

Abstract
Proteins that are destined for release outside the eukaryotic cell, insertion into the plasma membrane, or delivery to intracellular organelles are processed and folded in the endoplasmic reticulum (ER). An imbalance between the level of nascent proteins entering the ER and the organelle's ability to manage that load results in the accumulation of unfolded proteins. Terminally unfolded proteins are disposed of by ER-associated degradation (ERAD), a pathway that transports the aberrant proteins across the ER membrane into the cytosol for proteasomal degradation. The ERAD pathway was targeted in the mold pathogen Aspergillus fumigatus by deleting the hrdA gene, encoding the A. fumigatus ortholog of Hrd1, the E3 ubiquitin ligase previously shown to contribute to ERAD in other species. Loss of HrdA was associated with impaired degradation of a folding-defective ERAD substrate, CPY*, as well as activation of the unfolded-protein response (UPR). The ΔhrdA mutant showed resistance to voriconazole and reduced thermotolerance but was otherwise unaffected by a variety of environmental stressors. A double-deletion mutant deficient in both HrdA and another component of the same ERAD complex, DerA, was defective in secretion and showed hypersensitivity to ER, thermal, and cell wall stress. However, the ΔhrdA ΔderA mutant remained virulent in mouse and insect infection models. These data demonstrate that HrdA and DerA support complementary ERAD functions that promote survival under conditions of ER stress but are dispensable for virulence in the host environment.
INTRODUCTION
Aspergillus fumigatus is the predominant mold pathogen of humans, responsible for a life-threatening pneumonia that can progress to invasive aspergillosis (IA), a disseminated infection with a very poor prognosis (1). Predisposition to IA is largely a consequence of immunosuppression. Thus, the expansion of immunosuppressive therapies to support organ transplantation, as well as the implementation of aggressive treatments for hematologic malignancies, is contributing to a rise in the number of individuals at risk for acquiring the infection (2–4). Moreover, despite several improvements in diagnostic platforms and antifungal drugs, the morbidity and mortality rates associated with IA remain high, emphasizing the need for more detailed knowledge of A. fumigatus physiology and its relationship to virulence and antifungal drug susceptibility (5).
An emerging body of evidence suggests that the secretory pathway of filamentous fungi is a point of vulnerability that could be accessible to therapeutic intervention (6, 7). The genome of A. fumigatus, like those of many other environmental saprophytic fungi, is enriched for secreted hydrolytic enzymes. This feature allows the organism to extract nutrients from diverse extracellular polymers, including those found in mammalian tissues (8, 9). The unfolded-protein response (UPR) is an adaptive stress response that supports the ability of the endoplasmic reticulum (ER) to secrete large quantities of these enzymes. This is accomplished by remodeling gene expression to expand the protein-folding capacity of the ER in proportion to the accumulation of unfolded nascent proteins in the ER lumen. The A. fumigatus UPR is regulated by IreA (Ire1 in yeast and higher eukaryotes), a molecular sensor embedded in the ER membrane that is comprised of a lumenal sensing domain and a cytosolic effector domain (6). Studies in Saccharomyces cerevisiae have shown that unfolded proteins trigger oligomerization of this protein in the ER membrane, which activates the cytosolic domain and induces the expression of a bZIP transcription factor known as HacA in filamentous fungi (10). As the master transcriptional regulator of the UPR, HacA maintains ER homeostasis by orchestrating transcriptional changes to buffer fluctuations in ER stress caused by increased levels unfolded proteins. Despite UPR activation, a substantial fraction of newly synthesized proteins will ultimately fail to attain their proper conformation (11). These proteins can form toxic aggregates in the ER lumen and are thus disposed of by ER-associated degradation (ERAD), a pathway that works in conjunction with the UPR to restore ER homeostasis. ERAD recognizes unfolded proteins and escorts them across the ER membrane into the cytosol, where they are targeted for proteasomal degradation (12). Hrd1 is at the center of this process, forming an ER-membrane complex comprised of the E3-ubiquitin ligase Hrd1 in close association with Der1 and other regulatory and scaffold proteins (13–15).
Current evidence indicates that the fungal pathogens A. fumigatus (16, 17), Cryptococcus neoformans (18), and Alternaria brassicicola (19) rely heavily on the UPR for the expression of virulence. However, much less is known about the importance of ERAD to fungal pathogenesis. We have previously shown that the hrdA gene, encoding the A. fumigatus homolog of the ERAD protein Hrd1, is induced by the UPR during acute ER stress (6). In this study, we examined the contribution of HrdA to the biology and virulence of A. fumigatus by deleting the hrdA gene. The ΔhrdA mutant showed a reduction in ERAD efficiency that was associated with compensatory activation of the UPR. The loss of hrdA alone had minimal effect on the stress response of the fungus. However, a double-deletion mutant lacking both HrdA and another member of the same complex, DerA, was hypersensitive to environmental stress, although it remained virulent. These data demonstrate that HrdA and DerA act cooperatively to relieve ER stress in A. fumigatus but are dispensable for surviving stress in the host environment.
MATERIALS AND METHODS
Strains and culture conditions.
The A. fumigatus strains used in this study are listed in Table 1. The strains were maintained on Aspergillus Minimal Medium (AMM) (20). Ammonium tartrate was used as the nitrogen source for all studies involving solid or liquid AMM. The radial growth rate on solid medium was assessed by inoculating 2,000 conidia onto the center of a plate of rich (inhibitory mold agar [IMA]) or minimal (AMM) medium, as indicated, and radial growth was monitored for 3 days at different temperatures. Growth in liquid culture was assessed by inoculating 20 ml of AMM with 2 × 107 conidia and incubating it at 37°C for 16 h with shaking at 150 rpm. The biomass was then measuring by weighing the dried mycelium. Sensitivity to tunicamycin (TM), brefeldin A (BFA), calcofluor white (CFW), or Congo red (CR) was determined by spotting conidia onto the center of each well of a 24-well plate containing different concentrations of each compound and monitoring growth for 2 to 4 days at 37°C. Statistical significance for growth assays was assessed by Student's t test on the average value of triplicate experiments. A P value of <0.05 was considered significant. Conidial counts were estimated by scraping the surface of the colony and pipetting up and down with liquid AMM three times. The released conidia were then counted with a hemacytometer.
Table 1.
Strains used in this study
| Strain | Genotype | Source |
|---|---|---|
| Wt (AfS28) | ΔakuA::ptrA | S. Krappmann |
| ΔhrdA | ΔakuA::ptrA ΔhrdA::hph | This study |
| ΔhrdA-C′ | ΔakuA::ptrA ΔhrdA::hph::hrdA::ble | This study |
| ΔderA | ΔakuA::ptrA ΔderA::ble | 7 |
| ΔhrdA ΔderA | ΔakuA::ptrA ΔhrdA::hph ΔderA::ble | This study |
| Wt (CPY*) | ΔakuA::ptrA (Af-CPY*-ble) | This study |
| ΔhrdA (CPY*) | ΔakuA::ptrA (Af-CPY*-ble) | This study |
Disruption of ERAD by deletion of hrdA and derA.
