration was determined using the BCA assay kit (Pierce, Rockford, IL) with bovine serum albumin (BSA) as a standard.
SDS-PAGE and Western blot analysis.
The integrity and purity of the rSAG1 were checked using electrophoresis under reducing or nonreducing conditions. rSAG1 was denatured in Laemmli buffer with heating at 95°C for 5 min. Electrophoresis was then carried out in 10% SDS-PAGE gel, and the gel was stained with Coomassie brilliant blue R-250 (32). Two Western blots were obtained to reveal the recombinant protein band. For the first Western blot under reducing conditions, rSAG1 protein was transferred onto a nitrocellulose membrane (Amersham Biosciences, France). The membrane was saturated with 5% skim milk in phosphate-buffered saline (PBS) (pH 7.4) containing 0.1% Tween 20 (PBS-T) for 30 min and was then incubated with monoclonal antipolyhistidine-peroxidase conjugate antibody (A7058; Sigma-Aldrich, Inc., Germany) diluted 1:10,000 for 2 h. After extensive washing, the immune complex was detected by enhanced chemiluminescence (ECL) (Western blotting detection reagent; Amersham). The second Western blot was obtained under nonreducing conditions; the membrane was incubated overnight at 4°C in a human reference serum sample (titer of 240 IU/ml, diluted 1:100 in PBS-T). After a washing step, it was incubated with an anti-human IgG–alkaline phosphatase conjugate (1:10,000) (Sigma-Aldrich, Inc., St. Louis, MO) for 60 min at room temperature. Finally, the immunocomplex recognized by the sera was revealed by the corresponding substrate-chromogenic solution containing 0.3 g/liter nitroblue tetrazolium and 0.15 g/liter 5-bromo-4-chloro-3-indolylphosphate (NBT/BCIP). The reaction was stopped by washing with distilled water.
Detection of specific anti-T. gondii IgG. (i) Sample collection.
A collection of 91 serum samples from pregnant women was used for rSAG1 ELISA evaluation. These women were referred consecutively to the Laboratoire de Parasitologie Médicale, Institut Pasteur de Tunis (IPT), and the Laboratoire d'Analyses de Biologie Médicale, Aziza Othmana Hospital, Tunis, for systematic toxoplasmosis screening during their first prenatal consultation. Serum samples were tested routinely by the standard ELISA Platelia Toxo IgG kit (product no. 72840; Bio-Rad, France). Questionably positive samples (IgG titers in the 6-to-9 IU range) required confirmatory testing with the complementary methods used by the IPT laboratory, namely, sensitive agglutination (Toxo-Screen DA; bioMérieux, France) and indirect immunofluorescence (Toxo-Spot IF; bioMérieux).
During blood sampling and after informed consent, saliva was collected from each patient with the Oracol device (Malvern Medical Developments, Worcester, United Kingdom). Briefly, the foam swab was removed from the collection device, rubbed over the women's gums until saturated with saliva, and then replaced into the test tube. In the laboratory, 1 ml of preservative medium (PBS [pH 7.4], 10% fetal calf serum, 0.2% Tween 20, 0.5% gentamicin, 0.2% Fungizone) was added to the tube; the swab was removed with a twisted movement (in order to extract as much liquid as possible), inverted, and replaced into the tube to keep the pink foam at the top. The tube was centrifuged at 100 × g for 10 min. The inverted swab was then removed and discarded. Finally, the supernatant was divided into aliquots and conserved at −20°C until use.
(ii) rSAG1 ELISA.
