

Abstract
Mucosal vaccination, capable of inducing protective immune responses both in the mucosal and systemic immune compartments, has many advantages and is regarded as a blue ocean in the vaccine industry. Mucosal vaccines can offer lower costs, better accessability, needle-free delivery, and higher capacity of mass immunizations during pandemics. However, only very limited number of mucosal vaccines was approved for human use in the market yet. Generally, induction of immune responses following mucosal immunization requires the co-administration of appropriate adjuvants that can initiate and support the effective collaboration between innate and adaptive immunity. Classically, adjuvant researches were rather empirical than keenly scientific. However, during last several years, fundamental scientific achievements in innate immunity have been translated into the development of new mucosal adjuvants. This review focuses on recent developments in the concepts of adjuvants and innate immunity, mucosal immunity with special interest of vaccine development, and basic and applied researches in mucosal adjuvant.
Keywords: Mucosal, Vaccine, Adjuvant, Innate immunity
Introduction
Vaccination is the most successful application of immunological principles to the well being of human being. A numerous number of vaccines was developed and tested against many infectious diseases, and only a limited number of them came to the market. Effective vaccine should be safe, immunogenic, elicit the proper type of immunity, and induce long lasting immunological memory. The vast majority of infections occurs at, or originates from, mucosal areas such as respiratory, gastrointestinal, and urogenital tracts [1]. Globally, mucosa-associated infections (e.g., acquired immunodeficiency syndrome, tuberculosis, influenza, diarrheal diseases, pneumonia, gastric ulcer, and sexually transmitted diseases) are major causes of illness and serve big socio-economic burdens in both developed and developing countries. Traditional injected vaccines are generally poor inducers of mucosal immunity and are therefore less effective against infections at mucosal sites [2]. Mucosal vaccines, in contrast to injected vaccines, have been reported to provide additional secretory antibody-mediated protection against pathogens at the mucosal site of entry. One of most important virtues of mucosal vaccination is, while injected vaccines generally fails to do so, capable of inducing protective immune responses both in the mucosal and systemic immune compartments [3]. Mucosal vaccination offers other advantages such as lower costs, ease of administration, higher patient compliance, needle-free administration, and a higher capacity for mass immunizations during pandemic situations [4].
However, there are several very serious concerns about mucosal vaccines. Mucosa-administered antigens are generally less immunogenic and apt to induce tolerance since the host strives to maintain mucosal homeostasis by responding to mucosal antigens with tolerance. Indeed, only very limited number of mucosal vaccines was approved for human use in the market: the oral polio vaccine, oral killed whole-cell B subunit and live-attenuated cholera vaccines, an oral live-attenuated typhoid vaccine, an oral Bacillus Calmette-Guérin (BCG) live vaccine (used in Brazil for vaccination against tuberculosis) and an oral adenovirus vaccine (the latter vaccine being restricted to military personnel). Two recent additional mucosal vaccines, an oral live-attenuated rotavirus vaccine and a nasal enterotoxin-adjuvanted inactivated influenza vaccine, were withdrawn after a short time on the market because of potential serious adverse reactions (intussusception and facial paresis, respectively), thus illustrating the complexity of mucosal vaccine development [4-6]. In this regard, induction of mucosal immunity through vaccination is a rather difficult task, and potent mucosal adjuvants, vectors or other special delivery systems are required for successful mucosal vaccination [7].
Properties of Adjuvants
Adjuvants are generally defined as agents that can potentiate and modulate immune reactions against antigens. The term adjuvant is derived from the latin word adjuvare, meaning 'to help'. The idea that some materials added to antigens could improve immune responses was recognized many years ago with the works Ramon [8] and Glenny [9], who empirically used strange reagents such as tapioca and aluminum hydroxide to improve the responses of horses or guinea pigs to diphtheria and tetanus toxoids. Ramon is the very first person who ever introduced the scientific term adjuvant. However, the importance of adjuvant was rather neglected until recently when more refined vaccines appeared in the market. Since vaccines are administered to normal and younger people, the public has had very strict concerns about side effects of vaccinations. There had been very serious vaccine side effect scandals throughout history. To reduce reactogenicity, newer generation vaccines have been required to have more defined antigenic composition. Generally, the more given vaccines are purified/defined, the less immunogenicity they exert compared with older generation whole-cell or virus-based vaccines. In this context, adjuvants are critically required to help new vaccines to induce more potent and long-lasting protective immune responses. There are several required properties of good adjuvants. Successful adjuvant should 1) be non-toxic at the dose range for effective adjuvanticity; 2) stimulate a strong humoral and/or cell mediated immunity; 3) provide good immunological memory; 4) not induce autoimmunity and hypersensitivity; 5) be non-mutagenic, non-carcinogenic, or non-teratogenic; 6) non-pyrogenic; 7) be stable under broad range of storage time, temperature, and pH [10].
Adjuvants can act in several nonmutually exclusive ways to augment the adaptive immune response and to generate effective immunological memory. Successful protective immunity could be generated as the result of a harmonious interaction between innate and adaptive immunity. Innate immune signals modulate not only the magnitude of the adaptive response but also the repertoire and quality of this response. Many of their effects seem to be on antigen-presenting cells (APCs) such as dendritic cells (DCs). As is described in the Fig. 1, the different signals required to induce a potent immune response as signal 0 (antigen recognition and APC activation), signal 1 (antigen presentation), and signal 2 (co-stimulation) [11,12]. Thereby adjuvants can affect the migration, maturation, antigen presentation, and expression of costimulatory molecules by DCs, and these events in turn improve the responses to antigen of T and B cells. Adjuvants, apparently via DCs, can also affect the nature of CD4 T helper (Th), CD8 T cell, and B cell responses, with some adjuvants promoting Th1-related responses and others preferentially inducing Th2-biased effects. Furthermore, some adjuvants enhance by DCs of major histocompatibility complex (MHC) I-restricted antigens to CD8+ T cells. Adjuvants may also act directly on T or B cells, improving their proliferation and/or conversion into memory cells that are essential for the success of vaccines [13].
Fig. 1.

The target site of vaccine adjuvants. Most of the recently developed specific adjuvants, such as pattern recognition receptor (PRR) ligands act on signal 0 (antigen recognition and antigen-presenting cells [APCs] activation), and indirectly on signal 2 (co-stimulation). In addition, PRR ligands can act on signal 1 (efficient presentation of the co-administered antigen). Modified from Guy [12].
Innate Immunity and Adjuvants
Innate immunity is mediated by cells collectively called phagocytes: they are DCs, macrophages, leukocytes, etc. DC and its derivatives are proved to be the professional APCs. Innate immune cells were regarded being non-specific in recognizing pathogens, and do not confer long-lasting immunity based upon immunological memory. However, recent studies have shown that the innate immune system recognizes pathogens with higher specificity than expected. Phagocytes have the ability to discriminate molecular patterns of foreign pathogens from those of self cells and tissues. The receptors of phagocytes that recognize pathogen associated molecular patterns (PAMPs) were termed as pattern recognition receptors (PRRs). Microorganisms expressing PAMPs are recognized by innate immune cells through specific and non-polymorphic PRRs. PRRs are expressed either on the cell surface or intracellular compartments. Toll-like receptors (TLRs), nucleotide binding domain (NOD)-like receptors (NLRs), retinoic acid-inducible gene (RIG)-like receptors (RLRs), and C-type lectins consist important PRR families (Fig. 2) [14,15]. DCs activated through the recognition of pathogens via PRRs express high levels co-stimulatory molecules and cytokines, and migrate to regional lymph nodes to further activate T cells.
Fig. 2.

Pattern recognition receptor (PRR) and signaling (A) Toll-like receptors (TLRs). TLR receptors recognize different microbial associated molecular patterns: the heterodimer of TLR4 and MD-2 recognizes lipopolysaccharide (LPS); TLR2 recognizes triacyl and diacyl portions of lipoproteins together with TLR1 or TLR6, respectively; TLR5 recognizes flagellin; TLR3 recognizes double-stranded RNA; TLR7 recognizes single-stranded RNA and TLR9 recognizes bacterial and viral DNA, the so-called CpG DNA. The signaling pathways of TLRs are mediated by selective usage of adaptor molecules, MyD88, Toll-receptor-associated activator of interferon (TRIF), TIR-associated protein (TIRAP) and Toll-receptor-associated molecule (TRAM). (B) C-type lectins (CLRs), retinoic acid-inducible gene-like receptors (RLRs) and nucleotide binding domain (NOD)-like receptors (NLRs). CLRs recognize carbohydrates on microorganisms via the carbohydrate-binding domain. Dectin-1 is well studied. RLRs are composed of two N-terminal caspase-recruitment domains (CARDs), a central DEAD box helicase/ATPase domain, and a C-terminal regulatory domain (RD). They are localized in the cytoplasm and recognize the genomic RNA of dsRNA viruses, and dsRNA generated as the replication intermediate of ssRNA viruses. RLRs interact with IPS1 via their CARD domains, resulting in type 1 interferon production through IkB kinase, inducible (IKKi)/TANK-binding kinase 1 (TBK1). NLRs are composed of a central NOD and C-terminal leucine-rich repeats (LRRs). NODs activate caspase-1, resulting in processing of pro-interleukin-1β (IL-1β) to mature IL-1β. ASC, apoptosis-associated speck-like protein; IFN, interferon; IPS1, IFN-β promoter stimulator 1; IRF, interferon regulatory factor; MDA-5, melanoma-differentiation-associated gene 5; NF-kB, nuclear factor-kB; NLPR3, NLR family, pyrin domain-containing 3; RIG-1, retinoic acid-inducible gene I. Modified from Akira [15].
