

The Gp78 E3 ubiquitin ligase is shown to target the mitofusin mitochondrial fusion proteins for degradation, inducing mitochondrial fission and mitofusin 1–dependent mitophagy of uncoupled mitochondria. Mitophagy induced by endoplasmic reticulum–associated gp78 defines a distinct cellular pathway to eliminate damaged mitochondria.
Abstract
Glycoprotein 78 (Gp78) is a critical E3 ubiquitin ligase in endoplasmic reticulum–associated degradation. Overexpression of Flag-tagged Gp78 (Flag-gp78), but not Flag-gp78 mutated in its RING-finger domain (Flag-RINGmut) with deficient ubiquitin ligase activity, induces mitochondrial fragmentation and ubiquitination and proteasome-dependent degradation of the mitofusin (Mfn) mitochondrial fusion factors Mfn1/Mfn2. After mitochondrial depolarization with carbonyl cyanide m-chlorophenylhydrazone (CCCP), Flag-gp78 induced a threefold loss of depolarized mitochondria and significant loss of the inner mitochondrial protein OxPhosV. Flag-gp78–dependent loss of OxPhosV, but not Mfn1 or Mfn2, was prevented by small interfering RNA (siRNA) knockdown of the autophagy protein Atg5 in CCCP-treated cells. Gp78-induced mitophagy required ubiquitin ligase activity, as it is not observed upon transfection of Flag-RINGmut or cotransfection of Flag-gp78 with ubiquitin mutated at three critical lysine residues (K29, 48, 63R) involved in polyubiquitin chain elongation. Short hairpin RNA knockdown of Gp78 in HT-1080 fibrosarcoma cells increased mitofusin levels and reduced depolarization-induced mitophagy, whereas siRNA knockdown showed that Mfn1, but not Mfn2, was required for Gp78-dependent depolarization-induced mitophagy. Mitochondrial depolarization induced Gp78-dependent expression of the autophagic marker LC3II and recruitment of enhanced green fluorescent protein–LC3 to the Gp78- and calnexin-labeled, mitochondria-associated ER. Finally, Gp78-induced mitophagy is Parkin independent, as it occurs in Parkin-null HeLa cells and upon siRNA-mediated Parkin knockdown in HEK293 cells. This study therefore describes a novel role for the ER-associated Gp78 ubiquitin ligase and the Mfn1 mitochondrial fusion factor in mitophagy.
INTRODUCTION
Mitochondria are dynamic organelles that undergo frequent cycles of fusion and fission that allow intermixing of metabolites and mitochondrial DNA, the renewal, redistribution, and proliferation of mitochondria, and cellular adaptation to energy demands (Knott et al., 2008). Mitochondrial fusion maintains bioenergetic levels in case of injury, whereas fission is implicated in the maintenance of a healthy mitochondrial cellular pool by segregating damaged and inactive mitochondria for mitophagic elimination (Twig et al., 2008). Mitophagy is the selective autophagic elimination of mitochondria that results in engulfment of mitochondria by autophagosomes (Lemasters, 2005).
PINK1, a novel PTEN-induced putative kinase 1 (Unoki and Nakamura, 2001), functions upstream of the cytosolic E3 ubiquitin ligase Parkin to promote mitochondrial dysfunction (Clark et al., 2006; Park et al., 2006; Yang et al., 2008). PINK recruitment of Parkin to uncoupled mitochondria enables Parkin-mediated mitophagy (Narendra et al., 2008, 2010; Matsuda et al., 2010; Vives-Bauza et al., 2010). PINK1 and Parkin genetically interact with components of the mitochondrial fission and fusion machinery, and the mitofusin (Mfn1 and Mfn2) mitochondrial fusion factors were identified as novel Parkin substrates whose loss upon mitochondrial depolarization promotes mitophagy (Deng et al., 2008; Narendra et al., 2008; Dagda et al., 2009; Gegg et al., 2010; Tanaka et al., 2010; Ziviani et al., 2010). Once localized to damaged mitochondria, Parkin facilitates autophagic mitochondrial clearance by ubiquitylating specific mitochondrial proteins (VDAC1) and recruiting autophagic adapters (p62/SQSTM1; Geisler et al., 2010).
Glycoprotein 78 (gp78) is an endoplasmic reticulum (ER) membrane–anchored ubiquitin ligase (E3) that is a key component of the ER-associated degradation (ERAD) machinery involved in ubiquitination and degradation of various substrates, including CD3-delta, the T-cell receptor, ApoB lipoprotein, HMG CoA reductase, cystic fibrosis transmembrane conductance regulator, and the metastasis suppressor KAI1 (Fang et al., 2001; Liang et al., 2003; Zhong et al., 2004; Song et al., 2005; Tsai et al., 2007; Morito et al., 2008; Joshi et al., 2010). Gp78 has been localized to a mitochondria-associated ER domain (Wang et al., 2000; Goetz et al., 2007), and here we show that Gp78 induces mitochondrial fragmentation and targets the mitochondrial fusion proteins mitofusin 1 and 2 (Mfn1 and Mfn2, respectively) for degradation. Gp78 ubiquitin ligase activity is further shown to induce mitophagy upon mitochondrial depolarization and recruit the autophagosome marker enhanced green fluorescent protein (EGFP)–LC3 to the mitochondria-associated ER. Gp78-induced mitophagy requires Mfn1 but not Mfn2, defining a novel mechanism by which ER-associated Gp78 ubiquitin ligase activity regulates mitophagy.