A standard indirect ELISA was optimized with serum samples. Briefly, the wells of a 96-well MaxiSorp plate (Nunc) were coated with pure refolded rSAG1 diluted in 100 mM carbonate-bicarbonate buffer (pH 9.6) at an optimal concentration determined in preliminary experiments (100 μg/ml, corresponding to 5 μg/well). After overnight incubation at 4°C, the wells were emptied and blocked with 5% skim milk in PBS-T. Serum samples from the patients (in duplicate) and calibrators were diluted 1:20 in PBS-T and distributed (100 μl per well of microplate). After 1 h at 37°C, the reaction mixture was developed with goat anti-human IgG (Fc-specific)–horseradish peroxidase conjugate (product no. A0170; Sigma-Aldrich, St. Louis, MO) at a 1:5,000 dilution. After another hour of incubation, the substrate solution consisting of 50 mM phosphate-citrate buffer (pH 5.6) containing 0.02% o-phenylenediamine (OPD) (Sigma-Aldrich) and 0.005% H2O2 was added, and the immediate colorimetric reaction was stopped by the addition of 2 mol/liter H2SO4 (50 μl per well). The wells were washed three times with PBS-T between each intermediate step. The absorbance at 492/620 nm was measured using a microplate reader (Multiskan EX; Labsystems, Finland). The background was determined by coating the wells with the same antigen concentration but without adding sera. The calibrators were those supplied with the Platelia Toxo IgG kit, ready to use as 6, 60, and 240 IU/ml. These 6-, 60-, and 240-IU standards contain human serum reactive for T. gondii IgG antibodies calibrated in IU by Bio-Rad Laboratories against the TOXM 185 standard serum of the World Health Organization.
A biotin-streptavidin assay was used with saliva specimens. Briefly, a 96-well plate was coated with purified rSAG1 in an optimal concentration determined in preliminary experiments (10 μg/well) at 4°C overnight and then blocked with 5% skim milk in PBS-T. After washing, saliva samples from the patients (in duplicate) diluted 1:4 in PBS-T were distributed (100 μl per well of microplate) and incubated at 37°C for 1 h. For the amplification assay, each well was incubated with 100 μl of goat biotinylated anti-human IgG antibody (gamma chain specific) (diluted 1;5,000 in PBS-T) at 37°C for 1 h, followed by incubation with horseradish peroxidase-labeled streptavidin. Intermediate washing steps with PBS-T, incubation conditions, and developing were the same as those described above for the serum samples.
Statistical analysis.
Statistical analysis was done using the MedCalc statistical software (version 11.4.4.0). Quantitative variables are reported as means and standard deviations (SDs), or medians and interquartile ranges (IQRs) if the data did not show a normal distribution. The rSAG1-based ELISA evaluation consisted of testing intrinsic sensitivity and specificity for a collection of serum samples from patients immune to toxoplasmosis and appropriate negative controls. To assess the agreement between serum-based and saliva-based tests, positive percent agreement (PPA) and negative percent agreement (NPA) between the 2 tests were estimated.
RESULTS
Production and characterization of recombinant SAG1 antigen.
An 867-bp DNA fragment coding for the chosen sequence of T. gondii SAG1 antigen was recovered by PCR and subsequently cloned into the pET22b(+) expression vector in frame with a hexahistidine tail sequence for further purification. The rSAG1 antigen was expressed in the Origami(DE3) bacterial strain. After expression optimization, soluble and insoluble fractions of bacterial culture were analyzed with a 10% continuous SDS-PAGE gel stained with Coomassie blue. A very weak protein band corresponding to soluble rSAG1 was detected (data not shown). Conversely, the insoluble rSAG1 fraction generated from cell lysis and subsequent solubilization with 8 M guanidine-HCl was detected with an apparent molecular size of 38 kDa (Fig. 1A, lane 2), slightly higher than its predicted molecular mass (∼32 kDa). This band was absent in the noninduced cell culture (Fig. 1A, lane 1), and its identification as the rSAG1 antigen was confirmed by antipolyhistidine Western blotting (Fig. 1B). Solubilized inclusion body extracted proteins were purified under denaturing conditions (6 M urea), and the eluted fractions were collected and checked for the presence of the rSAG1 antigen. As shown by gel electrophoresis with Coomassie blue staining (Fig. 1A, lane 3), the purified solubilized rSAG1 migrated as a unique broad band around 38 kDa. The recombinant protein was further evaluated by performing antipolyhistidine Western blotting with equal amounts of the insoluble preparation before and after purification (Fig. 1B). No degradation products were detected under these conditions. The expression product underwent a refolding step, consisting of consecutive dialysis baths with decreasing urea concentrations. The recovery of immunoreactivity was confirmed by Western blot analysis with a human reference serum sample previously characterized for T. gondii-specific IgG reactivity. As shown in Fig. 1C (lane 3), the refolded purified rSAG1 is highly recognized by the human Toxoplasma serum. No degradation products, protein aggregates, or misfolded species were detected. However, the immunoblot profile of the earlier dialyzed crude insoluble fraction, revealed with the human reference serum, recognized the rSAG1 antigen but also all contaminating insoluble proteins (Fig. 1C, lane 2).