TLRs
TLRs are well conserved throughout the evolution from insects to humans. Toll, as the founder of the TLR family, was originally identified as an essential developmental protein of Drosophila and later proved to be playing an essential role in the anti-fungal resistance of the fly [16]. To date, 13 members of the TLR family have been identified in mammals [17-19]. TLRs are type 1 integral membrane glycoproteins having characteristic horseshoe-shaped leucine rich repeat (LRR) motif for PAMP recognition and cytoplasmic Toll/interleukin-1 receptor signaling domain [15]. TLRs 1, 2, 4, 5, 6, and 11 are surface-exposed, whereas TLRs 3, 7, 8, and 9 are located within endosomes. In addition to the cellular sub-compartmentalization, TLRs are differentially expressed in different cell types [20,21]. Each TLR recognize specific molecular patterns of TLR ligands. TLR ligands could be categorized into lipids, proteins, and nucleic acids. All TLR ligands are potent immune adjuvants and TLRs are also addressed as adjuvant receptors. Many TLR ligand adjuvants are under investigation for clinical applications. The first TLR ligands containing adjuvant AS04 was recently approved by the US Food and Drug Administration (US FDA) for the human papillomavirus vaccine Cervarix of GlaxoSmithKline. AS04 formulation is composed of alum with the TLR4 ligand monophosphoryl lipid A (MPL) [22]. MPL was reported to promote a Th1-biased immune response towards antigens [23]. Another most actively studied TLR ligand adjuvant is CpG oligonucleotides [24]. In human subjects, CpGs are mainly investigated for cancer, human immunodeficiency virus (HIV), and malarial vaccines. Clinical applicability of TLR ligand adjuvant seem to be very promising since existing successful vaccines contain adjuvants that are intrinsic to the immunogen. For example, vaccines that contain attenuated live or heat-killed viruses or bacteria include components (lipopolysaccharide [LPS] for TLR4, flagellin for TLR5, CpG for TLR9, etc.) that can engage TLRs (Fig. 2A) [25]. These components therefore act as natural adjuvants because TLR signaling has many of the effects on DC antigen presentation that one would wish for an adjuvant: improvement in antigen presentation and increases in co-stimulatory molecules and cytokine production, leading usually to improved Th responses. Such responses are well suited to defend against the organisms involved, probably because TLRs have been designed through evolution to respond in exactly the appropriate way to these infections and their attendant, intrinsic adjuvants [13].
NLRs
Bacterial components in the cytoplasm are recognized by NLR family PRRs. NLRs consist of a C-terminal LRR domain, a central nucleotide-binding domain and N-terminal protein-protein interaction caspase activation and recruitment domain (CARD) and pyrin or baculovirus inhibitor-of-apoptosis repeat (BIR) domains [26]. NOD1 and NOD2 differentially recognize the minimal components and lead to nuclear factor-κB (NF-κB) activation and inflammatory responses [27]. NOD1 recognized meso-diaminopimelic acid from Gram-negative bacteria and NOD2 senses muramyl dipeptide from Gram-negative and Gram-positive bacteria [28]. NLRs also detect danger-associated host components such as uric acid crystals [29]. NLRs are involved in the inflammasome formation and production of mature interleukin (IL)-1β and IL-18 (Fig. 2B) [30]. Expression of IL-1β and IL-18 is regulated at both transcriptional and post-translational levels. Transcriptional activation of these genes leads to the production of 1β and pro-IL-18. These inactive pro-proteins are further processed to mature forms by caspase-1 and secreted outside thereafter. Inflammasome formation is essential for the caspase-1 activation. To date, 4 different inflammasomes have been identified: they are nucleotide-binding domain and leucine-rich repeat containing family, pyrin domain containing 1 (NLRP1), nucleotide-binding domain and leucine-rich domain containing domain, CARD domain containing 4 (NLRC4), NLRP3 and absent in melanoma 2 (AIM2) inflammasomes. AIM2 does not belong to the NOD family. Flagellin (TLR5 ligand) and imidazoquinoline/RNA (TLR7 ligands) activate NLRC4 and NLRP3 inflammasomes, respectively. The NLRP3 inflammasome is involve in the recognition of crystallized molecules in the cytoplasm such as monosodium urate, asbestos and cholesterol crystals [15].
Despite its long and widespread use, the adjuvanticity mechanism of alum had been elusive until recently. One of the most widely accepted hypotheses is that alum, because it adsorbs antigens, serves as a depot, releasing the antigen slowly into the body, thereby allowing antigen-specific lymphocytes to be exposed to antigen for a longer time [13]. Although alum certainly extends the half-life of antigen in vivo, whether the depot theory accounts for its adjuvanticity has been challenged in several studies. Even when the alum depot at the site of injection was removed 1 week after immunization, the antibody response that developed against the co-injected antigen was not affected [31]. Moreover, stable adsorption of an antigen to alum was not necessary for alum's ability to potentiate antibody responses [32]. An important breakthrough appeared in 2007, when it was reported that alum induced IL-1β and IL-18 production in a caspase-1-dependent manner in LPS-primed human and murine DC in vitro [33]. Shortly after then, experimental evidence showing that the alum-mediated caspase-1 activation was NLRP3 dependent was reported by different groups [34-37]. Danger signals released from distressed and injured cells affected by alum seem to be sensed by NLRP3 inflammasome serve an important role in alarming the immune system [38,39]. After reporting the involvement of NLRP3 in alum-mediated immunostimulation, researchers studied whether inflammasomes could be targeted for the development of effective adjuvants and vaccine delivery systems. Poly(lactic-co-glycolic acid) and polystyrene microparticles were found to activate caspase-1 through NLRP3 in vitro as efficiently as alum [40]. Other experimental adjuvants have also been shown to mediate an NLRP-dependent IL-1 release, including QuilA, a saponin extracted from the bark of the Quillaria saponaria tree, and chitosan [41].
RLRs
RNA virus infection can induce type I interferon (IFN) genes in fibroblasts lacking both MyD88 and TRIF, which are crucial adapter molecules in TLR signaling, suggesting the existence of TLR-independent virus sensors [42]. An RNA helicase RIG-1 and melanoma differentiation-associated gene 5 (MDA5) are cytosolic sensor for viral infections. These two RNA helicases possess two N-terminal caspase-recruitment domains (CARDs) followed by an RNA helicase domain [43]. They are activated viral infections, and trigger cooperative activation of NF-κB and IFN regulatory factor 3 and 7 (IRF3/7) to induce antiviral type I IFNs (Fig. 2B). For more detailed signaling mechanisms of RLRs, please refer to recent specific review articles [42,44]. RIG-1 and MDA5 have differential functions in the antiviral immunity. RIG-1 detects a variety of RNA viruses by recognizing 5'-triphosphate short dsRNAs or long dsRNAs from several hundred to several thousand base pairs. MDA5 detects picornavirus family members by sensing ling dsRNAs longer than 2 K base pairs [45]. In addition, stimulator of IFN genes (STING) and DNA dependent activator of IRFs (DAI; also known as DLM1 and ZBP1) have been reported to induce type I IFNs in response to cytosolic DNA [46,47]. Because of relatively recent identification of RLRs, reports about RLR-targeting adjuvants are scarce yet. However, adjuvants targeting the RLRs-type I IFN axis would appear soon for certain diseases since the immunomodulatory mechanisms of type I IFNs are being highlighted recently [48]. Since type I IFNs have very pleiotropic activities, beneficiary effect in some diseases (inflammatory bowel diseases and multiple sclerosis) and aggravating effect in regulatory activities in other diseases (systemic lupus erythematosus and psoriasis), those adjuvant should be designed to act in a relatively narrower spectrum.
