

Abstract
The genomes of microsporidia in the genus Encephalitozoon have been extensively studied for their minimalistic features, but they have seldom been used to investigate basic characteristics of the biology of these organisms, such as their ploidy or their mode of reproduction. In the present study, we aimed to tackle this issue by mapping Illumina sequence reads against the genomes of four strains of E. cuniculi. This approach, combined with more conventional molecular biology techniques, resulted in the identification of heterozygosity in all strains investigated, a typical signature of a diploid nuclear state. In sharp contrast with similar studies recently performed on a distant microsporidian lineage (Nematocida spp.), the level of heterozygosity that we identified across the E. cuniculi genomes was found to be extremely low. This reductive intraindividual genetic variation could result from the long-term propagation of these strains under laboratory conditions, but we propose that it could also reflect an intrinsic capacity of these vertebrate pathogens to self-reproduce.
INTRODUCTION
Microsporidia are a medically and economically important group of obligate intracellular parasites that are known to infect a wide array of hosts, including protists, invertebrates, humans, and many other vertebrates. The cells of these eukaryotic organisms are atypical in that they lack conventional mitochondria and a Golgi apparatus and harbor prokaryote-like rRNA molecules (1). For some time, these simple cellular features were thought to reflect the primitive nature of microsporidian cells (2), but all recent phylogenetic studies now strongly support the association of microsporidia with the fungal kingdom, with current scientific disputes focusing on their exact phylogenetic placement within the kingdom itself (i.e., earliest branch versus more-derived lineage) (3–7).
Besides their intriguing evolutionary origin, microsporidia have drawn scientific attention for their reductive genome features. Specifically, their obligate intracellular parasitic lifestyle has resulted in dramatic gene losses in the biochemical repertoires of all known species, making them ideal models to study the processes involved in genome adaptation to obligate intracellular parasitism (8–12). The most compelling examples of genome reduction in microsporidia are known from species in the genus Encephalitozoon, where gene losses have been paralleled by a massive compression of the genomes themselves (13). To date, complete genomes from four species in the genus Encephalitozoon have been sequenced (Encephalitozoon cuniculi, E. intestinalis, E. hellem, and E. romaleae) (11–13), all of which have been found to be strikingly small and very similar in size, form, and content, carrying approximately 1,900 genes with a limited metabolic capacity. The few differences between these four genomes are notable, however, and include a significant variability in the number of genes acquired by means of horizontal gene transfer (HGT) (11, 14, 15).
To date, the acquisition of genome data from several Encephalitozoon spp. has helped us understand how these intracellular parasites have managed to offset their reduced metabolism and ultimately have benefited more efficiently from their hosts. However, this sequence information could have been better exploited to comprehend other aspects of their biology that are still poorly understood but vital for their survival and propagation, including their mode of reproduction or the ploidy of their nuclei (16, 17). In particular, claims of sexual reproduction in Encephalitozoon spp. have so far been based only on the presence of sexually related genomic regions (i.e., meiosis genes and the sex-related locus) (12, 18–20), and the presence of a diploid state for their nuclei has been inferred only from limited and unrelated observations (e.g., one heterozygous allele in E. cuniculi, and potential homologous chromosomes detected by pulse gel electrophoresis) (12, 18–20).
Here, we confirm the presence of a diploid state and the potential for sexual reproduction in the vertebrate pathogen Encephalitozoon cuniculi by identifying the presence of heterozygosity along the resequenced genomes of four strains of this species. Similar investigations recently performed on distant microsporidian relatives with small genomes (Nematocida spp.) have resulted in the identification of extensive heterozygosity (3), a typical hallmark of diploidy that also suggests the presence of sex (3, 21, 22). However, it remains to be seen if diploidy and elevated heterozygosity are genetic features that are also present in more-derived members of the group.
MATERIALS AND METHODS
Genome acquisition, sequence analysis, and polymorphism detection.
Strains of E. cuniculi are generally classified within three genotypes based on the number of “GTTT” repeats found within the internal transcribed spacer (ITS) of their RNA operon (23). In the present paper, these isolates are designated ECI (rabbit isolate, ATCC 50503 [24]), ECII (mouse isolate [25]), and ECIII (canine isolate, ATCC 50502 [26]). One-hundred-base-pair Illumina sequence reads from representatives of these genotypes have been mapped against their respective reference genomes to identify the presence of intraindividual genetic variation. In total, 7,805,454, 1,898,812, and 13,704,622 reads were found to map against the reference genomes of ECI, ECII, and ECIII, respectively. Three of these Illumina read sets have been obtained from the NCBI SRA database (accession numbers SRX002289, SRX002287, SRX002285; sequenced at the Broad Institute), and mapped against their respective reference genomes available from http://microsporidiadb.org/micro/ (27, 28) and NCBI (ECI, AEWD01; ECII, AEWQ01; and ECIII, AEWR01). Genome assembly procedures for the ECI, ECII, and ECIII strains are provided in an accompanying article (28). In parallel, reads were also newly obtained from one E. cuniculi strain of genotype II isolated from Czech Republic (referred to here as ECII-CZ) (29).