The PCR primers used in the study are listed in Table S1 in the supplemental material. For analysis of the cDNA sequence, total RNA was extracted from overnight cultures of wild-type (wt) A. fumigatus by crushing the mycelium in liquid nitrogen and resuspending it in TRI reagent (Sigma-Aldrich). The RNA was then reverse transcribed using the Superscript II reverse transcriptase first-strand synthesis system (Invitrogen), and the hrdA cDNA was PCR amplified using primers 730 and 731 and sequenced.
The hrdA gene was replaced with the phleomycin resistance gene using the split-marker method (21). The first two-thirds of the phleomycin resistance cassette was amplified from pAN7-1 using primers 398 and 408, creating PCR product 1. The second two-thirds of the phleomycin was then amplified with primers 410 and 409, creating PCR product 2. The left arm of the hrdA gene was amplified from wt DNA using primers 733 and 734, and the right arm was amplified with primers 735 and 736, generating PCR products 3 and 4, respectively. PCR products 1 and 3 were then combined in an overlap PCR with primers 733 and 408 to generate PCR product 5, and PCR products 2 and 4 were combined in an overlap reaction with primers 410 and 736 to generate PCR product 6. PCR products 5 and 6 were then cloned into pCR-Blunt II-TOPO (Invitrogen) to create plasmids p601 and p602, respectively. The p601 and p602 plasmids were linearized with XhoI/BamHI and EcoRI, respectively, and the bands corresponding to the inserts were gel extracted and transformed into A. fumigatus protoplasts as previously described (22). Loss of the hrdA gene was confirmed by Southern blotting of genomic DNA isolated from phleomycin-resistant monoconidial isolates using a probe located in the right arm of the deletion cassette (Fig. 1). Deletion of derA was accomplished as previously described (7), and the construction of the ΔhrdA ΔderA mutant was performed by deleting derA in the background of the ΔhrdA mutant. To construct the hrdA complementation plasmid, the entire hrdA locus was PCR amplified using primers 736 and 733 and cloned into PCR-Blunt II-TOPO. The resulting plasmid was manipulated further using a QuikChange II XL site-directed mutagenesis kit (Agilent) and primers 769 and 770 to introduce a new NcoI site downstream from the hrdA stop codon, generating plasmid p619. Digestion of p619 with NsiI released the insert, which was transformed into A. fumigatus protoplasts as previously described (22). Homologous reconstitution of the hrdA gene was demonstrated by genomic Southern blot analysis using a probe located in the right arm of the deletion cassette (Fig. 1), and the presence of the engineered NcoI site was confirmed by PCR amplification of genomic DNA and sequencing.
Fig 1.

Deletion of A. fumigatus hrdA and derA. The hrdA gene was replaced by a phleomycin resistance cassette (ble) and the derA gene by a hygromycin resistance cassette (hph), as shown. Homologous recombination with the hrdA gene was demonstrated by Southern blotting of XbaI-digested genomic DNA using a probe in the right arm of the deletion cassette (P1). Homologous reconstitution of the ΔhrdA mutant to create a ΔhrdA::hrdA strain was demonstrated using the same probe. Homologous recombination with the derA gene was shown by hybridizing NsiI-digested genomic DNA with a probe in the left arm of the deletion cassette (P2). A ΔhrdA ΔderA strain, lacking both genes, was constructed by deleting the derA gene in the background of the ΔhrdA mutant using the same derA deletion strategy. The ΔhrdA::hrdA strain has the hrdA deletion homologously reconstituted, as detailed in Materials and Methods. X, XbaI; N, NsiI.
Analysis of mRNA expression.
For Northern blot analysis, total RNA was extracted from overnight cultures in YG medium (0.5% yeast extract-2% glucose) by crushing the mycelium in liquid nitrogen and resuspending it in TRI reagent (Molecular Research Center, Cincinnati, OH). The RNA was fractionated by formaldehyde gel electrophoresis, and rRNA loading was visualized by SYBR Green II staining using a Storm PhosphorImager (Molecular Dynamics). Following transfer to a BioBond nylon membrane (Sigma), the RNA was hybridized to a 32P-labeled DNA probe for A. fumigatus bipA, as described previously (17).
For analysis of erg11A and erg11B mRNA expression by quantitative PCR (qPCR), 5 ml of AMM was inoculated with 5 × 106 conidia and incubated at 37°C for 16 h with shaking at 200 rpm. RNA was prepared by crushing the mycelium in liquid nitrogen and resuspending it in TRI reagent (Sigma-Aldrich). One microgram of the total RNA was reverse transcribed with avian myeloblastosis virus (AMV) reverse transcriptase using oligo(dT)18 as the primer. The qPCRs were performed using the SYBR green qPCR Super Mix (Invitrogen) according to the manufacturer's protocol, and tubulin was used as an internal control. The melting curve was monitored to verify the specificity of the amplification reaction. Control reactions in the absence of reverse transcriptase were used to verify the absence of DNA contamination. Relative RNA levels were then compared by the 2−ΔΔCT method using standard procedures. The erg11 primers are listed in Table S1 in the supplemental material.
Analysis of collagenase secretion.
Collagenase secretion was quantified using the Azocoll assay, as previously described (17). Briefly, conidia were inoculated at a concentration of 1 × 105/ml in 50 ml of AMM-FBS (AMM containing 10% heat-inactivated fetal bovine serum as the sole nitrogen and carbon source). The cultures were incubated at 37°C with gentle shaking at 150 rpm. After 72 h, a 1-ml aliquot of the culture was microcentrifuged at 15,000 × g for 5 min to remove residual biomass, and a 15-μl aliquot of the supernatant was added to 2.4 ml of a 5-mg/ml suspension of prewashed Azocoll (an insoluble collagen linked to an azo dye). The collagen-supernatant mixture was incubated at 37°C for 3 h with constant shaking at 350 rpm. The Azocoll-supernatant mixture was centrifuged at 13,000 × g for 5 min, and the amount of absorbance was normalized to the lyophilized weight of the 72-h biomass. The experiment was performed in triplicate, and the values represent the average and standard deviation (SD).
Analysis of ERAD activity.