Mucosal Immunity with Special Interest of Vaccine Development
Mucosal immunity consists of a complex network of innate and adaptive immune components. Mucosal surfaces maintains very dangerous equilibrium: they are exposed to extraneous pathogens and enormous constant antigenic load from commensal microorganisms and diets. Mucosal innate immune and parenchymal cells express PRRs and continuously monitor antigenic loads to maintain the integrity of epithelial barrier function and homeostatic interactions among different mucosal components [49]. Mucosal immunologic homeostasis is mediated by continuous antigen sampling by APCs in mucosal tissues: including DCs, macrophages, and B cells. Mucosal CXCR1+ CD11b+ DCs are most important initiators of adaptive immune responses and vaccine-induced protective immunity. However, CD103+ DCs in the lamina propria induce regulatory T cells, which are important for the induction and maintenance of immunological tolerance [50,51]. Antigen sampling in the mucosal surfaces are performed both by professional APCs like CXCR1+ CD11b+ DCs and through a specific microfold (M) cells overlying the mucosa associated lymphoid tissue (MALT) follicles such as nasopharynx-associated lymphoid tissue (NALT) and Peyer's patches [52]. Peyer's patches contain all of the immunocompetent cells that are required for the generation of an immune response and are the key inductive tissues for the mucosal immune system. Peyer's patches are interconnected with effector tissues (e.g., the lamina propria of the intestine) for the induction of IgA immune responses specific to ingested antigens [53]. NALT also contains all of the necessary lymphoid cells, including T cells, B cells, and APCs, for the induction and regulation of inhaled antigen-specific mucosal immune responses [52]. Recently, respiratory M cells were also identified and reported to be serving antigen sampling roles just like other M cells in the Peyer's patch and NALT [54]. In this context, M cells became a very important target in in designing new mucosal vaccines [55].
Common mucosal immune system
Different mucosal tissues are immunologically connected each other to form the 'common mucosal immune system' [56]. The mucosal immune system can act independently of the systemic immune system [52]. Although anatomically separated, different regions of the MALT are functionally connected and crosstalk each other, which permits T cells and B cells activated by antigen at one place to appear as effector cells in distant mucosal sites [52]. This functional connectivity is achieved through the induction of specific sets of mucosal homing receptors during the interaction on T and B cells with mucosal DCs. For example, oral and intranasal immunization can stimulate effector T and B cell responses in distant mucosal tissues such as the intestinal and urogenital tracts. In spite of this functional connectivity, NALT-targeted immunization preferentially induces antigen-specific immunity in respiratory and reproductive tissues, whereas gut associated lymphoid tissue-targeted immunization predominantly elicits protective responses in gastrointestinal tissues [57]. These findings will provide inspirations in designing mucosal vaccines. It might be sufficient to immunize antigens intranasally to prevent genitourinary infections rather than doing inconvenient vaginal administration.
Mucosal Vaccine Adjuvants
Since many pathogens infect the host via the mucosal route such as inhalation, ingestion, or sexual contact, development of vaccine that can both prevent the mucosal invasion of the pathogen at the infection stage and also neutralize the pathogen-derived toxin or inhibit replication of the pathogen within the body at later infection stages are essential to prevent infectious diseases [58]. The mucosal route of immunization could meet these requirements by stimulating immune responses in both mucosal and systemic compartment. Despite many above mentioned advantages of the mucosal vaccination strategies, still there exist many disadvantages: large dose due to instability of antigens in the mucosal surfaces, poor immunogenicity (tolerance induction), physical mucosal barriers, etc. Most of those disadvantages could be circumvented with effective adjuvants and vaccine delivery systems. Alum, the most common adjuvant used in human vaccines for several decades, is a poor inducer of mucosal immunity. In this context, we notice a rush in good review articles dealing with mucosal adjuvants and mucosal vaccine delivery strategies [1,4,59-64]. The most promising mucosal adjuvants are derived from bacterial toxins, TLR ligands, non-TLR immunostimulants, and novel small molecules (Table 1) [1,4,12,15,25,58-60,62,63,65-79].
Table 1.
Current mucosal vaccine adjuvants and vaccine delivery systems under clinical application or preclinical researches
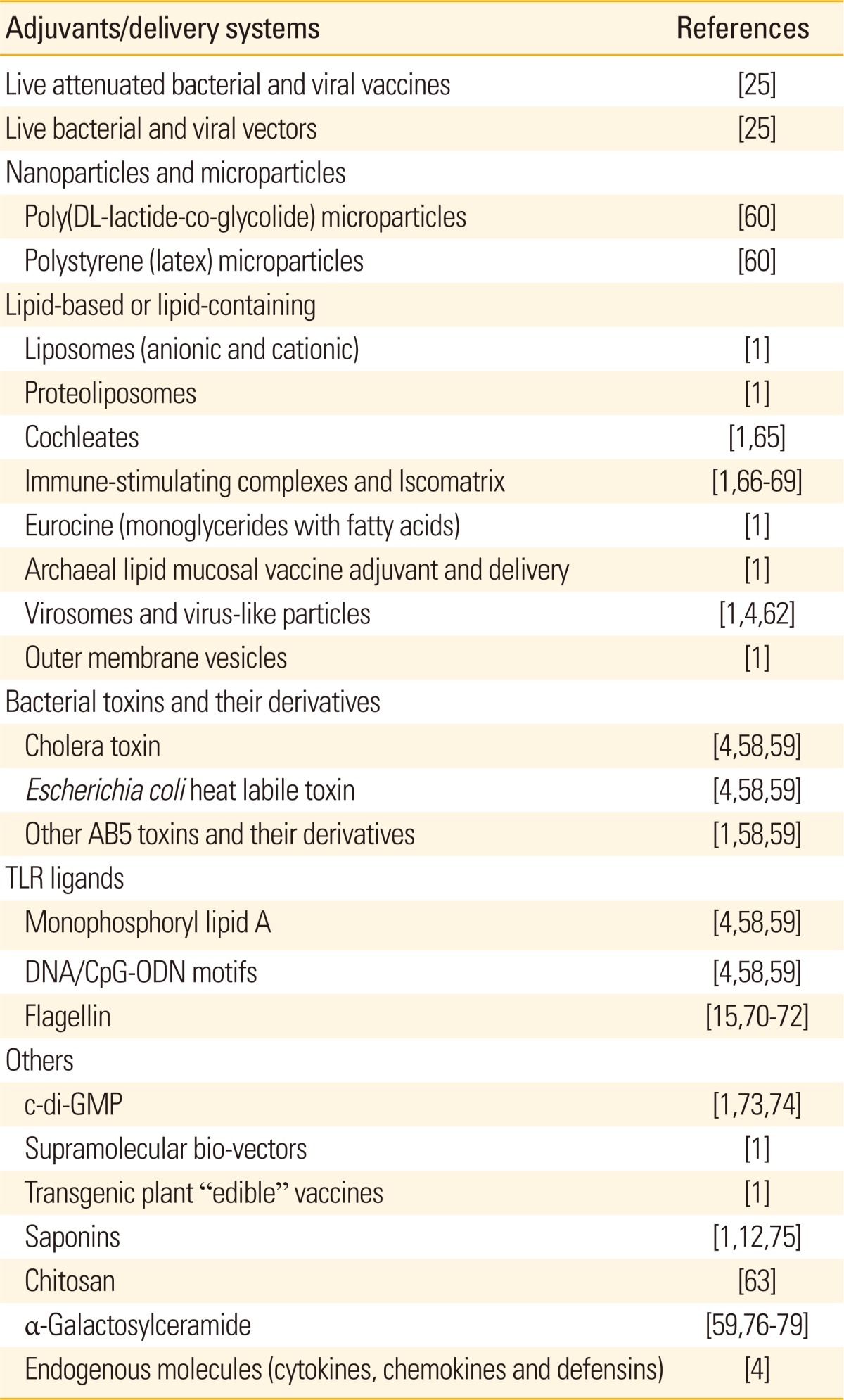
Modified from Chen et al. [1].
TLR, Toll-like receptor; ODN, oligodeoxynucleotide; c-di-GMP, 3',5'-cyclic diguanylic acid.
Bacterial toxins and derivatives
The most widely experimentally used mucosal adjuvants in animals are cholera toxin (CT) and closely related Escherichia coli heat labile enterotoxin (LT), and their mutants and subunits. These enterotoxins are composed of active (A) and binding (B) subunits. The A subunit has ADP-ribosylating activity and the B subunit binds to GM1 gangliosides expressed on the surface of many cell types [80]. CT and LT consist of a homo-pentamer B subunits and one A subunit. A subunit has enzyme activity that can ADP-ribosylate the Gs protein and subsequently activate adenylate cyclase to produce profuse amount cAMP in affected cells. CT and LT should be the ones of most mucosally active adjuvants reported to date. However, they are too toxic to be considered for human use. A nasal enterotoxin adjuvanted influenza vaccine had been withdrawn while a clinical trial was undertaken. A considerable number of Bell's palsy was reported in vaccinees, which was proposed to associated with the high affinity binding of the enterotoxin to GM1 ganglioside moieties of olfactory nerves [6,81]. Many research groups concentrated efforts to separate the adjuvant activity from toxicity of those enterotoxins to use them in human mucosal vaccination regimens. CT and LT have been found to affect several steps in the induction of mucosal immune responses: 1) increased permeability of the intestinal epithelium leading to enhanced uptake of co-administered antigens; 2) enhanced antigen presentation by a variety of cell types; 3) promotion of isotype differentiation in B cells leading to increased IgA production; and 4) complex stimulatory as well as inhibitory effects on T cell proliferation and cytokine production [4]. It has been claimed that CT primarily induces Th2 type immune responses characterized by CD4+ T cells producing IL-4, IL-5, IL-6, and IL-10 and by preferential production of IgA, IgG1, and IgE antibodies, while LT activates both Th1 and Th2 type immune responses. However, subsequent well controlled experiment showed that CT also induced mixed Th1 and Th2 type immune responses, in contrast to CT B subunit (CTB), which appeared to drive more restricted Th2 responses [82].
