

Abstract
Although alterations in mitochondrial dynamics are associated with cellular responses to injury, the functional role of these dynamic changes in ischemic neurons is underexplored. One of these dynamic responses to injury includes mitochondrial biogenesis. Various sublethal preconditioning stimuli that induce an ischemic tolerant state (e.g., lipopolysaccharide (LPS)) may also induce mitochondrial biogenesis. Using neuron-enriched cultures, we found that sublethal LPS preconditioning induced both ischemic tolerance and markers of mitochondrial biogenesis with overlapping dose response temporal kinetics. Sublethal LPS transiently increased the expression of critical components of the mitochondrial transcriptional machinery, including NRF1 and TFAM, as well as mtDNA copy number, mitochondrial protein levels and markers of functional mitochondria, such as increased cellular ATP content, citrate synthase activity and maximal respiration capacity. Importantly, knockdown of TFAM abrogated both the induction of mitochondrial biogenesis and the neuroprotective preconditioning effects of LPS. Several signaling pathways coordinated these events. AMPK inhibition suppressed NRF1 and TFAM expression by LPS, whereas PI3K/Akt signaling was necessary for the nuclear translocation of NRF1 and subsequent induction of TFAM. This is the first demonstration that LPS preconditioning initiates multiple signaling pathways leading to mitochondrial biogenesis in neurons and that these dynamic changes contribute to ischemic tolerance.
Keywords: Mitochondrial biogenesis, ischemia, neurons, preconditioning, ischemic tolerance
Introduction
Mitochondria are highly dynamic organelles capable of sensing and responding quickly to altered cellular environments. Not surprisingly, mitochondria dysfunctions are implicated in a wide variety of neurological disease states (Lin & Beal 2006). Until recently, their roles in cell death and survival were described primarily in terms of the various signaling pathways that converge on or emanate from mitochondria (Goldenthal & Marin-Garcia 2004). Extending this concept of mitochondria as signal processing centers, recent studies indicate that mitochondrial plasticity itself also contributes to cellular fate (Nunnari & Suomalainen 2012).
The phenomenon of ischemic tolerance was discovered over 20 years ago. Several cellular mechanisms underlying tolerance have been identified, including heat shock protein induction, ROS-sensitive mechanisms, and cellular signaling (Cadet & Krasnova 2009, Correia et al. 2011, Stevens et al. 2011). Although many of these mechanisms affect mitochondria (Busija et al. 2008, Correia et al. 2010), the response of the mitochondria as an organelle has not been fully examined. Interestingly, many of the preconditioning stimuli that induce tolerance may also induce mitochondrial biogenesis, or the induction of critical mitochondrial components (Gutsaeva et al. 2008, McLeod et al. 2004, Ning et al. 1998). For example, markers of mitochondrial biogenesis were induced following neonatal hypoxia/ischemia in regions that survived the injury (Yin et al. 2008). Resveratrol has been used as a preconditioning stimulus to confer ischemic tolerance (Raval et al. 2006) and has also been correlated with markers of mitochondrial biogenesis in several cell types (Biala et al. 2010, Zheng et al. 2012, Csiszar et al. 2009).
Non-lethal doses of lipopolysaccharide (LPS) induce markers of mitochondrial biogenesis in isolated neonatal cardiomyocytes in the absence of cell death (Hickson-Bick et al. 2008) and has also been used for a number of non-neuronal mechanistic studies for the induction of mitochondrial biogenesis (Piantadosi et al. 2011, Suliman et al. 2003, Suliman et al. 2010). In addition to effects on non-neural cells, sublethal LPS has been used as a preconditioning stimulus for cerebral ischemic tolerance (Tasaki et al. 1997, Lin et al. 2009, Yu et al. 2010, Vartanian et al. 2011). Most of these studies administer LPS into animals systemically and focus on the effects of LPS on microglia via actions on toll-like receptors (TLRs). Although microglia are well characterized as the predominant neural cell type expressing TLRs, neurons have recently been found to also express TLRs (Tang et al. 2007) and thus may also respond directly to LPS.
Using the model of sublethal LPS preconditioning, the present study tested the hypothesis that neuronal mitochondrial biogenesis contributes to and is necessary for establishing the ischemic tolerant state. We found that sublethal LPS exposure induced markers of mitochondrial biogenesis via the convergence of multiple signaling cascades. Markers of mitochondrial biogenesis were induced with the same temporal kinetics and dose-responsiveness as the induction of ischemic tolerance. Furthermore, shRNA-mediated knockdown of TFAM, a critical regulator of mitochondrial biogenesis, abolished both the induction of mitochondrial biogenesis as well as ischemic neuroprotection afforded by LPS. Thus, we conclude that mitochondrial biogenesis is a novel mechanism contributing to the neuroprotection afforded by sublethal LPS preconditioning.
Methods
Primary neuronal cultures and in vitro model of ischemia
Enriched cortical neurons (>97% purity) were derived from embryos of SD rats and maintained 10–12 d in vitro before experiments. To model ischemia-like conditions in vitro, primary cultures were exposed to transient oxygen and glucose deprivation (OGD) for 60 min as described previously (Stetler et al. 2010). Control glucose-containing cultures were incubated for the same periods of time in humidified 95% air and 5% CO2. LPS was added to cultures 24 h prior to OGD to induce a preconditioned state. Fluorescence of alamar blue, an indicator that changes from blue to red and fluoresces when reduced by cellular metabolic activity, was used to measure the viability of the cultured neurons at 24–48 h after OGD as described (Stetler et al. 2010). OGD-induced cell death was quantified by measuring lactate dehydrogenase (LDH) release from damaged cells into the culture medium as described previously (Stetler et al. 2010). In select experiments, cell death was also evaluated after OGD using Hoechst 33258 nuclear staining. The percentages of cells showing chromatin condensation or DNA damage were quantified by counting at least 3000 cells under each experimental condition (three randomly selected fields per well, four to six wells per condition per experiment, and three independent experiments). In inhibition studies, one of the kinase inhibitors SB203580 (10μM), polymyxin B (20U/ml), N-acetyl cysteine (NAC, 1mM), Compound C (20μM), LY294002 (10μM), U0126 (10μM), KN62 (10μM) and L-NAME (1mM) was applied to cultures 1 h prior to and during the course of LPS treatment.
Viral vectors
Lentiviral vectors carrying the TFAM-targeting shRNA or the scramble control shRNA were purchased (Santa Cruz Biotechnology Inc). Lentiviral vectors overexpressing human full-length TFAM were constructed using the procedures as described previously (Stetler et al. 2012). In brief, the cDNA (HA tagged) was inserted into the lentiviral transfer vector FSW under the control of neuron-specific Synapsin I promoter. A plasmid mixture containing 435 μg of pCMVΔR8.9 (packaging construct), 237 μg of pVSVG (envelope plasmid) and 675 μg of FSW (transfer vector) was suspended in 34.2 ml of CaCl2 (250 mM) and co-transfected into human kidney 293 FT cells. The supernatant was collected 72 h after transfection, filtered through the 0.45 μm filter flask and centrifuged at 21,000 rpm for 2 h using the SW28 rotor (Beckman Coulter). Viruses were further purified by sucrose gradient ultracentrifuge.
Adeno-associated virus (AAV) vectors carrying human cDNA for AKT or dominant negative AKT (AKTdn) were constructed using the procedures as previously described (Stetler et al. 2012). For neuronal transfection, cultures were infected with the AAV-AKT or AAV-AKTdn, or the control vector (AAV-GFP or empty AAV) for 6 h and then incubated in vector-free normal media for 48-72 h. The overexpression of AKT or AKTdn in neurons was confirmed by immunocytochemistry and Western blot, respectively, using the anti-hemagglutinin (HA) antibody. For gene transfection using lentivirus vectors, the neuronal cultures were infected for 3 d with the lenti-TFAMt, lenti-TFAMsc, and/or lenti-TFAM or the control vector (Lenti-GFP). Knockdown of TFAM in neurons by lentivirus vectors was assessed using Western blot.