Fig 1.
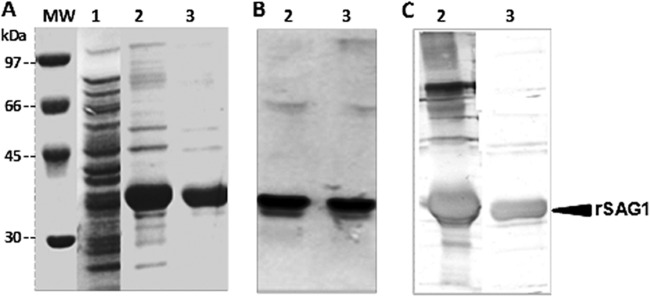
Expression and purification analysis. SDS-PAGE analysis of bacterial lysis extract and Coomassie blue staining of rSAG1 antigen (A) and Western blot treated with antipolyhistidine-peroxidase conjugate antibody (B) or a human reference serum sample (C). Numbers correspond to the noninduced cell culture (lane 1), the crude solubilized preparation with 8 M guanidine-HCl (lanes 2), and the purified solubilized preparation under denaturing conditions (6 M urea) (lanes 3). Molecular mass (MW) markers in kDa are indicated on the left.
Typically, 1 liter of the recombinant bacterial culture led to the production of about 20 mg purified rSAG1. This amount largely exceeds the requirement for conventional biological activity tests.
Detection of specific anti-T. gondii IgG by rSAG1 ELISA.
The routinely used techniques for diagnosis of Toxoplasma infection identified 49 seropositive and 42 seronegative pregnant women. Among the positive samples, the anti-T. gondii IgG titers varied from 7 to 210 IU (median, 51 IU; IQR, 32 to 70 IU), and only one serum sample was considered questionable by the Platelia commercial kit. This sample was confirmed positive by the complementary tests.
The analysis showed that the rSAG1 serum-based ELISA had an excellent ability to discriminate between nonimmune patients (lowest OD value, 0.03; highest OD value, 0.21; mean, 0.11; SD, 0.05) and immune patients (lowest OD value, 0.27; highest OD value, 0.67; mean, 0.45; SD, 0.09). The serum sample that had the lowest IgG titer by the Platelia kit (7 IU) was positive by the rSAG1 serum-based ELISA for a chosen OD cutoff in the range of 0.22 to 0.26 (Fig. 2A). At this cutoff value, both the diagnostic specificity and the sensitivity of IgG detection by rSAG1 ELISA were 100%.
Fig 2.
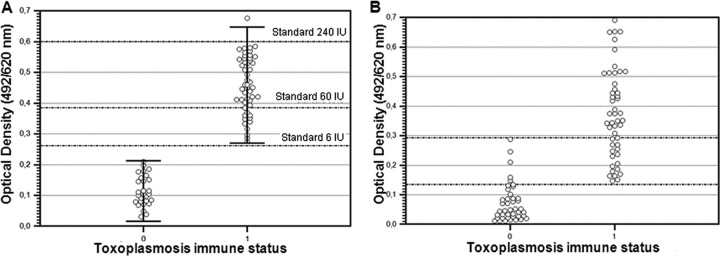
rSAG1 ELISA results according to the toxoplasmosis immune status of pregnant women. (A) Serum samples; markers correspond to mean ODs ± 2 SD. The dotted lines refer to OD values obtained by the rSAG1 serum-based ELISA for the Platelia kit-positive calibrators (6, 60, and 240 IU). (B) Saliva samples; 0 and 1 refer to nonimmune and immune toxoplasmosis status, respectively.