To avoid toxicity, isolated B subunits of CT and LT (CTB and LTB) have been explored for their adjuvanticity to co-administered antigens. However, their capacity as mucosal adjuvant has proven to be much less than that of holotoxins [4]. Adjuvant activity of CTB or LTB was much improved when couple to antigens, possibly because of efficient presentation of the coupled antigen DCs, macrophages, and naïve B cells [83]. Site directed mutagenesis of LT and CT generated mutant enterotoxins having reduced toxicity, but which retain significant adjuvanticity when given to the nasal route or, even though they perform less will through the oral administration route [84]. Another approach to avoid aforementioned toxicity is to link the A subunit with other protein or polypeptide having different cell-binding activity. One representative example is linking with the cell binding domain of Staphylococcus aureus protein A (CTA1-DD). CTA1-DD, like most other toxin derivatives, was effective when administered nasally but not when applied orally. This drawback was overcome by the incorporation of CTA1-DD into immune stimulating complexes (ISCOM). Oral vaccination with the ISCOM-CTA1-DD induced significant Th1 and Th2 immune responses in both mucosal and systemic immune compartments [85]. In addition to above efforts, attempts to compromise enterotoxicity/ADP-ribosylating activities of CTA by genetically modifying the protein, such as inserting peptide sequence to disrupt the active site structure. A mutant named eCT6, manifesting enterotoxicity decrease by 10-20 fold, displayed comparable adjuvanticity as the wild type CT [86]. Similar approaches were given to LT modification. Newly constructed double site-directed nontoxic mutant (R192G/L211A) appeared promising for oral, sublingual, rectal, or transcutaneous vaccination [87]. However, after several TLR ligands displaying less adverse effects had been reported to have comparable mucosal adjuvant activity as CT, efforts to modify enterotoxins become relatively scarce.
As for the immunological action mechanism of CT and LT, it has been shown that they activate both innate and adaptive immunity. Recent studies further elucidated the importance of APC modulation by the enterotoxins [59]. CT, LT, and their derivatives promote antigen capture by increasing DC migration from the subepithelial dome to follicle-associated epithelia after oral administration [88]. DC seems to be crucial for the CT-mediated adjuvanticity since DC ablation prevents antibody induction and antigen-specific T cell responses after oral or nasal immunization [89]. CT is reported to have remote mucosal immunomodulating activity that transcutaneous CT administration could affect intestinal through affecting the Ag presentation in mesenteric lymph nodes [90]. Th17 cells also seem to play important roles in the mucosal adjuvant activities of CT. Mice vaccinated intranasally with antigen plus CT produce antigen-specific Th17 cells with high IgA levels and can be protected against inhalational anthrax; while IgA levels are reduced and mice succumb to anthrax challenge in IL-17 neutralized or knockout mice [91]. In another study, sublingual, buccal and transcutaneous delivery of a pneumococcal whole-cell vaccine with the LT mutants LT (R192G) or LT (R192G/L211A) enhanced IL-17A expression and reduced nasopharynx and middle ear bacterial burden upon challenge, similar to that observed following intranasal delivery [92]. Intradermally administered CT was found to stimulate the production of IFN-γ and IL-17 by CD4+ T cells over IL-4 or IL-5 production through the activation of DCs in the injected area [93]. Sublingually administered CT stimulated co-administered Helicobacter pylori antigen-specific IFN-γ and IL-17 production in the stomach [94]. During last several years, roles of Th17 cells in mucosal immunity have been enlightened [95]. In this regard, most of previously documented CT-mediated adjuvant activities should have been related to Th17-mediated mechanisms.
TLR ligands
Recognition of PAMP by TLR triggers a signaling pathway resulting in the production of pro-inflammatory cytokines and up-regulation in expression of co-stimulatory molecules, thereby activating not only innate immunity but ultimately also acquired immune responses (Fig. 2A). In theory, cells in the mucosa have the potential to help immune responses to vaccine antigens when properly activated since they are expressing more or less substantial amount of TLRs. Thus, activation of the innate immune system through TLR by using TLR ligands may represent an attractive strategy to enhance immune responses against pathogens.
The first and only current usage of a TLR ligand mucosal adjuvant in US is AS04 incorporated in the recently FDA-approved human papillomavirus vaccine, Cervarix, by GlaxoSmithKline. The AS04 is composed of alum with the TLR4 ligand MPL [22,23,96,97]. MPL is monophosphoryl lipid A isolated from the lipopolysaccharide of Salmonella minnesota R595 and retains much of the immunostimulatory properties of the parent lipopolysaccharide without the inherent toxicity [64]. MPL promotes a Th1-biased response towards co-administered antigens [23]. Another MPL formulation for mucosal vaccines AS01, which consists of liposomes, MPL, and saponin, was reported to enhance systemic and mucosal immunity in a HIV vaccine study with non-human primates [98].
Another promising mucosal adjuvant is the TLR9 ligand CpG. These small oligodeoxynucleotide (ODN) sequences induce strong Th1 responses and have been effective in animal vaccine studies when delivered mucosally [99-101]; however, in human subjects CpGs have only been investigated for cancer and systemic immunity in HIV patients or malaria, but not for induction of mucosal immunity. Among ligands of 13 TLRs, CpG has been relatively well studied because of its general high efficacy and synthetic nature. The adjuvant activity of CpG ODN is due to several different effects it has on innate and adaptive immune responses [58]. First, it causes B cells to proliferate and secrete immunoglobulin, and these direct effects synergize strongly with antigen-specific effects mediated through the B cell. As well, CpG ODN causes up-regulation of expression of co-stimulatory molecules and MHC class II molecules, improving antigen presentation. CpG ODN also directly activates monocytes, macrophages and DCs to secrete IFN-γ, IL-6, IL-12, granulocyte-macrophage colony-stimulating factor (GM-CSF), chemokines, and tumor necrosis factor-α, which in turn stimulate T cells to secrete additional cytokines and natural killer (NK) cells to secrete IFN-γ. The mechanisms through which CpG ODN functions as an immunostimulator are not thoroughly documented yet. One hypothesis is that CpG-DNA (either as a free molecule or encapsulated in whole bacteria) is taken up by an immune cell, for instance a DC, and sensed by TLR9 in the endosomal compartment. As with the ADP-ribosylating enterotoxin adjuvants, CpG ODNs have been shown to function as effective adjuvants for vaccines against a variety of bacteria, viruses, fungi, and parasites [101]. Its propensity to stimulate Th1 polarization, researchers accept that CpG should be superior adjuvant for intracellular bacterial pathogens and viruses to other TLR ligands. Human clinical trials demonstrate CpG ODNs have a good safety profile, which provides a scientific ground to expand the CpG application towards mucosal vaccines [59].
