

Abstract
The high prevalence of type 2 diabetes mellitus in the world as well as the increasing reports about the adverse side effects of the existing diabetes treatment drugs have made developing new and effective drugs against the disease a very high priority. In this study, we report ten novel compounds found by targeting peroxisome proliferator-activated receptors (PPARs) using virtual screening and core hopping approaches. PPARs have drawn increasing attention for developing novel drugs to treat diabetes due to their unique functions in regulating glucose, lipid, and cholesterol metabolism. The reported compounds are featured with dual functions, and hence belong to the category of dual agonists. Compared with the single PPAR agonists, the dual PPAR agonists, formed by combining the lipid benefit of PPARα agonists (such as fibrates) and the glycemic advantages of the PPARγ agonists (such as thiazolidinediones), are much more powerful in treating diabetes because they can enhance metabolic effects while minimizing the side effects. This was observed in the studies on molecular dynamics simulations, as well as on absorption, distribution, metabolism, and excretion, that these novel dual agonists not only possessed the same function as ragaglitazar (an investigational drug developed by Novo Nordisk for treating type 2 diabetes) did in activating PPARα and PPARγ, but they also had more favorable conformation for binding to the two receptors. Moreover, the residues involved in forming the binding pockets of PPARα and PPARγ among the top ten compounds are explicitly presented, and this will be very useful for the in-depth conduction of mutagenesis experiments. It is anticipated that the ten compounds may become potential drug candidates, or at the very least, the findings reported here may stimulate new strategies or provide useful insights for designing new and more powerful dual-agonist drugs for treating type 2 diabetes.
Keywords: diabetes, PPAR-alpha, PPAR-gamma, dual-agonist drug, core hopping, molecular docking, ADME, binding pocket
Introduction
Type 2 diabetes mellitus (T2DM) is the most common type of diabetes mellitus, which is characterized by insulin resistance and combined with relatively reduced insulin secretion.1 It is a chronic metabolic disease that affects the body’s ability to turn food into energy. People developing T2DM may suffer from blindness, renal failure, coronary artery disease, and so forth.2 Because of its dramatic increase worldwide, T2DM has reached an epidemic scale that is anticipated to affect over 360 million people by 2030.3,4
Belonging to the nuclear hormone receptor family, peroxisome proliferator-activated receptors (PPARs) are the ligand-activated transcription factors.5,6 PPARs, particularly the subtypes PPARα and PPARγ, played a central role in regulating the expression of genes involved in the control of lipid and lipoprotein metabolism, glucose homeostasis, and inflammatory processes. Accordingly, activating these receptors has become a promising therapeutic strategy for designing drugs against T2DM.7
The activated PPARα receptors have a beneficial effect on lipid metabolism by regulating genes on fatty acid and cholesterol metabolism.8 PPARα agonists belong to the class of lipid-lowering drugs (such as fenofibrate and gemfibrozil), and are widely prescribed to reduce triglycerides.9 The PPARγ receptors, the most widely studied subtype of PPARs, are located in adipocytes, muscles, and macrophages, where they have a direct influence on T2DM, dyslipidemia, atherosclerosis, and cardiovascular diseases.10,11 Thiazolidinediones are a class of PPARγ agonists used in clinical practice to reduce plasma glucose level in type 2 diabetic patients, or to regulate glucose homeostasis by increasing insulin sensitivity and glucose disposal.12
Unfortunately, if the drug were used alone, the undesirable side effects such as weight gain, edema, and anemia would be caused;13 therefore, it is an urgent and challenging task to develop new dual agonists. In this regard, the novel PPARα/PPARγ dual agonist, formed by combining the lipid benefit of PPARα agonists (such as fibrates) with the glycemic advantages of the PPARγ agonists (such as thiazolidinediones), has drawn considerable attention.14 In developing the dual agonists, a critical problem surrounds how to identify the selectivity ratio of the receptor subtype,15 because it may provide useful insights for finding the new drug candidates. There is more information available about designing potential dual treatments for treating T2DM.16
Many studies have indicated that computational approaches, such as structural bioinformatics,17 molecular docking,18 pharmacophore modeling,19 Quantitative Structure–Activity Relationship (QSAR) techniques,20–24 and a series of user-friendly web server predictors developed recently, such as G-protein-coupled receptors-grey incident degree analysis (GPCR-GIA) and G-protein-coupled receptors -2-layer predictor (GPCR-2L) for identifying G protein-coupled receptors and their types,25,26 iLoc-Euk and iLoc-Hum for predicting subcellular localization of eukaryotic and human proteins,27,28 NR-2L for identifying nuclear receptors and their subfamilies,29 ProtIdent for identifying proteases and their types,30 and HIVcleave for predicting human immunodeficiency virus protease cleavage sites in proteins,31,32 can provide timely and very useful information, as well as insights for drug development. In addition to the aforementioned approaches, the Virtual Screening technique and Core Hopping technique are also very useful.33–36 The former can be used to screen for novel molecular scaffolds,37,38 while the latter can be used to generate new lead compounds with improved core properties.37,38
Encouraged by the aforementioned successful studies, the present study was initiated in an attempt to screen the fragment database in hopes to find a new antidiabetic compound. Meanwhile, the techniques of the core hopping associated with the glide docking and molecular dynamic simulation were utilized to analyze the binding interactions between the agonist and PPARs.39,40 The absorption, distribution, metabolism, and excretion (ADME) predictions were also used to evaluate whether the new agonist found to date possesses great potential to become a promising drug candidate for treating diabetes mellitus.41,42
Materials and methods
The representative complex crystal structures of PPARα (Protein Data Bank [PDB] ID 1k7l) and PPARγ (PDB ID 1k74) were downloaded from the PDB,43,44 and were to be used for the molecular modeling studies.
Our calculations are operated with the Schrödinger software package (version 9.0.111; Schrödinger, LLC, New York, NY, USA), which runs on a Dell Precision™ T5500 computer (Dell, Round Rock, TX, USA). The molecular dynamic simulation was performed using the Gromacs 4.0 package (www.gromacs.com) for the Linux system.
Protein structures and databases
We downloaded the crystal structures of PPARα (1k71.pdb) and PPARγ (1k74.pdb) from the Protein Data Bank,43,44 and used them as receptors for the current modeling. The reasons behind the selection of the two proteins are: (1) the two proteins contain the same ligand, GW409544, which is used to easily identify the binding site for comparison; (2) the resolutions of the two crystal structures are quite similar with one (1k7l.pdb) having 2.50 Å and the other (1k74.pdb) having 2.30 Å; (3) the source organism of both structures was humans.43
The protein-ligand binding site was identified by the SiteMap tool embedded in the Schrodinger Suite 2009 (Schrödinger, LLC), as described previously.45–47 The binding site encompassed the ligand GW409544, which was observed in the crystal structures of both PPARα (1k7l) and PPARγ (1k74). The binding site of PPARs is composed of three arms: arm 1, arm 2, and arm 3, as marked in the panels A and B of Figure 1. These three segments play an important role in the interactions between the receptor and ligand.
Figure 1.
