

Abstract
The immunosuppressive drug FK506 binding proteins (FKBPs), an immunophilin family with the immunosuppressive drug FK506 binding property, exhibit peptidylprolyl cis-trans isomerase (PPIase) activity. While the cyclophilin-catalyzed peptidylprolyl isomerization of X-Pro peptide bonds has been extensively studied, the mechanism of the FKBP-mediated peptidylprolyl isomerization remains uncharacterized. Thus, to investigate the binding of FKBP with its substrate and the underlying catalytic mechanism of the FKBP-mediated proline isomerization, here we employed the FK506 binding domain (FKBD) of the human malarial parasite Plasmodium vivax FK506 binding protein 35 (PvFKBP35) and examined the details of the molecular interaction between the isomerase and a peptide substrate. The crystallographic structures of apo PvFKBD35 and its complex with the tetrapeptide substrate succinyl-Ala-Leu-Pro-Phe-p-nitroanilide (sALPFp) determined at 1.4 Å and 1.65 Å resolutions, respectively, showed that the substrate binds to PvFKBD35 in a cis conformation. Nuclear magnetic resonance (NMR) studies demonstrated the chemical shift perturbations of D55, H67, V73, and I74 residues upon the substrate binding. In addition, the X-ray crystal structure, along with the mutational studies, shows that Y100 is a key residue for the catalytic activity. Taken together, our results provide insights into the catalytic mechanism of PvFKBP35-mediated cis-trans isomerization of substrate and ultimately might aid designing substrate mimetic inhibitors targeting the malarial parasite FKBPs.
INTRODUCTION
A member of the immunophilin family, the FK506 binding proteins (FKBPs) that bind to the immunosuppressive drugs FK506 and rapamycin with high affinity belongs to the peptidylprolyl cis-trans isomerase (PPIase) family (1), which also includes cyclophilins and parvulins (2, 3). PPIase proteins catalyze the cis-trans isomerization of the peptidylprolyl bond. The cis-trans isomerization of the peptidylprolyl bond is one of the rate-limiting steps in protein folding due to a large energy barrier of 14 to 24 kcal/mol (4). Conformation of the protein backbone is usually defined by three torsion angles, φ, ψ, and ω. Values of ω are 0° in cis conformation of the peptide bond and 180° in trans conformation. These cis (0°) and trans (180°) forms are separated by a rotational barrier of the perpendicular high-energy state where ω is 90°. Therefore, the transition state of cis-trans isomerization is approximated by a high energy, “twisted” syn-90 state (5, 6). FKBPs act as a catalyst and accelerate the cis-trans isomerization by reducing the energy barrier of the reaction (1) and help in correct folding of proteins (5). FK506 binding to FKBP inhibits its PPIase activity (7), while its immunosuppressive function is a result of the inhibition of calcineurin activity after the FKBP-FK506 binary complex binds to calcineurin (8, 9). Inhibition of PPIase activity by FK506 suggests that both the substrate and FK506 bind to the same binding pocket of the protein.
The physiological role of the PPIase activity was first established by showing that cyclosporine (CsA) slows down the folding of procollagen I into triple-helix collagen (10). Later it was found that Cyp20, a component of the mitochondrial folding machinery, cooperates with Hsp70 and Hsp60 in protein folding (11). However, a major physiological significance of the PPIase-catalyzed cis-trans isomerization was recognized after the discovery of the catalytic properties of Pin1 (6, 12). Unlike other PPIases, Pin1 isomerizes only phosphorylated Ser/Thr-Pro motifs, indicating its important role in cellular signaling pathways, where it can regulate the conformation of its substrates after phosphorylation to control protein function (6, 13). Recent studies showed that the peptidylprolyl cis-trans isomerization may function as an effective and reversible molecular switch that controls the kinetics of autoinhibition of a signaling molecule, Crk adaptor protein (14). It is also responsible for the opening of the pores of the neurotransmitter-gated ion channel (15). PPIases act as a catalyst for cis-trans isomerization and increase the reaction rate by several orders of magnitude.
While extensive studies have been done on cyclophilins (16, 17) and Pin1-mediated (18) cis-trans isomerization, to our knowledge, no structural study has been performed on FKBPs with their in vitro peptide substrate succinyl-Ala-Leu-Pro-Phe-p-nitroanilide (sALPFp). We previously showed that PvFKBD35 (the FK506 binding domain of Plasmodium vivax FKBP35) possesses a canonical PPIase activity like HsFKBP12 (human FKBP12) (19). A preliminary search in plasmoDB (20) using “Ala-X-Pro-Phe,” where X represents any hydrophobic amino acid, resulted in 629 proteins across seven different Plasmodium species (see Table S1 in the supplemental material), and the probable recognition of FKBPs by these proteins could be taken up for future interest. Further, a study on the identification of unique proline-containing motifs across proteins in the Plasmodium falciparum proteomes (21) has proposed that these motifs could serve as possible leads toward developing peptidomimetic antimalarial drugs. In this direction, we have performed X-ray crystallographic studies of both apo PvFKBD35 and its complex with the peptide sALPFp as well as nuclear magnetic resonance (NMR) titration and site-specific mutational studies. Our data presented here provide molecular insights into the FKBP-mediated peptidylprolyl cis-trans isomerization, the first of its kind.
MATERIALS AND METHODS
Sample preparation.