The most recently reported, but having many virtue, TLR ligand mucosal adjuvant is flagellin. Flagellin is the structural component of flagellar filament and a self-assembling protein subunit arranged in a helix to form a hollow tube [102]. Flagellin is the only cognate ligand reported so far for TLR5, which uses only MyD88 as the cytoplasmic adaptor molecule while other TLRs use multiple adaptor molecules and redundant signaling pathways [15]. This unique signaling characteristic of TLR5 might be useful designing a flagellin-based adjuvant since relatively simple pathway is employed for downstream signal relay. It has been very well understood, throughout the molecular bacterial pathogenesis study history, that flagellin is a very important virulence factor and highly immunogenic component of mucosal bacterial pathogens [103]. Our group first showed superior mucosal adjuvanticity of flagellin over other TLR ligands. A Vibrio vulnificus flagellin could efficaciously potentiate anti-tetanus toxin protective immunity when administered intranasally as a mixture with tetanus toxoid [70]. Intranasally administered FlaB was mostly trapped by DCs and mobilized to parafollicular areas of draining lymph nodes and stayed there up to 6 hours, which could partly explain the excellent mucosal adjuvanticity of flagellin [70]. Flagellin is one of the very limited numbers of TLR ligand that could be engineered genetically. Flagellin is one of highly expressing bacterial proteins and biochemically very stable. Many hard-to-express proteins could be well expressed when they were fused with flagellin. So we hypothesized that antigens fused with flagellin would more effectively stimulate protective immunity through more robust induction TLR5 signaling and the fusion proteins could be developed as mucosal vaccines. We showed that recombinant FlaB-pneumococcal surface protein A (PspA) fusion protein is able to elicit more efficient protective mucosal immune responses against pneumococcal infection than immunization with PspA alone or with a stoichiometric mixture of PspA and FlaB [71]. Flagellin seems to have the ability that can have any given antigen, which is unable to stimulate significant immune reactions in the mucosal compartment, convert to effective mucosal vaccines. In a recent study, we demonstrated the possibility that the mere combination of FlaB is capable of having conventional injectable inactivated influenza vaccine be converted into a needless nasal spray vaccine. When mice were intranasally immunized with inactivated trivalent influenza vaccines (TIV) in combination with FlaB, influenza-specific IgG and IgA responses in serum and mucosal secretions were significantly enhanced. The FlaB-induced potent antibody responses were fully functional in protecting vaccinees from natural virus infection. FlaB-TIV vaccine-immunized mice manifested remarkably higher serum haemagglutination inhibition titer and were significantly protected from lethal live virus challenge [91]. As a mucosal adjuvant, flagellin is almost as potent as CT or LT while it does not accumulate in olfactory nerve and bulb [72]. Flagellin will hopefully become the champion of proteinaceous mucosal adjuvant in the place of CT and LT-it is as potent as the latter but much safer and molecular biologically maneuverable.
Cytokines and chemokines
Most biological activities of adjuvants are mediated by cytokines and chemokines produced by immunocytes either directly or indirectly influenced by specific adjuvants. More potent adjuvant will stimulate more production of cytokines/chemokines in quantity and also more variety, which will inevitably cause more adverse effects and make it inappropriate for clinical use. One approach to circumvent the problem with overtly toxic adjuvants is to mimic the signals they induce in vivo by simply adding these signaling molecules either directly as proteins, or indirectly as coding DNA [4,66]. As discussed above, the most powerful mucosal adjuvants known to date are CT and LT, which promote strong mucosal IgA responses, systemic IgG responses and CTL to co-administered antigens. Several combinations of cytokines have been shown capable of recapitulating activities of CT (or LT) as a nasal adjuvant. The most important of these is IL-1, which in combination with Th1-inducing cytokines such as IL-12, IL-18, and GM-CSF, can bring about as strong mucosal and systemic responses as CT [104]. The combination of IL-1 and IL-12/IL-18/GM-CSF gives rise to a combined Th1 (CTL and IFN-γ)/Th2 (mucosal IgA) profile also against weak antigens such as synthetic peptides. The addition of genes coding for specific chemokines, e.g., the CCR7 ligands which are involved in directing DC to the T cell areas of secondary lymphoid organs, to a herpes simplex virus 2 DNA plasmid vaccine has stimulated the immune responses in mice following both nasal and intragastric vaccination [105]. The use of regulated upon activation, normal T cell expressed, and secreted (RANTES; a chemoattractant for monocytes, T cells and NK cells and a potent inducer of Th1/CTL responses) as a mucosal adjuvant has also given promising initial results. Nasal co-administration of RANTES and a protein antigen was shown to enhance Th1 and Th2 responses both at local and remote mucosal tissues as well as systemically [106]. The exact action mechanisms of mucosal cytokine/chemokine adjuvants are not thoroughly studied yet. One research group recently demonstrated that CD11c+ cells must be directly activated by nasally administered IL-1 alpha for maximal adjuvant activity and that, although stromal cells are required for maximal adjuvant-induced cytokine production, the adjuvant-induced stromal cell cytokine responses are not required for effective induction of adaptive immunity [107]. This result suggest that exogenously administered cytokines/chemokines will intervene into the preexisting cytokine/chemokine networks, which will give rise to more complicated interactions among mucosal immunocytes that simply be imagined. Because of cost effectiveness, using cytokine/chemokine protein products as mucosal adjuvant should not be realistic, and DNA vaccination would be the more optimal field where cytokine/chemokine adjuvant will have real impact.
Novel adjuvants and delivery systems
As for novel adjuvants (other than bacterial enterotoxin, TLR ligands, and cytokines/chemokines), please refer to two recent review articles that comprehensively summarized recent trends and achievements [1,59]. α-Galactosylceramide, a CD1d ligand and NKT cell activator, was recently reported to be able to be used as an efficacious mucosal adjuvant in various vaccines [97-100]. Alternatively, there are several fungal or bacterially derived small molecules that will bind to antimicrobial receptors on APCs. A dectin-1 agonist was reported to enhance Th17 responses concurrent with IgA induction [108]. Mast cell activators such as compound 48/80 can promote Th17 cell development when given intradermally [109] and enhance B cell proliferation and IgA production [110]. Lipid-based and archeal lipid mucosal vaccine adjuvants are also actively researched recently [1]. Iscomatrix (colloidal, spherical structures, comprising of saponin such as Quil A, cholesterol and a phospholipid) has potential as a mucosal adjuvant [67-69,92], but it may not be easily amenable for use with hydrophilic protein antigens, and concerns/perceptions regarding saponin toxicity remain [111,112]. Cochleate vaccines derived by interaction of multivalent cations proteoliposomes have been investigated for mucosal vaccination [65].
3',5'-Cyclic diguanylic acid (c-di-GMP), an intracellular signaling molecule, has recently shown some promise as a mucosal adjuvant. Although absent in higher eukaryotes, c-di-GMP is ubiquitously present in bacteria where it functions as a bacterial second messenger [113]. However, it was the recent finding that c-di-GMP can act as a danger signal to eukaryotic cells that prompted the evaluation of the immunomodulatory properties of c-di-GMP and its potential application as a vaccine adjuvant. It induces the activation and maturation of human immature DCs in vitro, and c-di-GMP-matured DCs also demonstrated enhanced T cell stimulatory activity [73]. Recent researches clearly show effective mucosal adjuvant activities of c-di-GMP in different experimental models [74,114].
Footnotes
No potential conflict of interest relevant to this article was reported.
References