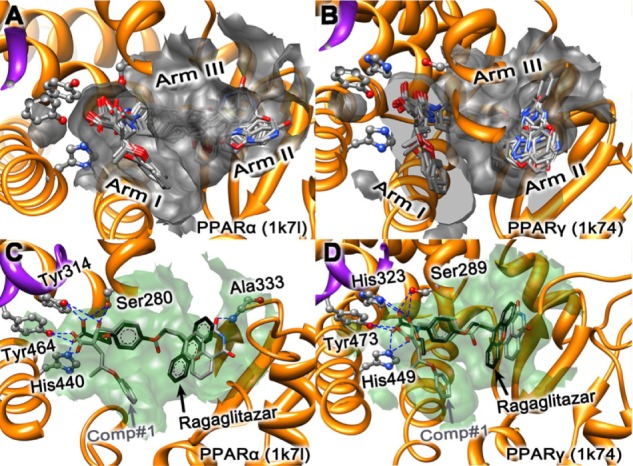
Illustration to show the superimposed conformations obtained by docking ragaglitazar and the ten derivative compounds (Comp#1–#10) to PPARα and PPARγ receptors, respectively. (A) Ragaglitazar and Comp#1–#10 to PPARα (1k7l). (B) Ragaglitazar and Comp#1–#10 to PPARγ (1k74). (C) Ragaglitazar and Comp#1 to PPARα (1k7l). (D) Ragaglitazar and Comp#1 to PPARγ (1k74).
Notes: The binding pocket is defined by those residues that have at least one heavy atom within a distance of 5Å from the ligand.92 The carbon atoms of ragaglitazar are in black, while the carbon atoms for Comp#1 are in gray. For the overlapping part between ragaglitazar and Comp#1, part of the Comp#1 was covered by ragaglitazar. The blue dotted lines indicate the H-bond interactions of the receptor with its ligands. The purple helix is a part of the AF2 function domain. See the text for further explanation.
Abbreviations: PPARα, peroxisome proliferator-activated receptor-alpha; PPARγ, peroxisome proliferator-activated receptor-gamma.
Several hydrophobic interactions between the three “arms” and the Y-shaped ligand were deemed as the key points for designing the new PPARs agonist.48
Arm 1 has a predominately polar character and includes the AF2-helix (shown as the purple ribbon in Figure 1), which is responsible for the function of transcriptional activation. The vital conformation of AF2-helix could generate a charge clamp formed by the network hydrogen bonds, thus significantly reducing the mobility of AF2 and regulating gene expression.7 The hydrophobic tail of the agonist interacts with either arm 2 or arm 3 to improve binding affinity.7
The lead compounds and the fragment databases derived from ZINC (Shoichet Laboratory, Department of Pharmaceutical Chemistry, University of California, San Francisco, CA, USA) were used for virtual screening and core hopping searching,49 as described below, respectively.
Virtual screening
ZINC (Shoichet Library, Department of Pharmaceutical Chemistry, University California) was a free database of commercially available compounds and contained over 13 million purchasable compounds in three-dimensional formats.49 The lead compound (or “lead-now”) database, one subdatabase of the ZINC, was chosen for virtual screening to find the optimal molecular scaffold by the Glide5 docking program (Schrödinger, LLC),35,36 which was interfaced with Schrödinger Suite 2009 (Schrödinger, LLC). The preparation and refinement protocols for the protein receptor and all compound structures were performed on the Protein Preparation Wizard and LigPrep modules embedded in the Schrödinger Suite (2009; Schrödinger, LLC),35 respectively. For protein preparation, the process included assigning bond orders, adding hydrogen, treating metals, treating disulfides, deleting waters, alleviating potential steric hindrance, adjusting bond order, and assigning the formal charges by protein minimization with the OPLS2005 force field;50 the constrained refinement value of RMSD (root mean square deviation) for the protein was limited to 0.3 Å. Meanwhile, for the compounds of the databases, the preparation consisted of generating possible states by ionization at a target pH of 7.0 ± 2.0, desalting, retaining specified chiralities from the three-dimensional structure, and completing geometry minimization with the OPLS2005 force field.50 When the above steps were accomplished, all the investigated compounds were docked into the receptor pocket through the rigid docking model with a high throughput virtual screening scoring function to estimate the binding affinities.39,40
Molecular docking with core hopping method
Many useful insights for drug design could be gained by conducting molecular docking studies.17,18,51–53 In this study, a new docking algorithm called “core hopping” was employed.35,36 Core hopping offers function of being able to perform both the fragment-based replacing and molecular docking,36 which makes it a very powerful and cutting-edge technique for de novo drug design because it allows for rapidly screening novel cores to help overcome unwanted properties by generating new lead compounds with improved core properties, as demonstrated by a series of previous studies.37,38
The procedures of the core hopping computation can be briefly described as follows. The first step was to define the possible points to which the cores were attached; this could be done by using the program “Define Combinations” from the Combinatorial Screening panel in Schrodinger 2009 (Schrödinger, LLC). The second step was to define the “receptor grid file,” which could be done in the “Receptor Preparation” panel. The third step was the cores preparation; this could be done by operating the “Protocore Preparation” module to find the cores attached to the scaffold and using the fragment database derived from ZINC.49
The fourth step was to align and dock the entire molecular structure built up by the core and scaffold. After all the above four steps were accomplished, all the investigated compounds were redocked into the receptor pocket via the rigid protein docking model with the Stand-Precision scoring function to estimate the binding affinities.39,40
Molecular dynamics simulations
Many marvelous biological functions in proteins and deoxyribonucleic acid (DNA) and their profound dynamic mechanisms, such as switching between active and inactive states,54,55 cooperative effects,56 allosteric transition,57–59 intercalation of drugs into DNA,60 and assembly of microtubules,61 can be revealed by studying their internal motions.62 Likewise, to really understand the interaction mechanism of a receptor with its ligand, we should consider not only the static structures concerned, but also the dynamic information obtained by simulating their internal motions or dynamic processes.63,64
To examine whether the designed agonist remains bound by an explicit solvent from a dynamic point of view, the molecular dynamic simulation was performed with the periodic boundary conditions using the GROMACS 4.0 package under GROMACS 96-53a6 force fields for proteins.65 The topology files and charges for the ligand atoms were generated by the Dundee PRODRG2.5 Server (beta).66 Before starting the simulations, the systems were solvated in explicit water with a flexible simple point charge model in a cubic box, under the condition that the surface of the protein was covered with a water shell of 1.0 nm (nanometer or 10−9 of a meter). The minimum number of sodium and chloride ions needed to balance the system charge was placed randomly in the solvated system. Subsequently, the energy minimization was performed for the complex system by using the steepest descent method.67,68 Then, the 10 ns (or 10−9 of a second) MD simulations were carried out with a time step of 2 fs (femtosecond, or 10−15 of a second). The corresponding coordinates are stored every 2ps (picosecond, or 10−12 of a second). The Particle Mesh Ewald algorithm was used to calculate the electrostatic interactions.69 All simulations were run under the periodic boundary condition with the Normal Pressure and Temperature (NPT) ensemble by using Berenson’s coupling algorithm for keeping the temperature at 310 K and the pressure at 1 atm. All bonds are constrained by using the Linear Constraint Solver algorithm.70
ADME prediction
ADME is a very useful program to eliminate unfavorable compounds with undesired properties in the early stage of drug development via evaluating the proposed structural refinements prior to expensive synthesis.27,42,71 Incorporating ADME predictions as a part of the drug development process can generate lead compounds that are more likely to exhibit satisfactory ADME performances during clinical trials.