RESULTS
Gp78 ubiquitin ligase activity induces mitochondrial fragmentation and proteasomal degradation of mitofusins
Overexpression of N-terminal Flag-tagged Gp78 (Flag-gp78-IRES-GFP) in Cos7 cells results in the fragmentation and perinuclear clustering of mitochondria labeled by transfection of pOct-dsRed (Figure 1). This effect depends on Gp78 ubiquitin ligase activity, as it is not observed in cells transfected with Gp78 mutated in the RING-finger domain (Flag-RINGmut-IRES-GFP). Based on four-dimensional (4D) movies of cells expressing both pOct-dsRed and GFP, and therefore Gp78, Gp78 overexpression significantly reduced the volume, elongation, and mobility of pOct-dsRed–labeled mitochondria. No significant effects were observed in Flag-RINGmut–transfected cells, suggesting that Gp78 ubiquitin ligase activity is responsible for mitochondrial fission and fragmentation (Figure 1 and Supplemental Videos S1–S3).
FIGURE 1:

Gp78 ubiquitin ligase activity induces mitochondrial fission. (A) Cos7 cells were cotransfected with pOct-dsRed and empty vector, Flag-gp78, or Flag-RINGmut, and 3D projections of mitochondrial pOct-dsRed fluorescence were generated from confocal stacks. (B) Bar graphs show the quantification of volume (μm3) and shape (percentage of mitochondria with an x/y-axis ratio of >2) of mitochondria from individual 3D projections and speed (μm/s) of individual mitochondria from 4D movies (mean ± SEM, 15–20 cells/experiment, n = 3; *p < 0.01, **p < 0.0001).
We investigated the effect of Gp78 ubiquitin ligase expression on mitofusin levels. In Cos7 cells, levels of endogenous Mfn1 and Mfn2 levels, but not the inner mitochondrial protein OxPhosV, were significantly reduced by expression of Flag-gp78 but not Flag-RINGmut (Figure 2A). Gp78-dependent loss of Mfn1 and Mfn2 was prevented by proteasomal inhibition with MG132. Mitochondrial uncoupling with 10 μM carbonyl cyanide m-chlorophenylhydrazone (CCCP) for 24 h enhanced Flag-gp78–dependent degradation of Mfn1 and Mfn2 and also resulted in loss of OxPhosV. CCCP did not affect Mfn1 or Mfn2 levels in untransfected cells or Flag-RINGmut–transfected cells. Immunoprecipitation of endogenous Mfn1 or Mfn2 shows that they are ubiquitylated in a Gp78-dependent manner (Figure 2B). Furthermore, a coimmunoprecipitation approach shows that Gp78 interacts directly with both Mfn1 and Mfn2 in a Ring finger–dependent manner (Figure 2C). Gp78 therefore targets both Mfn1 and Mfn2 for proteasomal degradation.
FIGURE 2:

Gp78 induces ubiquitylation and proteasomal degradation of Mfn1 and Mfn2. (A) Endogenous Mfn1 or Mfn2 expression was detected by Western blot in Cos7 cells cotransfected with pcDNA, Flag-gp78, or Flag-RINGmut and treated or not with 30 μM MG132 for 4 h. Flag-gp78 and Flag-RINGmut expression was determined by anti-Flag immunoprecipitation and Western blots. Endogenous Mfn1 and Mfn2 expression was quantified by densitometry relative to β-actin (n = 3, ±SEM; *p < 0.05, **p < 0.01). (B) Endogenous Mfn1, Mfn2, and OxPhosV expression was detected by Western blot in Cos7 cells cotransfected with pcDNA, Flag-gp78, or Flag-RINGmut and treated or not with 20 μM CCCP for 4 h. Immunoprecipitates of endogenous Mfn1 and Mfn2 were immunoblotted with antibodies to Mfn1 or Mfn2, respectively, as well as ubiquitin (Ub; n = 3, ±SEM; *p < 0.05, **p < 0.01). (C) Cos 7 cells were transfected with empty vector (EV), Flag-gp78 (Gp78), Flag-RINGmut (RF), myc-Mfn1 (Mfn1), and myc-Mfn2 alone or cotransfected with Gp78-Mfn1, Gp78-Mfn2, RF-Mfn1, or RF-Mfn2. Anti-Flag immunoprecipitates were probed with antibodies to Mfn1, Mfn2, and Flag. Flag-Gp78 but not Flag-RINGmut interacts with Mfn1 and Mfn2. Flag-gp78 interacts with, reduces expression of, and ubiquitylates Mfn1 and Mfn2, indicating that Gp78 regulates mitofusin levels by targeting these proteins for ERAD.