The A. fumigatus carboxypeptidase Y (Af-CPY) gene (GenBank accession no. XP_751230; CpyA/Prc1), was PCR amplified from genomic DNA using primers 659 and 664 (primer 664 adds a single FLAG tag to the C terminus of the protein). The fusion gene was then inserted downstream of the Aspergillus nidulans gpdA promoter in a plasmid containing a hygromycin resistance cassette, using the XbaI sites incorporated into each primer. The coding sequence of the Af-CPY gene was then modified by site-directed mutagenesis using primers 657 and 658 to create Af-CPY* (G264R), which creates the same point mutation that targets CPY* for degradation in S. cerevisiae (G255R) (23). The plasmid was linearized with KpnI and SacI and introduced into wt and ΔhrdA protoplasts as an ectopically integrated expression construct using standard transformation procedures, as previously described (6). For analysis of CPY* degradation efficiency, the indicated strains were grown in 5 ml of YG medium for 18 h at 37°C and then treated with cycloheximide (100 μg/ml) for 6 h before harvesting. Mycelium was collected through a Miracloth filter (EMD Millipore, USA) and crushed in liquid nitrogen, and total proteins were extracted in a protein extraction buffer: 25 mM Tris-HCl, pH 7.4, 1 mM EDTA, 1 mM dithiothreitol (DTT), and 150 μl protease inhibitor cocktail (Thermo Scientific, IL). Particulate material was removed by centrifugation (13,000 × g for 10 min at 4°C), and supernatants containing the proteins were collected. Protein concentrations were determined using a Bio-Rad protein assay kit (Bio-Rad, San Francisco, CA) with bovine serum albumin as the standard. For each sample, 40 μg of total protein was fractionated by SDS-PAGE and immobilized on a polyvinylidene difluoride (PVDF) membrane (Bio-Rad, CA). Nonspecific binding sites were blocked by incubating the membranes overnight at 4°C in 5% (wt/vol) skim dry milk in phosphate-buffered saline (PBS) containing 0.05% (vol/vol) Tween 20. The PVDF membrane was then incubated for 4 h at 22°C with horseradish peroxidase (HRP)-conjugated mouse anti-FLAG monoclonal antibody (Sigma, MO). Antibody binding was detected using the Immobilon Western Chemiluminescent HRP Substrate (Millipore, Billerica, MA) according to the manufacturer's instructions. To confirm equal protein loading, the membrane was probed with a mouse monoclonal anti-actin antibody (Millipore, Temecula, CA). Band intensity was quantified with a ChemiDocXRS+ imaging system using ImageLab software.
Analysis of sterol content.
A total of 5 × 106 conidia were inoculated into 5 ml of AMM in a 50-ml conical tube and incubated at 37°C for 18 h, with gentle shaking at 200 rpm. The biomass was washed with sterile distilled water and dried under vacuum. The dried mycelium was weighed prior to crushing under liquid nitrogen and then saponified in 1 ml of alcoholic KOH (3% KOH in ethanol). Cholesterol was added as an internal standard, and sterols were extracted into petroleum ether (primarily hexane). The sterol concentrations were analyzed by gas chromatography using a known ratio of ergosterol to cholesterol as an external standard. Values are presented as micrograms of ergosterol per milligram (dry weight).
Animal models of invasive aspergillosis.
Groups of 12 CF-1 outbred female mice were given a single dose of the synthetic corticosteroid triamcinolone acetonide (40 mg/kg of body weight injected subcutaneously) on day −1. On day 0, the mice were anesthetized with 3.5% isoflurane and inoculated intranasally with a 20-μl saline suspension containing 2 × 106 conidia or with 20 μl of saline for a mock infection control (4 mice). Mortality was monitored for 7 days, and statistical significance was assessed by the log-rank test using Sigma Stat 3.5. For histopathologic analysis, CF-1 outbred female mice were immunosuppressed as described above, infected with 2 × 106 conidia, and sacrificed on day +3. The lungs were fixed by inflation with 4% phosphate-buffered paraformaldehyde, dehydrated, embedded in paraffin, sectioned at 5 μm, and stained with hematoxylin and eosin (HE) or Grocott methenamine silver (GMS). Microscopic examinations were performed on an Olympus BH-2 microscope and imaging system using Spot software version 4.6. Animal experiments were carried out in accordance with the Guide for the Care and Use of Laboratory Animals, the Public Health Service Policy on the Humane Care and Use of Laboratory Animals, and all U.S. Animal Welfare Act regulations. The experiments were approved by the Institutional Animal Care and Use Committee of the University of Cincinnati (protocol 06-01-03-02).
For analysis of virulence using the wax moth larva model (24, 25), Galleria mellonella larvae in the final instar stages were obtained from Vanderhorst, Inc. (St. Marys, OH). Fifteen larvae per group, weighing between 200 and 300 mg, were inoculated with 5 × 105 conidia in a 10-μl saline suspension in the last left proleg of each larva using a Hamilton syringe. The larvae were placed in petri dishes and incubated in the dark at 37°C for 5 days. The larvae were examined daily, and mortality was defined as lack of movement upon physical stimulation. A Kaplan-Meier survival curve was constructed and analyzed using a log-rank test with pairwise analysis (the Holm-Sidak method). A P value of < 0.001 was considered significant.
Nucleotide sequence accession numbers.
The GenBank accession numbers for the A. fumigatus hrdA gene and A. fumigatus CPY are EDP48823 and XP_751230.1, respectively.
RESULTS
Deletion of the ERAD genes hrdA and derA in A. fumigatus.
The A. fumigatus hrdA cDNA was reverse transcribed from mRNA isolated from vegetative cultures, and the splicing of two predicted introns from the hrdA gene was confirmed by sequencing of the cloned cDNA. The predicted HrdA protein revealed a domain organization similar to that of S. cerevisiae Hrd1p, including multiple transmembrane helices in the N-terminal half of the protein that are closely followed by a zinc RING finger domain (see Fig. S1 in the supplemental material). A. fumigatus HrdA is comprised of 771 amino acids, which is longer than the corresponding proteins in both S. cerevisiae (551 amino acids) and humans (616 amino acids). The hrdA gene was deleted from A. fumigatus by homologous recombination, resulting in the loss of an XbaI site within the hrdA gene, which is evident from the disappearance of a 2.7-kb fragment and the appearance of a 10-kb fragment when cut with XbaI (Fig. 1). DerA is the ortholog of S. cerevisiae Der1p, an ER membrane-anchored ERAD protein that is found in the same complex as Hrd1p (26). A double-deletion mutant lacking both the hrdA and derA genes was constructed by replacing the derA gene with a hygromycin resistance cassette in the background of the ΔhrdA mutant, resulting in the addition of an NsiI site that truncated the wt 9-kb NsiI fragment containing the derA gene to 3.6 kb in the ΔhrdA ΔderA mutant (Fig. 1). Confirmation of a complete deletion was obtained using probes located within the respective open reading frames (data not shown).
Loss of hrdA impairs the degradation of an ERAD substrate and induces ER stress.