Long fragment PCR and mtDNA quantification
Long fragment PCR was used to quantify the relative abundance of intact mtDNA as previously described (Yin et al. 2008). Total DNA was purified using the genomic DNA isolation kit (Qiagen, Valencia, CA), and linearized by digestion with the restriction enzyme SacII (Promega, Madison, WI). An equal amount (0.4 ng) of total DNA was added to the polymerase chain reaction (PCR) reaction mixture to serve as an internal standard. The primers used for the amplification of 14.3 kbp mitochondrial genomes were:
59-ATATTTTCACTGCTGAGTCCCGTGG-39 (forward)
59-AATTTCGGTTGGGGTGACCTCGGAG-39 (reverse)
The PCR cycle proceeded by denaturation for 1 minute at 94°C followed by 26 cycles of denaturation at 94°C for 10 seconds, annealing and extension at 68°C for 15 minutes, and a final extension at 72°C for 10 minutes. The PCR products were then digested with the restriction enzyme NcoI (Promega). The reaction yielded DNA fragments of 14.3 kbp, representing the amplified rat mtDNA. The bands were semi-quantitatively measured using MCID Elite Image Analysis system (Image Research, Linton, England). Rat b-globin DNA was amplified as an internal control.
Real time RT-PCR
Total RNA was isolated from cell lysates using the RNeasy Mini kit according to the manufacturer’s instructions (Qiagen). The first strand of cDNA used random hexamer primers and the Superscript™ First-strand synthesis system for RT-PCR (Invitrogen, Carlsbad, CA). PCR was performed using SYBR green PCR Master Mix (Applied Biosystems, Foster City, CA). Fluorescence was quantified using SDS v1.2x system software (Applied Biosystems). The forward and reverse primers used were:
TFAM: GAAAGCACAAATCAAGAGGAG, CTGCTTTTCATCATGAGACAG.
NRF-1: TTACTCTGCTGTGGCTGATGG, CCTCTGATGCTTGCGTCGTCT.
PGC-1a: GTGCAGCCAAGACTCTGTATGG, GTCCAGGTCATTCACATCAAGTTC.
β-actin: GGGTCAGAAGGATTCCTATG, GGTCTCAAACATGATCTGGG.
Western blot analysis
Western blot was performed using the standard method and enhanced chemiluminescence detection reagents (GE Healthcare). The following antibodies were used: mouse monoclonal antibodies against HSP60 (H-1), NRF-1 and TFAM (from Santa Cruz Biotechnology Inc); COXIV, VDAC and TOM20 (from Molecular Probes, Invitrogen); rabbit monoclonal antibodies p-Akt (Ser473) (clone 193H12), total AKT and p-AMPKα(Thr172) (clone 40H9) from Cell Signaling.
Electron microscopy
Primary cortical neurons grown in 35-mm tissue culture dishes were fixed for 60 minutes in 2.5% gluteraldehyde at 4°C and postfixed in aqueous 1% OsO4 and 1% K3Fe(CN)6 for 1 hour. The cultures were dehydrated through a graded series of 30 to 100% ethanol and embedded in Polybed 812 epoxy resin (Polysciences, Warrington, PA). Ultra thin (60 nm) sections were collected on copper grids and stained with 2% uranyl acetate in 50% methanol for 10 minutes, followed by 1% lead citrate for 7 minutes. Sections were photographed using a Zeiss transmission electron microscope (Zeiss Inc., Thornwood, NY, USA) at 80 kV.
In vitro kinase activity
Akt kinase activity was assessed using an in vitro kinase activity kit as previously described (Stetler et al. 2012). Cell lysates were prepared under non-denaturing conditions. To assess Akt activity, an anti-Akt antibody was added to 150 μg of protein and then incubated with recombinant GSK3β in the presence of [γ-32P]ATP. GSK3β phosphorylation was visualized using electrophoresis and autoradiogram.
Oxygen consumption rate (OCR) measurements
Cellular respiration was analyzed using the Seahorse XF24 Extracellular Flux Analyzer as described (Dagda et al. 2011). OCR was assessed in living cells that were analyzed at baseline and following the sequential addition of culture medium, oligomycin and FCCP, according to the manufacturer’s protocol.
ATP measurements
ATP cellular content was assessed as previously described (Van Laar et al. 2011). Cultures were washed and harvested by scraping into ice-cold PBS, pelleted by centrifugation at 2000g for 5 min, resuspended in urea/CHAPS lysis buffer, cleared by centrifugation and stored at −80°C until analysis. Intracellular ATP levels were determined using an ATP Determination Kit (Molecular Probes) based on the reaction of luciferase with luciferin and measured using a luminometer, according to the procedure provided by the manufacturer. Data were normalized to the sample protein concentration as determined by the Bradford protein assay.
Citrate synthase activity
Citrate synthase activity was measured using the Citrate Synthase Assay Kit (Sigma). Arbitrary activity units were calculated per manufacturer’s instruction.
Mitochondrial length and movement
Cortical cultures were transfected using Lipofectamine2000 with dsRed2, which labels mitochondria, and eGFP for visualization of axonal and dendritic morphology. Mitochondrial length and movement was assessed using SimplePCI as described (Chang et al. 2006, Rintoul et al. 2003). Briefly, length was calculated from skeletonized projections. For movement, rectangular regions of interest (ROIs) were placed along the axonal or dendritic regions and the number of mitochondria moving past or remaining stopped in each ROI was counted over the entire course of each 2 min movie.
Statistical analyses
Each measurement was made in quadruplicate wells of a single culture dish, and each experiment was repeated at least three times using cultures prepared from different dams. All data were presented as mean ± standard error (SE). The difference in means between 2 groups was assessed by 2-tailed Student’s t test. Differences in means among multiple groups were analyzed using ANOVA with time or treatment as the independent factors. When ANOVA showed significant differences, pair-wise comparisons between means were tested by post hoc Bonferroni/Dunn tests. In all analyses, p < 0.05 was considered statistically significant.
Results
LPS preconditioning protects cultured neurons from ischemic insults
Neurons have recently been found to express TLRs (Tang et al. 2007). Thus, in order to examine whether LPS preconditioning induces alterations of mitochondrial dynamics in neurons, we used primary cortical neuron-enriched cultures pretreated with LPS prior to the induction of OGD. One hour of OGD induced robust cell death as measured by alamar blue uptake (a measure of cell viability), LDH release (a measure of loss of plasma membrane integrity), and condensation of nuclei (morphology typically associated with apoptotic cell death). Exposure to high concentrations of LPS (1-3ug/mL) 24 h prior to OGD significantly improved neuronal survival. Furthermore, using the lowest effective concentration in all cell death and viability assays (1ug/mL, Fig. 1A-C), we determined that the effective tolerance window ranged between 15-48 hours (Fig. 1D-F). Based on these results, we wished to determine if alterations in mitochondrial dynamics occurred within 24 h following a 1ug/mL LPS preconditioning stimulus.
Figure 1. LPS confers ischemic tolerance in neuron cultures in a dose- and time-dependent fashion.
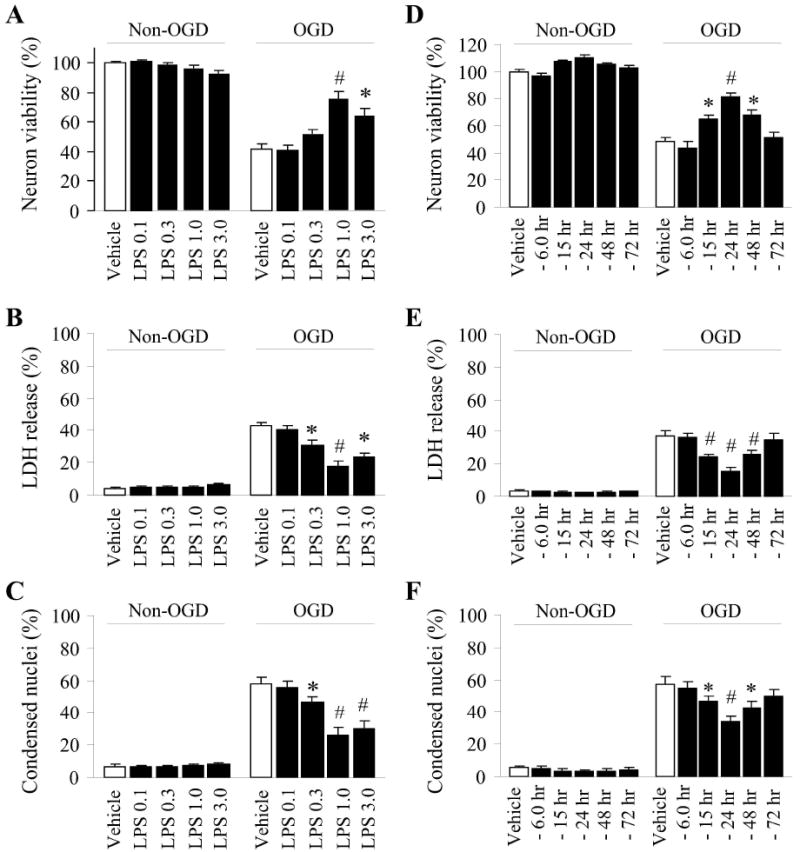
The dose-response effects (0.1-3ug/mL LPS applied prior to 1 h of OGD, A-C) and time window efficacy (1 μg/mL LPS applied 6 to 72 h prior to the induction of OGD, D-F) for LPS preconditioning afforded neuroprotection in neuron enriched cultures were established using cell viability assay (alamar blue uptake, A, D), plasma membrane permeability as a measure of cell death (LDH release, B, E), and assessment of nuclear morphology associated with apoptosis (condensed nuclei visualized by Hoescht staining, C, F). All data are mean±SE, from 3 independent experiments using cultures prepared from different dams, and each experiment was done in quadruplicate wells. *p<0.05; #p<0.01 versus vehicle controls of OGD neurons.