The immunoreactivity of saliva against the rSAG1 antigen was evaluated in a standard procedure and in a biotin-streptavidin procedure. The biotin-streptavidin assay improved the sensitivity of the detection of the rSAG1 antigen and was therefore preferred to the standard procedure for ELISA (data not shown). Figure 2B shows the absorbance values measured by the biotin-streptavidin rSAG1 ELISA applied to saliva samples according to toxoplasmosis immune status. OD values less than 0.14 (n = 37) were recorded only in nonimmune patients, OD values greater than 0.29 (n = 33) were recorded only in the immune group, and OD values in the 0.14 to 0.29 range (n = 21) were recorded in immune (n = 16) and nonimmune patients (n = 5). Thus, for an OD threshold of 0.14, the overall percent agreement between saliva-based and serum-based tests was 94.5%, the PPA was 100% (95% confidence interval [CI], 92.7 to 100%), and the NPA was 88.1% (95% CI, 74.3 to 96%) (Table 1), whereas for an OD threshold of 0.29, the overall percent agreement between the 2 tests was 82.4%, the PPA was 67.3% (95% CI, 52.4 to 80%), and the NPA was 100% (95% CI, 91.5 to 100%) (Table 1). If the rSAG1 serum-based ELISA is assumed to be a gold standard and the rSAG1 saliva-based ELISA is being measured against it, at a specificity of 100%, the sensitivity of the saliva-based test was 69.4%, and at a sensitivity of 100%, the specificity of the test was 88.1% (Fig. 3).
Table 1.
Agreements between serum-based and saliva-based rSAG1 ELISAs according to the selected OD thresholds in saliva samples
| rSAG1 saliva-based ELISA result at an OD threshold of: | rSAG1 serum-based ELISA result (no.) |
Total no. | |
|---|---|---|---|
| Positive | Negative | ||
| 0.14 | |||
| Positive | 49 | 5 | 54 |
| Negative | 0 | 37 | 37 |
| Total | 49 | 42 | 91 |
| 0.29 | |||
| Positive | 33 | 0 | 33 |
| Negative | 16 | 42 | 58 |
| Total | 49 | 42 | 91 |
Fig 3.
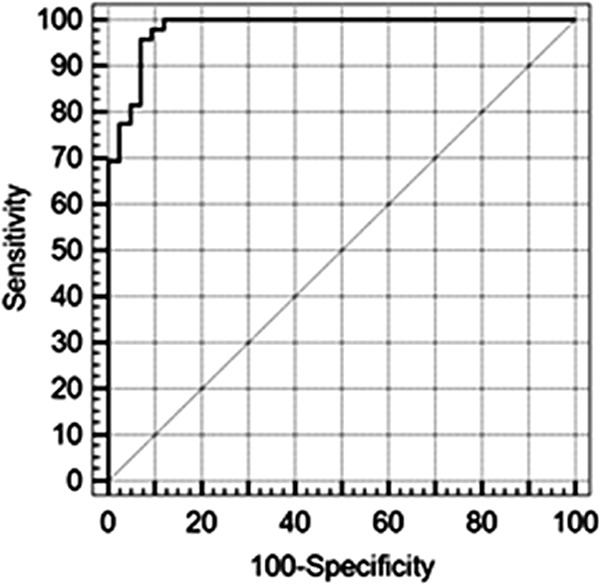
Receiver operating characteristic (ROC) curve for the rSAG1 saliva-based ELISA applied to positive versus negative individuals identified by the rSAG1 serum-based ELISA.
DISCUSSION
In order to overcome the interassay variability of the commercially available kits and to improve the serologic diagnosis of Toxoplasma infection, it is useful to identify antigens of potential diagnostic value and to use them as recombinant proteins that allow for more accurate, reliable, and specific tests (17). In this context, several Toxoplasma target genes coding for such antigens were previously cloned and expressed (17). rSAG1 was the most widely explored antigen and has been shown to be a good candidate for the diagnosis of Toxoplasma infection. However, reported studies on rSAG1-based assays showed variability in performance, which was probably due to differences in the selected gene fragments, the cloning strategies, or the panel of biological samples used in their evaluations (10, 14, 33). In the present study, we selected a gene fragment coding for the whole SAG1 protein of the T. gondii RH strain, corresponding to amino acid residues 47 to 336, because using a truncated form might decrease the immunoreactivity of the recombinant antigen. The sag1 gene was cloned into the pET22b(+) vector and highly expressed in the Origami(DE3) E. coli system. This system was chosen to overcome the metabolic burden related to high-level protein expression, since it contains double genomic mutations of the thioredoxin reductase (trxB) and glutathione reductase (gor) genes, resulting in marked enhancement of disulfide bond formation in the cell oxidative cytoplasm (34). In fact, the SAG1 protein adopts a native structure defined by six intramolecular cysteine bridges that give rise to immunologically relevant conformational epitopes (18, 35). However, the rSAG1 antigen was essentially expressed as insoluble inclusion bodies. This might be due to saturation of the host cell folding machinery or cofactor deficiency (36) or due to the impact of rare codons that might contribute to rapid mRNA decay, limiting the expression of foreign genes in