The program of QikProp embedded in Schrödinger 2009 (Schrödinger, LLC) is a quick, accurate, and easy-to-use program for predicting the ADME properties of the compounds.41,72,73 The QikProp program adopted here was featured by having the functions predict a total of 44 properties of compounds, including the principal descriptors and physiochemical properties.
All the investigated compounds need not be neutralized before using QikProp because it will be automatically done in QikProp. The normal mode of prediction of QikProp was used in this study. The property analysis for the molecular weight – partition coefficient (QP logP o/w) – predicted aqueous solubility (QP logS); in addition, apparent PMDCK permeability (QPP MDCK) was computed in the QikProp program to evaluate whether the compounds hold the potential to become drug candidates.74
Results and discussion
Virtual screening and core hopping
The detailed procedure of discovering the desired dual agonists is shown in Figure 2. The lead-now database from ZINC was screened by utilizing the Glide5 software for its optimal performance targeting the PPAR receptors (PPARα:1k7l and PPARγ:1k74).35,36,49 The top hit, ZINC36728034 (see Figure 2) was selected as the optimal lead compound for further modification according to the well known structures of ragaglitazar and GW409544, as shown in Figure 2.43,75 Consequently, the phenyl group rendered by red color in ZINC36728034 (see Figure 2) is modified into an acidic group colored with blue, as shown in Comp#0 (Figure 2). The functional acidic group also exits in the following structures muraglitazar,76 faraglitazar,77 GW6471,78 GW7845,77 CHEMBL166572,79 and so on. The new molecule Comp#0 was designed with the intention so it has stronger affinity than ZINC36728034. Subsequently, the core hopping method was used to search the fragment database for the desired chemical groups to replace the amide group (ie, the R0 group) (Figure 2). Doing this was to help lengthen the Comp#0 structure so as to make it better fit the roomy space in arm 2 (see Figure 1). As a result, the amide group (ie, R0 group) of Comp#0 in Figure 2 was replaced by each of the best ten R groups (ie, the R1 to R10, as marked on the right side of Figure 2). Subsequently, they were used to generate the whole molecules (ie, Comp#1 to Comp#10) through the core hopping approach (see Figure 2). The ten compounds displayed the same chiral center (S) of the α carbon atom of the acidic group with the ragaglitazar and the GW409544 (see Figure 2), and the other compounds of Comp#5 to Comp#10 with the other preferred chiral centers in R groups allowed them to match the active pocket.
Figure 2.
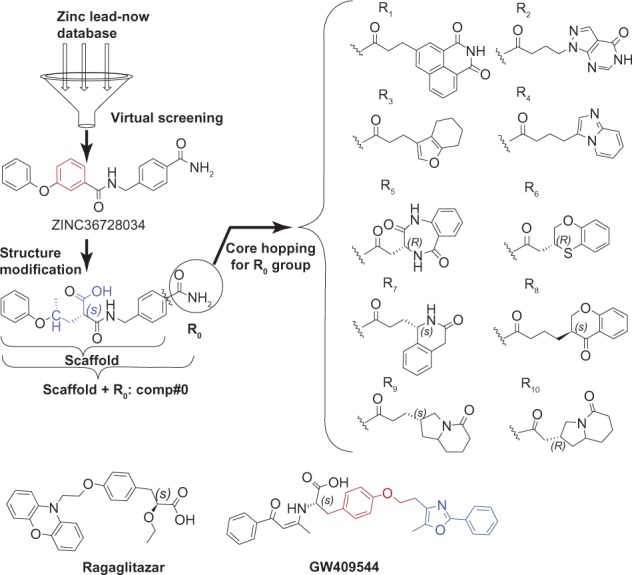
Illustration to show how to generate the best ten compounds from the ZINC36728034 structure through the core hopping method.
Notes: The top hit compound, ZINC36728034, screened out from the lead-now database was selected as the most potential lead compound for further modification, according to the vital importance of the acidic head of the ligand. Based on ZINC36728034, the new molecule Comp#0 was designed, as shown on the left bottom. Subsequently, the core hopping method was used to search the fragment database for replacing the amide group (ie, the R0 group) by the best ten R groups (ie, R1 to R10), as shown on the right side of the figure.
The above strategy of this study is very similar that in to our previous paper;38 however, the starting point at which the study was carried out is very different. This current study used virtual screening as the first operation to find the novel scaffold against PPARs; we also used a previous study for this same purpose,80 and we then developed new compounds with the core hopping method, while the previous study mainly modified the well known ligand GW409544 though the use of two steps of the core hopping method.38 At first glance, the current study is quite similar to a previous study,38 for we used the same protein targets, fragment database, core hopping method, and molecular dynamics modeling, but the detailed approaches are completely different. Therefore, it will be necessary to combine both studies that use the core hopping method for novel drug development in order to provide more comprehensive information.
Modeling of PPAR agonist complex
The best ten potential lead compounds or candidates obtained via the above step were redocked into the two receptors, PPARα (1k71) and PPARγ (1k74), respectively. As shown in the panels A and B of Figure 1, the best docked poses of the ten compounds are superposed together in the active pockets of the two receptors, PPARα (1k71) and PPARγ (1k74), respectively. From the docking simulation results, it can be seen that all the compounds form good steric complementarity with the hydrophobic pocket of each receptor and produce strong van der Waals contacts and hydrophobic interactions. As shown in the black in panels C and D of Figure 1, ragaglitazar (a novel dual agonist for PPARα and PPARγ that also includes the important polar acidic head group in its structure) was chosen as a positive control for its high affinity with the PPARα and PPARγ receptors in the previous research.75,81–83
Listed in Table 1 are the top ten compounds (ie, Comp#1 to Comp#10) that are much stronger than ragaglitazar and Comp#0 in binding to the two PPARα (1k71) and PPARγ (1k74) receptors, and these are ranked roughly according to their docking scores to the receptors. The best docked conformations of Comp#1 (carbon atoms colored in gray) and ragaglitazar (carbon atoms colored in black) are aligned together in panels C and D of Figure 1. The results of receptor–ligand interactions obtained from the docking simulation had proved that the key residues for the binding interactions between Comp#1 and the receptor were fully consistent with the previous reports.43,71,84 The conservative H-bonding network formed by the polar acidic head group in both ragaglitazar and Comp#1 to the four key residues of PPARα (or PPARγ), such as Ser280 (or Ser289), Tyr314 (or His323), Tyr464 (or Tyr473), and His440 (or His449) were observed in our docking simulation. It is noteworthy that Comp#1 has three oxygen atoms to serve as the polar acidic head, which allows Comp#1 to have more opportunities to interact with the AF2-helix. In addition, one more H-bond formed between the oxygen atom of the R1 group and Ala333 residue only in the PPARα (1k7l) receptor was observed in this study. The H-bonding networks made a major contribution in stabilizing the active conformation of AF2-helix in arm I1, which is extraordinarily important for the regulation of gene expression.7 The hydrophobic part of Comp#1 derived from ZINC36728034 played an important role in complementing the hydrophobic arm 1 formed by the hydrophobic residues, as shown by the light green surface (see panels C and D of Figure 1). Compared with ragaglitazar, the Comp#1 compound has an extra hydrophobic part, making it more fitted to the hydrophobic arm 1, and hence resulting in a much better binding affinity than ragaglitazar, as clearly indicated by the docking scores shown in Table 1. Among the compounds listed in Table 1, Comp#1 has the strongest binding affinity with the two receptors PPARα and PPARγ, and hence it was singled out as the representative compound used for further investigation.