The coding sequence for PvFKBD35 (M1-E126) was amplified and cloned into the pSUMO vector to generate the pSUMO-PvFKBD35 plasmid with N-terminal hexahistidine and SUMO protein. The resulting pSUMO-PvFKBD35 plasmid was used as the template to generate single-amino-acid-substitution mutants (Y100F, Y100A, Y100W, Y100R, Y100P, Y100E, Y100L) using the Quick Change site-directed mutagenesis kit (Stratagene, La Jolla, CA). The pSUMO-PvFKBD35 construct was transformed in Escherichia coli BL21(DE3) cells, cells were grown into LB medium and M9 medium containing 1 g/liter of 15NH4Cl for X-ray crystallography and NMR studies, respectively, and protein was purified as described before (19). In short, protein expression was induced by adding 1 mM isopropyl-β-d-thio-galactoside (IPTG) when the A600 of cell culture reached 0.6 to 0.8. The cells were harvested by centrifugation at 18,600 × g for 10 min, resuspended in the lysis buffer (20 mM NaPO4, 500 mM NaCl, pH 7.8), and broken by sonication for 20 to 30 min on ice. The cell lysate was cleared by centrifugation at 48,400 × g for 20 min and purified by Ni2+-nitrilotriacetic acid (NTA) resin. For a further purification, Ni2+-NTA column elution fractions were loaded onto a Superdex-200 filtration column (GE Healthcare, Singapore). The N-terminal SUMO tag was removed by sumo protease and passed through an Ni2+-NTA column to obtain the pure protein near homogeneity. The NMR samples were prepared in a buffer containing 20 mM NaPO4 (pH 6.8), 20 mM NaCl, 1 mM dithiothreitol (DTT), and 0.01% NaN3. For X-ray crystallography, the protein was concentrated to 10 mg/ml in 10 mM phosphate buffer, 137 mM NaCl, and 2.7 mM KCl (pH 6.8), as described before (19).
Peptide synthesis.
Solid-phase peptide synthesis method with 9-fluorenylmethoxy carbonyl (Fmoc) chemistry and Wang resin was used on a fully automated CEM microwave peptide synthesizer to synthesize the tetrapeptide Ala-Leu-Pro-Phe (ALPF) as described elsewhere (22, 23). After synthesis, the peptide was cleaved from the resin by hydrolysis. Purity of the peptide was checked by high-performance liquid chromatography (HPLC) (Shimazu, Tokyo, Japan), and the sequence of the peptide was confirmed by matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS) analysis (Thermo Scientific, Waltham, MA). The succinyl-Ala-Leu-Pro-Phe-p-nitroanilide (sALPFp) used for crystallographic studies was purchased from Peptide Institute, Osaka, Japan.
Crystallization of apo PvFKBD35 and cocrystallization of PvFKBD35 with peptide substrate sALPFp.
For apo PvFKBD35, an automated initial crystallization screen of 672 conditions was performed using the CyBio-Crystal Creator (Jena Biosciences). Crystal drops were prepared by mixing 0.2 μl protein solution (10 mg/ml in 10 mM phosphate buffer, 137 mM NaCl, 2.7 mM KCl, pH 6.8) with an equal volume of precipitant solution and equilibrated by sitting-drop vapor diffusion against 0.1 ml precipitant solution at 291 K. Crystals in Crystal Screen condition 6 (0.1 M Tris-HCl [pH 8.5], 30% [wt/vol] polyethylene glycol 4000, 0.2 M magnesium chloride hexahydrate; Hampton Research) showed suitable ordered diffraction for structural studies.
PvFKBD35 and sALPFp complex initial crystals were produced using the ammonium sulfate buffer grid screen from Hampton Research followed by optimization in 2.85 M ammonium sulfate and 0.1 M MES buffer, pH 5.5. The concentrated protein sample was mixed with the peptide in a molar ratio of 1:5 and incubated overnight at 4°C prior to crystallization. Crystals were set up by the hanging-drop vapor diffusion method using 2 μl of complex protein solution mixed with an equal volume of reservoir solution equilibrated against 1 ml reservoir solution. Crystals 0.4- by 0.4- by 0.2-mm3 in size appeared after 10 days at 18°C.
Data collection and structure determination.
A single crystal of PvFKBD35 was transferred into a cryoprotectant composed of 25% (vol/vol) glycerol in addition to the components of the reservoir solution. Diffraction intensities were collected at the National Synchrotron Radiation Research Center (NSRRC; Hsinchu, Taiwan) to a resolution of 1.42 Å. Similarly, a crystal of the sALPFp-bound form of PvFKBD35 was transferred into a cryo-protectant composed of 50% mineral oil and paratone mixture and flash cooled in liquid nitrogen. Single-wavelength data sets of the cocrystallized crystals were collected in-house at 100 K on a Rigaku RAXIS IV image plate detector with a Rigaku RA-Micro 7 HFM rotating copper anode generator (Rigaku/MSC). The crystals diffracted to 1.65 Å. Data sets of apo PvFKBD35 and the PvFKBD35 complex with sALPFp were indexed, integrated, and scaled using the HKL2000 suite of programs (24) and iMosflm (25), respectively. The data collection statistics are given in Table 1 for these two crystals. The monomer coordinates of the previously solved structure of FK506-bound PvFKBD35 (PDB identifier 3IHZ) was used as a search model (19) for structure solution by the molecular replacement method using the program PHASER (26). Difference Fourier syntheses were calculated after rigid-body refinement of the structure. Inspection of the FO-FC and 2FO-FC map of the protein-peptide complex clearly showed electron density corresponding to the bound peptide, albeit for few terminal atoms of the succinic acid and p-nitroanilide moieties. Upon closer investigation, it was observed that these two moieties are exposed to the surface (see Fig. S1 in the supplemental material), while the remaining residues of the peptide are sandwiched in the dimer interface. An occupancy refinement using the program PHENIX (27) could not help improve the density, indicating the high mobility of these groups. Iterative cycles of model building were carried out using the program COOT (28) followed by refinement using REFMAC5 (29) of the CCP4 suite (30) and PHENIX (27), for the apo- and peptide-bound structures, respectively. The geometry of the final model was checked with PROCHECK (31), and the figures are drawn using the program PyMOL (32). Atomic coordinates and structure factors for the apo PvFKBD35 and sALPFp-bound forms have been deposited in the RCSB Protein Data Bank under accession codes 3NI6 and 4ITZ, respectively.