heterologous systems (37). From elsewhere, aberrant interactions in the multiple disulfide bonds might lead to misfolded protein and insoluble aggregate formation (38). In addition, persistence of the rSAG1 C-terminal hydrophobic region (residues 308 to 336), which serves as an acceptor of the so-called GPI group (i.e., a phosphatidylinositol glycolipid) in the parasite, might have promoted the protein aggregation (10). After one-step chromatographic purification under denaturing conditions, we have optimized a cost-saving dialysis procedure for refolding (30) that yields milligrams of active rSAG1 antigen from 1 liter of bacterial culture. Under these conditions, the rSAG1 ELISA developed was more sensitive than assays reported in the literature (22, 39, 40) and as sensitive as the soluble rSAG1-based assays (10, 23, 33). This suggests that a correct refolding procedure might improve the specific immunoreactivity of this very complex molecule. Moreover, the rSAG1 serum-based ELISA was revealed to be an interesting tool for Toxoplasma screening in pregnant women. However, further evaluation on a larger series should be performed to validate the chosen cutoff value. Furthermore, it will be interesting to evaluate the accuracy of the method in detecting recent versus chronic Toxoplasma infection.
Previous works showed the usefulness of saliva-based methods in the diagnosis of toxoplasmosis using whole-cell extracts as the antigen (27, 28, 29). In the current study, we tested a recombinant antigen for antibody detection in saliva samples. The oral fluids were collected by the Oracol device for optimal IgG screening (26, 41), and a preservative medium was added to the saliva samples to help maintain IgG stability over time (42). Finally, a biotin-streptavidin system was used for signal amplification of the low expected antibody levels in the saliva. Under these conditions, positive and negative percent agreements between the saliva-based and serum-based rSAG1 ELISAs varied according to the selected threshold in saliva samples. In fact, for a selected cutoff of 0.14, the PPA was 100% and the NPA was 88.1%, whereas for a cutoff of 0.29, the PPA was 67.3% and the NPA was 100%. To avoid false-negative or false-positive results by saliva-based ELISA, the 0.14-to-0.29 OD range can be considered a “gray zone.” Under this condition, samples with OD values in this zone should be considered questionable and confirmed as negative or positive by complementary methods.
When the presence of a gray zone is taken into account, the rSAG1 saliva-based ELISA allowed immune status determination in 70 of 91 (76.9%) women; 37 (88.1%) were seronegative, and 33 (67.3%) were seropositive. This suggests that more research is needed to improve the sensitivity of the method for application in large-scale epidemiological surveys, where the noninvasive nature of the sampling is of considerable interest. In fact, the saliva-based ELISA seems to be less sensitive for specific IgG detection in toxoplasmosis than in other infectious diseases (26, 43, 44, 45). This is probably due to the low antibody level observed in this chronic infection. To improve the accuracy of diagnosis, sensitive antibody capture assays (46, 47, 48) or an immunoblotting procedure (49) can be optimized for IgG screening in saliva. Furthermore, because IgA is a major immunoglobulin isotype in oral fluid, it will be interesting to evaluate the accuracy of the rSAG1 saliva-based ELISA in detecting IgA in acute infection.
ACKNOWLEDGMENTS
We are grateful to Olfa Bahri (Laboratoire d'Analyses de Biologie Médicale, Aziza Othmana Hospital, Tunis, Tunisia) for providing us with the serum and saliva samples and for her technical assistance. We thank Julien Muzard (Center for Molecular Innovation & Drug Discovery, University College Dublin, Ireland) for critically reading the manuscript.
This work was supported by the Ministry of Higher Education and Scientific Research in Tunisia and carried out within the framework of the Laboratoire de Parasitologie Médicale, Biotechnologies et Biomolécules, Institut Pasteur de Tunis.