Table 1.
The compound ragaglitazar was used as a positive control, and the ten compounds (Comp#1–#10) were ranked roughly according to their docking scores to the receptors PPARα and PPARγ
| Compound | Docking scores (Kcal/mol)
|
ADME properties predicted
|
|||||
|---|---|---|---|---|---|---|---|
| PPARα (1k71) | PPARγ (1k74) | PSAa | logPo/wb | logSc | PPCacod | Human oral absorptione | |
| Ragaglitazar | −11.49 | −12.29 | 70.42 | 6.07 | −6.38 | 364.38 | 95.35 |
| Comp#0f | −10.33 | −11.79 | 126.98 | 3.27 | −4.65 | 258.17 | 71.34 |
| Comp#1 | −13.65 | −14.98 | 180.80 | 5.61 | −5.76 | 106.26 | 48.14 |
| Comp#2 | −13.64 | −14.58 | 185.78 | 4.16 | −6.05 | 114.87 | 37.69 |
| Comp#3 | −13.31 | −14.55 | 146.67 | 4.23 | −4.60 | 111.24 | 57.53 |
| Comp#4 | −13.22 | −14.41 | 121.81 | 6.42 | −6.59 | 176.39 | 73.58 |
| Comp#5 | −13.18 | −13.85 | 130.62 | 5.96 | −6.40 | 141.02 | 64.80 |
| Comp#6 | −13.01 | −13.68 | 183.70 | 4.55 | −6.47 | 106.99 | 55.75 |
| Comp#7 | −12.95 | −14.11 | 118.33 | 6.23 | −6.25 | 175.59 | 71.11 |
| Comp#8 | −13.85 | −13.07 | 152.69 | 5.01 | −6.15 | 105.27 | 43.31 |
| Comp#9 | −13.36 | −13.45 | 146.77 | 5.83 | −6.30 | 134.45 | 62.69 |
| Comp#10 | −13.04 | −14.89 | 145.04 | 4.58 | −5.00 | 112.41 | 60.36 |
Notes: Listed are also the corresponding physiochemical descriptors calculated with QP simulations.41,72,73
The van der Waals surface area of the polar nitrogen and oxygen atoms; the accepted region is (7.0 to 200.0);
the predicted octanol/water partition coefficient; the accepted region is (−2.0 to 6.5);
the predicted aqueous solubility, where S (mol dm−3) is the concentration of the solute in a saturated solution that is in equilibrium with the crystalline solid; the accepted region is (−6.5 to 0.5);
predicted apparent Caco-2 cell permeability in nm/second. Caco-2 cells are a model for the gut–blood barrier. QikProp predictions are for nonactive transport. The result of <25 is poor;
predicted percent of human oral absorption on a scale from 0% to 100%. The prediction is based on a quantitative multiple linear regression model. This property usually correlates well with human oral absorption. The result of <25% is poor;
Comp#0 was an initial structure for core hopping that was designed with the intention to make it have stronger affinity than ZINC36728034. See the bottom left of Figure 2 for its structure.
Abbreviations: PPARα, peroxisome proliferator-activated receptor-alpha; PPARγ, peroxisome proliferator-activated receptor-gamma; ADME, absorption, distribution, metabolism, and excretion; PSA, polar surface area; QP, QikProp.
Molecular dynamics trajectory analysis
To acquire the relevant information from a dynamic point of view, the 10 ns molecular dynamic simulations were carried out, respectively, for the crystal structures of PPARα (1k7l), PPARγ (1k74), as well as their complexes with ragaglitazar and Comp#1: PPARα–ragaglitazar, PPARγ–ragaglitazar, PPARα–Comp#1, and PPARγ–Comp#1. As we can see from Figure 3, all of the characters concerned reached the simulation equilibrium within the 5 ns.
Figure 3.
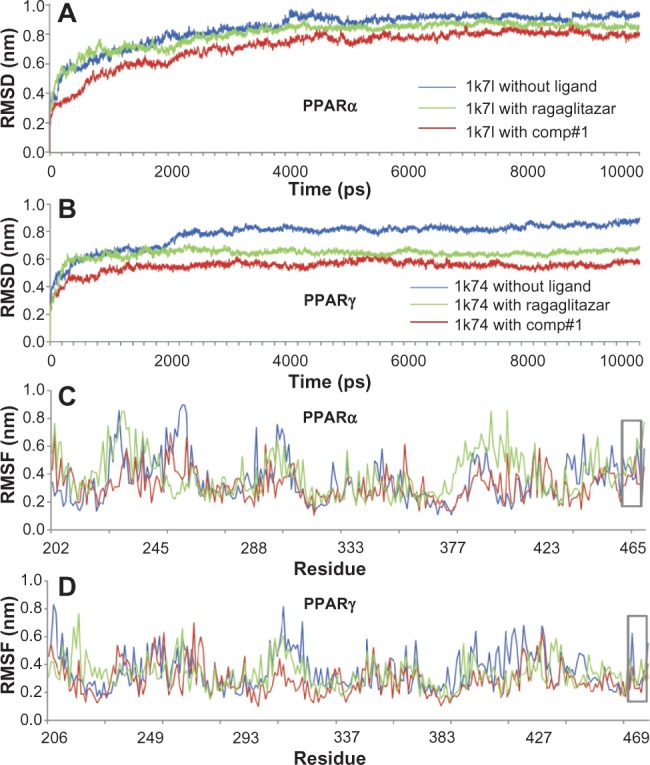
Illustration to show the outcomes of molecular dynamic simulations for the interactions of the receptors with Comp#1 – the best derivative found in this study, as shown in Table 1. (A) The RMSD of all backbone atoms for the receptor PPARα. (B) The RMSD of all backbone atoms for the receptor PPARγ. (C) The RMSF of the side-chain atoms for the receptor PPARα. (D) The RMSF of the side-chain atoms for the receptor PPARγ.
Notes: The blue line indicates the outcome for the system of the receptor alone without any ligand; the red line indicates the outcome for the system of the receptor with the ligand Comp#1; and the green line indicates the outcome for the system of the receptor with the ligand Comp#1. The curves involved with the AF2 helix region are framed with the grey box.
Abbreviations: RMSD, root mean square deviation; PPARα, peroxisome proliferator-activated receptor-alpha; PPARγ, peroxisome proliferator-activated receptor-gamma; RMSF, root mean square fluctuation.
The RMSD from the initial conformation is a major criterion used to evaluate the stability of a protein system. Shown in the panels A and B of Figure 3 are the backbone RMSD curves for PPARα, PPARγ, PPARα–ragaglitazar, PPARγ–ragaglitazar, PPARα–Comp#1, and PPARγ–Comp#1. It is interesting to see that the RMSD values of PPARα–Comp#1 and PPARγ–Comp#1 are remarkably smaller than those of PPARα–ragaglitazar and PPARγ–ragaglitazar, indicating that the complexes of PPARα–Comp#1 and PPARγ–Comp#1 are more stable than PPARα–ragaglitazar and PPARγ–ragaglitazar.
In order to perform an in-depth study of the interactions of the AF2 helix with the agonist (Figure 1), the root mean square fluctuations for all the side-chain atoms of the receptors were also computed, as shown in panels C and D of Figure 3. It can be seen from the figure that the side-chain of the root mean square fluctuations of PPARα–Comp#1 or PPARγ–Comp#1 were very similar to that of PPARα–ragaglitazar or PPARγ–ragaglitazar, respectively. This is particularly true for the residues 459–465 of PPARα–Comp#1 and the residues 469–477 of PPARγ–Comp#1 (see the gray framed box marked with AF2 in panels C and D of Figure 3), indicating that the new designed Comp#1 is very likely to perform exactly the same function in activating the AF2 helix as the compound ragaglitazar did.