Gp78 induction of mitophagy is Mfn1 dependent
Mitochondrial fission is required for mitophagy (Nowikovsky et al., 2007; Twig et al., 2008; Kanki and Klionsky, 2009), and the selective Gp78-dependent loss of mitochondrial OxPhosV suggests that Gp78 is inducing mitophagy. We therefore investigated whether Gp78 ubiquitin ligase regulates mitochondrial elimination upon depolarization. Cos7 cells transfected with Flag-gp78 or Flag-RINGmut and cotransfected with Flag-gp78 and hemagglutinin (HA)-Ub or HA-Ubmono, a triple lysine (K29, 48, 63R) Ub mutant that limits polyubiquitin chain extension, were incubated with CCCP for 24 h and mitochondria labeled with anti-OxPhosV. In cells transfected with Flag-gp78, mitochondrial mass was greatly reduced or even, in some cells, lost completely after CCCP treatment (Figure 3). Transfection with Flag-RINGmut did not affect mitochondrial mass after CCCP treatment. Furthermore, whereas cotransfection with HA-Ub had no effect on the ability of Flag-gp78 to induce mitochondrial loss, cotransfection with HA-Ubmono prevented Flag-gp78–induced mitophagy (Figure 3). Gp78-dependent loss of mitochondria is therefore dependent on its ubiquitin ligase activity.
FIGURE 3:

Gp78 ubiquitin ligase activity induces mitophagy of depolarized mitochondria. (A) Cos7 cells were transfected with Flag-gp78 or Flag-RINGmut or cotransfected with Flag-gp78 and either HA-Ub or HA-Ubmono, as indicated, and (B) HEK293 cells were transfected with Flag-gp78 or Flag-RINGmut. Cells were either untreated (CTL) or treated with CCCP for 24 h and labeled with anti-Flag and mitochondrial OxPhosV Abs. Bar graphs show the percentage loss of mitochondrial mass of CCCP-treated cells relative to untreated cells ((mitochondrial area[CTL − CCCP]/mitochondrial area[CTL]) ×100). For both A and B, mean ± SEM; 10–20 cells/experiment; n = 3, **p < 0.01; see Supplemental Table S1 for mitochondrial area values. (C) HEK293 cells were transfected for 24 h with either control siRNA or ATG5-specific siRNA and then transfected with Flag-gp78 for a further 24 h. Cell lysates were blotted with antibodies to Mfn1, Mfn2, OxPhosV, ATG5, and β-actin, and anti-Flag immunoprecipitates were probed with anti-Flag antibodies to reveal presence of Flag-Gp78.
Similarly, Gp78 induced mitochondrial loss upon CCCP treatment of HEK293 cells (Figure 3B). To define the contribution of autophagy to Gp78-induced Mfn1/2 degradation and mitochondrial loss, we knocked down autophagy-associated ATG5 in these cells. In the absence of CCCP, ATG5 knockdown did not affect Gp78 degradation of Mfn1 and Mfn2. Of importance, neither Gp78 expression nor ATG5 knockdown altered expression of OxPhosV. However, upon CCCP treatment, Gp78 expression induced loss of OxPhosV that was reversed by Atg5 knockdown (Figure 3C). Gp78 therefore induces ATG5-dependent mitophagy.
To demonstrate that Gp78 is a critical regulator of mitophagy, we used a lentiviral system to express Tet-inducible Gp78 short hairpin RNA (shRNA) in HT-1080 cells that express elevated levels of Gp78. After doxycycline treatment over 72 h, two Gp78-specific shRNA sequences (sh2 and sh6), but not a nontargeted control shRNA (shCN), were found to effectively knock down Gp78 protein expression, with sh6 showing a more robust reduction (∼60%) in Gp78 expression levels (Figure 4A). Reduction in Gp78 levels was associated with significant increases in Mfn1 and Mfn2 levels, confirming that expression of these mitochondrial fusion proteins is regulated by Gp78 (Figure 4A). After Gp78 knockdown with either sh2 or sh6 Gp78–targeted shRNAs, loss of mitochondria upon CCCP treatment was significantly reduced (Figure 4B). Similarly, reduced expression of the mitochondrial protein OxPhosV was partially reversed by Gp78 knockdown (Figure 4C). Endogenous Gp78 therefore regulates mitophagy in HT-1080 cells.