LPS preconditioning stimulus induces molecular markers of mitochondrial biogenesis in neurons
We next wished to examine if LPS is capable of inducing hallmarks of mitochondrial biogenesis when delivered at a concentration and in a timeframe that confers ischemic tolerance. The field of assessing mitochondrial biogenesis is still in its infancy. Therefore, we examined several aspects of mitochondria that have been associated with the induction of biogenesis. The coordinated upregulation of critical nuclear- and mitochondrial-encoded genes is necessary for both the maintenance and biogenesis of mitochondria (Garesse & Vallejo 2001, Scarpulla 2008). For example, NRF1 is a nuclear-encoded nuclear transcription factor, which can bind to and transactivate the TFAM promoter region upon translocation to the nucleus (Scarpulla 2008). TFAM is a nuclear-encoded mitochondrial transcription/replication factor. The TFAM protein is imported into mitochondria and leads to increased transcription of the mitochondrial genome (Scarpulla 2008). TFAM is also positively correlated and appears to contribute to the control of mtDNA copy number (Scarpulla 2008). We found that the mRNA content of the critical transcriptional factors NRF1 and TFAM rapidly increased 3-6 h following LPS exposure (Fig. 2A). Interestingly, PGC1-α, a major upstream transcription factor for NRF1 promoter transactivation, was not significantly upregulated at the mRNA level until 24h following LPS exposure (Fig. 2A). However, the function of PGC1-α is controlled by a wide variety of post-translational modifications and has overlapping function with PGC1-β (Wareski et al. 2009). Thus, the delayed induction of PGC1-α mRNA may represent a secondary transcriptional feedback response to LPS, and may not be necessary in the establishment of ischemic tolerance.
Figure 2. Preconditioning by LPS induces molecular markers of mitochondrial biogenesis in neuron cultures.
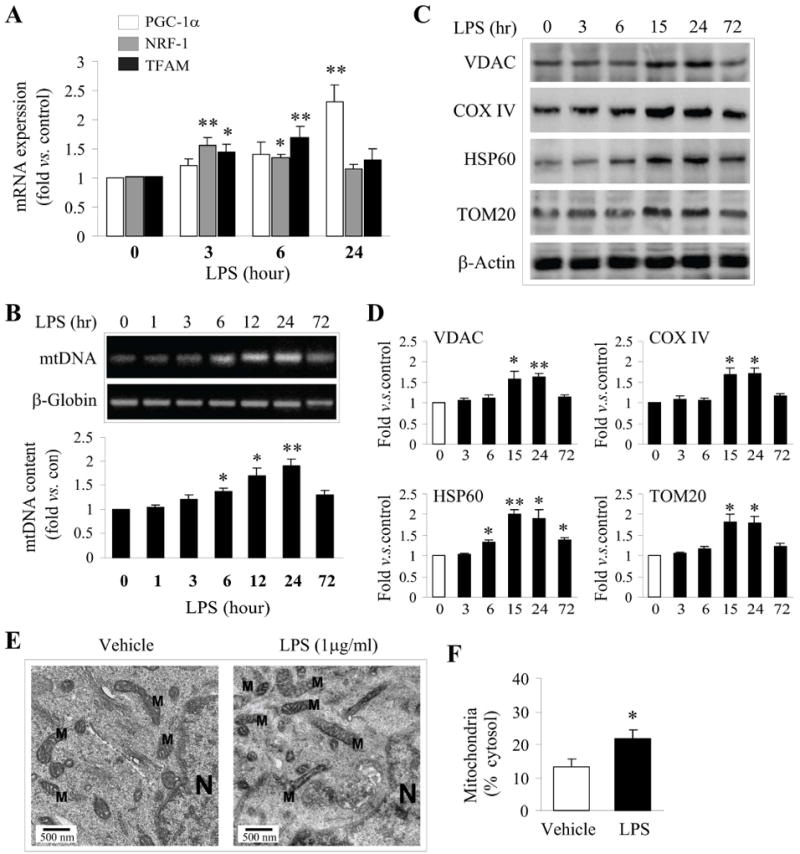
A. The mRNA levels of critical transcription factors (PGC-1a, NRF-1 and TFAM) involved in mitochondrial biogenesis were assessed by real-time PCR over 24 h following 1ug/mL of LPS addition to cultures. B. Mitochondrial DNA (mtDNA) content was assessed by long chain PCR following exposure to 1μg/mL of LPS. Bands were semiquantified and expressed as fold increase over controls. C. Representative Western blots showing increased protein expression of VDAC, COXIV, HPS60 and TOM20 after LPS exposure in a time-dependent manner. D. The semi-quantified results (fold increase over controls) of C are illustrated in the graphs. All quantitative data (A-D) are mean±SE, from 3-4 independent experiments using cultures of different dams. *p<0.05, **p<0.01 compared to controls (timepoint “0”). E. Representative electron microscopic images (EM) from neurons exposed to vehicle and 1ug/mL LPS for 24 h, respectively, showing structures for mitochondria (M) and nucleus (N). F. Semiquantitative analysis of mitochondrial mass by assessing the percentages of cytosol occupied by mitochondrial structures in randomly selected EM images. Data were derived from 5 consecutive sections per cell, using a minimum of 3 cells per condition and 3 independent experiments. Statistical analysis was based on a paired student t-test, *p<0.05 versus vehicle.
One major function of increased NRF1 and TFAM in mitochondrial biogenesis is the upregulation of the mitochondrial respiratory chain (MRC), such as COXIV, and other proteins critical for mitochondrial function, such as the voltage-dependent anion channel VDAC and TOM20 (Dhar et al. 2008). Consistent with the upregulation of NRF1 and TFAM, we found that both COXIV and VDAC protein products were significantly increased 24 h following LPS exposure (Fig. 2C, D). Likewise, TOM20, an essential protein in the mitochondrial protein import system, and HSP60, a mitochondrial-associated heat shock protein, were also upregulated following LPS exposure. Consistent with the upregulation of VDAC and the nuclear-encoded COXIV, we found an increase in the abundance of MRC complexes following 24 h of LPS exposure (Suppl. Fig. 1). These data indicate that the sublethal LPS preconditioning stimulus increases critical components of the transcriptional machinery and functional downstream MRC proteins, consistent with an increase in mitochondrial biogenesis in cortical neurons. Importantly, the timeframe of the increased expression of mitochondrial proteins correlates with the timing of the neuroprotective effects of LPS against OGD.
Induction of mtDNA has often been used as a marker of mitochondrial biogenesis. Using long fragment PCR, we found a significant increase in mtDNA 6 to 24 h following exposure to 1ug/mL LPS (Fig. 2B). mtDNA levels returned to control levels within 72 h following LPS exposure, suggesting the induction of control mechanisms targeting either mtDNA copy number or mitochondrial quantity. The early induction of mtDNA is consistent with the early induction of TFAM, which functions not only as a major mtDNA transcription factor, but also simultaneously controls the replication and copy number of mtDNA (Scarpulla 2008). Furthermore, we examined whether the concentrations of LPS required to induce ischemic tolerance correlated with the concentrations required to increase mtDNA copy number. Supporting a correlation between neuroprotective concentrations of LPS preconditioning and induction of mitochondrial biogenesis, the same concentrations that conferred ischemic tolerance effectively increased mtDNA copy number (data not shown). To correlate the observation of increased mitochondrial proteins and mtDNA with increased mitochondrial mass within cells, we evaluated electron microscopic images and semi-quantified the mitochondria as the percent of area of cytosol. Following 24 h of LPS exposure, a significant increase in the mitochondrial content in the cytosol was detected (Fig. 2E,F). Taken together, these data indicate that exposure of neuron-enriched cortical cultures to LPS induces multiple molecular hallmarks of mitochondrial biogenesis in a timeframe and with a dose-response curve consistent with LPS-induced ischemic tolerance.