Footnotes
Published ahead of print 23 January 2013
REFERENCES
- 1. Montoya JG, Remington JS. 2008. Management of Toxoplasma gondii infection during pregnancy. Clin. Infect. Dis. 47:554–566 [DOI] [PubMed] [Google Scholar]
- 2. Wong SY, Remington JS. 1994. Toxoplasmosis in pregnancy. Clin. Infect. Dis. 18:853–861 [DOI] [PubMed] [Google Scholar]
- 3. Boughattas S, Ben-Abdallah R, Siala E, Souissi O, Aoun K, Bouratbine A. 2010. Direct genotypic characterization of Toxoplasma gondii strains associated with congenital toxoplasmosis in Tunisia (North Africa). Am. J. Trop. Med. Hyg. 82:1041–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bouratbine A, Siala E, Chahed MK, Aoun K, Ben Ismail R. 2001. Sero-epidemiologic profile of toxoplasmosis in northern Tunisia. Parasite 8:61–66 [DOI] [PubMed] [Google Scholar]
- 5. Kravetz JD, Federman DG. 2005. Toxoplasmosis in pregnancy. Am. J. Med. 118:212–216 [DOI] [PubMed] [Google Scholar]
- 6. Gilbert RE, Dunn DT, Wallon M, Hayde M, Prusa A, Lebech M, Kortbeek T, Peyron F, Pollak A, Petersen E. 2001. Ecological comparison of the risks of mother to child transmission and clinical manifestations of congenital toxoplasmosis according to prenatal treatment protocol. Epidemiol. Infect. 127:113–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wallon M, Liou C, Garner P, Peyron F. 1999. Congenital toxoplasmosis: systematic review of evidence of efficacy of treatment in pregnancy. BMJ 318:1511–1514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Montoya JG. 2002. Laboratory diagnosis of Toxoplasma gondii infection and toxoplasmosis. J. Infect. Dis. 185:S73–S82 [DOI] [PubMed] [Google Scholar]
- 9. Remington JS, Thulliez P, Montoya JG. 2004. Recent developments for diagnosis of toxoplasmosis. J. Clin. Microbiol. 42:941–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wu K, Chen XG, Li H, Yan H, Yang PL, Lun ZR, Zhu XQ. 2009. Diagnosis of human toxoplasmosis by using the recombinant truncated surface antigen 1 of Toxoplasma gondii. Diagn. Microbial. Infect. Dis. 64:261–266 [DOI] [PubMed] [Google Scholar]
- 11. Hassl A, Müller WA, Aspöck H. 1991. An identical epitope in Pneumocystis carinii and Toxoplasma gondii causing serological cross reactions. Parasitol. Res. 77:351–352 [DOI] [PubMed] [Google Scholar]
- 12. Taylor DW, Evans CB, Aley SB, Barta JR, Danforth HD. 1990. Identification of an apically-located antigen that is conserved in sporozoan parasites. J. Protozool. 37:540–545 [DOI] [PubMed] [Google Scholar]
- 13. Beghetto E, Spadoni A, Bruno L, Buffolano W, Gargano N. 2006. Chimeric antigens of Toxoplasma gondii: toward standardization of toxoplasmosis serodiagnosis using recombinant products. J. Clin. Microbiol. 44:2133–2140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Buffolano W, Beghetto E, Del Pezzo M, Spadoni A, Di Cristina M, Petersen E, Gargano N. 2005. Use of recombinant antigens for early postnatal diagnosis of congenital toxoplasmosis. J. Clin. Microbiol. 43:5916–5924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hofgärtner WT, Swanzy SR, Bacina RM, Condon J, Gupta M, Matlock PE, Bergeron DL, Plorde JJ, Fritsche TR. 1997. Detection of immunoglobulin G (IgG) and IgM antibodies to Toxoplasma gondii: evaluation of four commercial immunoassay systems. J. Clin. Microbiol. 35:3313–3315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wilson M, Remington JS, Clavet C, Varney G, Press C, Ware D, FDA Toxoplasmosis Ad Hoc Working Group. 1997. Evaluation of six commercial kits for detection of human immunoglobulin M antibodies to Toxoplasma gondii. J. Clin. Microbiol. 35:3112–3115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kotresha D, Noordin R. 2010. Recombinant proteins in the diagnosis of toxoplasmosis. APMIS 118:529–542 [DOI] [PubMed] [Google Scholar]