CPY is a monomeric soluble enzyme that is delivered to the vacuole via the secretory pathway (27). The degradation of a folding-defective mutant version of the protein, CPY*, is commonly used as a molecular marker to monitor ERAD degradation activity in S. cerevisiae (28), but an analogous system has not been developed for filamentous fungi. To address this, the A. fumigatus homolog of S. cerevisiae CPY was PCR amplified from genomic DNA, incorporating a FLAG tag at the C terminus. Site-directed mutagenesis was then used to create a G264A mutation (Af-CPY*), which corresponds to the G255A mutation in S. cerevisiae CPY* (23). Following the introduction of the Af-CPY* expression construct into the wt and ΔhrdA strains, the efficiency of Af-CPY* degradation over time was monitored by arresting protein synthesis with cycloheximide (CHX) and monitoring Af-CPY* levels by Western blot analysis using an anti-FLAG tag antibody. As shown in Fig. 2, the steady-state levels of CPY* were similar between the two strains at the zero time point. However, in contrast to the wt, where the majority of the CPY* protein was degraded within 6 h of the CHX chase, the levels of CPY* failed to decline in the ΔhrdA mutant (Fig. 2), confirming that ERAD function is impaired in the absence of HrdA.
Fig 2.
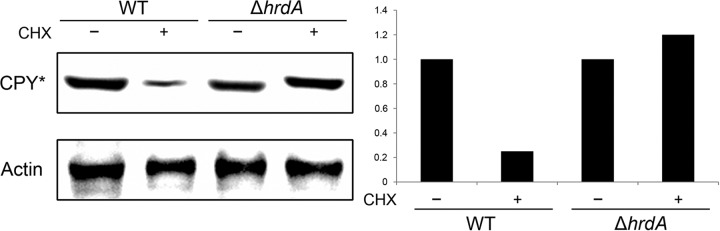
Loss of hrdA reduces ERAD efficiency. A CHX chase assay was used to monitor the degradation efficiency of a folding-defective mutant of A. fumigatus CPY (Af-CPY*). Overnight cultures of the indicated strains were treated with CHX to arrest protein synthesis, and total protein was extracted from the crushed mycelium after 6 h of incubation at 37°C. Equal amounts of protein were fractionated by SDS-PAGE and transferred to a PVDF membrane. The expected 62-kDa Af-CPY* protein was detected by immunoblot analysis using an anti-FLAG tag antibody, and actin was detected with an anti-actin antibody, as described in Materials and Methods. Quantitation of Af-CPY* levels was accomplished by measuring the relative band intensity normalized to actin levels. The values shown are relative to the wt in the absence of CHX.
The incomplete degradation of Af-CPY* in the ΔhrdA mutant suggests that the clearance of other misfolded proteins will be deficient in that strain, resulting in the accumulation of unfolded proteins and activation of the UPR. To test this, Northern blot analysis was used to monitor the expression of the UPR target gene bipA in the absence of any externally applied ER stress (17). The levels of bipA mRNA were elevated in the ΔhrdA mutant relative to the wt (Fig. 3), consistent with a compensatory response of the UPR to the loss of HrdA function. However, the ΔhrdA ΔderA mutant had levels of bipA mRNA comparable to those of the ΔhrdA single mutant, suggesting that the level of UPR signaling required to compensate for loss of HrdA is similar to what is required when both ERAD genes are absent, at least under normal conditions in the absence of exogenous ER stress.
Fig 3.
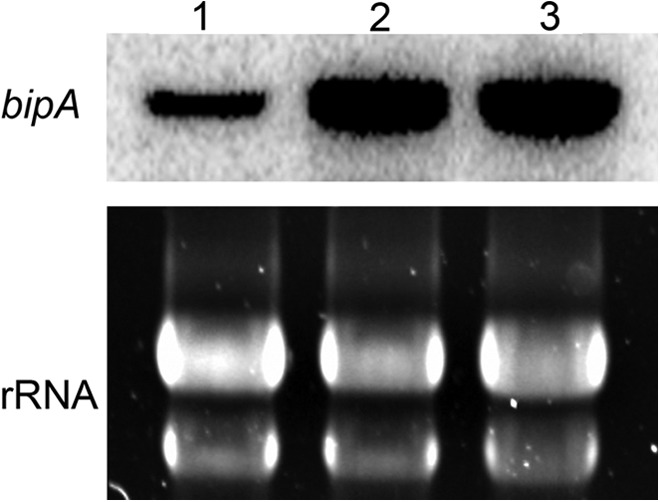
Loss of hrdA induces the UPR. Total RNA was extracted from overnight cultures in the absence of ER stress-inducing conditions, and the expression of the UPR target gene bipA was determined by Northern blotting. Lane 1, wt; lane 2, ΔhrdA; lane 3, ΔhrdA ΔderA. Equivalent RNA loading was confirmed by staining rRNA with SYBR Green.
HrdA synergizes with DerA to support radial growth under conditions of acute ER stress.
To determine the importance of hrdA to growth under conditions of ER stress, the ability of the ΔhrdA mutant to withstand conditions that disrupt ER-Golgi apparatus transport (BFA treatment) or impair N-linked glycosylation (TM treatment) (29) was tested. The radial growth rates of the ΔhrdA and ΔderA mutants were indistinguishable from wt in the presence of BFA or TM, although colony morphology was altered in the mutants due to reduced conidiation (approximately 3-fold less than the wt) and increased aerial hyphae at the higher levels of BFA. In contrast, the radial growth of the ΔhrdA ΔderA mutant was reduced on medium containing either of these compounds, indicating that the loss of both hrdA and derA caused a more severe growth defect than the loss of either gene alone under these conditions (Fig. 4). However, the growth of all three mutant strains was indistinguishable from that of the wt in the absence of added ER stress on either solid AMM (Fig. 4) or liquid AMM (data not shown), suggesting that hrdA and/or derA is dispensable in the absence of stress but provides synergistic functions that support growth under conditions of acute ER stress.
Fig 4.
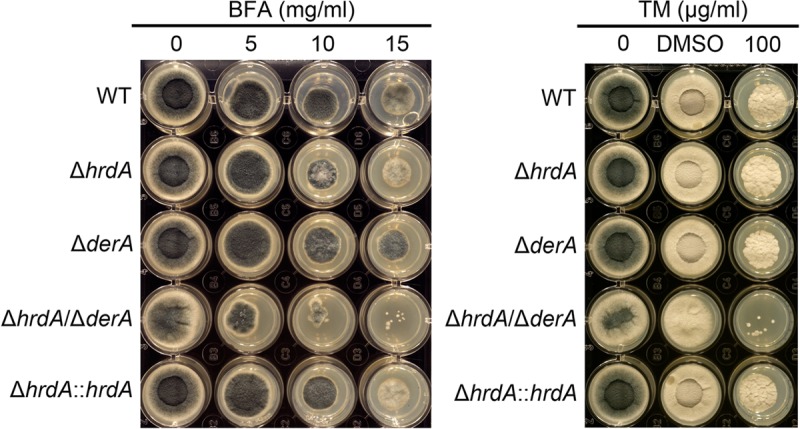
Loss of ERAD increases sensitivity to acute ER stress. Conidia from the indicated strains were inoculated onto solid medium in a multiwell plate containing AMM and the indicated concentrations of BFA or TM. The plates were incubated at 37°C for 2 days. Dimethyl sulfoxide (DMSO) was used as the vehicle control for TM.