The mapping procedure was performed as follows. Low-quality reads were either removed or trimmed prior to alignment in order to eliminate biases induced by sequencing errors. Variation was subsequently searched using the “Find Variations/SNPs” function available in the Geneious software package. Heterozygosity was scored in all cases where reference and alternate alleles were found to map to one genomic region at a similar frequency (between 35% and 65% respective frequency; n loci = 89). All these heterozygous loci were verified using PCR, followed by direct Sanger sequencing of the resulting products and manual inspection of the sequencing chromatograms (see Fig. S1 in the supplemental material). We found that heterozygous loci located within highly covered regions (i.e., within the 35/65% cutoff but within regions with coverage twice as large as the average coverage of the genome) all represented false positives following PCR and direct Sanger sequencing (i.e., variation was due to paralogy rather than allelic variation). For this reason, these highly covered genome regions, which typically include the subtelomeres of Encephalitozoon spp. and the rRNA operons of these strains, were discarded for downstream analyses (i.e., no evidence of heterozygosity in vitro; 23 loci tested [data not shown]). Finally, variation that deviated from the 35/65% cutoff in silico was always found to be the result of sequencing or assembly errors following PCR and Sanger sequencing (no evidence of heterozygosity both in silico and in vitro; 14 loci tested) (data not shown).
PCRs were performed in 25 μl containing a final concentration of 1× EconoTaq DNA polymerase (Lucigen, Middleton, WI), 0.5 mM each primer, and 0.3 μl of DNA. The thermal cycling conditions included an initial step of 94°C for 3 min, followed by 35 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 2 min and final step of 72°C for 12 min.
ECII-CZ genome acquisition and annotation.
Spores from one strain of E. cuniculi isolated from a wild mouse in the Czech Republic (29) were grown in the Vero E6 cell line, and the resulting spores were subjected to deep sequencing to further confirm the presence of reduced heterozygosity in Encephalitozoon cuniculi. The strain belongs to genotype II, as suggested by a specific tandem repeat present in its internal transcribed spacer (23), and is referred in the present paper as ECII-CZ. Both strains of genotype II investigated in the present study (i.e., ECII and ECII-CZ) were originally isolated from mice within a close geographical region in the Czech Republic (25, 29), and our genome analyses confirmed that both isolates indeed represent two very closely related strains of the same genotype. However, the exact time at which ECII-CZ was isolated and the overall history of its propagation under laboratory environment are currently unknown (29). For these reasons, we will refrain from speculating about the evolutionary relationship between ECII and ECII-CZ and will exclusively use their genome sequences to confirm the methodology that we have used to detect heterozygous sites (i.e., the same methodology used on two independently acquired genomes of one genotype leads to very similar results).
Genomic DNA was extracted from ECII-CZ spores, and deep-sequencing shotgun libraries were prepared as previously described (13). The ECII-CZ library was subjected to one round of deep sequencing using 1/10th of a channel on the Illumina GA-IIx instrument, resulting in 331,449,750 bp of unique DNA sequence (50-bp-long sequence reads). Reads were assembled using the reassembly tool available in the Geneious package and the ECII genome as a reference (AEWQ0100000), resulting in 54 scaffolds, with an average size of 13,355 bp and an average coverage of 46×. A total of 2,224,725 reads were mapped against this newly acquired genome sequence using the same procedure described above for other strains. The ECII-CZ reference genome was annotated as previously described (13).
Accession numbers.
The Illumina reads for ECII-CZ have been deposited in the NCBI Sequence Read Archive (SRA) database under accession number SRA059442. The scaffolds have been deposited in GenBank under accession numbers KC513604 to KC513657.
RESULTS AND DISCUSSION
Identification of low levels of heterozygosity in four genetically different strains of Encephalitozoon cuniculi.
Aligning Illumina reads against the reference genomes of ECI, ECII, and ECIII resulted in the detection of a total of 66 regions that were mapped by both reference and alternate reads at similar frequency (i.e., within the expected threshold). The amount of heterozygosity identified is far lower than that recently reported for distant microsporidia (Fig. 1; Table 1) (3), with a maximum of 23 nucleotide sites (less than 0.00001% of the total genome sequence) being affected by heterozygosity in E. cuniculi, compared to over 42,175 (1.035% of the complete genome) for more basal species such as Nematocida spp. (Fig. 1) (3). In all cases, heterozygous loci were confirmed using PCR and direct Sanger sequencing, with the resulting chromatograms showing the presence of double peaks of similar intensity irrespective of the genotype (Fig. S1 in the supplemental material). Additional confirmation of the presence of reduced heterozygosity in these strains was also obtained by mapping reads onto the genome sequence from a second strain of genotype II (ECII-CZ) that we sequenced independently. Specifically, ECII and ECII-CZ were found to differ by only 9 homozygous substitutions (Fig. 1; Table 2) but were otherwise found to harbor an almost identical pattern of heterozygosity (ECII-CZ has two heterozygous indels that are absent in ECII) (Tables 1 and 2). Given the elevated divergence that is generally present between strains of E. cuniculi (28), the great sequence similarity between ECII and ECII-CZ suggests that they represent two very closely related strains.