Table 1.
X-ray crystallographic data collection and refinement statistics for the apo- and sALPFp-bound forms of PvFKBD35
| Statistic | Value fora: |
|
|---|---|---|
| Apo PvFKBD35 | PvFKBD35-sALPFp complex | |
| Data collection | ||
| Space group | P 1 21 1 | P 1 21 1 |
| Unit cell a, b, c (Å) | 54.9, 41.8, 55.3 | 53.12, 45.79, 54.78 |
| α, β, γ (°) | 90.0, 106.4, 90.0 | 90.0, 112.47, 90.0 |
| Resolution (Å) | 22.66–1.42 (1.47–1.42) | 33.5–1.65 (1.68–1.65) |
| Completeness (%) | 97.9 (88.3) | 92.7 (88.8) |
| Redundancy | 5.8 (4.4) | 10.7 (10.9) |
| Rmerge (%) | 5.5 (53.2) | 5.2 (34.7) |
| I/σ(I) | 40.3 (2.0) | 25.4 (5.0) |
| Refinement | ||
| No. of reflections (working set/test set) | 42,266/2,249 | 27,158/954 |
| R factor (Rwork/Rfree) | 0.171/0.219 | 0.174/0.206 |
| No. of atoms of protein/water | 1,947/278 | 1,930/301 |
| Average B factor (Å2) | 22.02 | 31.31 |
| RMS deviation | ||
| Bond length (Å) | 0.014 | 0.013 |
| Bond angle (°) | 1.502 | 1.458 |
| Ramachandran (%) | ||
| Most favored | 94.6 | 99.59 |
| Additionally allowed | 5.4 | 0.41 |
| Generously allowed | 0.0 | 0.0 |
| Disallowed | 0.0 | 0.0 |
Values in parentheses refer to the corresponding values of the highest-resolution shell.
NMR titration of PvFKBD35 with ALPF tetrapepetide.
Interaction between PvFKBD35 and the ALPF peptide was studied by a two-dimensional (2D) 1H-15N heteronuclear single quantum coherence (HSQC) NMR titration experiment at 298 K on a Bruker Avance700. The 15N-labeled PvFKBD35 (0.35 mM) was titrated with various concentrations of the peptide up to 1:20 (protein/peptide ratio). Chemical shift perturbations were analyzed after adding the peptide. Spectra were processed with NMRPipe (33) and analyzed using SPARKY (34).
PPIase assay.
PPIase activities of the wild-type and mutant PvFKBD35 were determined by protease-coupled assay as described previously (35). Reaction was performed at 4°C, and the change in the absorbance was measured at 390 nm for 5 min in a Shimadzu UV-1601PC UV-Vis spectrophotometer.
Protein structure accession numbers.
The coordinates and structure factors of apo PvFKBD35 and the PvFKBD35 complex with sALPFp have been deposited in Protein Data Bank with PDB identifiers of 3NI6 and 4ITZ, respectively.
RESULTS
Crystal structure of apo PvFKBD35.
The crystallographic structure of apo PvFKBD35 shows two molecules per asymmetric unit. A total of 245 residues, eight glycerol molecules, and 278 water molecules were included in the final model. The N-terminal residues 1 and 2 of molecule A and 1 to 5 of molecule B are missing from the final model, presumably due to their high mobility in the crystal. Details of data collection and refinement statistics are given in Table 1. The structure of the monomer consists of six antiparallel β-strands and a short central α-helix (Fig. 1A). The two molecules in the asymmetric unit have nearly identical structure (root mean square [RMS] deviation is 0.46 Å for 121 α-carbon atoms). The two monomers of PvFKBD35 assemble to form a dimer through interactions between β3-β4 loops and β5-β6 loops (Fig. 1A). There are hydrogen bonds between the main chain N and O atoms and involve residues R60, E103, G104, and G106 of one PvFKBD35 molecule and residues Q70, G111, G106, and G104 of the other molecule (Fig. 1B). The tertiary structure of PvFKBD35 shows one disulfide bond formed by Cys105-Cys105 of both molecules (Fig. 1A and B). But native polyacrylamide gel electrophoresis (PAGE) and gel filtration chromatography of the protein sample and dissolved crystals revealed only the presence of monomeric species, indicating that the disulfide bond between two molecules could be a crystallization artifact (data not shown).