ADME prediction
Also listed in Table 1 are the corresponding ADME properties that are directly relevant to pharmaceutical potentials. It can be seen from the table that, compared with ragaglitazar, the predicted ADME properties, such as the polar surface area, logPo/w, logS, PPCaco, and percent of human oral absorption for the newly designed agonists are either much better than or quite close to that of ragaglitazar. Actually, these percentages are all within the acceptable ranges for humans, indicating that the ten compounds found in this study, particularly the Comp#1 compound, hold very high potential to become a new drug candidate.
Binding pocket
In drug design, particularly for conducting mutagenesis studies,17 the information of the binding pocket of a targeted receptor with its ligand is crucially important. According to a previous study,85 such binding pockets were defined by those residues of the receptor that have at least one heavy atom (ie, an atom other than hydrogen) with a distance ≤ 5 Å from a heavy atom of the ligand. Similar criteria were also used to define the binding pockets of many other receptor–ligand interactions.37,51,86–91 The same criterion used in a previous study was also used in this study for defining the binding pockets of receptors PPARα and PPARγ with the ligand, GW409544.85 From this point of view, the residues involved in forming the binding pocket of PPARα and PPARγ for the docked ligand Comp#1 are listed in Table 2, which are identical to the other nine compounds, where those marked by an asterisk are the key residues for forming the H-bonding network, as shown in Figure 1.
Table 2.
The residues involved in forming the binding pocket of PPARα and PPARγ for the ligand Comp#1a
| PPARα (1k7l) | PPARγ (1k74) | ||||
|---|---|---|---|---|---|
| Leu-254 | Glu-269 | Ile-272 | Pro-269 | Ala-278 | Arg-280 |
| Phe-273 | Cys-275 | Cys-276 | Ile-281 | Phe-282 | Gly-284 |
| Gln-277 | Cys278 | Thr-279 | Cys-285 | Gln-286 | Phe-287 |
| *Ser-280 | *Tyr-314 | Ile-317 | Arg-288 | *Ser-289 | *His-323 |
| Phe-318 | Leu-321 | Val-324 | Ile-326 | Tyr-327 | Leu-330 |
| Met-330 | Leu-331 | Val-332 | Leu-333 | Val-339 | Leu-340 |
| *Ala-333 | Tyr-334 | Leu-344 | Ile-341 | Ser-342 | Met348 |
| Leu-347 | Phe-351 | Ile-354 | Leu-353 | Phe-360 | Phe-363 |
| Met-355 | Lys-358 | *His-440 | Met-364 | *His-449 | Leu-453 |
| Val-444 | Ile-447 | Leu-456 | Leu-465 | Leu-469 | *Tyr-473 |
| Leu-460 | *Tyr-464 | ||||
Notes:
See the text or the study by Chakrabarti et al83 for the definition of binding pockets used in this study. Those residues marked with an asterisk are the key residues for forming the H-bonding network, as shown in Figure 1.
Abbreviations: PPARα, peroxisome proliferator-activated receptor-alpha; PPARγ, peroxisome proliferator-activated receptor-gamma.
Acknowledgments
The authors thank the three anonymous reviewers and the editor for their valuable comments, which were very helpful for strengthening the presentation of this study.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Chehade JM, Mooradian AD. A rational approach to drug therapy of type 2 diabetes mellitus. Drugs. 2000;60(1):95–113. doi: 10.2165/00003495-200060010-00006. [DOI] [PubMed] [Google Scholar]
- 2.Carpino PA, Goodwin B. Diabetes area participation analysis: a review of companies and targets described in the 2008–2010 patent literature. Expert Opin Ther. 2010;20(12):1627–1651. doi: 10.1517/13543776.2010.533171. [DOI] [PubMed] [Google Scholar]
- 3.Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27(5):1047–1053. doi: 10.2337/diacare.27.5.1047. [DOI] [PubMed] [Google Scholar]
- 4.DeFronzo RA. Pathogenesis of type 2 diabetes mellitus. Med Clin North Am. 2004;88(4):787–835. ix. doi: 10.1016/j.mcna.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 5.Berger J, Moller DE. The mechanisms of action of PPARs. Annu Rev Med. 2002;53:409–435. doi: 10.1146/annurev.med.53.082901.104018. [DOI] [PubMed] [Google Scholar]
- 6.Berger JP, Akiyama TE, Meinke PT. PPARs: therapeutic targets for metabolic disease. Trends Pharmacol Sci. 2005;26(5):244–251. doi: 10.1016/j.tips.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 7.Markt P, Schuster D, Kirchmair J, Laggner C, Langer T. Pharmacophore modeling and parallel screening for PPAR ligands. J Comput Aided Mol Des. 2007;21(10–11):575–590. doi: 10.1007/s10822-007-9140-0. [DOI] [PubMed] [Google Scholar]
- 8.van Raalte DH, Li M, Pritchard PH, Wasan KM. Peroxisome proliferator-activated receptor (PPAR)-alpha: a pharmacological target with a promising future. Pharm Res. 2004;21(9):1531–1538. doi: 10.1023/b:pham.0000041444.06122.8d. [DOI] [PubMed] [Google Scholar]
- 9.Issemann I, Prince RA, Tugwood JD, Green S. The peroxisome proliferator-activated receptor:retinoid X receptor heterodimer is activated by fatty acids and fibrate hypolipidaemic drugs. J Mol Endocrinol. 1993;11(1):37–47. doi: 10.1677/jme.0.0110037. [DOI] [PubMed] [Google Scholar]
- 10.Evans RM, Barish GD, Wang YX. PPARs and the complex journey to obesity. Nat Med. 2004;10(4):355–361. doi: 10.1038/nm1025. [DOI] [PubMed] [Google Scholar]
- 11.Semple RK, Chatterjee VK, O’Rahilly S. PPAR gamma and human metabolic disease. J Clin Invest. 2006;116(3):581–589. doi: 10.1172/JCI28003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Wilson TM, Klieweer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma) J Biol Chem. 1995;270(22):12953–12956. doi: 10.1074/jbc.270.22.12953. [DOI] [PubMed] [Google Scholar]
- 13.Rosen ED, Spiegelman BM. PPARgamma: a nuclear regulator of metabolism, differentiation, and cell growth. J Biol Chem. 2001;276(41):37731–37734. doi: 10.1074/jbc.R100034200. [DOI] [PubMed] [Google Scholar]
- 14.Henke BR. Peroxisome proliferator-activated receptor alpha/gamma dual agonists for the treatment of type 2 diabetes. J Med Chem. 2004;47(17):4118–4127. doi: 10.1021/jm030631e. [DOI] [PubMed] [Google Scholar]
- 15.Mochizuki K, Suruga K, Yagi E, Takase S, Goda T. The expression of PPAR-associated genes is modulated through postnatal development of PPAR subtypes in the small intestine. Biochim Biophys Acta. 2001;1531(1–2):68–76. doi: 10.1016/s0167-4889(01)00071-4. [DOI] [PubMed] [Google Scholar]
- 16.Jones RM, editor. New Therapeutic Strategies for Type 2 Diabetes: Small Molecule Approaches. London, UK: RSC Publishing; 2012. [Google Scholar]