- 1.Chen W, Patel GB, Yan H, Zhang J. Recent advances in the development of novel mucosal adjuvants and antigen delivery systems. Hum Vaccin. 2010;6:706–714. doi: 10.4161/hv.6.9.11561. [DOI] [PubMed] [Google Scholar]
- 2.Lamm ME. Interaction of antigens and antibodies at mucosal surfaces. Annu Rev Microbiol. 1997;51:311–340. doi: 10.1146/annurev.micro.51.1.311. [DOI] [PubMed] [Google Scholar]
- 3.Neutra MR, Kozlowski PA. Mucosal vaccines: the promise and the challenge. Nat Rev Immunol. 2006;6:148–158. doi: 10.1038/nri1777. [DOI] [PubMed] [Google Scholar]
- 4.Holmgren J, Czerkinsky C, Eriksson K, Mharandi A. Mucosal immunisation and adjuvants: a brief overview of recent advances and challenges. Vaccine. 2003;21(Suppl 2):S89–S95. doi: 10.1016/s0264-410x(03)00206-8. [DOI] [PubMed] [Google Scholar]
- 5.Murphy TV, Gargiullo PM, Massoudi MS, et al. Intussusception among infants given an oral rotavirus vaccine. N Engl J Med. 2001;344:564–572. doi: 10.1056/NEJM200102223440804. [DOI] [PubMed] [Google Scholar]
- 6.Mutsch M, Zhou W, Rhodes P, et al. Use of the inactivated intranasal influenza vaccine and the risk of Bell's palsy in Switzerland. N Engl J Med. 2004;350:896–903. doi: 10.1056/NEJMoa030595. [DOI] [PubMed] [Google Scholar]
- 7.Fujkuyama Y, Tokuhara D, Kataoka K, et al. Novel vaccine development strategies for inducing mucosal immunity. Expert Rev Vaccines. 2012;11:367–379. doi: 10.1586/erv.11.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ramon G. Sur l'augmentation anormale de l'antitoxine chez les chevaux producteurs de serum antidiphtherique. Bull Soc Cent Med Vet. 1925;101:227–234. [Google Scholar]
- 9.Glenny AT. Insoluble precipitates in diphtheria and tetanus immunization. Br Med J. 1930;2:244–245. doi: 10.1136/bmj.2.3632.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marciani DJ. Vaccine adjuvants: role and mechanisms of action in vaccine immunogenicity. Drug Discov Today. 2003;8:934–943. doi: 10.1016/s1359-6446(03)02864-2. [DOI] [PubMed] [Google Scholar]
- 11.Schijns VE. Induction and direction of immune responses by vaccine adjuvants. Crit Rev Immunol. 2001;21:75–85. [PubMed] [Google Scholar]
- 12.Guy B. The perfect mix: recent progress in adjuvant research. Nat Rev Microbiol. 2007;5:505–517. doi: 10.1038/nrmicro1681. [DOI] [PubMed] [Google Scholar]
- 13.McKee AS, Munks MW, Marrack P. How do adjuvants work? Important considerations for new generation adjuvants. Immunity. 2007;27:687–690. doi: 10.1016/j.immuni.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 14.Creagh EM, O'Neill LA. TLRs, NLRs and RLRs: a trinity of pathogen sensors that co-operate in innate immunity. Trends Immunol. 2006;27:352–357. doi: 10.1016/j.it.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 15.Akira S. Innate immunity and adjuvants. Philos Trans R Soc Lond B Biol Sci. 2011;366:2748–2755. doi: 10.1098/rstb.2011.0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA. The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell. 1996;86:973–983. doi: 10.1016/s0092-8674(00)80172-5. [DOI] [PubMed] [Google Scholar]
- 17.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 18.Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449:819–826. doi: 10.1038/nature06246. [DOI] [PubMed] [Google Scholar]
- 19.Shi Z, Cai Z, Sanchez A, et al. A novel Toll-like receptor that recognizes vesicular stomatitis virus. J Biol Chem. 2011;286:4517–4524. doi: 10.1074/jbc.M110.159590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Flacher V, Bouschbacher M, Verronèse E, et al. Human Langerhans cells express a specific TLR profile and differentially respond to viruses and Gram-positive bacteria. J Immunol. 2006;177:7959–7967. doi: 10.4049/jimmunol.177.11.7959. [DOI] [PubMed] [Google Scholar]
- 21.Jarrossay D, Napolitani G, Colonna M, Sallusto F, Lanzavecchia A. Specialization and complementarity in microbial molecule recognition by human myeloid and plasmacytoid dendritic cells. Eur J Immunol. 2001;31:3388–3393. doi: 10.1002/1521-4141(200111)31:11<3388::aid-immu3388>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 22.Schwarz TF. Clinical update of the AS04-adjuvanted human papillomavirus-16/18 cervical cancer vaccine, Cervarix. Adv Ther. 2009;26:983–998. doi: 10.1007/s12325-009-0079-5. [DOI] [PubMed] [Google Scholar]
- 23.Didierlaurent AM, Morel S, Lockman L, et al. AS04, an aluminum salt- and TLR4 agonist-based adjuvant system, induces a transient localized innate immune response leading to enhanced adaptive immunity. J Immunol. 2009;183:6186–6197. doi: 10.4049/jimmunol.0901474. [DOI] [PubMed] [Google Scholar]
- 24.Vollmer J, Krieg AM. Immunotherapeutic applications of CpG oligodeoxynucleotide TLR9 agonists. Adv Drug Deliv Rev. 2009;61:195–204. doi: 10.1016/j.addr.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 25.Duthie MS, Windish HP, Fox CB, Reed SG. Use of defined TLR ligands as adjuvants within human vaccines. Immunol Rev. 2011;239:178–196. doi: 10.1111/j.1600-065X.2010.00978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fritz JH, Ferrero RL, Philpott DJ, Girardin SE. Nod-like proteins in immunity, inflammation and disease. Nat Immunol. 2006;7:1250–1257. doi: 10.1038/ni1412. [DOI] [PubMed] [Google Scholar]
- 27.Inohara, Chamaillard, McDonald C, Nuñez G. NOD-LRR proteins: role in host-microbial interactions and inflammatory disease. Annu Rev Biochem. 2005;74:355–383. doi: 10.1146/annurev.biochem.74.082803.133347. [DOI] [PubMed] [Google Scholar]
- 28.Traub S, von Aulock S, Hartung T, Hermann C. MDP and other muropeptides: direct and synergistic effects on the immune system. J Endotoxin Res. 2006;12:69–85. doi: 10.1179/096805106X89044. [DOI] [PubMed] [Google Scholar]
- 29.Meylan E, Tschopp J, Karin M. Intracellular pattern recognition receptors in the host response. Nature. 2006;442:39–44. doi: 10.1038/nature04946. [DOI] [PubMed] [Google Scholar]
- 30.Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 31.Holt LB. Quantitative studies in diphtheria prophylaxis: the second response. Br J Exp Pathol. 1950;31:233–241. [PMC free article] [PubMed] [Google Scholar]
- 32.Iyer S, HogenEsch H, Hem SL. Relationship between the degree of antigen adsorption to aluminum hydroxide adjuvant in interstitial fluid and antibody production. Vaccine. 2003;21:1219–1223. doi: 10.1016/s0264-410x(02)00556-x. [DOI] [PubMed] [Google Scholar]
- 33.Li H, Nookala S, Re F. Aluminum hydroxide adjuvants activate caspase-1 and induce IL-1beta and IL-18 release. J Immunol. 2007;178:5271–5276. doi: 10.4049/jimmunol.178.8.5271. [DOI] [PubMed] [Google Scholar]
- 34.Eisenbarth SC, Colegio OR, O'Connor W, Sutterwala FS, Flavell RA. Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature. 2008;453:1122–1126. doi: 10.1038/nature06939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kool M, Pétrilli V, De Smedt T, et al. Cutting edge: alum adjuvant stimulates inflammatory dendritic cells through activation of the NALP3 inflammasome. J Immunol. 2008;181:3755–3759. doi: 10.4049/jimmunol.181.6.3755. [DOI] [PubMed] [Google Scholar]
- 36.Hornung V, Bauernfeind F, Halle A, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 2008;9:847–856. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Franchi L, Nunez G. The Nlrp3 inflammasome is critical for aluminium hydroxide-mediated IL-1beta secretion but dispensable for adjuvant activity. Eur J Immunol. 2008;38:2085–2089. doi: 10.1002/eji.200838549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matzinger P. The danger model: a renewed sense of self. Science. 2002;296:301–305. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 39.Spreafico R, Ricciardi-Castagnoli P, Mortellaro A. The controversial relationship between NLRP3, alum, danger signals and the next-generation adjuvants. Eur J Immunol. 2010;40:638–642. doi: 10.1002/eji.200940039. [DOI] [PubMed] [Google Scholar]
- 40.Sharp FA, Ruane D, Claass B, et al. Uptake of particulate vaccine adjuvants by dendritic cells activates the NALP3 inflammasome. Proc Natl Acad Sci U S A. 2009;106:870–875. doi: 10.1073/pnas.0804897106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li H, Willingham SB, Ting JP, Re F. Cutting edge: inflammasome activation by alum and alum's adjuvant effect are mediated by NLRP3. J Immunol. 2008;181:17–21. doi: 10.4049/jimmunol.181.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yoneyama M, Fujita T. RNA recognition and signal transduction by RIG-I-like receptors. Immunol Rev. 2009;227:54–65. doi: 10.1111/j.1600-065X.2008.00727.x. [DOI] [PubMed] [Google Scholar]