FIGURE 4:

Gp78 knockdown restricts mitophagy in HT-1080 fibrosarcoma cells. (A) pTRIPZ lentiviral vectors expressing nontargeted shRNA (shCN) or Gp78-targeted shRNAs (sh2 and sh6) were used to generate inducible Gp78-knockdown HT-1080 cells. shRNA expression was induced by doxycycline treatment for 72 h, and cell lysates were probed by Western blot for 3F3A, Mfn1, Mfn2 and β-actin. (B) shCN, sh2, and sh6 pTRIPZ HT-1080 cells were induced with doxycycline for 72 h and then either left untreated (CTL) or treated with CCCP for 24 h in the continued presence of doxycycline. The cells were then labeled with antibodies to mitochondrial OxPhosV (Mito) and to the ER marker calnexin (Cal) to identify the cells. Bar graphs show the percentage loss of mitochondrial mass of CCCP-treated cells relative to untreated cells ((mitochondrial area[CTL − CCCP]/mitochondrial area[CTL]) × 100; see Supplemental Table S1 for mitochondrial area values). (C) Western blots show anti-Gp78 (3F3A), mitochondrial OxPhosV, and β-actin expression in the cells. 3F3A and OxPhosV band density were quantified by densitometry relative to β-actin (n = 3, ±SEM; *p < 0.05, **p < 0.01).
To determine whether mitofusin degradation mediates Gp78 regulation of mitophagy, we knocked down Mfn1 and Mfn2 by small interfering RNA (siRNA) transfection (48 h) in HEK293 cells (Figure 5A). siRNA knockdown of either Mfn1 or Mfn2 did not affect CCCP-induced mitophagy in cells transfected with empty vector. However, whereas overexpression of Flag-gp78 enhanced CCCP-induced mitophagy in Mfn2-knockdown cells, it was no longer able to induce mitophagy in Mfn1-knockdown cells (Figure 5B). This suggests that Gp78 induction of mitophagy depends on Mfn1.
FIGURE 5:

Mfn1 is required for Gp78-dependent mitophagy upon mitochondrial depolarization. (A) Western blots show knockdown of endogenous Mfn1 and Mfn2 expression in HEK293 cells transfected with siControl or siMfn1 (left) or siMfn2 (right) for 24 and 48 h. Endogenous Mfn1/Mfn2 expression was quantified by densitometry relative to β-actin (n = 3, ±SEM; *p < 0.05, **p < 0.01). (B) siMfn1- or siMfn2-treated HEK293 cells were transfected with empty vector or Flag-gp78. Cells were either untreated (CTL) or treated with CCCP for 24 h and labeled with antibodies to Flag and mitochondrial OxPhosV. Bar graphs show the percentage loss of mitochondrial mass of CCCP-treated cells relative to untreated cells ((mitochondrial area[CTL − CCCP]/mitochondrial area[CTL]) × 100; see Supplemental Table S1 for mitochondrial area values).
Gp78 recruits EGFP-LC3 to the mitochondria-associated ER
We then assessed expression of the autophagosomal marker LC3 upon mitochondrial depolarization. After CCCP treatment of Cos7 cells transfected with Flag-gp78, but not Flag-RINGmut or empty vector, expression of the autophagosomal membrane-associated LC3-II is elevated. The limited, although significant, expression of LC3-II is likely a consequence of the low transfection efficiency of only 15–20% of the cell population. Similarly, expression of the mitochondrial marker OxPhosV was significantly reduced in CCCP-treated, Gp78-transfected cells (Figure 6A).
FIGURE 6:

Gp78 recruits the autophagosomal marker LC3 to the ER at sites of mitochondrial degradation. (A) Western blots show expression of LC3-I and LC3-II isoforms and of mitochondrial OxPhosV in Cos7 cells transfected with pcDNA (empty vector), Flag-gp78, or Flag-RINGmut and treated or not with 10 μM CCCP for 2 h. OxPhosV expression was quantified by densitometry relative to β-actin and LC3-II relative to LC3-I (n = 3, ±SEM; *p < 0.05, **p < 0.01). (B) Cos7 cells were transfected with EGFP-LC3 alone or cotransfected with EGFP-LC3 and either Flag-gp78 or Flag-RINGmut and treated with 10 μM CCCP for 24 h. Paraformaldehyde (PFA)-fixed cells were labeled with anti-Flag (red) and anti-calnexin (blue) antibodies and EGFP-LC3 fluorescence (black and white; green) imaged directly. Nuclear EGFP-LC3 label is saturated to highlight cytoplasmic and ER-localized EGFP-LC3. Bar graphs show colocalization of EGFP-LC3 and either Flag-gp78 or calnexin (mean ± SEM, n = 3, ±SEM; **p < 0.01; ***p < 0.005; bar, 10 μm). (C) Cos7 cells cotransfected with EGFP-LC3 and Flag-gp78 were treated with CCCP for 24 h and after PFA fixation labeled with anti-Flag (green) and anti-OxPhosV (blue) antibodies. EGFP-LC3 fluorescence (green) was imaged directly. Residual mitochondria in Flag-gp78 transfected cells are closely associated with the EGFP-LC3–labeled ER. Bar, 10 μm.