Fig 1.

Crystal structure of apo PvFKBD35. (A) The overall structure of the FKBD35 domain of P. vivax is shown as a cartoon diagram. The disulfide bond between Cys 105 of both chains is displayed in stick mode with the sulfur atoms colored in yellow. The loops corresponding to chains A (left) and B (right) are colored blue and green, respectively, while the helices are colored red and sheets are in yellow. (B) Close view of active-site residues, in stick mode, that line up the dimer interface formed between the two PvFKBD35 (chain A, blue; and chain B, green) molecules in the asymmetric unit. The disulfide bond is shown as a stick, and hydrogen bonds are displayed as dashed lines. (C) Shown is the superimposition of the backbone traces of apo PvFKBD35 colored in blue (β3-β4, β4-α1, and β5-β6 loops colored red) and FK506-bound PvFKBD35 colored in white (β3-β4, β4-α1, and β5-β6 loops colored purple). The FK506 inhibitor is shown as yellow-colored sticks.
A superimposition of the crystal structures of the apo form of PvFKBD35 and FK506-bound PvFKBD35 (PBB identifier 3IHZ) gives an overall RMS deviation of 0.743 Å for 121 equivalent α-carbon atoms. Most of the secondary structural elements of the apo enzyme are superimposed in FK506-bound PvFKBD35 without major differences, except differences observed in the loops that connect strand β3 to strand β4 and strand β5 to strand β6 (Fig. 1C).
Crystal structure of PvFKBD35 complexed with substrate sALPFp.
PvFKBD35 cocrystallized with the substrate tetrapeptide, sALPFp, diffracted to a 1.65 Å resolution with two molecules in the asymmetric unit. The initial phases of the peptide-bound structure of PvFKBD35 were determined using the molecular replacement technique. The detailed statistics of data collection, phasing, and refinement are given in Table 1.
The final refined crystallographic structure of the sALPFp-bound PvFKBD35 complex from P. vivax contains a total of 242 amino acids belonging to two independent molecules present in the asymmetric unit; a tetrapeptide, sALPFp (Fig. 2A); 301 water molecules; and a sulfate ion. The two PvFKBD35 monomers present in the asymmetric unit can be superimposed with an average RMS deviation of 0.278 Å for the 121 Cα atoms, indicating that the molecules in the asymmetric unit have very similar molecular conformations. Their interface is formed by active-site residues similar to that found in the apo form (Fig. 1B) and the FK506-bound form (19). In addition, this crystallographic dimer interface can accommodate only one substrate or inhibitor (19). In solution, the protein exists as a monomer confirmed by the NMR structure (19) and the lack of disulfide bonds in dissolved crystals. The final electron density map for the structure shows good density for all the residues except for the first five residues in the N terminus, which are very flexible in both the chains. The final model has good stereochemistry, as can be seen from the Ramachandran plot, where 99.59% of the residues are in the most favored region and 0.41% are in the additionally allowed region. No residues are found either in the generously allowed region or in the disallowed region. The binding of the tetrapeptide sALPFp shows a canonical substrate binding mode in the binding pocket formed within α1 and the β-sheet platform, which is similar to that of FK506 (Fig. 2B and C; see also Fig. 4A and 6). During the structure refinement procedure, the peptide density could be clearly observed by the positive peaks in the difference Fourier map in the dimer interface (Fig. 3A), but the density for the terminal atoms of the succinic and p-nitroanilide moieties was weak. The electron density was found in the active-site pocket, a shallow cavity located within α1 and the β-sheet platform (Fig. 2C). The 2FO-FC electron density map of the peptide at the 1.0 σ level is shown in Fig. 3A. The tetrapeptide interacts mainly with the β5-β6 loop and also with residues in the β4-α1 loop (Fig. 2B and 4A). The proline residue in the substrate peptide sALPFp adopts the cis conformation when bound to PvFKBD35 (Fig. 3A and 4A). The conformation of this proline residue can be clearly distinguished from the electron density map (Fig. 3A) owing to the high resolution of the crystal data. To examine whether the substrate binding causes a conformational change in the binding interface, the sALPFp-bound PvFKBD35 structure was compared with apo PvFKBD35 (Fig. 3B). The RMS deviation calculated by superposition of the main chains of both structures is 0.677 Å, suggesting there is no major structural change due to the substrate binding, though subtle changes are observed in the ligand-flanking loops, namely the β3-β4 loop, β4-α1 loop, and β5-β6 loop.
Fig 2.

Crystal structure of sALPFp-bound PvFKBD35. (A) The cartoon diagram of the two monomers of PvFKBD35-sALPFp in the asymmetric unit, with color code and orientation similar to those described in Fig. 1A. The sALPFp tetrapeptide is represented as sticks within the pink-colored circle. (B) Cartoon representation of the complex formed between PvFKBD35 and sALPFp (in green sticks), with the secondary structures labeled in black. (C) Electrostatic surface potential representation of PvFKBD35, with sALPFp shown as green-colored sticks. It could be seen that the proline of sALPFp docks itself deep into the hydrophobic pocket.
Fig 4.