LPS preconditioning increases hallmarks of functional mitochondria
Although increased gene products essential to mitochondrial function may indicate mitochondrial biogenesis, we wished to further investigate whether LPS preconditioning could affect the overall capacity and/or function of mitochondria in neurons. Critical indicators of functional mitochondria include the capacity to generate ATP and maintain their membrane potential. To assess energetic function following LPS exposure, we first measured cellular ATP using luciferase activity in cell lysates. Cellular ATP levels were significantly increased 12-24 h following LPS exposure (Fig. 3A). In order to further explore whether the increased ATP could be associated with increased activity of the Krebs cycle, the major aerobic cycle that generates ATP within the mitochondrial matrix and feeds into the MRC, we assessed the activity of citrate synthase, the initiating enzyme in the Krebs cycle whose activity is closely linked to the output of ATP and indicates the presence of intact mitochondria. As expected, citrate synthase activity was significantly increased 12-24 h following LPS exposure (Fig. 3B), which closely correlated with our observations that protein expression of MRC subunits and ATP were both increased within 24h following LPS exposure.
Figure 3. LPS preconditioning increases mitochondrial functional capacity and axonal movement.
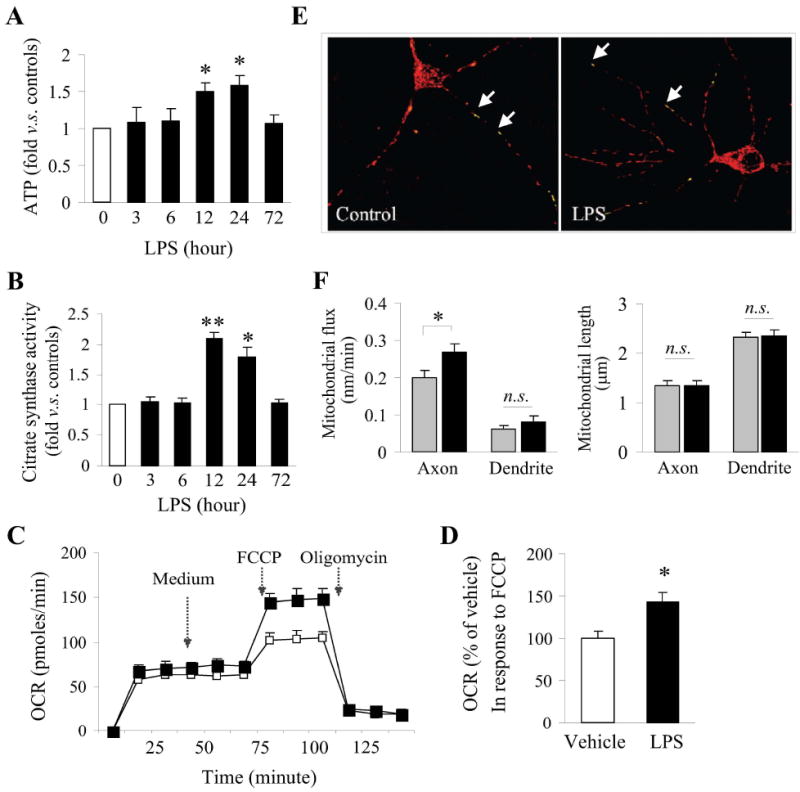
Cellular ATP content (A) and citrate synthase activity (B) were assessed in lysates obtained from neuronal cultures exposed to 1ug/mL LPS at the given timepoints. Data were obtained from 3 separate cultures and statistically analyzed using ANOVA and post hoc Bonferroni/Dunn tests. *p<0.05, **p<0.01 versus controls (0). C. Mitochondrial respiration was measured in live cultures using the Seahorse analyzer. Cultures were treated with medium as a vehicle control, FCCP or oligomycin at the indicated time and the rate of oxygen consumption was recorded. D. The data were normalized in vehicle controls and statistically compared using two-tail Student’s t-test, *p<0.05 versus vehicle. E-F. Mitochondrial movement along axonal and dendritic regions was assessed in live cultures under time-lapse imaging. Data were from 3 separate cultures and each experiment was done in quadruplicate wells. *p<0.05 between the indicated groups; n.s., not significant.
Although increased ATP content and citrate synthase activity are indicators of increased mitochondrial function, a more direct assessment of mitochondrial functional capacity is the measurement of the oxygen consumption rate (OCR). Using a direct method of assessing mitochondrial respiration, we measured OCR in the presence of FCCP, an uncoupler of electron transport and oxidative phosphorylation, in live cultures. Application of FCCP reveals the maximal respiration in a given sample. In LPS-treated cultured neurons, a significant increase in the OCR following FCCP application was observed (Fig. 3C, D). These results indicate not only that mitochondrial protein expression and ATP content is increased in LPS-treated neurons, but also that LPS-treated neurons have a higher capacity for oxygen respiration. In sum, LPS appears to increase both mitochondrial content and subsequently, mitochondrial function.
LPS affects mitochondrial organelle movement
Both mitochondrial movement and length have been used in determinations of mitochondrial morphology and associated health. For example, mitochondrial movement within the cell may be related to membrane potential and sensitivity to stress (Miller & Sheetz 2004). We therefore examined both the movement of labeled mitochondria across defined regions of interest over time (flux) and length of mitochondria in the axon and dendrites of neurons exposed to LPS for 24 h. We found that mitochondrial flux along axons was increased 24 h following LPS exposure (Fig. 3E, F). In contrast, movement in dendrites and overall mitochondrial length remained unchanged.
Mitochondrial biogenesis contributes to ischemic neuroprotection afforded by LPS
According to our data, the transcriptional control of mitochondrial biogenesis by LPS is correlated with its protective effects. As mentioned above, TFAM leads to increased transcription and replication of the mitochondrial genome. To address the functional role of the TFAM pathway in LPS preconditioning, we transduced cultured neurons with lentivirus encoding shRNA constructs targeting TFAM (shRNAt) or a control scramble sequence (shRNAc), and/or in combination with a construct overexpressing human TFAM cDNA. Neurons were transduced 3 d prior to LPS exposure and assessed for targeted knockdown (Fig. 4A), hallmarks of mitochondrial biogenesis (Fig. 4B,C) and ischemic tolerance (Fig. 4D,E). Knockdown of TFAM significantly attenuated the rise in mtDNA content observed following LPS exposure, as well as the rise in ATP production and citrate synthase activity (Fig. 4C). Co-transduction with the human TFAM construct, which is resistant to the shRNA targeting rat TFAM, restored the ability of LPS to increase mtDNA and ATP content and citrate synthase activity.
Figure 4. TFAM is required LPS preconditioning afforded neuroprotection.