- 17.Chou KC. Structural bioinformatics and its impact to biomedical science. Curr Med Chem. 2004;11(16):2105–2134. doi: 10.2174/0929867043364667. [DOI] [PubMed] [Google Scholar]
- 18.Chou KC, Wei DQ, Zhong WZ. Binding mechanism of coronavirus main proteinase with ligands and its implication to drug design against SARS. Biochem Biophys Res Commun. 2003;308(1):148–151. doi: 10.1016/S0006-291X(03)01342-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sirois S, Wei DQ, Du QS, Chou KC. Virtual Screening for SARS-CoV protease based on KZ7088 pharmacophore points. J Chem Inf Comput Sci. 2004;44(3):1111–1122. doi: 10.1021/ci034270n. [DOI] [PubMed] [Google Scholar]
- 20.Dea-Ayuela MA, Pérez-Castillo Y, Meneses-Marcel A, et al. HP-Lattice QSAR for dynein proteins: experimental proteomics (2D-electrophoresis, mass spectrometry) and theoretic study of a Leishmania infantum sequence. Bioorg Med Chem. 2008;16(16):7770–7776. doi: 10.1016/j.bmc.2008.07.023. [DOI] [PubMed] [Google Scholar]
- 21.Prado-Prado FJ, González-Díaz H, de la Vega OM, Ubeira FM, Chou KC. Unified QSAR approach to antimicrobials. Part 3: First multi-tasking QSAR model for input-coded prediction, structural back-projection, and complex networks clustering of antiprotozoal compounds. Bioorg Med Chem. 2008;16(11):5871–5880. doi: 10.1016/j.bmc.2008.04.068. [DOI] [PubMed] [Google Scholar]
- 22.Prado-Prado FJ, Martinez de la Vega O, Uriarte E, Ubeira FM, Chou KC, González-Díaz H. Unified QSAR approach to antimicrobials. 4. Multitarget QSAR modeling and comparative multi-distance study of the giant components of antiviral drug-drug complex networks. Bioorg Med Chem. 2009;17(2):569–575. doi: 10.1016/j.bmc.2008.11.075. [DOI] [PubMed] [Google Scholar]
- 23.Du QS, Huang RB, Wei YT, Pang ZW, Du LQ, Chou KC. Fragment-based quantitative structure-activity relationship (FB-QSAR) for fragment-based drug design. J Comput Chem. 2009;30(2):295–304. doi: 10.1002/jcc.21056. [DOI] [PubMed] [Google Scholar]
- 24.Hou X, Du J, Fang H, Li M. 3D-QSAR study on a series of Bcl-2 protein inhibitors using comparative molecular field analysis. Protein Pept Lett. 2011;18(5):440–449. doi: 10.2174/092986611794927992. [DOI] [PubMed] [Google Scholar]
- 25.Lin WZ, Xiao X, Chou KC. GPCR-GIA: a web-server for identifying G-protein coupled receptors and their families with grey incidence analysis. Protein Eng Des Sel. 2009;22(11):699–705. doi: 10.1093/protein/gzp057. [DOI] [PubMed] [Google Scholar]
- 26.Xiao X, Wang P, Chou KC. GPCR-2L: predicting G protein-coupled receptors and their types by hybridizing two different modes of pseudo amino acid compositions. Mol Biosyst. 2011;7(3):911–919. doi: 10.1039/c0mb00170h. [DOI] [PubMed] [Google Scholar]
- 27.Chou KC, Wu ZC, Xiao X. iLoc-Euk: a multi-label classifier for predicting the subcellular localization of singleplex and multiplex eukaryotic proteins. PLoS One. 2011;6(3):e18258. doi: 10.1371/journal.pone.0018258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chou KC, Wu ZC, Xiao X. iLoc-Hum: using accumulation-label scale to predict subcellular locations of human proteins with both single and multiple sites. Mol Biosyst. 2012;8(2):629–641. doi: 10.1039/c1mb05420a. [DOI] [PubMed] [Google Scholar]
- 29.Wang P, Xiao X, Chou KC. NR-2L: a two-level predictor for identifying nuclear receptor subfamilies based on sequence-derived features. PLoS One. 2011;6(8):e23505. doi: 10.1371/journal.pone.0023505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chou KC, Shen HB. ProtIdent: a web server for identifying proteases and their types by fusing functional domain and sequential evolution information. Biochem Biophys Res Commun. 2008;376(2):321–325. doi: 10.1016/j.bbrc.2008.08.125. [DOI] [PubMed] [Google Scholar]
- 31.Shen HB, Chou KC. HIVcleave: a web-server for predicting human immunodeficiency virus protease cleavage sites in proteins. Anal Biochem. 2008;375(2):388–390. doi: 10.1016/j.ab.2008.01.012. [DOI] [PubMed] [Google Scholar]
- 32.Chou KC. Prediction of human immunodeficiency virus protease cleavage sites in proteins. Anal Biochem. 1996;233(1):1–14. doi: 10.1006/abio.1996.0001. [DOI] [PubMed] [Google Scholar]
- 33.Richards WG. Virtual screening using grid computing: the screensaver project. Nat Rev Drug Discov. 2002;1(7):551–555. doi: 10.1038/nrd841. [DOI] [PubMed] [Google Scholar]
- 34.Roche O, Schneider P, Zuegge J, et al. Development of a virtual screening method for identification of “frequent hitters” in compound libraries. J Med Chem. 2002;45(1):137–142. doi: 10.1021/jm010934d. [DOI] [PubMed] [Google Scholar]
- 35.Senger S. Using Tversky similarity searches for core hopping: finding the needles in the haystack. J Chem Inf Model. 2009;49(6):1514–1524. doi: 10.1021/ci900092y. [DOI] [PubMed] [Google Scholar]
- 36.Henke BR, Consler TG, Go N, et al. A new series of estrogen receptor modulators that display selectivity for estrogen receptor beta. J Med Chem. 2002;45(25):5492–5505. doi: 10.1021/jm020291h. [DOI] [PubMed] [Google Scholar]
- 37.Li XB, Wang SQ, Xu WR, Wang RL, Chou KC. Novel inhibitor design for hemagglutinin against H1N1 influenza virus by core hopping method. PLoS One. 2011;6(11):e28111. doi: 10.1371/journal.pone.0028111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ma Y, Wang SQ, Xu WR, Wang RL, Chou KC. Design novel dual agonists for treating type-2 diabetes by targeting peroxisome proliferator-activated receptors with core hopping approach. PLoS One. 2012;7(6):e38546. doi: 10.1371/journal.pone.0038546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eldridge MD, Murray CW, Auton TR, Paolini GV, Mee RP. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J Comput Aided Mol Des. 1997;11(5):425–445. doi: 10.1023/a:1007996124545. [DOI] [PubMed] [Google Scholar]
- 40.Halgren TA, Murphy RB, Friesner RA, et al. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem. 2004;47(7):1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 41.Jorgensen WL, Duffy EM. Prediction of drug solubility from structure. Adv Drug Deliv Rev. 2002;54(3):355–366. doi: 10.1016/s0169-409x(02)00008-x. [DOI] [PubMed] [Google Scholar]