HrdA supports thermotolerant growth of A. fumigatus.
Since elevated temperatures can have adverse effects on protein folding (30), the contribution of HrdA to growth under conditions of thermal stress was tested. The radial growth rate of the ΔhrdA mutant was impaired at 37°C and above (Fig. 5), suggesting a role for HrdA in the well-known thermotolerance of the fungus (31). As previously reported, the ΔderA mutant did not show this increase in thermosensitivity (7). Moreover, the ΔhrdA ΔderA mutant showed no further increase in temperature sensitivity relative to the ΔhrdA mutant, suggesting that HrdA function is more vital to thermotolerance than DerA function.
Fig 5.
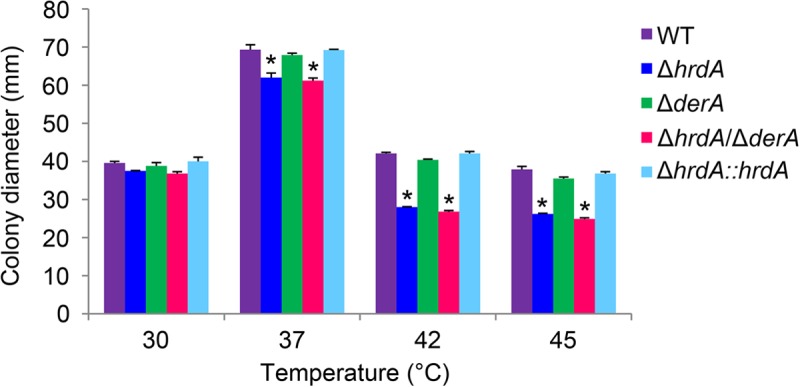
HrdA promotes thermotolerant growth. Conidia from the indicated strains were spotted onto the center of a plate containing rich medium (IMA), and colony diameter was measured after 3 days of growth at the indicated temperatures. The experiment was performed in triplicate, and the values represent the average ± SD. *, statistically significant by Student's t test (P < 0.001).
HrdA synergizes with DerA to support growth during cell wall stress.
We previously demonstrated that the UPR has an important role in the maintenance of cell wall integrity (6, 17). The extent to which HrdA contributes to cell wall homeostasis was determined by examining growth in the presence of CFW or CR, agents that are known to induce cell wall stress in fungi (32). The radial growth of the ΔhrdA and ΔderA single mutants was comparable to that of the wt in the presence of CFW or CR (Fig. 6), indicating that loss of either of these proteins had minimal effects on the growth rate under these conditions. Although the ΔhrdA mutant exhibited abnormal colony morphology (due to an approximately 3-fold reduction in conidiation), the extent of radial growth was equivalent to that of the wt. In contrast, the radial growth of the ΔhrdA ΔderA mutant was impaired in the presence of CR and, to a lesser extent, CFW, indicating that the combined loss of HrdA and DerA creates a more severe growth defect than the loss of either gene alone under these conditions (Fig. 6).
Fig 6.
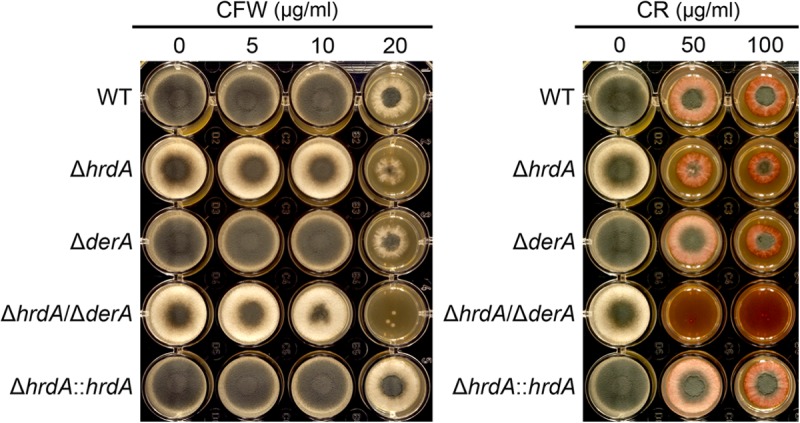
ERAD protects against cell wall stress. A total of 2,000 conidia were inoculated into the center of each well of a 24-well plate containing 2 ml of IMA agar supplemented with the indicated concentrations of CFW or CR and incubated for 2 days at 37°C.
HrdA cooperates with DerA to support secretion of collagenolytic enzymes.
To determine the effects of HrdA deficiency on the secretory pathway, extracellular collagenase activity was used as an indirect measure of secretory function (33). The ΔhrdA mutant showed a slight reduction in secreted collagenase activity relative to the wt (Fig. 7). The ΔderA mutant did not show this decrease, consistent with a previous study on this strain (7). In contrast, the ΔhrdA ΔderA mutant showed a 90% reduction in secreted collagenolytic activity, indicating that the combined loss of hrdA and derA is more detrimental to this process than the loss of either gene alone. We speculate that failure to eliminate unfolded proteins in the ΔhrdA ΔderA mutant results in ER dysfunction, which interferes with secretory efficiency. However, the mutant retains sufficient secretory capacity to degrade a complex substrate of proteins, because it grew normally on plates containing bovine serum albumin as the sole nutrient source or on explants of mouse lung tissue (data not shown).
Fig 7.

Loss of ERAD impairs the secretion of collagenolytic activity. Conidia from the indicated strains were inoculated in liquid cultures of AMM containing fetal bovine serum as the sole carbon/nitrogen source. After incubation for 72 h at 37°C, the collagenolytic activity of culture supernatants was quantified using the Azocoll assay, as described in Materials and Methods. The experiment was performed in triplicate, and the values represent the mean A520/g (dry weight) plus SD. *, statistically significant by Student's t test (P < 0.001).
Loss of HrdA decreases voriconazole sensitivity.