- 18. Burg JL, Perelman D, Kasper LH, Ware PL, Boothroyd JC. 1988. Molecular analysis of the gene encoding the major surface antigen of Toxoplasma gondii. J. Immunol. 141:3584–3591 [PubMed] [Google Scholar]
- 19. Bülow R, Boothroyd JC. 1991. Protection of mice from fatal Toxoplasma gondii infection by immunization with p30 antigen in liposomes. J. Immunol. 147:3496–3500 [PubMed] [Google Scholar]
- 20. Hartati S, Kusumawati A, Wuryastuti HH, Widada JS. 2006. Primary structure of mature SAG1 gene of an Indonesian Toxoplasma gondii and comparison with other strains. J. Vet. Sci. 7:263–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sibley LD, Boothroyd JC. 1992. Virulent strains of Toxoplasma gondii comprise a single clonal lineage. Nature 359:82–85 [DOI] [PubMed] [Google Scholar]
- 22. Aubert D, Maine GT, Villena I, Hunt JC, Howard L, Sheu M, Brojanac S, Chovan LE, Nowlan SF, Pinon JM. 2000. Recombinant antigens to detect Toxoplasma gondii-specific immunoglobulin G and immunoglobulin M in human sera by enzyme immunoassay. J. Clin. Microbiol. 38:1144–1150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pietkiewicz H, Hiszczyńska-Sawicka E, Kur J, Petersen E, Nielsen HV, Stankiewicz M, Andrzejewska I, Myjak P. 2004. Usefulness of Toxoplasma gondii-specific recombinant antigens in serodiagnosis of human toxoplasmosis. J. Clin. Microbiol. 42:1779–1781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Santoro F, Afchain D, Pierce R, Cesbron JY, Ovlaque G, Capron A. 1985. Serodiagnosis of Toxoplasma infection using a purified parasite protein (P30). Clin. Exp. Immunol. 62:262–269 [PMC free article] [PubMed] [Google Scholar]
- 25. Mortimer PP, Parry JV. 1994. Detection of antibody to HIV in saliva: a brief review. Clin. Diagn. Virol. 2:231–243 [DOI] [PubMed] [Google Scholar]
- 26. Galaï Y, Chabchoub N, Ben-Abid M, Ben-Abda I, Ben-Alaya-Bouafif N, Amri F, Aoun K, Bouratbine A. 2011. Diagnosis of Mediterranean visceral leishmaniasis by detection of Leishmania antibodies and Leishmania DNA in oral fluid samples collected using an ORACOL device. J. Clin. Microbiol. 49:3150–3153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hajeer AH, Balfour AH, Mostratos A, Crosse B. 1994. Toxoplasma gondii: detection of antibodies in human saliva and serum. Parasite Immunol. 16:43–50 [DOI] [PubMed] [Google Scholar]
- 28. Loyola AM, Durighetto, Silva AFDA, Mineo JR. 1997. Anti-Toxoplasma gondii immunoglobulins A and G in human saliva and serum. J. Oral Pathol. Med. 26:187–191 [DOI] [PubMed] [Google Scholar]
- 29. Stroehle A, Schmid K, Heinzer I, Naguleswaran A, Hemphill A. 2005. Performance of a western immunoblot assay to detect specific anti-Toxoplasma gondii IgG antibodies in human saliva. J. Parasitol. 91:561–563 [DOI] [PubMed] [Google Scholar]
- 30. Burgess RR. 2009. Refolding solubilized inclusion body proteins. Methods Enzymol. 17:259–282 [DOI] [PubMed] [Google Scholar]
- 31. Cabrita LD, Bottomley SP. 2004. Protein expression and refolding—a practical guide to getting the most out of inclusion bodies. Biotechnol. Annu. Rev. 10:31–54 [DOI] [PubMed] [Google Scholar]
- 32. Sambrook J, Russel DW. 2001. Molecular cloning: a laboratory manual, 3rd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 33. Pfrepper KI, Enders G, Gohl M, Krczal D, Hlobil H, Wassenberg D, Soutschek E. 2005. Seroreactivity to and avidity for recombinant antigens in toxoplasmosis. Clin. Diagn. Lab. Immunol. 12:977–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Prinz WA, Aslund F, Holmgren A, Beckwith J. 1997. The role of the thioredoxin and glutaredoxin pathways in reducing protein disulfide bonds in the Escherichia coli cytoplasm. J. Biol. Chem. 272:15661–15667 [DOI] [PubMed] [Google Scholar]