EGFP-LC3 exhibits, in untreated cells, a predominant cytosolic and nuclear localization. However, upon Flag-gp78 transfection, EGFP-LC3 shows a partial recruitment to the Flag-gp78 and calnexin-labeled ER that is dramatically enhanced upon CCCP treatment. CCCP-dependent EGFP-LC3 recruitment to the ER is not observed in Flag-RINGmut–transfected cells or in cells transfected only with EGFP-LC3. Quantification showed a significant increase in EGFP-LC3/Flag-gp78 and EGFP-LC3/calnexin colocalization selectively in Flag-gp78–transfected cells after CCCP treatment (Figure 6B). In CCCP-treated cells, Flag-gp78–transfected cells show reduced levels of mitochondria relative to untransfected cells (Figure 6C). The residual fragmented mitochondria are closely associated with EGFP-LC3–labeled ER domains (Figure 6C). These results are consistent with the recruitment of LC3 to a Gp78-positive ER domain that is closely associated with depolarized mitochondria.
Gp78 induced mitophagy is Parkin independent
As previously reported (Narendra et al., 2008; Geisler et al., 2010; Matsuda et al., 2010), Parkin expression could not be detected by Western blot in HeLa cells. Parkin was expressed in HEK293 cells but not detected in Cos7 or HT-1080 cells (Figure 7A). As observed for Cos7 and HEK293 cells (Figure 3), Flag-gp78 induced mitochondrial loss upon CCCP treatment in HeLa and HT-1080 cells (Figure 7B). Furthermore, Parkin knockdown in HEK293 cells did not affect the extent of Gp78-induced mitochondrial loss (Figure 8). Gp78-induced mitophagy is therefore Parkin independent.
FIGURE 7:
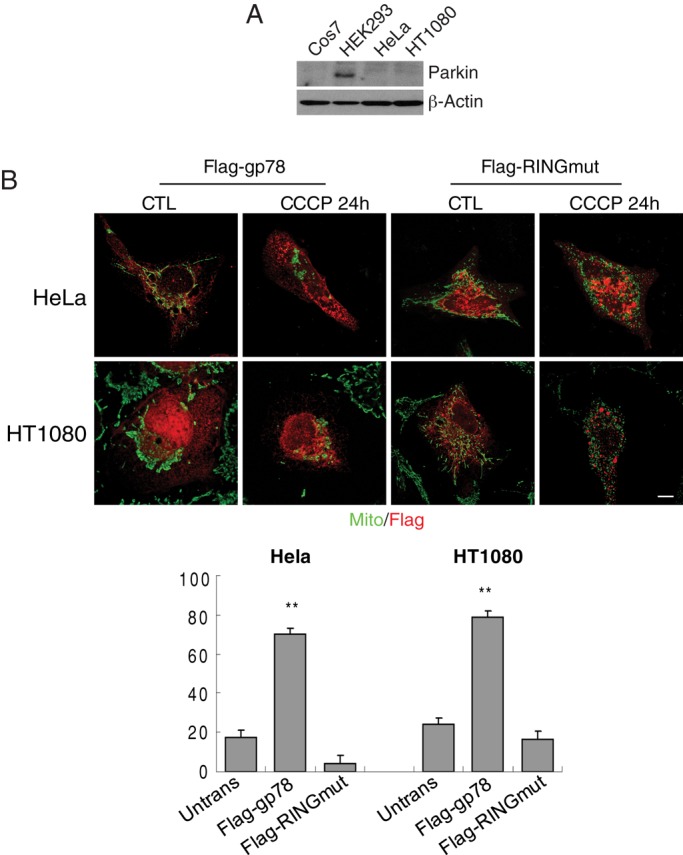
Gp78 promotes CCCP-induced mitophagy in Parkin-null HeLa cells. (A) Cos7, HeLa, HEK293, and HT-1080 cells were western blotted for Parkin and β-actin. (B) Parkin-deficient HeLa and HT-1080 cells were transfected with Flag-gp78 or Flag-RINGmut and either untreated (CTL) or treated with CCCP for 24 h, as indicated, and labeled with antibodies to Flag and OxPhosV (mito). Bar graphs show the percentage loss of mitochondrial mass of CCCP-treated cells relative to untreated cells ((mitochondrial area[CTL − CCCP]/mitochondrial area[CTL]) × 100). Mean ± SEM; 10–20 cells/experiment; n = 3, **p < 0.01; bar, 10 μM; see Supplemental Table S1 for mitochondrial area values.