Interaction of sALPFp with PvFKBD35. (A) The hydrogen bonded (blue dashes) and nonbonded (gray dashes) interactions made by sALPFp (green sticks) with PvFKBD35, revealing the binding of the proline (P3) region into the deep active-site pocket. The three ω angles corresponding to the peptide bonds are also labeled and shown by pink dots. (B) Chemical shift perturbations of PvFKBD35 (0.35 mM) upon adding the substrate peptide. Residues showing perturbations upon addition of the peptide are labeled.
Fig 6.

Comparison of FK506 and sALPFp binding to PvFKBD35. (A) Shown is a superposition of the Cα traces of FK506-bound PvFKBD35 (colored in white) with the sALPFp complex (colored in brown). The ligand-flanking loops of the FK506- and sALPFp-bound structures are colored in purple and green, respectively. The bound FK506 and sALPFp are represented in yellow and green sticks, respectively. (B) A closer view of the superposition of the FK506 (yellow sticks) over the sALPFp (green sticks) shows that the pipecolic moiety of FK506 overlays itself on the proline residue of substrate (within the blue circle). The cartoon representation of the PvFKBD35-sALPFp (in orange) and W77 (in stick mode), which forms the base of the pocket, is shown for reference. (C) The common residues that are involved in the binding of FK506 (in cyan) and sALPFp (in orange) in the PvFKBD35 structure are shown in stick mode, while those of the apo structure are shown in white. The figure enables us to visualize the subtle changes in the active-site residues due to ligand binding.
Fig 3.

Stereoview of sALPFp peptide and overlay of apo PvFKBD35 and sALPFp-bound PvFKBD35. (A) Stereoview of the electron density map (2FO-FC) contoured at the 1.0 σ level for the peptide molecule. It could be clearly seen that the Leu-Pro peptide bond adopts a cis-isomer conformation. (B) Superposition of the Cα traces of apo PvFKBD35 colored in blue and PvFKBD35 bound to sALPFp colored in brown. The ligand-flanking loops of the apo- and sALPFp-bound structures are colored in red and green, respectively. The sALPFp peptide is shown as green sticks for reference.
Residues of PvFKBD35 responsible for substrate binding and PPIase activity.
The active site has a network of hydrogen bonds between amino acid side chains and main chains and solvent molecules. The sALPFp tetrapeptide has hydrogen bonding interactions with D55, I74, and Y100 and three water molecules (Fig. 4A and Table 2). Specifically, the main chain nitrogen and carbonyl oxygen atom of proline interact with the side chain hydroxyl group of Y100, the leucine carbonyl oxygen interacts with the main chain nitrogen of I74, and the nitrogen of p-nitroanilide interacts with the side chain oxygen of D55. The Y100 forms a strong hydrogen bond (36) with a distance of 2.26 Å from the carbonyl oxygen of proline, while the D55 forms a weak one (36) at 3.6 Å from the nitrogen of p-nitroanilide (Table 2). Such short, strong hydrogen bonds observed in protein structures (37) are believed to play an important role in the enzyme catalysis, implying the importance of Y100. In addition, main chain nitrogen of alanine and carbonyl oxygen of phenylalanine is stabilized by three water molecules. Nonpolar contacts (Fig. 4A and Table 2) with other key active-site residues, such as Y43, F54, D55, V73, W77, C105, I109, and F117, also stabilize the substrate interactions. The D55 OD2 of PvFKBD35 is just 3.14 Å away from the beta carbon of the substrate proline and 3.6 Å from the nitrogen atom of p-nitroanilide, which could have probably resulted in their peak shifts during NMR titration (Fig. 4B). The residues of PvFKBD35 involved in sALPFp interactions are mostly coming from the β4-α1 loop (V73 and I74) and β5-β6 loop (C105, I109, Y100) (Fig. 4A). The leucine and proline, the two key residues of the substrate, form strong hydrogen bonding interaction with Y100 and I74 (Fig. 4A) and are buried deep inside the nucleus of the FKBP active-site pocket (Fig. 2C). We further investigated the molecular interaction between the active-site residues and the substrate peptide by NMR spectroscopy. To this end, the 15N-labeled protein was titrated with various concentrations of the peptide, and amide chemical shifts of PvFKBD35 were investigated on 2D 1H-15N HSQC. The chemical shift perturbations were detected mainly in four residues, D55, H67, V73, and I74 (Fig. 4B; see also Fig. S2 in the supplemental material). The chemical shift perturbations of D55, V73, and I74 are due to substrate binding and consistent with the crystallographic structure of the protein-substrate complex. However, in the crystal structure, the residue H67 is ∼10 Å away from the substrate located at the C-terminal end of the β4 strand. Thus, one possibility for the chemical shift perturbation of H67 might be an indirect effect of the substrate binding-induced conformational changes in the β4-α1 loop (Fig. 3B).
Table 2.
Interactions between PvFKBD35 and sALPFp
| Type of contact | PvFKBD35 atoms | Donor-acceptor distance (Å) |
|---|---|---|
| Polar | ||
| A1amino | H2O | 2.77 |
| L2 carbonyl oxygen | I74 N | 2.92 |
| P3 carbonyl oxygen | Y100 OH | 2.26 |
| P3 amino | Y100 OH | 3.43 |
| F4 carbonyl oxygen | H2O | 2.65 |
| F4 carbonyl oxygen | H2O | 2.51 |
| p-Nitroanilide N1 | D 55 OD2 | 3.60 |
| Nonpolar | ||
| L2 | V73, I74, Y100 | |
| P3 | Y43, D55, W77, Y100, F117 | |
| F4 | C105, I109 | |
| p-Nitroanilide | F54 |
Role of Y100 in catalysis.