- 35. Cesbron-Delauw MF, Tomavo S, Beauchamps P, Fourmaux MP, Camus D, Capron A, Dubremetz JF. 1994. Similarities between the primary structures of two distinct major surface proteins of Toxoplasma gondii. J. Biol. Chem. 269:16217–16222 [PubMed] [Google Scholar]
- 36. Idicula-Thomas S, Balaji PV. 2005. Understanding the relationship between the primary structure of proteins and its propensity to be soluble on over expression in Escherichia coli. Protein Sci. 14:582–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rosano GL, Ceccarelli EA. 2009. Rare codon content affects the solubility of recombinant proteins in a codon bias-adjusted Escherichia coli strain. Microb. Cell Fact. 24:8–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lilie H, Schwarz E, Rudolph R. 1998. Advances in refolding of proteins produced in E. coli. Curr. Opin. Biotechnol. 9:497–501 [DOI] [PubMed] [Google Scholar]
- 39. Chen XG, Gong Y, Hua-Li Lun ZR, Fung MC. 2001. High level expression and purification of immunogenic recombinant SAG1 (P30) of Toxoplasma gondii in Escherichia coli. Protein Expr. Purif. 23:33–37 [DOI] [PubMed] [Google Scholar]
- 40. Kotresha D, Poonam D, Muhammad Hafiznur Y, Saadatnia G, Nurulhasanah O, Sabariah O, Tan SY, Izzati Zahidah AK, Rahmah N. 2012. Recombinant proteins from new constructs of SAG1 and GRA7 sequences and their usefulness to detect acute toxoplasmosis. Trop. Biomed. 29:129–137 [PubMed] [Google Scholar]
- 41. Lehner T. 1992. Immunology of oral diseases, 3rd ed, p 18–27 Blackwell Scientific Publications, Oxford, United Kingdom [Google Scholar]
- 42. Malamud D. 1997. Oral diagnosis testing for detecting human immune-deficiency virus-1 antibodies: a technology whose time has come. Am. J. Med. 102:9–14 [DOI] [PubMed] [Google Scholar]
- 43. Oba IT, Spina AMM, Saraceni CP, Lemos MF, Senhoras RCFA, Moreira RC, Granato CFH. 2000. Detection of hepatitis A antibodies by ELISA using saliva as clinical samples. Rev. Inst. Med. Trop. Sao Paulo 42:197–200 [DOI] [PubMed] [Google Scholar]
- 44. Parry JV. 1993. Simple and reliable salivary tests for HIV and hepatitis A and B virus diagnosis and surveillance. Ann. N. Y. Acad. Sci. 694:216–233 [DOI] [PubMed] [Google Scholar]
- 45. White DA, Scribner AN, Huang JV. 2009. A comparison of patient acceptance of fingerstick whole blood and oral fluid rapid HIV screening in an emergency department. J. Acquir. Immune Defic. Syndr. 52:75–78 [DOI] [PubMed] [Google Scholar]
- 46. de Azevedo Neto RS, Richards A, Nokes DJ, Silveira ASB, Cohen BJ, Passos SD, de Souza VAUF, Brown DWG, Pannuti CS, Massad E. 1995. Salivary antibody detection in epidemiological surveys: a pilot study after a mass vaccination campaign against rubella in Sao Paulo, Brazil. Trans. R. Soc. Trop. Med. Hyg. 89:115–118 [DOI] [PubMed] [Google Scholar]
- 47. Parisi MR, Soldini L, Di Perri G, Tiberi S, Lazzarin A, Lillo FB. 2009. Offer of rapid testing and alternative biological samples as practical tools to implement HIV screening programs. New Microbiol. 32:391–396 [PubMed] [Google Scholar]
- 48. Vyse AJ, Brown DWG, Cohen BJ, Samuel R, Nokes DJ. 1999. Detection of rubella virus-specific immunoglobulin G in saliva by an amplification-based enzyme-linked immunosorbent assay using monoclonal antibody to fluorescein isothiocyanate. J. Clin. Microbiol. 37:391–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Grant RM, Piwowar EM, Katongole-Mbidde E, Muzawalu W, Rugera S, Abima J, Stramer SL, Kataaha P, Jackson B. 1996. Comparison of saliva and serum for human immunodeficiency virus type 1 antibody testing in Uganda using a rapid recombinant assay. Clin. Diagn. Lab. Immunol. 3:640–644 [DOI] [PMC free article] [PubMed] [Google Scholar]