FIGURE 8:
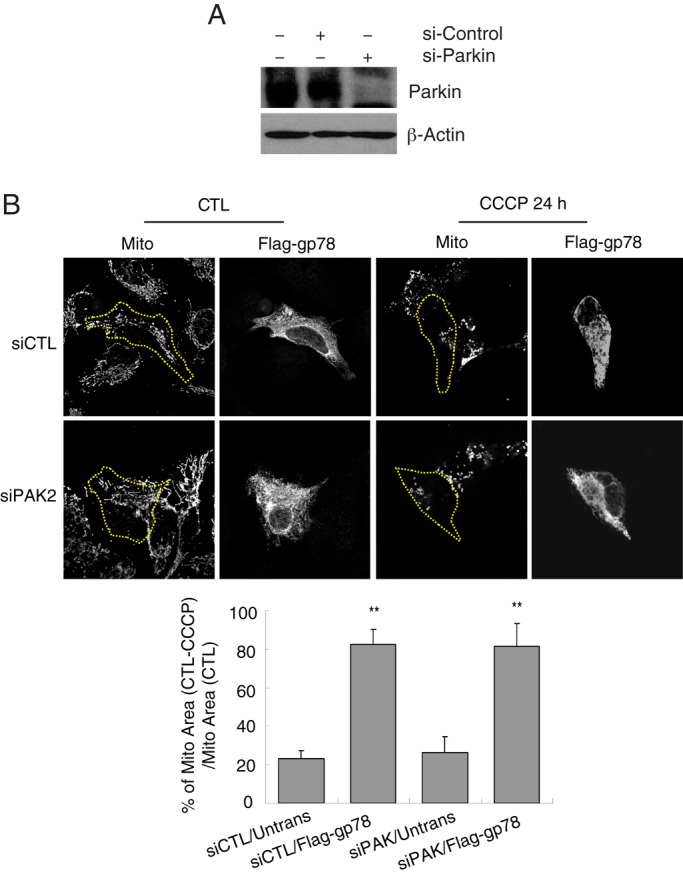
Gp78 induces mitophagy independently of Parkin. (A) Western blots show endogenous Parkin expression in cells transfected with siCTL or siParkin for 48 h. (B) Images of siControl and siParkin HEK293 cells transfected with empty vector or Flag-gp78. Cells were either untreated (UT) or treated with 10 μM CCCP for 24 h (+CCCP) and labeled with Flag and OxPhosV (mito) Abs. Bar graphs show the percentage loss of mitochondrial mass of CCCP-treated cells relative to untreated cells ((mitochondrial area[CTL − CCCP]/mitochondrial area[CTL]) × 100). Mean ± SEM; 10–20 cells/experiment; n = 3, **p < 0.01; see Supplemental Table S1 for mitochondrial area values.
DISCUSSION
Gp78 is a key component of the ERAD machinery, a process involving recognition of misfolded proteins, ubiquitylation, deglycosylation, retrotranslocation to the cytosol, and targeting to the proteasome (Fang et al., 2003; Fairbank et al., 2009). In this study, we show that Gp78 ubiquitin ligase activity induces ubiquitylation and proteasome-dependent degradation of Mfn1 and Mfn2 and regulates mitochondrial fission/fusion and mobility, as well as mitochondrial autophagy. Gp78 interacts directly with the mitofusins and induces their ubiquitylation and degradation in a RING finger–dependent manner. Gp78-induced mitofusin degradation is prevented by the proteasomal inhibitor MG132 but not inhibition of autophagy by ATG5 knockdown. In contrast, Gp78-dependent loss of inner mitochondrial OxPhosV occurs selectively after mitochondrial depolarization and is prevented by ATG5 knockdown. In HT-1080 fibrosarcoma cells, Gp78 knockdown results in elevated mitofusin levels and reduced mitochondria loss upon CCCP treatment. The mitofusins therefore represent substrates of Gp78 ubiquitin ligase activity whose loss results in mitochondrial fission and, upon mitochondrial depolarization, mitophagy.
Inhibition of mitochondrial fusion through loss of mitofusins may represent an essential element of autophagic elimination of depolarized mitochondria by segregating damaged mitochondria from the mitochondrial network (Nowikovsky et al., 2007; Twig et al., 2008; Kanki and Klionsky, 2009). Indeed, mitochondrial fusion to form tubular networks protects mitochondria from starvation-induced autophagy (Rambold et al., 2011). Induction of mitophagy by Parkin upon mitochondrial depolarization is also associated with ubiquitylation and degradation of Mfn1 and Mfn2 (Tanaka et al., 2010). However, Gp78-induced mitophagy is Parkin independent, as it is still observed in Parkin-null HeLa cells and not affected by Parkin knockdown in HEK293 cells. This identifies Gp78 as an ER-associated ubiquitin ligase capable of inducing mitophagy.