Fig 1.
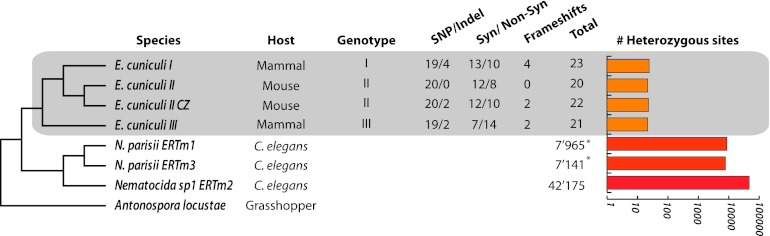
Amount, location, and nature of heterozygous loci in E. cuniculi and comparison with Nematocida. (A) Phylogenetic reconstruction of the studied microsporidian species based on the nucleotide sequences of the actin and alpha-tubulin genes. Known genotype, parasite's host, amount of variation, and nature of variation are indicated. SNPs and indels are included in the count of (non)synonymous mutations. Antonospora locustae was used as an outgroup. The total number of heterozygous sites is presented in a bar graph on a logarithmic (log10) scale. An asterisk indicates that variation was found using 454 sequencing. The Encephalitozoon genus is highlighted by a gray square. A detailed list of heterozygous loci is shown in Table 1.
Table 1.
Heterozygous loci in E. cuniculi genotypes I, II, and III
| Genotype | Gene | Product | Polymorphism type | Protein effect | Nucleotide change | Amino acid change | Position in coding sequence |
|---|---|---|---|---|---|---|---|
| I | ECU02_0050 | Hypothetical protein | SNP (transversion) | None | C/A | 1738 | |
| ECU02_0050 | Hypothetical protein | SNP (transition) | Substitution | G/A | R → W | 1736 | |
| ECU02_0260 | GATA zinc finger transcription factor 3 | SNP (transition) | Truncation | C/T | 60 | ||
| ECU02_0720 | Histone H2A | SNP (transition) | None | A/G | 221 | ||
| ECU05_1190 | Belongs to the ABC transporter superfamily | SNP (transition) | None | A/G | 381 | ||
| ECU06_0470 | Nuclear pore complex protein nup155 | SNP (transition) | None | A/G | 2656 | ||
| Intergenic_ch6 | SNP (transversion) | A/C | |||||
| ECU06_0590 | Hypothetical protein | Indel | Frameshift | C/- | 655 | ||
| ECU06_0700 | Hypothetical protein | Indel | Frameshift | CC/- - | 429 | ||
| ECU06_0730 | Ribonucleoside diphosphate reductase small chain | SNP (transition) | None | G/A | 719 | ||
| ECU06_0830 | Hypothetical protein | SNP (transversion) | Substitution | C/A | M → I | 1711 | |
| ECU07_1005 | 60S ribosomal protein l37a | SNP (transition) | None | A/G | 252 | ||
| ECU11_1160 | Possible protein of nuclear scaffold | SNP (transition) | None | C/T | 609 | ||
| ECU11_1330.6 | Transcription factor (forkhead domain) | Indel | Frameshift | A/- | 692 | ||
| ECU11_1330.1 | Transcription factor (forkhead domain) | SNP (transition) | None | G/A | 511 | ||
| ECU11_1330.2 | Transcription factor (forkhead domain) | SNP (transition) | None | T/C | 457 | ||
| ECU11_1330.2 | Transcription factor (forkhead domain) | SNP (transition) | Substitution | C/T | D → N | 452 | |
| ECU11_1330.3 | Transcription factor (forkhead domain) | SNP (transition) | None | A/G | 368 | ||
| ECU11_1330.4 | Transcription factor (forkhead domain) | SNP (transition) | None | A/G | 232 | ||
| ECU11_1330.5 | Transcription factor (forkhead domain) | SNP (transition) | Substitution | C/T | A → T | 108 | |
| Intergenic_ch11 | SNP (transition) | C/T | |||||
| ECU11_1340 | ABC transporter-like protein | Indel | Frameshift | -/A | 1890 | ||
| ECU11_1340 | ABC transporter-like protein | SNP (transversion) | Substitution | T/A | I → F | 1020 | |
| II | ECU04_0440 | Metal-dependent protease of the Pad1 Jab1 | SNP (transition) | None | T/C | 908 | |
| ECU05_0720 | Splicing factor U2AF large subunit b | SNP (transition) | Substitution | A/G | R → C | 1012 | |
| ECU06_0700 | Hypothetical protein | SNP (transition) | None | C/T | 413 | ||
| ECU07_0680 | Chromosome segregation ATPase | SNP (transition) | None | G/A | 564 | ||