- 43.Yoneyama M, Kikuchi M, Natsukawa T, et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 44.Takeuchi O, Akira S. Innate immunity to virus infection. Immunol Rev. 2009;227:75–86. doi: 10.1111/j.1600-065X.2008.00737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kato H, Takeuchi O, Sato S, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 46.Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Takaoka A, Wang Z, Choi MK, et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature. 2007;448:501–505. doi: 10.1038/nature06013. [DOI] [PubMed] [Google Scholar]
- 48.Gonzalez-Navajas JM, Lee J, David M, Raz E. Immunomodulatory functions of type I interferons. Nat Rev Immunol. 2012;12:125–135. doi: 10.1038/nri3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lavelle EC, Murphy C, O'Neill LA, Creagh EM. The role of TLRs, NLRs, and RLRs in mucosal innate immunity and homeostasis. Mucosal Immunol. 2010;3:17–28. doi: 10.1038/mi.2009.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Varol C, Vallon-Eberhard A, Elinav E, et al. Intestinal lamina propria dendritic cell subsets have different origin and functions. Immunity. 2009;31:502–512. doi: 10.1016/j.immuni.2009.06.025. [DOI] [PubMed] [Google Scholar]
- 51.Coombes JL, Siddiqui KR, Arancibia-Carcamo CV, et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med. 2007;204:1757–1764. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kiyono H, Fukuyama S. NALT-versus Peyer's-patch-mediated mucosal immunity. Nat Rev Immunol. 2004;4:699–710. doi: 10.1038/nri1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yuki Y, Kiyono H. New generation of mucosal adjuvants for the induction of protective immunity. Rev Med Virol. 2003;13:293–310. doi: 10.1002/rmv.398. [DOI] [PubMed] [Google Scholar]
- 54.Kim DY, Sato A, Fukuyama S, et al. The airway antigen sampling system: respiratory M cells as an alternative gateway for inhaled antigens. J Immunol. 2011;186:4253–4262. doi: 10.4049/jimmunol.0903794. [DOI] [PubMed] [Google Scholar]
- 55.Yamamoto M, Pascual DW, Kiyono H. M cell-targeted mucosal vaccine strategies. Curr Top Microbiol Immunol. 2012;354:39–52. doi: 10.1007/82_2011_134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen K, Cerutti A. Vaccination strategies to promote mucosal antibody responses. Immunity. 2010;33:479–491. doi: 10.1016/j.immuni.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kunisawa J, Nochi T, Kiyono H. Immunological commonalities and distinctions between airway and digestive immunity. Trends Immunol. 2008;29:505–513. doi: 10.1016/j.it.2008.07.008. [DOI] [PubMed] [Google Scholar]
- 58.Holmgren J, Czerkinsky C. Mucosal immunity and vaccines. Nat Med. 2005;11(4 Suppl):S45–S53. doi: 10.1038/nm1213. [DOI] [PubMed] [Google Scholar]
- 59.Lawson LB, Norton EB, Clements JD. Defending the mucosa: adjuvant and carrier formulations for mucosal immunity. Curr Opin Immunol. 2011;23:414–420. doi: 10.1016/j.coi.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 60.Mishra N, Goyal AK, Tiwari S, et al. Recent advances in mucosal delivery of vaccines: role of mucoadhesive/biodegradable polymeric carriers. Expert Opin Ther Pat. 2010;20:661–679. doi: 10.1517/13543771003730425. [DOI] [PubMed] [Google Scholar]
- 61.Pavot V, Rochereau N, Genin C, Verrier B, Paul S. New insights in mucosal vaccine development. Vaccine. 2012;30:142–154. doi: 10.1016/j.vaccine.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 62.Eriksson K, Holmgren J. Recent advances in mucosal vaccines and adjuvants. Curr Opin Immunol. 2002;14:666–672. doi: 10.1016/s0952-7915(02)00384-9. [DOI] [PubMed] [Google Scholar]
- 63.Jabbal-Gill I, Watts P, Smith A. Chitosan-based delivery systems for mucosal vaccines. Expert Opin Drug Deliv. 2012 Jun 19; doi: 10.1517/17425247.2012.697455. [Epub] http://dx.doi.org/10.1517/17425247.2012.697455. [DOI] [PubMed] [Google Scholar]
- 64.Freytag LC, Clements JD. Mucosal adjuvants. Vaccine. 2005;23:1804–1813. doi: 10.1016/j.vaccine.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 65.Bracho G, Lastre M, del Campo J, et al. Proteoliposome derived cochleate as novel adjuvant. Vaccine. 2006;24(Suppl 2):S2-30–S2-31. doi: 10.1016/j.vaccine.2005.01.108. [DOI] [PubMed] [Google Scholar]
- 66.Cox E, Verdonck F, Vanrompay D, Goddeeris B. Adjuvants modulating mucosal immune responses or directing systemic responses towards the mucosa. Vet Res. 2006;37:511–539. doi: 10.1051/vetres:2006014. [DOI] [PubMed] [Google Scholar]
- 67.Coulter A, Harris R, Davis R, et al. Intranasal vaccination with ISCOMATRIX adjuvanted influenza vaccine. Vaccine. 2003;21:946–949. doi: 10.1016/s0264-410x(02)00545-5. [DOI] [PubMed] [Google Scholar]
- 68.Sanders MT, Deliyannis G, Pearse MJ, McNamara MK, Brown LE. Single dose intranasal immunization with ISCOMATRIX vaccines to elicit antibody-mediated clearance of influenza virus requires delivery to the lower respiratory tract. Vaccine. 2009;27:2475–2482. doi: 10.1016/j.vaccine.2009.02.054. [DOI] [PubMed] [Google Scholar]
- 69.Vujanic A, Snibson KJ, Wee JL, et al. Long-term antibody and immune memory response induced by pulmonary delivery of the influenza Iscomatrix vaccine. Clin Vaccine Immunol. 2012;19:79–83. doi: 10.1128/CVI.05265-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee SE, Kim SY, Jeong BC, et al. A bacterial flagellin, Vibrio vulnificus FlaB, has a strong mucosal adjuvant activity to induce protective immunity. Infect Immun. 2006;74:694–702. doi: 10.1128/IAI.74.1.694-702.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nguyen CT, Kim SY, Kim MS, Lee SE, Rhee JH. Intranasal immunization with recombinant PspA fused with a flagellin enhances cross-protective immunity against Streptococcus pneumoniae infection in mice. Vaccine. 2011;29:5731–5739. doi: 10.1016/j.vaccine.2011.05.095. [DOI] [PubMed] [Google Scholar]
- 72.Hong SH, Byun YH, Nguyen CT, et al. Intranasal administration of a flagellin-adjuvanted inactivated influenza vaccine enhances mucosal immune responses to protect mice against lethal infection. Vaccine. 2012;30:466–474. doi: 10.1016/j.vaccine.2011.10.058. [DOI] [PubMed] [Google Scholar]
- 73.Chen W, Kuolee R, Yan H. The potential of 3',5'-cyclic diguanylic acid (c-di-GMP) as an effective vaccine adjuvant. Vaccine. 2010;28:3080–3085. doi: 10.1016/j.vaccine.2010.02.081. [DOI] [PubMed] [Google Scholar]
- 74.Yan H, KuoLee R, Tram K, et al. 3',5'-Cyclic diguanylic acid elicits mucosal immunity against bacterial infection. Biochem Biophys Res Commun. 2009;387:581–584. doi: 10.1016/j.bbrc.2009.07.061. [DOI] [PubMed] [Google Scholar]
- 75.Mowat AM, Smith RE, Donachie AM, Furrie E, Grdic D, Lycke N. Oral vaccination with immune stimulating complexes. Immunol Lett. 1999;65:133–140. doi: 10.1016/s0165-2478(98)00136-9. [DOI] [PubMed] [Google Scholar]
- 76.Noda K, Kodama S, Umemoto S, Abe N, Hirano T, Suzuki M. Nasal vaccination with P6 outer membrane protein and alpha-galactosylceramide induces nontypeable Haemophilus influenzae-specific protective immunity associated with NKT cell activation and dendritic cell expansion in nasopharynx. Vaccine. 2010;28:5068–5074. doi: 10.1016/j.vaccine.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 77.Lee YS, Lee KA, Lee JY, et al. An alpha-GalCer analogue with branched acyl chain enhances protective immune responses in a nasal influenza vaccine. Vaccine. 2011;29:417–425. doi: 10.1016/j.vaccine.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 78.Courtney AN, Nehete PN, Nehete BP, Thapa P, Zhou D, Sastry KJ. Alpha-galactosylceramide is an effective mucosal adjuvant for repeated intranasal or oral delivery of HIV peptide antigens. Vaccine. 2009;27:3335–3341. doi: 10.1016/j.vaccine.2009.01.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lindqvist M, Persson J, Thorn K, Harandi AM. The mucosal adjuvant effect of alpha-galactosylceramide for induction of protective immunity to sexually transmitted viral infection. J Immunol. 2009;182:6435–6443. doi: 10.4049/jimmunol.0900136. [DOI] [PubMed] [Google Scholar]
- 80.de Haan L, Hirst TR. Cholera toxin and related enterotoxins: a cell biological and immunological perspective. J Nat Toxins. 2000;9:281–297. [PubMed] [Google Scholar]
- 81.van Ginkel FW, Jackson RJ, Yoshino N, et al. Enterotoxin-based mucosal adjuvants alter antigen trafficking and induce inflammatory responses in the nasal tract. Infect Immun. 2005;73:6892–6902. doi: 10.1128/IAI.73.10.6892-6902.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Eriksson K, Fredriksson M, Nordström I, Holmgren J. Cholera toxin and its B subunit promote dendritic cell vaccination with different influences on Th1 and Th2 development. Infect Immun. 2003;71:1740–1747. doi: 10.1128/IAI.71.4.1740-1747.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.George-Chandy A, Eriksson K, Lebens M, Nordström I, Schön E, Holmgren J. Cholera toxin B subunit as a carrier molecule promotes antigen presentation and increases CD40 and CD86 expression on antigen-presenting cells. Infect Immun. 2001;69:5716–5725. doi: 10.1128/IAI.69.9.5716-5725.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pizza M, Giuliani MM, Fontana MR, et al. Mucosal vaccines: non toxic derivatives of LT and CT as mucosal adjuvants. Vaccine. 2001;19:2534–2541. doi: 10.1016/s0264-410x(00)00553-3. [DOI] [PubMed] [Google Scholar]