Analysis of antifungal drug susceptibility showed that the loss of HrdA, either alone or in combination with DerA, did not affect the MIC for either amphotericin B or caspofungin (data not shown). However, the ΔhrdA mutant was slightly more resistant to voriconazole, with an MIC of 0.38 μg/ml, which was approximately 3-fold higher than that of the wt (0.125 μg/ml) (Fig. 8). Ergosterol levels did not differ between the wt and the ΔhrdA mutant in the presence or absence of a concentration of voriconazole that was subinhibitory in liquid medium (0.125 μg/ml), suggesting that this voriconazole resistance is not due to alterations in ergosterol levels (Fig. 9). However, inhibition of Erg11 function by voriconazole is known to cause the accumulation of sterol intermediates (34), and we found that two sterols, presumably intermediates of the ergosterol pathway, accumulated to higher levels in the ΔhrdA mutant than in the wt (Fig. 9).
Fig 8.
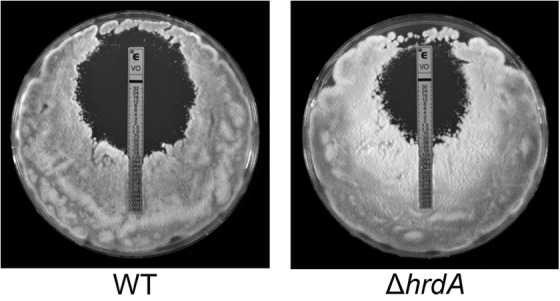
Loss of HrdA reduces voriconazole sensitivity. Conidia from the indicated strains were spread onto the surface of a plate of IMA, and an Etest strip containing voriconazole was applied to the inoculated agar surface. The plates were incubated at 37°C for 24 h. The two strains here are shown for clarity. The voriconazole sensitivities of the additional strains in this study are shown in Fig. S2 in the supplemental material.
Fig 9.

The ΔhrdA mutant accumulates higher levels of sterol intermediates in response to voriconazole treatment. Analysis of sterol content (ergosterol and two unidentified intermediates) was performed in the presence (+) or absence (−) of voriconazole (VCZ) (0.125 μg/ml) by gas chromatography. The values represent the averages of three replicates, expressed as μg sterol per mg dry fungal weight plus SD (*, P < 0.005; **, P < 0.0005).
Loss of HrdA and/or DerA does not affect virulence.
The virulence levels of the ERAD mutants were compared in a mouse model of aspergillosis (16). As shown in Fig. 10, the virulence levels of all three mutants were indistinguishable from that of the wt, indicating that both of these ERAD proteins are dispensable for virulence. Since inflammation has been reported to contribute to pathogenesis in this model (35), we considered the possibility that the reconstituting immune system could potentially mask an attenuated virulence phenotype for the ΔhrdA ΔderA mutant if the inflammatory response mounted against it was exaggerated. However, the mutant revealed no decrease in fungal growth relative to the wt in histopathologic sections of infected lung tissue, and comparable levels of inflammation were observed between strains (see Fig. S3 in the supplemental material). We also found that the ΔhrdA ΔderA mutant had wt virulence in a G. mellonella infection model (see Fig. S4 in the supplemental material). This contrasted with the attenuated virulence of the ΔhacA UPR mutant in the G. mellonella model (see Fig. S4 in the supplemental material), providing further evidence that an intact UPR is more important to virulence than a fully functional ERAD pathway.
Fig 10.

HrdA is dispensable for virulence. Groups of 12 CF-1 outbred mice were immunosuppressed with triamcinolone on day −1 and infected intranasally with conidia from the indicated strains on day 0. Mortality was monitored for 7 days. The virulence levels of all mutant strains were statistically indistinguishable from that of the wt.
DISCUSSION
In this study, we demonstrate that deletion of HrdA in A. fumigatus reduces the ability of the fungus to degrade the ERAD substrate CPY*, and potentially many other proteins that fail to accurately fold in the ER environment. This is likely to cause unfolded-protein stress in the ER, which is consistent with our finding that the UPR is constitutively upregulated in mutants that lack hrdA. The defect in ERAD function caused by the absence of hrdA was associated with very mild phenotypes under normal growth conditions, even when it was combined with the deletion of a second ERAD gene, derA. This indicates that A. fumigatus can tolerate the absence of two major components of the major ER membrane complex required for the retrotranslocation and degradation of unfolded ER proteins (13–15). The dispensability of a functional ERAD pathway for growth under normal growth conditions was also observed in the filamentous fungus Aspergillus niger (36). However, in this study, we also demonstrate that a fully operational ERAD pathway was necessary for optimal growth of A. fumigatus under environmental conditions that disrupt the folding capacity of the ER, including thermal stress, disruption of vesicular trafficking (BFA treatment), disruption of N-linked glycosylation (TM treatment), and cell wall stress.
An intriguing observation from this study was that loss of hrdA was associated with increased voriconazole resistance. Voriconazole inhibits the cytochrome P-450-dependent 14-α-lanosterol demethylation step in ergosterol biosynthesis (Erg11), resulting in the accumulation of 14-α-methyl sterol intermediates and a consequent depletion of plasma membrane ergosterol (37). Hrd1 also has a role in ergosterol synthesis, contributing to a negative-feedback mechanism that degrades the committed step enzyme, 3-hydroxy 3-methylglutaryl coenzyme A reductase (HMG-CoA reductase [HMGR]) (38). Two sterols, presumably intermediates of the ergosterol pathway, accumulated to higher levels in the ΔhrdA mutant than in the wt when treated with voriconazole (Fig. 9). We speculate that these intermediates accumulate more rapidly in ΔhrdA because the absence of HrdA reduces HMGR turnover, thereby increasing flux through the ergosterol pathway until it is blocked by voriconazole at the Erg11 step. However, since ergosterol levels were ultimately unaffected by loss of HrdA in the absence of voriconazole, it appears that any increase in flow through the beginning of the pathway is likely to be countered by feedback inhibition from subsequent sites in the pathway (39). Although increased expression of erg11 is a well-known mechanism of azole resistance in this fungus, analysis of erg11A and erg11B levels by qPCR showed no increased expression in the ΔhrdA mutant relative to the wt (data not shown). Further analysis of how azole antifungal drugs affect sterol biosynthesis in the absence of HrdA could provide important new insights into mechanisms of antifungal drug resistance in clinical isolates of the fungus.
We have previously shown that two UPR mutants, ΔhacA and ΔireA, show dramatically reduced virulence in a mouse model of invasive aspergillosis that employs transient immunosuppression with triamcinolone to predispose the mice to infection (6, 17). This finding is in striking contrast to the ΔhrdA and ΔhrdA ΔderA mutants in this study, which we found to be fully virulent in the same mouse model. The ΔhrdA and ΔhrdA ΔderA mutants also had wt virulence in the G. mellonella insect infection model, consistent with current evidence showing that virulence assessments in G. mellonella correlate well with data generated using mice (25).