| ECU07_0770 | Npl4 family protein | SNP (transition) | Substitution | A/G | H → Y | 433 | |
| ECU07_1490 | Copper amine oxidase N-terminal domain protein | SNP (transition) | None | C/T | 267 | ||
| ECU07_1500 | Calcium-transporting ATPase | SNP (transition) | None | A/G | 1857 | ||
| ECU08_0080 | Dihydrofolate reductase | Indel | Frameshift | T/- | 94 | ||
| ECU08_0230 | Cell division protein kinase | SNP (transition) | None | T/C | 186 | ||
| ECU08_1010 | Proteophosphoglycan Ppg4 | SNP (transition) | Substitution | G/A | I → M | 768 | |
| ECU08_1730 | Hypothetical protein | SNP (transversion) | Substitution | T/G | H → N | 241 | |
| ECU09_0360 | Similarity to hypothetical proteins (Upf0129 family) | SNP (transversion) | None | A/C | 272 | ||
| ECU09_0410 | DNA excision repair protein Ercc-6 | SNP (transition) | Substitution | C/T | F → L | 1417 | |
| ECU09_0730 | Bem46 family protein | SNP (transition) | Substitution | A/G | G → D | 738 | |
| ECU09_1120 | Hypothetical protein | SNP (transition) | None | C/T | 35 | ||
| ECU09_1820 | snRNP-like protein | Indel | Frameshift | G/- | 149 | ||
| ECU10_1210 | DNA polymerase catalytic subunit a | SNP (transition) | Substitution | A/G | S → F | 857 | |
| ECU11_0410 | Hypothetical protein | SNP (transition) | Substitution | C/T | N → S | 461 | |
| Intergenic_ch11 | SNP (transition) | C/T | |||||
| Intergenic_ch8.1 | SNP (transition) | G/A | |||||
| Intergenic_ch8.2 | SNP (transversion) | T/C | |||||
| Intergenic_ch10 | SNP (transition) | A/G | |||||
| III | ECU01_0410 | Hypothetical protein | SNP (transition) | Substitution | A/G | R → G | 674 |
| ECU01_0750 | Hypothetical protein | SNP (transition) | Substitution | T/C | F → S | 333 | |
| ECU03_0320 | 60S ribosomal protein l13 | SNP (transition) | None | C/T | 264 | ||
| ECU03_1180 | Subtilisin-related endopeptidase K | SNP (transition) | Substitution | G/A | P → S | 1109 | |
| ECU04_0470 | Hypothetical protein | SNP (transition) | None | A/G | 754 | ||
| ECU04_1460 | Cyclin b-like guanine nucleotide binding protein | Indel | Frameshift | T/- | 2100 | ||
| ECU04_1470 | Na+/H+ antiporter | SNP (transversion) | None | T/G | 1073 | ||
| ECU04_1490 | Hypothetical protein | SNP (transversion) | Substitution | C/A | D → Y | 1237 | |
| ECU05_0700 | Hypothetical protein | SNP (transition) | Substitution | G/A | G → R | 762 | |
| ECU05_0850 | Hypothetical protein | SNP (transition) | Substitution | C/T | G → S | 41 | |
| Intergenic_ch7.1 | SNP (transition) | T/C | |||||
| Intergenic_ch7.2 | SNP (transition) | A/G | |||||
| ECU08_1150 | Hypothetical oxidoreductase short-chain dehydrogenases/reductases (SDR) family | SNP (transversion) | Substitution | T/G | W → G | 549 | |
| ECU08_1680 | Hypothetical protein | SNP (transition) | Substitution | A/G | E → G | 597 | |
| ECU10_0540 | Similarity to ADP/ATP carrier protein | SNP (transition) | Substitution | C/T | R → K | 5 | |
| ECU10_0960 | Similarity to hypothetical proteins Ycd7_human | SNP (transition) | Substitution | A/G | S → P | 68 | |
| ECU10_1030 | Hypothetical protein | Indel | Frameshift | G/- | 1938 | ||
| ECU11_1120 | Translation elongation factor 2 | SNP (transition) | Substitution | G/A | V → M | 433 | |
| ECU11_1410 | Hypothetical protein | SNP (transition) | None | T/C | 632 | ||
| Intergenic_ch11 | SNP (transversion) | G/T | |||||
| ECU06_0630 | Hypothetical protein | SNP (transition) | Substitution | G/A | C → Y | 1472 |
Table 2.