The structural analysis of PvFKBD35 in complex with sALPFp reveals the importance of Y100 in the cis-trans isomerization of the peptidylprolyl bond. It could be observed (Fig. 4A, Table 2) that Y100 is the only residue in the PvFKBD35 active site that plays a key role by anchoring the leucine and proline, the two key residues of the substrate peptide. Interestingly, Y100 did not show as much perturbations as D55, V73, and I74 from NMR titration (Fig. 4B), consistent with a comparison of the apo with the sALPFp-bound crystal structures (see Fig. 6C). The average RMS deviation of Y100 is 0.22 Å, while that of D55, V72, and I74 is 0.44 (0.17) Å. This indicates that Y100 might hold on to its position in the active site and need not require much perturbation upon substrate binding. To further validate the role of the Y100 side chain on PPIase activity, mutagenesis studies were performed using Y100F, Y100W, Y100R, Y100P, Y100E, Y100L, and Y100A mutants. The data showed that the Y100F and Y100W mutants maintain activity similar to that of the wild type. The Y100R mutant retains 70% activity, while the Y100P and Y100E mutants have 60% and 50% of the wild-type activity, respectively. The Y100L and Y100A mutants show 20 to 30% activity (Fig. 5), substantiating the fact that the aromatic residue at the Y100 position plays an important role for the PPIase activity, corroborating with the short hydrogen bond (2.26 Å) observed in the crystal structure.
Fig 5.
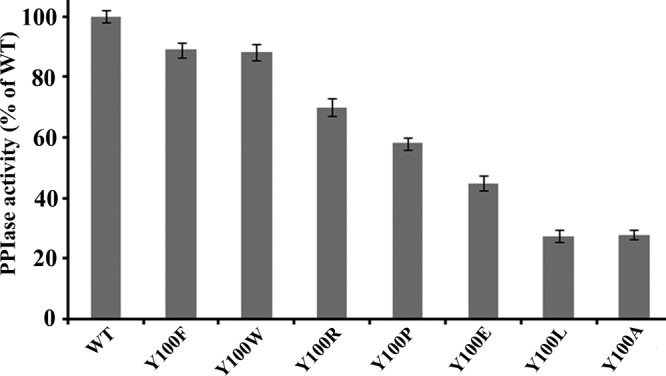
PPIase activity of wild-type PvFKBD35 and the Y100 mutants. PPIase assay was performed at 4°C as described in Materials and Methods, and change in the absorbance was measured at 390 nm for 5 min.
Comparison of the substrate- and inhibitor-bound forms of PvFKBD35.
The substrate-bound state of the PvFKBD35-sALPFp complex enables, for the first time, a comparison with the inhibitor (FK506)-bound state of FKBP (19). The three PvFKBD35 structures in apo-, FK506-, and sALPFp-bound forms possess similar resolutions, 1.42 Å, 1.67 Å, and 1.65 Å, respectively, and cell parameters which validate the comparison. The RMS deviation values calculated by superposition of the main chain atoms of the PvFKBD35-sALPFp-bound structure with the apo- and FK506-bound forms are 0.360 Å and 0.645 Å, respectively, for the 121 equivalent α-carbon atoms (Fig. 3B and 6A). Minor differences could be observed in the ligand-flanking loop regions of β3-β4, β4-α1, and β5-β6. The comparison reveals that both the substrate and the inhibitor FK506 are located in the ligand binding pocket, formed within α1 and the β-sheet platform (Fig. 6B and C). The proline of the substrate superimposes over the pipecolic ring of FK506 (Fig. 6B) just above W77, which forms the platform for this hydrophobic active site. A comparison of the active-site residues in the apo-, FK506-, and sALPFp-bound forms (Fig. 6C) indicates subtle changes in W77 and C105, the two residues positioned at the perpendicular ends of the active site apart from other residues, notably V73 and I109. A directional motion, though negligible, can be observed in these four residues, where it closes in toward the pocket in the sALPFp structure and moves away in the FK506 structure (Fig. 6C).
DISCUSSION
In our present study, we examined the binding of PvFKBD35 with its peptide substrate sALPFp and also compared the modes of binding between the substrate and its inhibitor, FK506. Our structural studies together with NMR titration and mutational data highlight the roles of D55, I74, and Y100 in the substrate binding in cis conformation. Previous studies demonstrated that most of the mutations on Y82 in HsFKBP12, which is equivalent to Y100 in PvFKBD35, resulted in a decrease in the PPIase activity of HsFKBP12, except aromatic, arginine, and proline residues (38). Similarly, in our studies, Y100A and Y100L mutations showed significant reductions in the PPIase activity. Other mutants, like Y100R, Y100P, and Y100E, also showed a decline in PPIase activity. However, Y100F and Y100W retained significant activity similar to that of the wild type, suggesting an important role of aromatic residues in isomerization by the parasite FKBP. The importance of Y100 in PPIase activity is reaffirmed from the short, strong hydrogen bond with the substrate proline, observed in the crystal structure. In the structure presented, the cis peptide binds with an ω angle of −1.91°, which confirms that the substrate binds to the enzyme at a ground-state conformation instead of transition state (39). Structural analysis of the PvFKBD35-sALPFp complex suggests that upon moving to the trans conformation, due to lack of any interaction with the protein, the N terminus of the peptide might rotate, because proline and phenylalanine are stabilized by a majority of the polar and nonpolar interactions with the active-site residues (Table 2), which stabilize the C terminus of the peptide; moreover, rotation of the C terminus could generate steric clashes. On the other hand, comparison between FK506 and substrate-bound PvFKBD35 revealed that they share the common residues. This also explains the molecular basis of FK506 binding and FK506-mediated inhibition of PPIase activity of PvFKBD35. Although FK506 inhibits the growth of the Plasmodium parasite (40), the potent immunosuppressive activity of FK506 excludes this from use as an antimalarial drug. It has been suggested that FK506 analogs, devoid of immunosuppressive activity, could be developed as antimalarial drugs (unpublished data), causing selective misfolding of proteins and parasite inhibition. Insightful information gained from our PvFKBD35-sALPFp crystal structure might also provide a strategy for developing antimalarial therapeutics targeting the parasite chaperone.