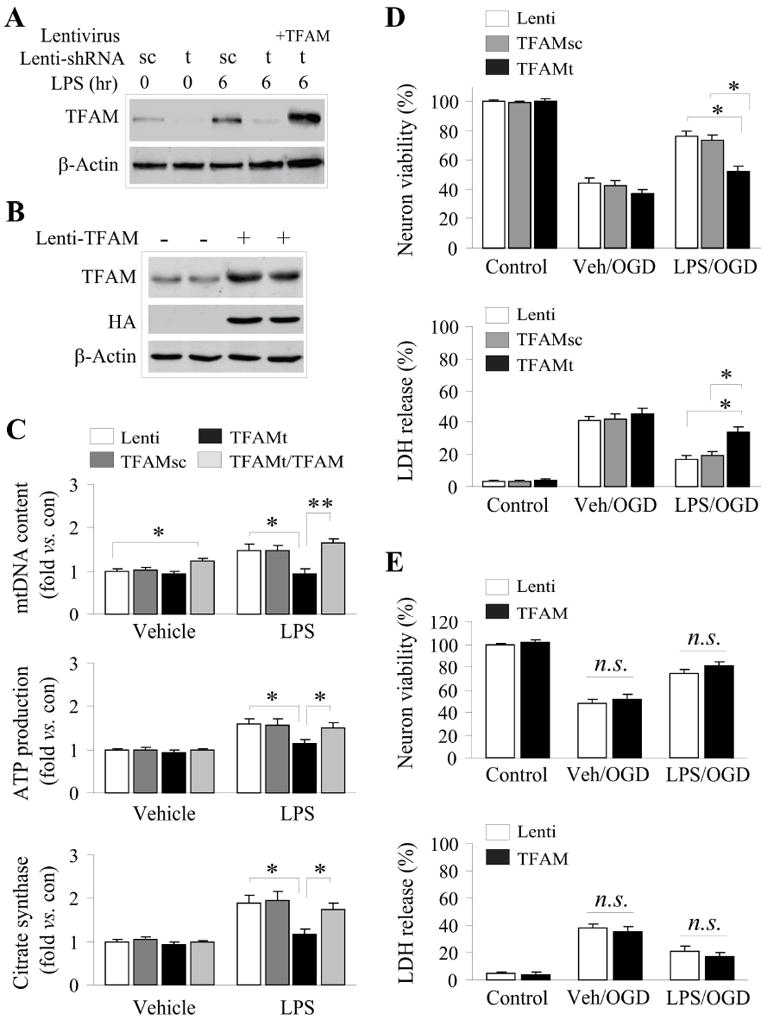
Enriched cortical cultures were transduced with lentiviral vectors containing either shRNA constructs or a human TFAM cDNA 3 days prior to exposure to LPS or vehicle. A. The TFAM-targeting shRNA (t), but not the scrambled control shRNA (sc), effectively suppressed TFAM protein expression without or with LPS (1μg/mL) challenge (6h). Overexpression of human TFAM (resistant to the rat-targeted shRNA) effectively restored TFAM expression, and was detected by the TFAM or HA antibody (B). C. Targeted knockdown of TFAM (TFAMt) but not the scrambled control (TFAMsc) blocked the induction of mitochondrial biogenesis (increased mtDNA, ATP content and citrate synthase activity) following LPS exposure (1μg/mL, 24h). Overexpression of the human TFAM effectively restored the induction of mitochondrial biogenesis in LPS-treated cultures. D. Targeted knockdown of TFAM significantly attenuated the neuroprotective effect of LPS preconditioning against OGD. E. Overexpression of TFAM alone was insufficient to protect neurons against OGD. All data were mean±SEM, from 3 independent experiments using cultures prepared from different dams, and each experiment was done in quadruplicate wells. *p<0.05; #p<0.01 between the indicated groups.
We next determined the functional significance of mitochondrial biogenesis in LPS-induced ischemic tolerance. ShRNA knockdown of TFAM significantly decreased the ischemic neuroprotection afforded by LPS preconditioning, as measured by two independent assays of cellular viability (Fig. 4D). Importantly, no loss of cell viability was observed in shRNA-transfected cells under basal, untreated conditions (Fig. 4D), nor were any decreases in mtDNA, ATP content or citrate synthase activity observed when compared to control levels in untreated conditions (Fig. 4C). Overexpression of TFAM itself did not improve neuronal survival following OGD with or without LPS preconditioning (Fig. 4E). This suggests that LPS preconditioning likely elicits a coordinated program by multiple players to induce biogenesis-related neuroprotection and that TFAM appears to be only one of such critical factors in this program. In other words, TFAM overexpression alone cannot improve neuronal survival against OGD because other elements involved in related processes (e.g., mitochondrial transport) may be equally essential. Furthermore, due to the absence of synergy when LPS stimulation was combined with TFAM overexpression, it is likely that LPS stimulates the maximal response available for ischemic tolerance that can be derived from TFAM induction. Taken together, these data suggest that TFAM regulates LPS-induced mitochondrial biogenesis in cultured cortical neurons, and that TFAM-dependent mitochondrial biogenesis contributes to ischemic neuroprotection afforded by LPS preconditioning.
AMPK contributes to the induction of the transcriptional program related to mitochondrial biogenesis
The signaling pathways leading to changes in mitochondrial biogenesis are still unclear and appear to be highly cell type and context specific. Using a panel of inhibitors, we thus explored the potential upstream signaling pathways that may be involved in our model of LPS-stimulated mitochondrial biogenesis in neurons, using TFAM mRNA induction as an endpoint. The increase in both TFAM expression (Suppl. Fig. 2A) and mtDNA (Suppl. Fig. 2B) was significantly attenuated by inhibitors of TLR4 (polymixin B), ROS (N-acetyl cysteine, NAC), AMPK (Compound C) and PI3K/Akt (LY294002) at non-neurotoxic concentration of each of these compounds (data not shown). In contrast, inhibitors of p38 (SB203580), MEK1/2 (U0126), CamKII (KN62) and NOS (L-NAME) were ineffective at inhibiting either the induction of TFAM mRNA expression or increased mtDNA following LPS (Suppl. Fig. 2A,B).
AMPK is an upstream kinase that can lead to the activation of PGC1α and the induction of mitochondrial biogenesis in some cellular contexts (Mihaylova & Shaw 2011). Indeed, we found a significant increase in the phosphorylation of AMPK following LPS stimulus (Suppl. Fig. 2C). Inhibition of AMPK phosphorylation with the pharmacological inhibitor Compound C was effective at blocking both the induction of NRF1 and TFAM mRNA and protein, suggesting that AMPK lies upstream of the transcriptional activation of mitochondrial biogenesis (Fig. 5A,B). Interestingly, inhibition of TLR4 receptor or ROS (using polymixin B or NAC, respectively) suppressed the activation of AMPK following LPS exposure, but inhibition of PI3K/Akt (using LY294002) had no effect on the activation of AMPK (Fig. 5C). These results suggest that the activation of AMPK is dependent on classical LPS TLR activation and ROS generation, and is either upstream or independent of PI3K/Akt activity.
Figure 5. Activation of AMPK contributes to LPS-induced biogenesis.
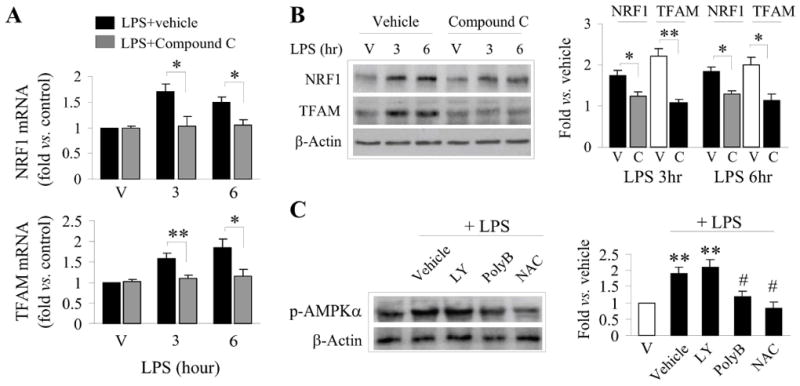
A,B. LPS induced mRNA (A) and protein (B) expression of NRF1 and TFAM in the presence or absence of the AMPK inhibitor, Compound C (20 μM). V=cultures exposed to vehicle. Semiquantitative data of protein expression are illustrated in the graph (right panel of B). C. Inhibition of TLR (polymixin B, polyB, 10μM) and ROS (NAC, 1mM) block LPS-stimulated AMPK activation. Semiquantitative data of p-AMPKα are illustrated in the graph (right panel of C). Data were from 3-4 independent cultures, expressed as fold change versus vehicle (non-LPS) treated cultures, and statistically analyzed using ANOVA and post hoc Bonferroni/Dunn tests. *p<0.05, **p<0.01 compared to vehicle, #p<0.5 compared to LPS-only challenged cultures.
PI3K/Akt is required for nuclear translocation of NRF1
We were particularly interested in exploring the role of PI3K/Akt in mitochondrial biogenesis. Several reports have reported a role for Akt at various points in this process (Carraway et al. 2010, Echave et al. 2009), and Akt has been largely found to be neuroprotective in terms of cell survival. Because LPS stimulates both markers of mitochondrial biogenesis as well as neuroprotection against OGD (Figs. 1-3), we hypothesized that in this model, Akt would be required for induction of mitochondrial biogenesis. As expected, Akt was rapidly phosphorylated following LPS exposure, with significantly increased levels of p-Akt (Fig. 6A, Suppl. Fig. 2D,E) and Akt activity (Fig. 6B) within 3 h following LPS exposure. Using an AAV construct encoding a dominant-negative version of Akt (Akt-dn), neurons were transduced prior to the addition of LPS. Akt-dn effectively blocked the activity of Akt, and overexpression of human Akt in the presence of the Akt-dn construct was able to restore Akt activity levels following LPS exposure (Fig. 6B). Interestingly, the inhibition of Akt by Akt-dn overexpression inhibited the induction of mtDNA by LPS. However, overexpression of human Akt by itself was insufficient to induce mtDNA. Thus, the expression of Akt alone cannot recapitulate the effects of LPS in inducing mitochondrial biogenesis, consistent with the hypothesis that multiple signaling pathways must be concurrently activated to achieve mitochondrial biogenesis. To further support the concept that multiple signaling pathways must be coordinated for LPS to induce biogenesis, the activation of Akt was independent of AMPK activity, as inhibition of AMPK with Compound C had no effect on the phosphorylation of Akt (Suppl. Fig. 3).