Substitution variation between two independent isolates of E. cuniculi genotype II, ECII and ECII-CZ
| Gene | Product | Protein ID | Polymorphism type | Protein effect | Nucleotide change (CZ → II) | Amino acid change |
|---|---|---|---|---|---|---|
| Intergenic-CH1 | SNP (transversion) | G → C | ||||
| ECU02_0080 | Hypothetical protein | CAD25039.1 | SNP (transversion) | Substitution | T → A | R → S |
| ECU02_0080 | Hypothetical protein | CAD25039.1 | SNP (transition) | Substitution | A → G | F → L |
| ECU02_0650 | Hypothetical protein | CAD25095.1 | SNP (transition) | Substitution | T → C | D → G |
| ECU03_0450 | Hypothetical protein | CAD26191.1 | SNP (transition) | None | C → T | |
| ECU03_0710 | 60S ribosomal protein L34 | CAD26217.1 | SNP (transversion) | None | T → A | |
| ECU08_1210 | Hypothetical protein | CAD26427.1 | SNP (transition) | Substitution | G → A | S → N |
| ECU08_2120 | Hypothetical protein | CAD26514.1 | SNP (transversion) | Substitution | T → A | I → N |
| ECU08_2120 | Hypothetical protein | CAD26514.1 | SNP (transversion) | Substitution | C → A | L → I |
On average, about 86% of the total heterozygous variation was found to affect coding regions of the genome, possibly because of the elevated gene density of these genomes (i.e., an average density of approximately 0.9 gene per kb [12]). Between 22% and 57% of substitutions resulted in an amino acid change at one allele, affecting a total of 31 genes with known function and 19 genes encoding hypothetical proteins (Table 1). Several heterozygous deletion and insertions events (indels) were also identified, all of which were found to affect coding regions. In total, 9 genes were found to have undergone pseudogenization at one allele, with ECI showing the largest number of heterozygous frameshifts mutations (Fig. 1; Table 1). The slightly more elevated heterozygosity of ECI mirrors an unusually large number of substitutions at the locus ECU11_1130 (n = 7), which encodes a transcription factor. While it is intriguing to speculate about a potential for this gene to represent a mating type locus in E. cuniculi (i.e., allelic variation within transcription factors is linked with sexual identity in other fungi [30–32]), it seems that the elevated number of mutations in this particular case results from the pseudogenization of one allele (i.e., relaxation of selective pressures at one allele).
Intriguingly, all strains investigated harbor similarly low levels of heterozygosity, but this genetic variation was always found to affect different portions of the proteome among genotypes. Finally, sequence coverage was never found to vary among different chromosomes in all genotypes, suggesting the absence of aneuploidy in all strains of this species.
Strains of Encephalitozoon are likely to be diploid.
Species in the genus Encephalitozoon have been proposed to be diploid on a few occasions, most notably following observations of potential homologous chromosomes using hybridization procedures (18–20) and reports of potential heterozygosity at one specific locus (reported for the CTP synthase but data not shown [12]). However, these reports have long remained speculative, warranting further explorations across available genomes of these pathogens.
By identifying the presence of several heterozygous loci across the genomes of four genetically divergent strains of E. cuniculi, the present study fills an important gap in our understanding of the cellular biology of these vertebrate-infecting parasites. In particular, the identification of heterozygosity in all strains analyzed strongly supports the notion that members of this lineage are diploid, harboring pairs of homologous chromosomes that almost certainly help these obligate intracellular parasites offset the deleterious mutational load harbored by their nuclei. Specifically, an average of 3 heterozygous loci per genome were found to harbor one allele with frameshift mutations, so having one functional allele at these loci (i.e., resulting from diploidy) was certainly essential for the survival of these parasites with ultrareduced genome contents.
The presence of two sets of chromosomes per nucleus may also have additional beneficial effects for E. cuniculi, particularly within natural populations of these parasites, as diploidy has been linked with the capacity of some vertebrate parasites to escape the immune system of their host (33). This speculative hypothesis, however, is not currently supported by our data, as most heterozygous loci did not appear to encode proteins known to be involved in such evading mechanisms (Tables 1 and 2). Certainly, future analyses based on natural populations of E. cuniculi will be a key to understand whether the presence of heterozygosity is relevant for epidemiological studies of these important pathogens of vertebrates.
Opportunistic sex in Encephalitozoon cuniculi?
Recently, mapping of Illumina reads against the genomes of different Nematocida strains (a basal microsporidian lineage that is evolutionarily very distant from Encephalitozoon spp.) has resulted in the identification of extremely high levels of heterozygosity. Such elevated levels typically result from the combination of a diploid nuclear state, frequent recombinational events (i.e., genetic variation resulting from meiosis), and genetic exchange. These are hallmarks of sexual reproduction in eukaryotes, and their identification in Nematocida spp. has been obviously linked with the presence of sexuality in those species (3).
Here, we propose that sexual reproduction may also occur in the more divergent microsporidian species E. cuniculi, for two main reasons. First, regardless of its frequency, the presence of heterozygosity represents compelling evidence for a diploid nuclear state in E. cuniculi and an essential prerequisite for the production of haploid cells through meiosis. This mechanism could allow E. cuniculi to complete a sexual cycle similar to those found in many other eukaryotes (Fig. 2A and B) or other diploid fungi (e.g., Candida albicans) (Fig. 2C) (3, 21, 22). Second, the low levels of heterozygosity that we have identified in E. cuniculi do not typically mirror the absence of sexual reproduction, because exclusively clonal diploids are expected to harbor many, highly divergent alleles in the absence of a mechanism that allows gene shuffling (i.e., meiosis) (22, 34–39). For this reason, the over 1,500-fold difference in the level of heterozygosity between strains of E. cuniculi and Nematocida spp. cannot be simply explained by the presence of different modes of reproduction in those lineages (i.e., exclusively clonal versus exclusively sexual).