In conclusion, we investigated the binding of PvFKBD35 with sALPFp, demonstrated the critical residues involved in the binding, and revealed that the substrate tetrapeptide adopted a cis-proline isomer mode. Comparison studies of FK506 and substrate binding confirmed the common residues essential for binding of these ligands.
ACKNOWLEDGMENTS
We thank Malathy Sony S. Manimekalai for her help in the structure determination of PvFKBD35 with the peptide substrate. We also thank the National Synchrotron Radiation Research Center (NSRRC) staff at beamline 13B1 for expert help with data collection. (NSRRC is a national user facility supported by the National Science Council of Taiwan, Republic of China. The Synchrotron Radiation Protein Crystallography Facility is supported by the National Research Program for Genomic Medicine.)
This study was supported by a Ministry of Health Singapore NMRC IRG grant (NMRC/1245/2010).
Footnotes
Published ahead of print 22 February 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/EC.00016-13.
REFERENCES
- 1. Fischer G, Aumuller T. 2003. Regulation of peptide bond cis/trans isomerization by enzyme catalysis and its implication in physiological processes. Rev. Physiol. Biochem. Pharmacol. 148:105–150 [DOI] [PubMed] [Google Scholar]
- 2. Lu KP, Hanes SD, Hunter T. 1996. A human peptidyl-prolyl isomerase essential for regulation of mitosis. Nature 380:544–547 [DOI] [PubMed] [Google Scholar]
- 3. Ou W, Luo W, Park YD, Zhou HM. 2001. Chaperone-like activity of peptidyl-prolyl cis-trans isomerase during creatine kinase refolding. Protein Sci. 10:2346–2353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lu KP, Finn G, Lee TH, Nicholson LK. 2007. Prolyl cis-trans isomerization as a molecular timer. Nat. Chem. Biol. 3:619–629 [DOI] [PubMed] [Google Scholar]
- 5. Fischer G. 2000. Chemical aspects of peptide bond isomerization. Chem. Soc. Rev. 29:119–127 [Google Scholar]
- 6. Yaffe MB, Schutkowski M, Shen M, Zhou XZ, Stukenberg PT, Rahfeld JU, Xu J, Kuang J, Kirschner MW, Fischer G, Cantley LC, Lu KP. 1997. Sequence-specific and phosphorylation-dependent proline isomerization: a potential mitotic regulatory mechanism. Science 278:1957–1960 [DOI] [PubMed] [Google Scholar]
- 7. Albers MW, Walsh CT, Schreiber SL. 1990. Substrate specificity for the human rotamase FKBP: a view of FK506 and rapamycin as leucine-(twisted amide)-proline mimics. J. Org. Chem. 55:4984–4986 [Google Scholar]
- 8. Feske S, Okamura H, Hogan PG, Rao A. 2003. Ca2+/calcineurin signalling in cells of the immune system. Biochem. Biophys. Res. Commun. 311:1117–1132 [DOI] [PubMed] [Google Scholar]
- 9. Liu J, Farmer JD, Jr, Lane WS, Friedman J, Weissman I, Schreiber SL. 1991. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 66:807–815 [DOI] [PubMed] [Google Scholar]
- 10. Steinmann B, Bruckner P, Superti-Furga A. 1991. Cyclosporin A slows collagen triple-helix formation in vivo: indirect evidence for a physiologic role of peptidyl-prolyl cis-trans-isomerase. J. Biol. Chem. 266:1299–1303 [PubMed] [Google Scholar]
- 11. Rassow J, Mohrs K, Koidl S, Barthelmess IB, Pfanner N, Tropschug M. 1995. Cyclophilin 20 is involved in mitochondrial protein folding in cooperation with molecular chaperones Hsp70 and Hsp60. Mol. Cell. Biol. 15:2654–2662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wulf G, Finn G, Suizu F, Lu KP. 2005. Phosphorylation-specific prolyl isomerization: is there an underlying theme? Nat. Cell Bio. 7:435–441 [DOI] [PubMed] [Google Scholar]
- 13. Zhou XZ, Lu PJ, Wulf G, Lu KP. 1999. Phosphorylation-dependent prolyl isomerization: a novel signaling regulatory mechanism. Cell. Mol. Life Sci. 56:788–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sarkar P, Reichman C, Saleh T, Birge RB, Kalodimos CG. 2007. Proline cis-trans isomerization controls autoinhibition of a signaling protein. Mol. Cell 25:413–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lummis SC, Beene DL, Lee LW, Lester HA, Broadhurst RW, Dougherty DA. 2005. cis-trans isomerization at a proline opens the pore of a neurotransmitter-gated ion channel. Nature 438:248–252 [DOI] [PubMed] [Google Scholar]
- 16. Ke H, Mayrose D, Cao W. 1993. Crystal structure of cyclophilin A complexed with substrate Ala-Pro suggests a solvent-assisted mechanism of cis-trans isomerization. Proc. Natl. Acad. Sci. U. S. A. 90:3324–3328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhao Y, Ke H. 1996. Crystal structure implies that cyclophilin predominantly catalyzes the trans to cis isomerization. Biochemistry 35:7356–7361 [DOI] [PubMed] [Google Scholar]