Figure 6. AKT activity is critical for NRF1 nuclear translocation, TFAM expression and mtDNA increase following LPS preconditioning.
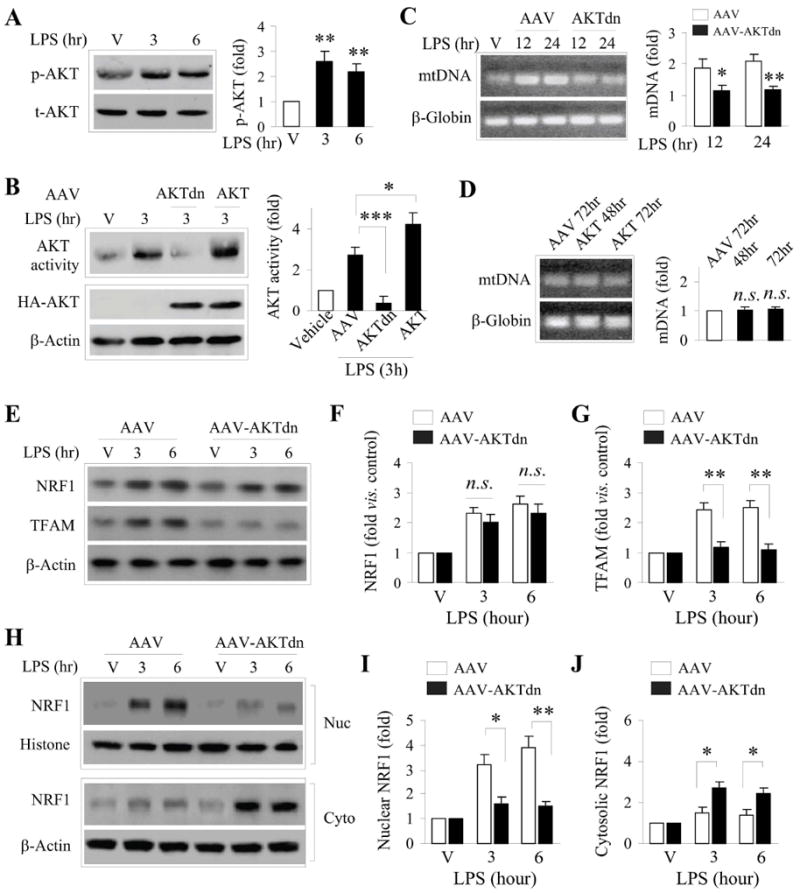
A. LPS preconditioning leads to a rapid increase in Akt phosphorylation (p-AKT) in neurons. Data was from 3 independent cultures, and normalized to total Akt (t-AKT) levels. B. Akt activity, as determined by an in vitro kinase activity assay, was rapidly increased in cell lysates from LPS treated-cultures, and was inhibited by overexpression of the dominant negative AKT (AKTdn). Overexpression of AKT cDNA further increased AKT activity following LPS exposure. C. Overexpression of AKTdn, but not the empty AAV (AAV), suppressed the induction of mtDNA following LPS exposure for 12 or 24 hr. D. Overexpression of AKT alone (without LPS) was insufficient to increase mtDNA content. E-G. Suppression of AKT by AKTdn (but not the empty AAV) overexpression failed to affect NRF1 protein expression (E, F), but significantly reduced TFAM protein expression (E, G) following LPS exposure. H-J. Subcellular fractionation and Western blotting for NFR1 show that inhibition of AKT by AKTdn prevented LPS-induced NRF1 protein levels in the nucleus (H, I) with a concomitant increase of NRF1 protein in the cytosol (H, J). Data were from 3-4 independent cultures, expressed as fold change versus vehicle (non-LPS) treated cultures, and statistically analyzed using ANOVA and post hoc Bonferroni/Dunn tests. *p<0.05, **p<0.01 compared to empty AAV (AAV) or between the indicated groups.
Interestingly, we found that overexpression of Akt-dn suppressed TFAM protein expression, but had no effect on the expression of its upstream transcription factor NRF1 (Fig. 6E-G). NRF1 must translocate to the nucleus following translation to induce expression of TFAM (Scarpulla 2008). Accordingly, we examined the intracellular localization of NRF1 in the presence of Akt-dn. Overexpression of Akt-dn effectively inhibited NRF1 translocation out of the cytosol into the nucleus following LPS stimulation (Fig. 6H-J). These data support a role for Akt in the transcriptional induction of mitochondrial biogenesis following LPS preconditioning, and describe a novel role for Akt in the control of the nuclear translocation of NRF1 in neuronal cells.
Discussion
Previous to the present report, the mechanisms of preconditioning against cerebral ischemia and the phenomenon of mitochondrial dynamics were both well documented, but the relationship between the two was not defined. Therefore, we explored the impact of mitochondrial biogenesis on ischemic tolerance in neurons. In doing so, we demonstrated several novel findings. First, a sublethal LPS preconditioning stimulus that can elicit an ischemic tolerant state in enriched neuronal cultures also induces hallmarks of mitochondrial biogenesis in a concurrent timeframe and concentration associated with ischemic neuroprotection. Secondly, the activation of mitochondrial biogenesis by LPS occurs through the coordinated activity of multiple signaling pathways, and none of which alone act as a “master switch.” Finally, suppression of mitochondrial biogenesis significantly attenuates the protective effects of LPS preconditioning. These data provide novel evidence for a critical role of mitochondrial biogenesis in protection against neuronal ischemic injury.
In addition to identifying a functional role for mitochondrial biogenesis in ischemic neuroprotection, we further identified several signaling pathways in the transcriptional regulation of mitochondrial biogenesis. AMPK activation was critical for both the induction of NRF1 and TFAM, whereas Akt was not necessary for the upstream initiation of a biogenesis cascade. Rather, Akt activity was required for downstream NRF-1 translocation to the nucleus in preconditioned neurons and for subsequent TFAM expression. Consistent with these results but in a different cellular context, NRF1 nuclear translocation under oxidative conditions was dependent upon Akt (Piantadosi & Suliman 2006). Akt has also been implicated in the upregulation of NRF1 transcription by the activation of Nfe2l2 (Piantadosi et al. 2011). However, we did not observe significant changes in NRF1 protein levels in the presence of Akt-dn. Despite this, Nfe2l2 could still be implicated in the current model, as the induction of mitochondrial biogenesis was also dependent on reactive oxygen species. The divergence of mechanisms that appear to exist between cell and model systems underscores the multifaceted nature of mitochondrial dynamics. Further investigation into the molecular regulation of NRF1 nuclear translocation and its dependence on Akt activation might expand the well-known role of Akt in ischemic neuroprotection.
Importantly, the overexpression of either TFAM or Akt alone (in the absence of LPS) was insufficient to induce mitochondrial biogenesis, as assessed by mtDNA content. This finding underscores the probability that several processes, including but not limited to the induction of the transcriptional cascade, lead to mitochondrial biogenesis. These processes are likely to include mitochondrial flux and fission or fusion events or synthesis of other critical factors leading to the full induction of mitochondrial biogenesis. The coordination of these processes with the NRF1/TFAM transcriptional cascade will lead to further insights on the biogenesis process itself, as well as the necessary pathways needed to target for a therapeutic effect.
Although the current study provides support for the concept that mitochondrial biogenesis contributes to ischemic neuroprotection, the means by which newly generated mitochondria promote cell survival remain unclear. We demonstrated that ATP content and citrate synthase activity are increased within the tolerant timeframe, which could provide additional energetic reserves for surviving an ischemic challenge. We also demonstrate that exposure to the LPS preconditioning stimulus increases mitochondrial flux specifically in axons. Mitochondria with lower potential were noted to undergo retrograde transport back toward the soma (Miller & Sheetz 2004), although in another report this was not observed (Verburg & Hollenbeck 2008). In addition, anterograde transport has been hypothesized to be required to shuttle newly synthesized mitochondrial elements to distal regions along the axon for replacement. However, given the recent findings of axonal mitochondrial mRNAs and localized translation, the requirement for transport in mitochondrial biogenesis is still unresolved (Saxton & Hollenbeck 2012).