Fig 2.

Selfing, inbreeding, and outcrossing in Encephalitozoon cuniculi. (A) Proposed modes of reproduction and mating systems present within laboratory strains of Encephalitozoon cuniculi. (B) Hypothesis regarding the modes of reproduction and mating systems present within natural populations of Encephalitozoon cuniculi. (C) Same mechanisms shown in panels A and B but involving the fusion of two diploid mating type cells to increase ploidy, followed by meiosis to generate the progeny.
So where are these sharp differences in heterozygosity between the two lineages coming from? One possibility is that the extremely low heterozygosity found in E. cuniculi is the result of self-reproduction (selfing), a mechanism that is known to heavily reduce the amount of heterozygosity in many other vertebrate parasites (22, 34, 35, 40) and to produce clones that are optimally adapted to certain hosts (i.e., highly homozygous individuals) but that can still maintain the capacity to undergo outcrossing and create new variation following rare changes in environmental conditions (i.e., opportunistic sexuals). However, given the source of the E. cuniculi strains we have analyzed, the extreme reduction in heterozygosity we have observed may have been exacerbated by the methodology used to originate and propagate these strains under laboratory conditions. Specifically, if those strains originated from few individuals, then founder effects combined with selfing or inbreeding (e.g., fusion of haploid cells produced by one individual or by genetically similar individuals) would quickly eradicate most genetic variation from those cultures, resulting in the highly homozygous individuals that we studied here (Fig. 2).
E. cuniculi could also possibly be clonal and yet find a way to produce genetic variation by a number of different mechanisms that are rarely observed, including the “asexual ploidy cycle” that is found in some amoebozoans and radiolarians (41) or through frequent mitotic recombination. In the asexual ploidy cycle, the ploidy level is increased by endomitosis (i.e., replication without cellular division) and subsequently reduced through cell divisions. This alternation of ploidy levels (2N → 4N → 2N in this case) could mirror a sexual cycle without necessitating the fusion of gametes.
In any case, a number of additional studies are still required to determine whether selfing represents the major cause of reduced heterozygosis in E. cuniculi and whether gene exchange between genetically different members of one species can actually occur in the field. One such experiment could involve coinfection of the same host cells with different genotypes (coinfection with ECI and ECII or with ECI and ECIII), followed by genomic analysis of the resulting spores after a number of generations in search of gene exchanges. Certainly, the results from such analyses would provide important clues about the sexual mechanisms that are used by these organisms to produce genetic variation in the first place (i.e., frequent versus rare sex).
Similarly, investigations of heterozygosity and presence of recombination along the genomes of E. cuniculi strains isolated from the field (i.e., natural populations) will be essential to determine whether selfing is common in these vertebrate pathogens or whether it is simply a side effect of their long-term propagation under laboratory conditions. In particular, the identification of an elevated number of heterozygous sites within natural populations of this species would be more in line with what was recently observed within natural strains of other species such as Nematocida spp. (3) and would suggest the capacity of E. cuniculi to outcross on a more regular basis in the field. Studying natural populations of E. cuniculi could also provide insights into the apparent genetic disparity that is found within and among genotypes of this species. Specifically, the ECI, ECII, and ECIII genomes have been recently found to diverge quite extensively at the sequence level (i.e., elevated homozygous divergence between individuals) (28) while being simultaneously able to maintain very reduced intragenomic diversity. The absence of shared heterozygosity between ECI, ECII, and ECIII certainly supports the notion that these strains are quite diverged and unlikely to have experienced genetic exchange between each other for quite some time. It now remains to be seen whether selfing has allowed these strains to accumulate a large amount of homozygous divergence without leaving many traces of heterozygosity behind.
ACKNOWLEDGMENTS
We thank Soo-Chan Lee, Timothy James, and three anonymous reviewers for their comments on a previous version of the manuscript and Christina Cuomo and the Broad Institute for the use of unpublished Encephalitozoon cuniculi genome data from the “Microsporidian Comparative Genomes Consortium” and MicrosporidiaDB.
N.C. is a Fellow of the Integrated Microbial Biodiversity program of the Canadian Institute for Advanced Research (CIFAR-IMB). This work was supported by grants from the Natural Sciences and Engineering Research Council of Canada to N.C. (NSERC-Discovery), the National Institutes of Health to L.M.W. (5R01AI031788-19), and the Czech Science Foundation (505/11/1163).
Footnotes
Published ahead of print 2 February 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/EC.00307-12.