Although the UPR and ERAD are tightly linked branches of the ER stress response (40), the findings from this study highlight fundamental differences between the two pathways with respect to A. fumigatus virulence: although ERAD has a clear role in protecting the fungus from high levels of ER stress, it is largely dispensable in the host environment. This contrasts with the UPR, which has a major role in the regulation of virulence (6, 17). Ostensibly, this would suggest that pharmacologic disruption of ERAD activity alone would not be an effective antifungal strategy. However, combination therapy using an ERAD-targeting drug together with a compound that prevents the UPR from responding could be an innovative approach to generate critical levels of ER stress and induce fungal death. In this regard, it is worth noting that a similar two-pronged attack on UPR and ERAD functions is being explored as a promising treatment to disrupt the viability of secretory tumor cells that rely extensively on the UPR and ERAD for ER homeostasis (41).
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grant R01AI072297 to D.S.A.
We thank Stephanie White and Thomas Jones for technical assistance and Jay Card for photography and illustration.
Footnotes
Published ahead of print 25 January 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/EC.00319-12.
REFERENCES
- 1. Segal BH. 2009. Aspergillosis. N. Engl. J. Med. 360:1870–1884 [DOI] [PubMed] [Google Scholar]
- 2. Marr KA. 2010. Fungal infections in oncology patients: update on epidemiology, prevention, and treatment. Curr. Opin. Oncol. 22:138–142 [DOI] [PubMed] [Google Scholar]
- 3. Shoham S, Marr KA. 2012. Invasive fungal infections in solid organ transplant recipients. Future Microbiol. 7:639–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Steinbach WJ, Marr KA, Anaissie EJ, Azie N, Quan SP, Meier-Kriesche HU, Apewokin S, Horn DL. 2012. Clinical epidemiology of 960 patients with invasive aspergillosis from the PATH Alliance registry. J. Infect. 65:453–464 [DOI] [PubMed] [Google Scholar]
- 5. Erjavec Z, Kluin-Nelemans H, Verweij PE. 2009. Trends in invasive fungal infections, with emphasis on invasive aspergillosis. Clin. Microbiol. Infect. 15:625–633 [DOI] [PubMed] [Google Scholar]
- 6. Feng X, Krishnan K, Richie DL, Aimanianda V, Hartl L, Grahl N, Powers-Fletcher MV, Zhang M, Fuller KK, Nierman WC, Lu LJ, Latge JP, Woollett L, Newman SL, Cramer RA, Jr, Rhodes JC, Askew DS. 2011. HacA-independent functions of the ER stress sensor IreA synergize with the canonical UPR to influence virulence traits in Aspergillus fumigatus. PLoS Pathog. 7:e1002330 doi:10.1371/journal.ppat.1002330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Richie DL, Feng X, Hartl L, Aimanianda V, Krishnan K, Powers-Fletcher MV, Watson DS, Galande AK, White SM, Willett T, Latge JP, Rhodes JC, Askew DS. 2011. The virulence of the opportunistic fungal pathogen Aspergillus fumigatus requires cooperation between the endoplasmic reticulum-associated degradation pathway (ERAD) and the unfolded protein response (UPR). Virulence 2:12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Galagan JE, Calvo SE, Cuomo C, Ma LJ, Wortman JR, Batzoglou S, Lee SI, Basturkmen M, Spevak CC, Clutterbuck J, Kapitonov V, Jurka J, Scazzocchio C, Farman M, Butler J, Purcell S, Harris S, Braus GH, Draht O, Busch S, D'Enfert C, Bouchier C, Goldman GH, Bell-Pedersen D, Griffiths-Jones S, Doonan JH, Yu J, Vienken K, Pain A, Freitag M, Selker EU, Archer DB, Penalva MA, Oakley BR, Momany M, Tanaka T, Kumagai T, Asai K, Machida M, Nierman WC, Denning DW, Caddick M, Hynes M, Paoletti M, Fischer R, Miller B, Dyer P, Sachs MS, Osmani SA, Birren BW. 2005. Sequencing of Aspergillus nidulans and comparative analysis with A. fumigatus and A. oryzae. Nature 438:1105–1115 [DOI] [PubMed] [Google Scholar]
- 9. Machida M, Asai K, Sano M, Tanaka T, Kumagai T, Terai G, Kusumoto K, Arima T, Akita O, Kashiwagi Y, Abe K, Gomi K, Horiuchi H, Kitamoto K, Kobayashi T, Takeuchi M, Denning DW, Galagan JE, Nierman WC, Yu J, Archer DB, Bennett JW, Bhatnagar D, Cleveland TE, Fedorova ND, Gotoh O, Horikawa H, Hosoyama A, Ichinomiya M, Igarashi R, Iwashita K, Juvvadi PR, Kato M, Kato Y, Kin T, Kokubun A, Maeda H, Maeyama N, Maruyama J, Nagasaki H, Nakajima T, Oda K, Okada K, Paulsen I, Sakamoto K, Sawano T, Takahashi M, Takase K, Terabayashi Y, Wortman JR, Yamada O, Yamagata Y, Anazawa H, Hata Y, Koide Y, Komori T, Koyama Y, Minetoki T, Suharnan S, Tanaka A, Isono K, Kuhara S, Ogasawara N, Kikuchi H. 2005. Genome sequencing and analysis of Aspergillus oryzae. Nature 438:1157–1161 [DOI] [PubMed] [Google Scholar]
- 10. Mori K. 2009. Signalling pathways in the unfolded protein response: development from yeast to mammals. J. Biochem. 146:743–750 [DOI] [PubMed] [Google Scholar]
- 11. Schubert U, Anton LC, Gibbs J, Norbury CC, Yewdell JW, Bennink JR. 2000. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature 404:770–774 [DOI] [PubMed] [Google Scholar]
- 12. Smith MH, Ploegh HL, Weissman JS. 2011. Road to ruin: targeting proteins for degradation in the endoplasmic reticulum. Science 334:1086–1090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schafer A, Wolf DH. 2009. Sec61p is part of the endoplasmic reticulum-associated degradation machinery. EMBO J. 28:2874–2884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Horn SC, Hanna J, Hirsch C, Volkwein C, Schutz A, Heinemann U, Sommer T, Jarosch E. 2009. Usa1 functions as a scaffold of the HRD-ubiquitin ligase. Mol. Cell 36:782–793 [DOI] [PubMed] [Google Scholar]
- 15. Carvalho P, Goder V, Rapoport TA. 2006. Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell 126:361–373 [DOI] [PubMed] [Google Scholar]