The concept of mitochondrial replacement rather than a sustained increase in number is an important distinction. Given the return of critical mitochondrial endpoints (e.g., TFAM mRNA expression, COXIV and VDAC protein expression, mtDNA content, ATP content and citrate synthase activity) to baseline levels at later timepoints following LPS exposure (e.g., 72 h), quantity control mechanisms are likely to be activated by a transient induction of mitochondrial biogenesis. It is likely that the induction of mitochondrial biogenesis – producing newer, healthier mitochondria – occurs in concert with the clearance of older mitochondria, such as those that remain depolarized under sublethal stress. Indeed, both the observations of increased mitochondrial flux along the axon as well as increased markers of autophagic processes in the current model (data not shown) suggest that mitochondrial clearance (mitophagy) may be a critical element of ischemic neuroprotection. These findings warrant further investigation of mitochondrial replacement in the preconditioned state.
Mechanisms of LPS preconditioning in neuron-enriched cultures may differ significantly from brain. LPS is best characterized for its action on TLR4 receptors. Indeed, our data demonstrate that polymyxin B, a TLR4 antagonist, effectively inhibits markers of mitochondrial biogenesis (e.g., TFAM upregulation and increases in mtDNA) following LPS exposure. These findings support the concept that LPS relies on the presence of functional TLR4 receptors in our model. TLRs are predominantly expressed in glial populations, and the concentration of LPS required to induce ischemic tolerance in our enriched neuronal model is significantly higher than the dose required stimulate cultured microglia (personal observation). The requirement for a higher concentration in the enriched neuronal culture may be because the minor fraction of glia present (2%) in the neuron-enriched cultures needs to be overstimulated to elicit an effect on the neurons. Alternatively, LPS may exert a direct effect on neurons themselves, as recent reports suggest that TLR4 (and other TLRs) are also expressed in neurons (Okun et al. 2009). However, the binding affinity of neuronal TLRs and the downstream mechanisms have not been investigated, and would be interesting to determine in light of the present study.
A schematic of our major findings is depicted in Figure 7. In neuron-enriched cortical cultures, LPS acts on TLR4 and elicits sublethal oxidative stress. The reactive oxygen species in turn contributes to the activation of AMPK. Phosphorylated AMPK, perhaps indirectly through other mediators, leads to the increased transcription of NRF1. NRF1 protein is then translocated into the nucleus via the activation of Akt. Once in the nucleus, Nrf1 increases transcription of the TFAM gene. TFAM protein is subsequently synthesized in the cytoplasm and then enters mitochondria to contribute to the overall effect of mitochondrial biogenesis. Our hypothesis is thus that a healthier mitochondrial population may possess greater energy reserves or other buffering capacity and effectively combat the ischemic challenge that follows LPS preconditioning. Future studies are needed to address the role of mitochondrial biogenesis in cerebral ischemic tolerance using in vivo models, where the functional effects of mitochondrial biogenesis in establishing ischemic tolerance are still unknown. Furthermore, the induction of mitochondrial biogenesis as a neuroprotective strategy in and of itself may lead to potentially safer prophylactic treatments compared to LPS stimulation. However, the range of molecular events involved in and exerting control over the mitochondrial organellar response needs to be further explored under different contexts and stimuli.
Figure 7. Schematic of LPS-induced mitochondrial biogenesis.
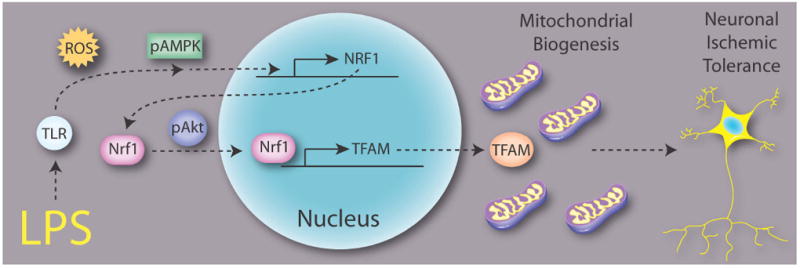
LPS activates TLR and ROS, which lead to the activation of AMPK. p-AMPK stimulates the upregulation of NRF1 and TFAM mRNA expression. Akt is also activated by LPS stimulation, independently of AMPK activation. p-Akt facilitates the nuclear translocation of NRF1 from the cytosol and subsequent upregulation of TFAM mRNA expression. These events are coordinated to induce mitochondrial biogenesis and contribute to ischemic tolerance.
In summary, our present study argues that induction of mitochondrial biogenesis is highly correlated with and serves as a critical mediator in LPS-induced ischemic tolerance. Furthermore, the induction of mitochondrial biogenesis in this model is mediated by the coordinated action of multiple signaling pathways that affect a range of processes, including but not limited to transcriptional induction of NRF1 and TFAM. The endogenous induction of mitochondrial biogenesis may thus be a pluripotent novel mechanism to confer ischemic neuroprotection.
Supplementary Material
Acknowledgments
This project was supported by NIH/NINDS grants NS43802, NS45048, NS36736, and NS56118 (to J.C.); and Chinese Natural Science Foundation grants 30670642, 30870794, and 81020108021 (to Y.G.). J.C. is the RK Mellon Endowed Chair of UPMC and a recipient of the VA Career Scientist Award. We thank Pat Strickler for excellent secretarial support.
Footnotes
The authors have no conflicts of interest to declare.
References
- Biala A, Tauriainen E, Siltanen A, et al. Resveratrol induces mitochondrial biogenesis and ameliorates Ang II-induced cardiac remodeling in transgenic rats harboring human renin and angiotensinogen genes. Blood Press. 2010;19:196–205. doi: 10.3109/08037051.2010.481808. [DOI] [PubMed] [Google Scholar]
- Busija DW, Gaspar T, Domoki F, Katakam PV, Bari F. Mitochondrial-mediated suppression of ROS production upon exposure of neurons to lethal stress: mitochondrial targeted preconditioning. Adv Drug Deliv Rev. 2008;60:1471–1477. doi: 10.1016/j.addr.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadet JL, Krasnova IN. Cellular and molecular neurobiology of brain preconditioning. Mol Neurobiol. 2009;39:50–61. doi: 10.1007/s12035-009-8051-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carraway MS, Suliman HB, Jones WS, Chen CW, Babiker A, Piantadosi CA. Erythropoietin activates mitochondrial biogenesis and couples red cell mass to mitochondrial mass in the heart. Circ Res. 2010;106:1722–1730. doi: 10.1161/CIRCRESAHA.109.214353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang DT, Honick AS, Reynolds IJ. Mitochondrial trafficking to synapses in cultured primary cortical neurons. J Neurosci. 2006;26:7035–7045. doi: 10.1523/JNEUROSCI.1012-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correia SC, Cardoso S, Santos RX, Carvalho C, Santos MS, Perry G, Smith MA, Moreira PI. New insights into the mechanisms of mitochondrial preconditioning-triggered neuroprotection. Curr Pharm Des. 2011;17:3381–3389. doi: 10.2174/138161211798072490. [DOI] [PubMed] [Google Scholar]
- Correia SC, Santos RX, Perry G, Zhu X, Moreira PI, Smith MA. Mitochondria: the missing link between preconditioning and neuroprotection. J Alzheimers Dis. 2010;20(Suppl 2):S475–485. doi: 10.3233/JAD-2010-100669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csiszar A, Labinskyy N, Pinto JT, et al. Resveratrol induces mitochondrial biogenesis in endothelial cells. Am J Physiol Heart Circ Physiol. 2009;297:H13–20. doi: 10.1152/ajpheart.00368.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagda RK, Gusdon AM, Pien I, Strack S, Green S, Li C, Van Houten B, Cherra SJ, 3rd, Chu CT. Mitochondrially localized PKA reverses mitochondrial pathology and dysfunction in a cellular model of Parkinson’s disease. Cell Death Differ. 2011;18:1914–1923. doi: 10.1038/cdd.2011.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar SS, Ongwijitwat S, Wong-Riley MT. Nuclear respiratory factor 1 regulates all ten nuclear-encoded subunits of cytochrome c oxidase in neurons. J Biol Chem. 2008;283:3120–3129. doi: 10.1074/jbc.M707587200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echave P, Machado-da-Silva G, Arkell RS, Duchen MR, Jacobson J, Mitter R, Lloyd AC. Extracellular growth factors and mitogens cooperate to drive mitochondrial biogenesis. J Cell Sci. 2009;122:4516–4525. doi: 10.1242/jcs.049734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garesse R, Vallejo CG. Animal mitochondrial biogenesis and function: a regulatory cross-talk between two genomes. Gene. 2001;263:1–16. doi: 10.1016/s0378-1119(00)00582-5. [DOI] [PubMed] [Google Scholar]