REFERENCES
- 1. Keeling PJ, Fast NM. 2002. Microsporidia: biology and evolution of highly reduced intracellular parasites. Annu. Rev. Microbiol. 56:93–116 [DOI] [PubMed] [Google Scholar]
- 2. Corradi N, Keeling PJ. 2009. Microsporidia: a journey through radical taxonomical revisions. Fungal Biol. Rev. 23:1–8 [Google Scholar]
- 3. Cuomo CA, Desjardins CA, Bakowski MA, Goldberg J, Ma AT, Becnel JJ, Didier ES, Fan L, Heiman DI, Levin JZ, Young S, Zeng Q, Troemel ER. 2012. Microsporidian genome analysis reveals evolutionary strategies for obligate intracellular growth. Genome Res. 22:2478–2488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Capella-Gutierrez S, Marcet-Houben M, Gabaldon T. 2012. Phylogenomics supports microsporidia as the earliest diverging clade of sequenced fungi. BMC Biol. 10:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lee SC, Corradi N, Byrnes EJ, III, Torres-Martinez S, Dietrich FS, Keeling PJ, Heitman J. 2008. Microsporidia evolved from ancestral sexual fungi. Curr. Biol. 18:1675–1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lee SC, Corradi N, Doan S, Dietrich FS, Keeling PJ, Heitman J. 2010. Evolution of the sex-related locus and genomic features shared in microsporidia and fungi. PLoS One 5:e10539 doi:10.1371/journal.pone.0010539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Keeling PJ. 2003. Congruent evidence from alpha-tubulin and beta-tubulin gene phylogenies for a zygomycete origin of microsporidia. Fungal Genet. Biol. 38:298–309 [DOI] [PubMed] [Google Scholar]
- 8. Akiyoshi DE, Morrison HG, Lei S, Feng X, Zhang Q., Corradi N, Mayanja H, Tumwine JK, Keeling PJ, Weiss LM, Tzipori S. 2009. Genomic survey of the non-cultivatable opportunistic human pathogen, Enterocytozoon bieneusi. PLoS Pathog. 5:e1000261 doi:10.1371/journal.ppat.1000261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Corradi N, Haag KL, Pombert JF, Ebert D, Keeling PJ. 2009. Draft genome sequence of the Daphnia pathogen Octosporea bayeri: insights into the gene content of a large microsporidian genome and a model for host-parasite interactions. Genome Biology 10:R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Keeling PJ, Corradi N, Morrison HG, Haag KL, Ebert D, Weiss LM, Akiyoshi DE, Tzipori S. 2010. The reduced genome of the parasitic microsporidian Enterocytozoon bieneusi lacks genes for core carbon metabolism. Genome Biol. Evol. 2:304–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pombert JF, Selman M, Burki F, Bardell FT, Farinelli L, Solter LF, Whitman DW, Weiss LM, Corradi N, Keeling PJ. 2012. Gain and loss of multiple functionally related, horizontally transferred genes in the reduced genomes of two microsporidian parasites. Proc. Natl. Acad. Sci. U. S. A. 109:12638–12643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Katinka MD, Duprat S, Cornillot E, Metenier G, Thomarat F, Prensier G, Barbe V, Peyretaillade E, Brottier P, Wincker P, Delbac F, El Alaoui H, Peyret P, Saurin W, Gouy M, Weissenbach J, Vivares CP. 2001. Genome sequence and gene compaction of the eukaryote parasite Encephalitozoon cuniculi. Nature 414:450–453 [DOI] [PubMed] [Google Scholar]
- 13. Corradi N, Pombert JF, Farinelli L, Didier ES, Keeling PJ. 2010. The complete sequence of the smallest known nuclear genome from the microsporidian Encephalitozoon intestinalis. Nat. Commun. 1:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Selman M, Pombert JF, Solter L, Farinelli L, Weiss LM, Keeling P, Corradi N. 2011. Acquisition of an animal gene by microsporidian intracellular parasites. Curr. Biol. 21:R576–R577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Selman M, Corradi N. 2011. Microsporidia: horizontal gene transfers in vicious parasites. Mob Genet. Elements 1:251–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schottelius J, Schmetz C, Kock NP, Schuler T, Sobottka I, Fleischer B. 2000. Presentation by scanning electron microscopy of the life cycle of microsporidia of the genus Encephalitozoon. Microbes Infect. 2:1401–1406 [DOI] [PubMed] [Google Scholar]
- 17. Bigliardi E, Sacchi L. 2001. Cell biology and invasion of the microsporidia. Microbes Infect. 3:373–379 [DOI] [PubMed] [Google Scholar]
- 18. Biderre C, Mathis A, Deplazes P, Weber R, Metenier G, Vivares CP. 1999. Molecular karyotype diversity in the microsporidian Encephalitozoon cuniculi. Parasitology 118:439–445 [DOI] [PubMed] [Google Scholar]
- 19. Biderre C, Pages M, Metenier G, Canning EU, Vivares CP. 1995. Evidence for the smallest nuclear genome (2.9 Mb) in the microsporidium Encephalitozoon cuniculi. Mol. Biochem. Parasitol. 74:229–231 [DOI] [PubMed] [Google Scholar]