Autophagy is a lysosomal pathway involved in the turnover of cellular macromolecules and organelles. However, the origin of the initial autophagic membrane, phagophore or isolation membrane, remains uncertain and ER, mitochondria, and plasma membrane have been proposed to be potential sites of autophagosome formation (Tooze and Yoshimori, 2010; Weidberg et al., 2011). Dunn (1990) originally showed that autophagic vacuoles were not formed from plasma membrane, Golgi apparatus, or endosomal constituents but from ribosome-free regions of the rough ER. Recently 3D electron microscopic tomography described direct contacts between the phagophore and the ER, and autophagosomes were shown to derive from phosphatidylinositol 3-phosphate–enriched compartments connected to the ER (Axe et al., 2008; Hayashi-Nishino et al., 2009; Ylä-Anttila et al., 2009). Electron microscopy studies with the Gp78-specific 3F3A monoclonal antibody (mAb) localized Gp78 to the plasma membrane and smooth mitochondria-associated and peripheral ER domains (Benlimame et al., 1995, 1998; Wang et al., 1997, 2000; Goetz et al., 2007; St-Pierre et al., 2012). Recruitment of LC3 to a Gp78/calnexin-positive ER domain that is closely associated with depolarized mitochondria supports a role for the ER in mitophagy.
The basis for the selective requirement of Mfn1 and not Mfn2 for Gp78-mediated mitophagy is not clear. Both mitofusins (Mfn1 and Mfn2) are transmembrane GTPases embedded in the outer membrane of mitochondria, and Mfn1 is selectively required to mediate inner mitochondrial membrane fusion through interaction with the dynamin-related protein OPA1 (Cipolat et al., 2004). A mitochondrial-associated ubiquitin ligase, MARCH5, selectively degrades Mfn1 and not Mfn2, and loss of MARCH5 results in mitochondrial elongation and stress-induced senescence (Park et al., 2010). Ectopically expressed Mfn1 inhibits amino-terminal activation, but not mitochondrial translocation, of Bax during staurosporine-induced apoptosis, whereas overexpression of Mfn2 has no effect (Ryu et al., 2012). Our data describe a novel role for Mfn1 as a regulator of Gp78-dependent mitophagy.
These results therefore describe Gp78 as a novel regulator of mitochondrial fission and fusion and, upon mitochondrial uncoupling, mitophagy. Mitofusin degradation and mitochondrial fission are common elements associated with mitophagy induced by both the Gp78 and Parkin ubiquitin ligases. However, Gp78-induced mitophagy is associated with recruitment of LC3 to the ER and requires Mfn1, defining a distinct cellular pathway to eliminate damaged mitochondria.
MATERIALS AND METHODS
Antibodies and chemicals
3F3A anti-gp78 mAb is as described (Nabi et al., 1990). Anti-OxPhosV is from Molecular Probes (Eugene, OR), M2 anti-Flag mAb, anti-Flag pAb, and anti-calnexin polyclonal antibody (pAb) are from Sigma-Aldrich (St. Louis, MO), anti–mitofusin 2 and anti-Parkin (PRK8) mAbs are from Santa Cruz Biotechnology (Santa Cruz, CA), anti-ubiquitin pAb is from Chemicon (Temecula, CA), anti–mitofusin 1 mAb is from Abcam (Cambridge, MA), anti-LC3 is from NanoTools (Teningen, Germany), and anti-ATG5 is from Novus Biologicals (Littleton, CO). Anti-rat immunoglobulin G (IgM) is from Jackson ImmunoResearch Laboratories (West Grove, PA), and anti-mouse and rabbit IgG are from Molecular Probes. Anti-Flag M2 agarose beads are from Sigma-Aldrich, and protein A–Sepharose beads are from GE Healthcare (Piscataway, NJ). CCCP and other chemicals are from Sigma-Aldrich.
Cell culture, constructs/transfection, and treatments
Cos7 and HEK293 cells were grown in complete DMEM and transfected using Effectene (Qiagen, Valencia, CA). HA-Ub and HA-Ubmono (K29, 48, 63R) expression plasmids were obtained from Tony Morielli (University of Vermont, Burlington, VT), pOct-dsRed from Heidi McBride (University of Ottawa, Ottawa, Canada), and pEGFP-LC3 (Kabeya et al., 2000) from Noboru Mizushima (University of Tokyo, Tokyo, Japan). Myc-tagged Mfn1 and Mfn2 cDNA were purchased from Addgene (Cambridge, MA). Flag-gp78 in pcDNA3.1(+) was as described (Goetz et al., 2007), and Flag-RINGmut (C356S) was generated by point mutation using the QuikChange mutagenesis kit (Stratagene, Santa Clara, CA). For 4D analysis, Flag-gp78 and Flag-RINGmut were cloned in pIRES2-EGFP and GFP used to identify transfected cells. For CCCP treatments, cells were incubated for 24 h with complete fresh media containing 10 μM CCCP for mitophagy measurement, 2 h with 10 μM CCCP for measurement of LC3 expression and EGFP-LC3 distribution, or 4 h with 20 μM CCCP for measurement of mitofusin degradation. Specific siRNAs against Parkin, Atg5, mitofusin 1, and mitofusin 2, as well as control siRNA, were from Thermo Scientific Dharmacon (Lafayette, CO) and were transfected using Lipofectamine according to the manufacturer's instructions.