- 18. Ranganathan R, Lu KP, Hunter T, Noel JP. 1997. Structural and functional analysis of the mitotic rotamase Pin1 suggests substrate recognition is phosphorylation dependent. Cell 89:875–886 [DOI] [PubMed] [Google Scholar]
- 19. Alag R, Qureshi IA, Bharatham N, Shin J, Lescar J, Yoon HS. 2010. NMR and crystallographic structures of the FK506 binding domain of human malarial parasite Plasmodium vivax FKBP35. Protein Sci. 19:1577–1586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Aurrecoechea C, Brestelli J, Brunk BP, Dommer J, Fischer S, Gajria B, Gao X, Gingle A, Grant G, Harb OS, Heiges M, Innamorato F, Iodice J, Kissinger JC, Kraemer E, Li W, Miller JA, Nayak V, Pennington C, Pinney DF, Roos DS, Ross C, Stoeckert CJ, Jr, Treatman C, Wang H. 2009. PlasmoDB: a functional genomic database for malaria parasites. Nucleic Acids Res. 37:D539–D543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ravi CB, Gowthaman R, Raj AR, Gupta D, Sharma A. 2004. Distribution of proline-rich (PxxP) motifs in distinct proteomes: functional and therapeutic implications for malaria and tuberculosis. Protein Eng. Des. Sel. 17:175–182 [DOI] [PubMed] [Google Scholar]
- 22. Alag R, Bharatham N, Dong A, Hills T, Harikishore A, Widjaja AA, Shochat SG, Hui R, Yoon HS. 2009. Crystallographic structure of the tetratricopeptide repeat domain of Plasmodium falciparum FKBP35 and its molecular interaction with Hsp90 C-terminal pentapeptide. Protein Sci. 18:2115–2124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brinker A, Weber E, Stoll D, Voigt J, Müler A, Sewald N, Jung G, Wiesmüller KH, Bohley P. 2000. Highly potent inhibitors of human cathepsin L identified by screening combinatorial pentapeptide amide collections. Eur. J. Biochem. 267:5085–5092 [DOI] [PubMed] [Google Scholar]
- 24. Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276:307–328 [DOI] [PubMed] [Google Scholar]
- 25. Battye TG, Kontogiannis L, Johnson O, Powell HR, Leslie AG. 2011. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 67:271–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J. Appl. Crystallogr. 40:658–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66:213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60:2126–2132 [DOI] [PubMed] [Google Scholar]
- 29. Murshudov GN, Vagin AA, Dodson EJ. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53:240–257 [DOI] [PubMed] [Google Scholar]
- 30. Collaborative Computational Project N 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50:760–765 [DOI] [PubMed] [Google Scholar]
- 31. Laskowski RA, MacArthur MW, Moss DS, Thornton JM. 1993. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26:283–293 [Google Scholar]
- 32. DeLano WL. 2002. The PyMol molecular graphics system. DeLano Scientific, Palo Alto, CA [Google Scholar]
- 33. Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. 1995. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6:277–293 [DOI] [PubMed] [Google Scholar]
- 34. Goddard TD, Kneller DG. 1997. SPARKY 3. University of California, San Francisco, CA [Google Scholar]
- 35. Harrison RK, Stein RL. 1990. Substrate specificities of the peptidyl prolyl cis-trans isomerase activities of cyclophilin and FK-506 binding protein: evidence for the existence of a family of distinct enzymes. Biochemistry 29:3813–3816 [DOI] [PubMed] [Google Scholar]
- 36. Jeffrey GA. 1997. An introduction to hydrogen bonding. Oxford University Press, New York, NY [Google Scholar]
- 37. Rajagopal S, Vishveshwara S. 2005. Short hydrogen bonds in proteins. FEBS J. 272:1819–1832 [DOI] [PubMed] [Google Scholar]
- 38. Ikura T, Ito N. 2007. Requirements for peptidyl-prolyl isomerization activity: a comprehensive mutational analysis of the substrate-binding cavity of FK506-binding protein 12. Protein Sci. 16:2618–2625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hur S, Bruice TC. 2002. The mechanism of cis-trans isomerization of prolyl peptides by cyclophilin. J. Am. Chem. Soc. 124:7303–7313 [DOI] [PubMed] [Google Scholar]
- 40. Bell A, Wernli B, Franklin RM. 1994. Roles of peptidyl-prolyl cis-trans isomerase and calcineurin in the mechanisms of antimalarial action of cyclosporine, FK506, and rapamycin. Biochem. Pharmacol. 48:495–503 [DOI] [PubMed] [Google Scholar]