- 85.Mowat AM, Donachie AM, Jagewall S, et al. CTA1-DD-immune stimulating complexes: a novel, rationally designed combined mucosal vaccine adjuvant effective with nanogram doses of antigen. J Immunol. 2001;167:3398–3405. doi: 10.4049/jimmunol.167.6.3398. [DOI] [PubMed] [Google Scholar]
- 86.Sanchez J, Wallerstrom G, Fredriksson M, Angstrom J, Holmgren J. Detoxification of cholera toxin without removal of its immunoadjuvanticity by the addition of (STa-related) peptides to the catalytic subunit. A potential new strategy to generate immunostimulants for vaccination. J Biol Chem. 2002;277:33369–33377. doi: 10.1074/jbc.M112337200. [DOI] [PubMed] [Google Scholar]
- 87.Norton EB, Lawson LB, Freytag LC, Clements JD. Characterization of a mutant Escherichia coli heat-labile toxin, LT(R192G/L211A), as a safe and effective oral adjuvant. Clin Vaccine Immunol. 2011;18:546–551. doi: 10.1128/CVI.00538-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Anosova NG, Chabot S, Shreedhar V, Borawski JA, Dickinson BL, Neutra MR. Cholera toxin, E. coli heat-labile toxin, and non-toxic derivatives induce dendritic cell migration into the follicle-associated epithelium of Peyer's patches. Mucosal Immunol. 2008;1:59–67. doi: 10.1038/mi.2007.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fahlen-Yrlid L, Gustafsson T, Westlund J, et al. CD11c (high) dendritic cells are essential for activation of CD4+ T cells and generation of specific antibodies following mucosal immunization. J Immunol. 2009;183:5032–5041. doi: 10.4049/jimmunol.0803992. [DOI] [PubMed] [Google Scholar]
- 90.Chang SY, Cha HR, Igarashi O, et al. Cutting edge: Langerin+ dendritic cells in the mesenteric lymph node set the stage for skin and gut immune system cross-talk. J Immunol. 2008;180:4361–4365. doi: 10.4049/jimmunol.180.7.4361. [DOI] [PubMed] [Google Scholar]
- 91.Datta SK, Sabet M, Nguyen KP, et al. Mucosal adjuvant activity of cholera toxin requires Th17 cells and protects against inhalation anthrax. Proc Natl Acad Sci U S A. 2010;107:10638–10643. doi: 10.1073/pnas.1002348107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lu YJ, Yadav P, Clements JD, et al. Options for inactivation, adjuvant, and route of topical administration of a killed, unencapsulated pneumococcal whole-cell vaccine. Clin Vaccine Immunol. 2010;17:1005–1012. doi: 10.1128/CVI.00036-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Meza-Sanchez D, Perez-Montesinos G, Sanchez-Garcia J, Moreno J, Bonifaz LC. Intradermal immunization in the ear with cholera toxin and its non-toxic beta subunit promotes efficient Th1 and Th17 differentiation dependent on migrating DCs. Eur J Immunol. 2011;41:2894–2904. doi: 10.1002/eji.201040997. [DOI] [PubMed] [Google Scholar]
- 94.Raghavan S, Ostberg AK, Flach CF, et al. Sublingual immunization protects against Helicobacter pylori infection and induces T and B cell responses in the stomach. Infect Immun. 2010;78:4251–4260. doi: 10.1128/IAI.00536-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dubin PJ, Kolls JK. Th17 cytokines and mucosal immunity. Immunol Rev. 2008;226:160–171. doi: 10.1111/j.1600-065X.2008.00703.x. [DOI] [PubMed] [Google Scholar]
- 96.Paavonen J, Naud P, Salmeron J, et al. Efficacy of human papillomavirus (HPV)-16/18 AS04-adjuvanted vaccine against cervical infection and precancer caused by oncogenic HPV types (PATRICIA): final analysis of a double-blind, randomised study in young women. Lancet. 2009;374:301–314. doi: 10.1016/S0140-6736(09)61248-4. [DOI] [PubMed] [Google Scholar]
- 97.Schwarz TF, Spaczynski M, Schneider A, et al. Immunogenicity and tolerability of an HPV-16/18 AS04-adjuvanted prophylactic cervical cancer vaccine in women aged 15-55 years. Vaccine. 2009;27:581–587. doi: 10.1016/j.vaccine.2008.10.088. [DOI] [PubMed] [Google Scholar]
- 98.Cranage MP, Fraser CA, Cope A, et al. Antibody responses after intravaginal immunisation with trimeric HIV-1 CN54 clade C gp140 in Carbopol gel are augmented by systemic priming or boosting with an adjuvanted formulation. Vaccine. 2011;29:1421–1430. doi: 10.1016/j.vaccine.2010.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Huang CF, Wu TC, Chu YH, Hwang KS, Wang CC, Peng HJ. Effect of neonatal sublingual vaccination with native or denatured ovalbumin and adjuvant CpG or cholera toxin on systemic and mucosal immunity in mice. Scand J Immunol. 2008;68:502–510. doi: 10.1111/j.1365-3083.2008.02172.x. [DOI] [PubMed] [Google Scholar]
- 100.Pesce I, Monaci E, Muzzi A, et al. Intranasal administration of CpG induces a rapid and transient cytokine response followed by dendritic and natural killer cell activation and recruitment in the mouse lung. J Innate Immun. 2010;2:144–159. doi: 10.1159/000254948. [DOI] [PubMed] [Google Scholar]
- 101.Bode C, Zhao G, Steinhagen F, Kinjo T, Klinman DM. CpG DNA as a vaccine adjuvant. Expert Rev Vaccines. 2011;10:499–511. doi: 10.1586/erv.10.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Samatey FA, Imada K, Nagashima S, et al. Structure of the bacterial flagellar protofilament and implications for a switch for supercoiling. Nature. 2001;410:331–337. doi: 10.1038/35066504. [DOI] [PubMed] [Google Scholar]
- 103.Ramos HC, Rumbo M, Sirard JC. Bacterial flagellins: mediators of pathogenicity and host immune responses in mucosa. Trends Microbiol. 2004;12:509–517. doi: 10.1016/j.tim.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 104.Staats HF, Bradney CP, Gwinn WM, et al. Cytokine requirements for induction of systemic and mucosal CTL after nasal immunization. J Immunol. 2001;167:5386–5394. doi: 10.4049/jimmunol.167.9.5386. [DOI] [PubMed] [Google Scholar]
- 105.Eo SK, Lee S, Kumaraguru U, Rouse BT. Immunopotentiation of DNA vaccine against herpes simplex virus via co-delivery of plasmid DNA expressing CCR7 ligands. Vaccine. 2001;19:4685–4693. doi: 10.1016/s0264-410x(01)00241-9. [DOI] [PubMed] [Google Scholar]
- 106.Lillard JW, Jr, Boyaka PN, Taub DD, McGhee JR. RANTES potentiates antigen-specific mucosal immune responses. J Immunol. 2001;166:162–169. doi: 10.4049/jimmunol.166.1.162. [DOI] [PubMed] [Google Scholar]
- 107.Thompson AL, Johnson BT, Sempowski GD, et al. Maximal adjuvant activity of nasally delivered IL-1alpha requires adjuvant-responsive CD11c(+) cells and does not correlate with adjuvant-induced in vivo cytokine production. J Immunol. 2012;188:2834–2846. doi: 10.4049/jimmunol.1100254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Agrawal S, Gupta S, Agrawal A. Human dendritic cells activated via dectin-1 are efficient at priming Th17, cytotoxic CD8 T and B cell responses. PLoS One. 2010;5:e13418. doi: 10.1371/journal.pone.0013418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.McGowen AL, Hale LP, Shelburne CP, Abraham SN, Staats HF. The mast cell activator compound 48/80 is safe and effective when used as an adjuvant for intradermal immunization with Bacillus anthracis protective antigen. Vaccine. 2009;27:3544–3552. doi: 10.1016/j.vaccine.2009.03.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Merluzzi S, Frossi B, Gri G, Parusso S, Tripodo C, Pucillo C. Mast cells enhance proliferation of B lymphocytes and drive their differentiation toward IgA-secreting plasma cells. Blood. 2010;115:2810–2817. doi: 10.1182/blood-2009-10-250126. [DOI] [PubMed] [Google Scholar]
- 111.Hu KF, Lovgren-Bengtsson K, Morein B. Immunostimulating complexes (ISCOMs) for nasal vaccination. Adv Drug Deliv Rev. 2001;51:149–159. doi: 10.1016/s0169-409x(01)00165-x. [DOI] [PubMed] [Google Scholar]
- 112.Kersten G, Hirschberg H. Antigen delivery systems. Expert Rev Vaccines. 2004;3:453–462. doi: 10.1586/14760584.3.4.453. [DOI] [PubMed] [Google Scholar]
- 113.Romling U, Amikam D. Cyclic di-GMP as a second messenger. Curr Opin Microbiol. 2006;9:218–228. doi: 10.1016/j.mib.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 114.Pedersen GK, Ebensen T, Gjeraker IH, et al. Evaluation of the sublingual route for administration of influenza H5N1 virosomes in combination with the bacterial second messenger c-di-GMP. PLoS One. 2011;6:e26973. doi: 10.1371/journal.pone.0026973. [DOI] [PMC free article] [PubMed] [Google Scholar]