- Goldenthal MJ, Marin-Garcia J. Mitochondrial signaling pathways: a receiver/integrator organelle. Mol Cell Biochem. 2004;262:1–16. doi: 10.1023/b:mcbi.0000038228.85494.3b. [DOI] [PubMed] [Google Scholar]
- Gutsaeva DR, Carraway MS, Suliman HB, Demchenko IT, Shitara H, Yonekawa H, Piantadosi CA. Transient hypoxia stimulates mitochondrial biogenesis in brain subcortex by a neuronal nitric oxide synthase-dependent mechanism. J Neurosci. 2008;28:2015–2024. doi: 10.1523/JNEUROSCI.5654-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickson-Bick DL, Jones C, Buja LM. Stimulation of mitochondrial biogenesis and autophagy by lipopolysaccharide in the neonatal rat cardiomyocyte protects against programmed cell death. J Mol Cell Cardiol. 2008;44:411–418. doi: 10.1016/j.yjmcc.2007.10.013. [DOI] [PubMed] [Google Scholar]
- Lin HY, Huang CC, Chang KF. Lipopolysaccharide preconditioning reduces neuroinflammation against hypoxic ischemia and provides long-term outcome of neuroprotection in neonatal rat. Pediatr Res. 2009;66:254–259. doi: 10.1203/PDR.0b013e3181b0d336. [DOI] [PubMed] [Google Scholar]
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- McLeod CJ, Jeyabalan AP, Minners JO, Clevenger R, Hoyt RF, Jr, Sack MN. Delayed ischemic preconditioning activates nuclear-encoded electron-transfer-chain gene expression in parallel with enhanced postanoxic mitochondrial respiratory recovery. Circulation. 2004;110:534–539. doi: 10.1161/01.CIR.0000136997.53612.6C. [DOI] [PubMed] [Google Scholar]
- Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13:1016–1023. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KE, Sheetz MP. Axonal mitochondrial transport and potential are correlated. J Cell Sci. 2004;117:2791–2804. doi: 10.1242/jcs.01130. [DOI] [PubMed] [Google Scholar]
- Ning XH, Xu CS, Song YC, Xiao Y, Hu YJ, Lupinetti FM, Portman MA. Hypothermia preserves function and signaling for mitochondrial biogenesis during subsequent ischemia. Am J Physiol. 1998;274:H786–793. doi: 10.1152/ajpheart.1998.274.3.H786. [DOI] [PubMed] [Google Scholar]
- Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148:1145–1159. doi: 10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okun E, Griffioen KJ, Lathia JD, Tang SC, Mattson MP, Arumugam TV. Toll-like receptors in neurodegeneration. Brain Res Rev. 2009;59:278–292. doi: 10.1016/j.brainresrev.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piantadosi CA, Suliman HB. Mitochondrial transcription factor A induction by redox activation of nuclear respiratory factor 1. J Biol Chem. 2006;281:324–333. doi: 10.1074/jbc.M508805200. [DOI] [PubMed] [Google Scholar]
- Piantadosi CA, Withers CM, Bartz RR, MacGarvey NC, Fu P, Sweeney TE, Welty-Wolf KE, Suliman HB. Heme oxygenase-1 couples activation of mitochondrial biogenesis to anti-inflammatory cytokine expression. J Biol Chem. 2011;286:16374–16385. doi: 10.1074/jbc.M110.207738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raval AP, Dave KR, Perez-Pinzon MA. Resveratrol mimics ischemic preconditioning in the brain. J Cereb Blood Flow Metab. 2006;26:1141–1147. doi: 10.1038/sj.jcbfm.9600262. [DOI] [PubMed] [Google Scholar]
- Rintoul GL, Filiano AJ, Brocard JB, Kress GJ, Reynolds IJ. Glutamate decreases mitochondrial size and movement in primary forebrain neurons. J Neurosci. 2003;23:7881–7888. doi: 10.1523/JNEUROSCI.23-21-07881.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton WM, Hollenbeck PJ. The axonal transport of mitochondria. J Cell Sci. 2012;125:2095–2104. doi: 10.1242/jcs.053850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev. 2008;88:611–638. doi: 10.1152/physrev.00025.2007. [DOI] [PubMed] [Google Scholar]
- Stetler RA, Gao Y, Zhang L, et al. Phosphorylation of HSP27 by protein kinase D is essential for mediating neuroprotection against ischemic neuronal injury. J Neurosci. 2012;32:2667–2682. doi: 10.1523/JNEUROSCI.5169-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetler RA, Gao Y, Zukin RS, Vosler PS, Zhang L, Zhang F, Cao G, Bennett MV, Chen J. Apurinic/apyrimidinic endonuclease APE1 is required for PACAP-induced neuroprotection against global cerebral ischemia. Proc Natl Acad Sci U S A. 2010;107:3204–3209. doi: 10.1073/pnas.1000030107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens SL, Leung PY, Vartanian KB, Gopalan B, Yang T, Simon RP, Stenzel-Poore MP. Multiple preconditioning paradigms converge on interferon regulatory factor-dependent signaling to promote tolerance to ischemic brain injury. J Neurosci. 2011;31:8456–8463. doi: 10.1523/JNEUROSCI.0821-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suliman HB, Carraway MS, Welty-Wolf KE, Whorton AR, Piantadosi CA. Lipopolysaccharide stimulates mitochondrial biogenesis via activation of nuclear respiratory factor-1. J Biol Chem. 2003;278:41510–41518. doi: 10.1074/jbc.M304719200. [DOI] [PubMed] [Google Scholar]
- Suliman HB, Sweeney TE, Withers CM, Piantadosi CA. Co-regulation of nuclear respiratory factor-1 by NFkappaB and CREB links LPS-induced inflammation to mitochondrial biogenesis. J Cell Sci. 2010;123:2565–2575. doi: 10.1242/jcs.064089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang SC, Arumugam TV, Xu X, et al. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc Natl Acad Sci U S A. 2007;104:13798–13803. doi: 10.1073/pnas.0702553104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasaki K, Ruetzler CA, Ohtsuki T, Martin D, Nawashiro H, Hallenbeck JM. Lipopolysaccharide pre-treatment induces resistance against subsequent focal cerebral ischemic damage in spontaneously hypertensive rats. Brain Res. 1997;748:267–270. doi: 10.1016/s0006-8993(96)01383-2. [DOI] [PubMed] [Google Scholar]
- Van Laar VS, Arnold B, Cassady SJ, Chu CT, Burton EA, Berman SB. Bioenergetics of neurons inhibit the translocation response of Parkin following rapid mitochondrial depolarization. Hum Mol Genet. 2011;20:927–940. doi: 10.1093/hmg/ddq531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vartanian KB, Stevens SL, Marsh BJ, Williams-Karnesky R, Lessov NS, Stenzel-Poore MP. LPS preconditioning redirects TLR signaling following stroke: TRIF-IRF3 plays a seminal role in mediating tolerance to ischemic injury. J Neuroinflammation. 2011;8:140. doi: 10.1186/1742-2094-8-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verburg J, Hollenbeck PJ. Mitochondrial membrane potential in axons increases with local nerve growth factor or semaphorin signaling. J Neurosci. 2008;28:8306–8315. doi: 10.1523/JNEUROSCI.2614-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wareski P, Vaarmann A, Choubey V, Safiulina D, Liiv J, Kuum M, Kaasik A. PGC-1{alpha} and PGC-1{beta} regulate mitochondrial density in neurons. J Biol Chem. 2009;284:21379–21385. doi: 10.1074/jbc.M109.018911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin W, Signore AP, Iwai M, Cao G, Gao Y, Chen J. Rapidly increased neuronal mitochondrial biogenesis after hypoxic-ischemic brain injury. Stroke. 2008;39:3057–3063. doi: 10.1161/STROKEAHA.108.520114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JT, Lee CH, Yoo KY, et al. Maintenance of anti-inflammatory cytokines and reduction of glial activation in the ischemic hippocampal CA1 region preconditioned with lipopolysaccharide. J Neurol Sci. 2010;296:69–78. doi: 10.1016/j.jns.2010.06.004. [DOI] [PubMed] [Google Scholar]
- Zheng J, Chen LL, Zhang HH, Hu X, Kong W, Hu D. Resveratrol improves insulin resistance of catch-up growth by increasing mitochondrial complexes and antioxidant function in skeletal muscle. Metabolism. 2012;61:954–965. doi: 10.1016/j.metabol.2011.11.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.