- 20. Brugere JF, Cornillot E, Metenier G, Bensimon A, Vivares CP. 2000. Encephalitozoon cuniculi (Microspora) genome: physical map and evidence for telomere-associated rDNA units on all chromosomes. Nucleic Acids Res. 28:2026–2033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ni M, Feretzaki M, Sun S, Wang X, Heitman J. 2011. Sex in fungi. Annu. Rev. Genet. 45:405–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Billiard S, Lopez-Villavicencio M, Hood ME, Giraud T. 2012. Sex, outcrossing and mating types: unsolved questions in fungi and beyond. J. Evol. Biol. 25:1020–1038 [DOI] [PubMed] [Google Scholar]
- 23. Didier ES, Vossbrinck CR, Baker MD, Rogers LB, Bertucci DC, Shadduck JA. 1995. Identification and characterization of three Encephalitozoon cuniculi strains. Parasitology 111:411–421 [DOI] [PubMed] [Google Scholar]
- 24. Shadduck JA. 1969. Nosema cuiculi: in vitro isolation. Science 166:516–517 [DOI] [PubMed] [Google Scholar]
- 25. Vavra J, Bedrnik P, Cinatl J. 1972. Isolation and in vitro cultivation of the mammalian microsporidian Encephalitozoon cuniculi. Folia Parasitol. 19:349–354 [PubMed] [Google Scholar]
- 26. Shadduck JA, Bendele R, Robinson GT. 1978. Isolation of the causative organism of canine encephalitozoonosis. Vet. Pathol. 15:449–460 [DOI] [PubMed] [Google Scholar]
- 27. Aurrecoechea C, Barreto A, Brestelli J, Brunk BP, Caler EV, Fischer S, Gajria B, Gao X, Gingle A, Grant G, Harb OS, Heiges M, Iodice J, Kissinger JC, Kraemer ET, Li W, Nayak V, Pennington C, Pinney DF, Pitts B, Roos DS, Srinivasamoorthy G, Stoeckert CJ, Jr, Treatman C, Wang H. 2011. AmoebaDB and MicrosporidiaDB: functional genomic resources for Amoebozoa and Microsporidia species. Nucleic Acids Res. 39:D612–D619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pombert J-F, Xu J, Smith DR, Heiman D, Young S, Cuomo CA, Weiss LM, Keeling PJ. 2013. Complete genome sequences from three genetically distinct strains reveal high intraspecies genetic diversity in the microsporidian Encephalitozoon cuniculi. Eukaryot. Cell 12:496–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Koudela B, Vítovec J, Kučerová Z, Ditrich O, Trávniček J. 1993. The severe combined immunodeficient mouse as a model for Encephalitozoon cuniculi microsporidiosis. Folia Parasitol. 40:279–286 [PubMed] [Google Scholar]
- 30. Gerke J, Lorenz K, Cohen B. 2009. Genetic interactions between transcription factors cause natural variation in yeast. Science 323:498–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Idnurm A. 2011. Sex determination in the first-described sexual fungus. Eukaryot. Cell 10:1485–1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wong Sak Hoi J, Dumas B. 2010. Ste12 and Ste12-like proteins, fungal transcription factors regulating development and pathogenicity. Eukaryot. Cell 9:480–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xu X, Sun J, Zhang J, Wellems D, Qing X, McCutchan T, Pan W. 2012. Having a pair: the key to immune evasion for the diploid pathogen Schistosoma japonicum. Sci. Rep. 2:346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Heitman J. 2006. Sexual reproduction and the evolution of microbial pathogens. Curr. Biol. 16:R711–R725 [DOI] [PubMed] [Google Scholar]
- 35. Heitman J. 2010. Evolution of eukaryotic microbial pathogens via covert sexual reproduction. Cell Host Microbe 8:86–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Barraclough TG, Birky CW, Jr, Burt A. 2003. Diversification in sexual and asexual organisms. Evolution 57:2166–2172 [DOI] [PubMed] [Google Scholar]
- 37. Balloux F, Lehmann L, de Meeus T. 2003. The population genetics of clonal and partially clonal diploids. Genetics 164:1635–1644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Balloux F, Amos W, Coulson T. 2004. Does heterozygosity estimate inbreeding in real populations? Mol. Ecol. 13:3021–3031 [DOI] [PubMed] [Google Scholar]
- 39. De Meeus T, Lehmann L, Balloux F. 2006. Molecular epidemiology of clonal diploids: a quick overview and a short DIY (do it yourself) notice. Infect. Genet. Evol. 6:163–170 [DOI] [PubMed] [Google Scholar]
- 40. Billiard S, Lopez-Villavicencio M, Devier B, Hood ME, Fairhead C, Giraud T. 2011. Having sex, yes, but with whom? Inferences from fungi on the evolution of anisogamy and mating types. Biol. Rev. Camb. Philos. Soc. 86:421–442 [DOI] [PubMed] [Google Scholar]
- 41. Kondrashov AS. 1994. The asexual ploidy cycle and the origin of sex. Nature 370:213–216 [DOI] [PubMed] [Google Scholar]