- 16. Powers-Fletcher MV, Jambunathan K, Brewer JL, Krishnan K, Feng X, Galande AK, Askew DS. 2011. Impact of the lectin chaperone calnexin on the stress response, virulence and proteolytic secretome of the fungal pathogen Aspergillus fumigatus. PLoS One 6:e28865 doi:10.1371/journal.pone.0028865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Richie DL, Hartl L, Aimanianda V, Winters MS, Fuller KK, Miley MD, White S, McCarthy JW, Latge JP, Feldmesser M, Rhodes JC, Askew DS. 2009. A role for the unfolded protein response (UPR) in virulence and antifungal susceptibility in Aspergillus fumigatus. PLoS Pathog. 5:e1000258 doi:10.1371/journal.ppat.1000258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cheon SA, Jung KW, Chen YL, Heitman J, Bahn YS, Kang HA. 2011. Unique evolution of the UPR pathway with a novel bZIP transcription factor, Hxl1, for controlling pathogenicity of Cryptococcus neoformans. PLoS Pathog. 7:e1002177 doi:10.1371/journal.ppat.1002177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Joubert A, Simoneau P, Campion C, Bataille-Simoneau N, Iacomi-Vasilescu B, Poupard P, Francois JM, Georgeault S, Sellier E, Guillemette T. 2011. Impact of the unfolded protein response on the pathogenicity of the necrotrophic fungus Alternaria brassicicola. Mol. Microbiol. 79:1305–1324 [DOI] [PubMed] [Google Scholar]
- 20. Cove DJ. 1966. The induction and repression of nitrate reductase in the fungus Aspergillus nidulans. Biochim. Biophys. Acta 113:51–56 [DOI] [PubMed] [Google Scholar]
- 21. Catlett N, Lee B, Yoder O, Turgeon B. 2002. Split-marker recombination for efficient targeted deletion of fungal genes. Fungal Genet. Newsl. 50:9–11 [Google Scholar]
- 22. Bhabhra R, Miley MD, Mylonakis E, Boettner D, Fortwendel J, Panepinto JC, Postow M, Rhodes JC, Askew DS. 2004. Disruption of the Aspergillus fumigatus gene encoding nucleolar protein CgrA impairs thermotolerant growth and reduces virulence. Infect. Immun. 72:4731–4740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mancini R, Aebi M, Helenius A. 2003. Multiple endoplasmic reticulum-associated pathways degrade mutant yeast carboxypeptidase Y in mammalian cells. J. Biol. Chem. 278:46895–46905 [DOI] [PubMed] [Google Scholar]
- 24. Mylonakis E. 2008. Galleria mellonella and the study of fungal pathogenesis: making the case for another genetically tractable model host. Mycopathologia 165:1–3 [DOI] [PubMed] [Google Scholar]
- 25. Slater JL, Gregson L, Denning DW, Warn PA. 2011. Pathogenicity of Aspergillus fumigatus mutants assessed in Galleria mellonella matches that in mice. Med. Mycol. 49( Suppl. 1): S107–S113 [DOI] [PubMed] [Google Scholar]
- 26. Knop M, Finger A, Braun T, Hellmuth K, Wolf DH. 1996. Der1, a novel protein specifically required for endoplasmic reticulum degradation in yeast. EMBO J. 15:753–763 [PMC free article] [PubMed] [Google Scholar]
- 27. Jung G, Ueno H, Hayashi R. 1999. Carboxypeptidase Y: structural basis for protein sorting and catalytic triad. J. Biochem. 126:1–6 [DOI] [PubMed] [Google Scholar]
- 28. Hiller MM, Finger A, Schweiger M, Wolf DH. 1996. ER degradation of a misfolded luminal protein by the cytosolic ubiquitin-proteasome pathway. Science 273:1725–1728 [DOI] [PubMed] [Google Scholar]
- 29. Back SH, Schroder M, Lee K, Zhang K, Kaufman RJ. 2005. ER stress signaling by regulated splicing: IRE1/HAC1/XBP1. Methods 35:395–416 [DOI] [PubMed] [Google Scholar]
- 30. Matsumoto R, Akama K, Rakwal R, Iwahashi H. 2005. The stress response against denatured proteins in the deletion of cytosolic chaperones SSA1/2 is different from heat-shock response in Saccharomyces cerevisiae. BMC Genomics 6:141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bhabhra R, Askew DS. 2005. Thermotolerance and virulence of Aspergillus fumigatus: role of the fungal nucleolus. Med. Mycol. 43( Suppl. 1): S87–S93 [DOI] [PubMed] [Google Scholar]
- 32. Ram AF, Klis FM. 2006. Identification of fungal cell wall mutants using susceptibility assays based on Calcofluor white and Congo red. Nat. Protoc. 1:2253–2256 [DOI] [PubMed] [Google Scholar]
- 33. Gifford AH, Klippenstein JR, Moore MM. 2002. Serum stimulates growth of and proteinase secretion by Aspergillus fumigatus. Infect. Immun. 70:19–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xiong Q, Hassan SA, Wilson WK, Han XY, May GS, Tarrand JJ, Matsuda SP. 2005. Cholesterol import by Aspergillus fumigatus and its influence on antifungal potency of sterol biosynthesis inhibitors. Antimicrob. Agents Chemother. 49:518–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Grahl N, Puttikamonkul S, Macdonald JM, Gamcsik MP, Ngo LY, Hohl TM, Cramer RA. 2011. In vivo hypoxia and a fungal alcohol dehydrogenase influence the pathogenesis of invasive pulmonary aspergillosis. PLoS Pathog. 7:e1002145 doi:10.1371/journal.ppat.1002145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Carvalho ND, Arentshorst M, Kooistra R, Stam H, Sagt CM, van den Hondel CA, Ram AF. 2011. Effects of a defective ERAD pathway on growth and heterologous protein production in Aspergillus niger. Appl. Microbiol. Biotechnol. 89:357–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Johnson LB, Kauffman CA. 2003. Voriconazole: a new triazole antifungal agent. Clin. Infect. Dis. 36:630–637 [DOI] [PubMed] [Google Scholar]
- 38. Hampton RY, Gardner RG, Rine J. 1996. Role of 26S proteasome and HRD genes in the degradation of 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Mol. Biol. Cell 7:2029–2044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bard M, Downing JF. 1981. Genetic and biochemical aspects of yeast sterol regulation involving 3-hydroxy-3-methylglutaryl coenzyme A reductase. J. Gen. Microbiol. 125:415–420 [DOI] [PubMed] [Google Scholar]
- 40. Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. 2000. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 101:249–258 [DOI] [PubMed] [Google Scholar]
- 41. Mimura N, Fulciniti M, Gorgun G, Tai YT, Cirstea D, Santo L, Hu Y, Fabre C, Minami J, Ohguchi H, Kiziltepe T, Ikeda H, Kawano Y, French M, Blumenthal M, Tam V, Kertesz NL, Malyankar UM, Hokenson M, Pham T, Zeng Q, Patterson JB, Richardson PG, Munshi NC, Anderson KC. 2012. Blockade of XBP1 splicing by inhibition of IRE1alpha is a promising therapeutic option in multiple myeloma. Blood 119:5772–5781 [DOI] [PMC free article] [PubMed] [Google Scholar]