- 42.Tian S, Li Y, Wang J, Zhang J, Hou T. ADME evaluation in drug discovery. 9. Prediction of oral bioavailability in humans based on molecular properties and structural fingerprints. Mol Pharm. 2011;8(3):841–851. doi: 10.1021/mp100444g. [DOI] [PubMed] [Google Scholar]
- 43.Xu HE, Lambert MH, Montana VG, et al. Structural determinants of ligand binding selectivity between the peroxisome proliferator-activated receptors. Proc Natl Acad Sci USA. 2001;98(24):13919–13924. doi: 10.1073/pnas.241410198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Berman HM, Battistuz T, Bhat TN, et al. The Protein Data Bank. Acta Crystallogr D Biol Crystallogr. 2002;58(Pt 61):899–907. doi: 10.1107/s0907444902003451. [DOI] [PubMed] [Google Scholar]
- 45.Husslein T, Moore PB, Zhong Q, Newns DM, Pattnaik PC, Klein ML. Molecular dynamics simulation of a hydrated diphytanol phosphatidylcholine lipid bilayer containing an alpha-helical bundle of four transmembrane domains of the influenza A virus M2 protein. Faraday Discuss. 1998;111:201–208. 225–246. doi: 10.1039/a806675b. discussion. [DOI] [PubMed] [Google Scholar]
- 46.Kochendoerfer GG, Salom D, Lear JD, Wilk-Orescan R, Kent SB, DeGrado WF. Total chemical synthesis of the integral membrane protein influenza A virus M2: role of its C-terminal domain in tetramer assembly. Biochemistry. 1999;38(37):11905–11913. doi: 10.1021/bi990720m. [DOI] [PubMed] [Google Scholar]
- 47.Gandhi CS, Shuck K, Lear JD, et al. Cu(II) inhibition of the proton translocation machinery of the influenza A virus M2 protein. J Biol Chem. 1999;274(9):5474–5482. doi: 10.1074/jbc.274.9.5474. [DOI] [PubMed] [Google Scholar]
- 48.Feige JN, Gelman L, Tudor C, Engelborghs Y, Wahli W, Desvergne B. Fluorescence imaging reveals the nuclear behavior of peroxisome proliferator-activated receptor/retinoid X receptor heterodimers in the absence and presence of ligand. J Biol Chem. 2005;280(18):17880–17890. doi: 10.1074/jbc.M500786200. [DOI] [PubMed] [Google Scholar]
- 49.Irwin JJ, Schoichet BK. ZINC – a free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45(1):177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Banks JL, Beard HS, Cao Y, et al. Integrated Modeling Program, Applied Chemical Theory (IMPACT) J Comput Chem. 2005;26(16):1752–1780. doi: 10.1002/jcc.20292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liao QH, Gao QZ, Wei J, Chou KC. Docking and molecular dynamics study on the inhibitory activity of novel inhibitors on epidermal growth factor receptor (EGFR) Med Chem. 2011;7(1):24–31. doi: 10.2174/157340611794072698. [DOI] [PubMed] [Google Scholar]
- 52.Cai L, Wang Y, Wang JF, Chou KC. Identification of proteins interacting with human SP110 during the process of viral infections. Med Chem. 2011;7(2):121–126. doi: 10.2174/157340611794859343. [DOI] [PubMed] [Google Scholar]
- 53.Liu XY, Wang RL, Xu WR, Tang LD, Wang SQ, Chou KC. Docking and molecular dynamics simulations of peroxisome proliferator activated receptors interacting with pan agonist sodelglitazar. Protein Pept Lett. 2011;18(10):1021–1027. doi: 10.2174/092986611796378701. [DOI] [PubMed] [Google Scholar]
- 54.Chou KC. The biological functions of low-frequency vibrations (phonons). 4. Resonance effects and allosteric transition. Biophys Chem. 1984;20(1–2):61–71. doi: 10.1016/0301-4622(84)80005-8. [DOI] [PubMed] [Google Scholar]
- 55.Wang JF, Gong K, Wei DQ, Li YX, Chou KC. Molecular dynamics studies on the interactions of PTP1B with inhibitors: from the first phosphate-binding site to the second one. Protein Eng Des Sel. 2009;22(6):349–355. doi: 10.1093/protein/gzp012. [DOI] [PubMed] [Google Scholar]
- 56.Chou KC. Low-frequency resonance and cooperativity of hemoglobin. Trends Biochem Sci. 1989;14(6):212–213. doi: 10.1016/0968-0004(89)90026-1. [DOI] [PubMed] [Google Scholar]
- 57.Chou KC. The biological functions of low-frequency vibrations (phonons). III. Helical structures and microenvironment. Biophys J. 1984;45(5):881–889. doi: 10.1016/S0006-3495(84)84234-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lian P, Wei DQ, Wang JF, Chou KC. An allosteric mechanism inferred from molecular dynamics simulations on phospholamban pentamer in lipid membranes. PLoS One. 2011;6(4):e18587. doi: 10.1371/journal.pone.0018587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chou KC. The biological functions of low-frequency vibrations (phonons): VI. A possible dynamic mechanism of allosteric transition in antibody molecules. Biopolymers. 1987;26(2):285–295. doi: 10.1002/bip.360260209. [DOI] [PubMed] [Google Scholar]
- 60.Chou KC, Mao B. Collective motion in DNA and its role in drug intercalation. Biopolymers. 1988;27(11):1795–1815. doi: 10.1002/bip.360271109. [DOI] [PubMed] [Google Scholar]
- 61.Chou KC, Zhang CT, Maggiora GM. Solitary wave dynamics as a mechanism for explaining the internal motion during microtubule growth. Biopolymers. 1994;34(1):143–153. doi: 10.1002/bip.360340114. [DOI] [PubMed] [Google Scholar]
- 62.Chou KC. Low-frequency collective motion in biomacromolecules and its biological functions. Biophys Chem. 1988;30(1):3–48. doi: 10.1016/0301-4622(88)85002-6. [DOI] [PubMed] [Google Scholar]
- 63.Schnell JR, Chou JJ. Structure and mechanism of the M2 proton channel of influenza A virus. Nature. 2008;451(7178):591–595. doi: 10.1038/nature06531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Huang RB, Du QS, Wang CH, Chou KC. An in-depth analysis of the biological functional studies based on the NMR M2 channel structure of influenza A virus. Biochem Biophys Res Commun. 2008;377(4):1243–1247. doi: 10.1016/j.bbrc.2008.10.148. [DOI] [PubMed] [Google Scholar]
- 65.Oostenbrink C, Soares TA, van der Vegt NF, van Gunsteren WF. Validation of the 53A6 GROMOS force field. Eur Biophys J. 2005;34(4):273–284. doi: 10.1007/s00249-004-0448-6. [DOI] [PubMed] [Google Scholar]
- 66.Schüttelkopf AW, van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 8):1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 67.Bhaya A, Kaszkurewicz E. Steepest descent with momentum for quadratic functions is a version of the conjugate gradient method. Neural Netw. 2004;17(1):65–71. doi: 10.1016/S0893-6080(03)00170-9. [DOI] [PubMed] [Google Scholar]
- 68.Deift P, Venakides S, Zhou X. An extension of the steepest descent method for Riemann-Hilbert problems: the small dispersion limit of the Korteweg-de Vries (KdV) equation. Proc Natl Acad Sci USA. 1998;95(2):450–454. doi: 10.1073/pnas.95.2.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cerutti DS, Duke RE, Darden TA, Lybrand TP. Staggered Mesh Ewald: An extension of the Smooth Particle-Mesh Ewald method adding great versatility. J Chem Theory Comput. 2009;5(9):2322. doi: 10.1021/ct9001015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hess B, Bekker H, Berendsen HJC, Fraaije JGEM. LINCS: a linear constraint solver for molecular simulations. J Comput Chem. 1998;18(12):1463–1472. [Google Scholar]
- 71.Liu XY, Wang RL, Xu WR, Tang LD, Wang SQ, Chou KC. Docking and molecular dynamics simulations of peroxisome proliferator activated receptors interacting with pan agonist sodelglitazar. Protein Pept Lett. 2011;18(10):1021–1027. doi: 10.2174/092986611796378701. [DOI] [PubMed] [Google Scholar]
- 72.Liu Y, Hong L, Yu LS, et al. The role of ADME evaluation in translation research of innovative drug. Yao Xue Xue Bao. 2011;46(1):19–29. Chinese. [PubMed] [Google Scholar]
- 73.Jorgensen WL, Duffy EM. Prediction of drug solubility from Monte Carlo simulations. Bioorg Med Chem Lett. 2000;10(11):1155–1158. doi: 10.1016/s0960-894x(00)00172-4. [DOI] [PubMed] [Google Scholar]
- 74.Singh KHD, Kirubakaran P, Nagarajan S, et al. Homology modeling, molecular dynamics, e-pharmacophore mapping and docking study of Chikungunya virus nsP2 protease. J Mol Model. 2012;18(1):39–51. doi: 10.1007/s00894-011-1018-3. [DOI] [PubMed] [Google Scholar]
- 75.Pickavance LC, Brand CL, Wassermann K, Wilding JP. The dual PPARalpha/gamma agonist, ragaglitazar, improves insulin sensitivity and metabolic profile equally with pioglitazone in diabetic and dietary obese ZDF rats. Br J Pharmacol. 2005;144(3):308–316. doi: 10.1038/sj.bjp.0706041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Waites CR, Dominick MA, Sanderson TP, Schilling BE. Nonclinical safety evaluation of muraglitazar, a novel PPARalpha/gamma agonist. Toxicol Sci. 2007;100(1):248–258. doi: 10.1093/toxsci/kfm193. [DOI] [PubMed] [Google Scholar]
- 77.Heppner TJ, Bonev AD, Eckman DM, Gomez MF, Petkov GV, Nelson MT. Novel PPARgamma agonists GI 262570, GW 7845, GW 1929, and pioglitazone decrease calcium channel function and myogenic tone in rat mesenteric arteries. Pharmacology. 2005;73(1):15–22. doi: 10.1159/000081070. [DOI] [PubMed] [Google Scholar]
- 78.Ding L, Liang XG, Lou YJ. Time-dependence of cardiomyocyte differentiation disturbed by peroxisome proliferator-activated receptor alpha inhibitor GW6471 in murine embryonic stem cells in vitro. Acta Pharmacol Sin. 2007;28(5):634–642. doi: 10.1111/j.1745-7254.2007.00558.x. [DOI] [PubMed] [Google Scholar]
- 79.Sauerberg P, Pettersson I, Jeppesen L, et al. Novel tricyclic-alpha-alkyloxyphenylpropionic acids: dual PPARalpha/gamma agonists with hypolipidemic and antidiabetic activity. J Med Chem. 2002;45(4):789–804. doi: 10.1021/jm010964g. [DOI] [PubMed] [Google Scholar]
- 80.Scarsi M, Podvinec M, Roth A, et al. Sulfonylureas and glinides exhibit peroxisome proliferator-activated receptor gamma activity: a combined virtual screening and biological assay approach. Mol Pharmacol. 2007;71(2):398–406. doi: 10.1124/mol.106.024596. [DOI] [PubMed] [Google Scholar]
- 81.Pavankuamr VV, Vinu CA, Mullangi R, Srinivas NR. Preclinical pharmacokinetics and interspecies scaling of ragaglitazar, a novel biliary excreted PPAR dual activator. Eur J Drug Metab Pharmacokinet. 2007;32(1):29–37. doi: 10.1007/BF03190987. [DOI] [PubMed] [Google Scholar]
- 82.Skrumsager BK, Nielsen KK, Müller M, Pabst G, Drake PG, Edsberg B. Ragaglitazar: the pharmacokinetics, pharmacodynamics, and tolerability of a novel dual PPAR alpha and gamma agonist in healthy subjects and patients with type 2 diabetes. J Clin Pharmacol. 2003;43(11):1244–1256. doi: 10.1177/0091270003257230. [DOI] [PubMed] [Google Scholar]
- 83.Chakrabarti R, Vikramadithyan RK, Misra P, et al. Ragaglitazar: a novel PPAR alpha PPAR gamma agonist with potent lipid-lowering and insulin-sensitizing efficacy in animal models. Br J Pharmacol. 2003;140(3):527–537. doi: 10.1038/sj.bjp.0705463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ebdrup S, Pettersson I, Rasmussen HB, et al. Synthesis and biological and structural characterization of the dual-acting peroxisome proliferator-activated receptor alpha/gamma agonist ragaglitazar. J Med Chem. 2003;46(8):1306–1317. doi: 10.1021/jm021027r. [DOI] [PubMed] [Google Scholar]
- 85.Chou KC, Watenpaugh KD, Heinrikson RL. A model of the complex between cyclin-dependent kinase 5 and the activation domain of neuronal Cdk5 activator. Biochem Biophys Res Commun. 1999;259(2):420–428. doi: 10.1006/bbrc.1999.0792. [DOI] [PubMed] [Google Scholar]
- 86.Chou KC. Molecular therapeutic target for type-2 diabetes. J Proteome Res. 2004;3(6):1284–1288. doi: 10.1021/pr049849v. [DOI] [PubMed] [Google Scholar]
- 87.Pielak RM, Schnell JR, Chou JJ. Mechanism of drug inhibition and drug resistance of influenza A M2 channel. Proc Natl Acad Sci USA. 2009;106(18):7379–7384. doi: 10.1073/pnas.0902548106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang JF, Chou KC. Insights from modeling the 3D structure of New Delhi metallo-β-lactamase and its binding interactions with antibiotic drugs. PLoS One. 2011;6(4):e18414. doi: 10.1371/journal.pone.0018414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Housaindokht MR, Bozorgmehr MR, Bahrololoom M. Analysis of ligand binding to proteins using molecular dynamics simulations. J Theor Biol. 2008;254(2):294–300. doi: 10.1016/j.jtbi.2008.04.036. [DOI] [PubMed] [Google Scholar]
- 90.Wang JF, Chou KC. Insights into the mutation-induced HHH syndrome from modeling human mitochondrial ornithine transporter-1. PLoS One. 2012;7(1):e31048. doi: 10.1371/journal.pone.0031048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chou KC. Insights from modeling the 3D structure of the extracellular domain of alpha7 nicotinic acetylcholine receptor. Biochem Biophys Res Commun. 2004;319(2):433–438. doi: 10.1016/j.bbrc.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 92.Chou KC, Watenpaugh KD, Heinrikson RL. A model of the complex between cyclin-dependent kinase 5 and the activation domain of neuronal Cdk5 activator. Biochem Biophys Res Commun. 1999;259(2):420–428. doi: 10.1006/bbrc.1999.0792. [DOI] [PubMed] [Google Scholar]