Doxycycline-inducible, Gp78-knockdown, HT-1080–stable cell lines
pGIPZ clones carrying 30–base pair shRNA-mir sequences (RHS4531-NM_001144, V2LHS_11875, V2LHS_14183, V2LHS_208794, V2LHS_206751, V3LHS_361162, V3LHS_361167, V3LHS_361166) specifically targeting Gp78 (NM_001144, AF124145, AK023874, BC017043, BC056869, BC069197, CR593778, CR625924) were purchased (Open Biosystems, ThermoFisher, Huntsville, AL) and used to establish stable Gp78-knockdown HT-1080 cell lines (St-Pierre et al., 2012). To generate doxycycline-inducible Gp78 knockdown cell lines, we used pTRIPZ vector (Open Biosystems) and transferred the effective shRNA mir sequences (#2, #3, #6, and #8) from pGIPZ to pTRIPZ by MluI–XhoI cloning strategy and verified the sequences. A second-generation packaging strategy was used to generate lentivirus particles. Briefly, ∼5 × 107 HEK293T cells, acting as a host, were plated in DMEM complete medium for 24 h and transfected on the next day with various pTRIPZ vectors by calcium phosphate method, along with GagPol Rev (M334) and Env (M5) packaging plasmids following standard protocol. At 6 h postincubation, the medium was changed, and after 48 h retrovirus particles were cleared through a 0.45-μm filter, collected, and assessed for their infective ability. HT-1080 human fibrosarcoma cancer cells were plated and incubated 24 h before subjecting them to spinoculation with retrovirus particles. Cells were selected against puromycin (2 μg/ml; Invitrogen, Carlsbad, CA) for approximately 1 wk, and resistant cells were induced with doxycycline (1 μg/ml) for 48 h and analyzed for turbo-RFP expression and also for Gp78 knockdown by both immunolabeling and Western blotting. Where indicated, CCCP was added for the final 24 h in the continued presence of doxycycline.
Immunofluorescence and live-cell imaging
Cells were fixed by precooled methanol–acetone (80–20% [vol/vol], −80°C) or, for EGFP-LC3–transfected cells, fixed with 3% paraformaldehyde, permeabilized with 0.5% Triton X-100, and labeled with 3F3A, OxPhosV, calnexin, and Flag antibodies as described previously (Goetz et al., 2007). Confocal images were obtained with the 100×/numerical aperture 1.4 UPlanApo objective of an Olympus FV1000 (Olympus, Tokyo, Japan) or the 60× PlanApo objective of a Nikon A300 confocal microscope (Nikon, Melville, NY). Colocalization was analyzed using ImagePro image analysis software (Media Cybernetics, Bethesda, MD). The 4D analysis and tracking of pOct-dsRed mitochondria was performed using a III-Zeiss spinning disk confocal microscope (Carl Zeiss, Jena, Germany) and Sharpstack image analysis software (Intelligent Imaging Innovations). Stacks were acquired every 5 s over 2 min and mitochondria movement tracked over at least five frames. To measure mitochondrial loss after CCCP treatment, cells were circumscribed and the area of mitochondrial labeling determined per cell before (CTL) and after CCCP treatment to generate a value reflecting the percentage mitochondrial loss ((mitochondrial area[CTL – CCCP]/mitochondrial area[CTL]) × 100).
Western blots and immunoprecipitation
Western blots and immunoprecipitations were as described (Fu et al., 2011; St-Pierre et al., 2012). To assess Gp78-dependent ubiquitylation, we transfected pcDNA, Flag-gp78, or Flag-RINGmut mutant for 24 h and, where indicated, cotransfected them with myc-tagged Mfn1 or Mfn2. Cell lysates were immunoprecipitated with anti-Flag M2 agarose beads (300 μg protein) or with anti-Mfn1 or Mfn2 antibodies, followed by protein A–Sepharose beads, and the immunoprecipitates were blotted with anti-Flag mAb, anti-ubiquitin pAb, or anti–mitofusin 1 and 2 mAbs. To assess expression of Gp78, LC3 isoforms, and mitochondrial markers, we probed cell lysates with 3F3A, LC3, and anti-OxPhosV antibodies, respectively. All antibodies were revealed using appropriate horseradish peroxidase–conjugated secondary antibodies and detected with ECL (GE Healthcare Bio-Sciences, Piscataway, NJ). Densitometry was performed using Quantity One V4.62 software (Bio-Rad, Hercules, CA).
Supplementary Material
Acknowledgments
This study was supported by a grant from the Canadian Institutes for Health Research (CIHR MT-15132). P.S-P. is the recipient of a Canadian Institutes for Health Research Banting and Best Graduate Scholarship.
Abbreviations used:
- ATG5
autophagy protein 5
- CCCP
carbonyl cyanide m-chlorophenylhydrazone
- CTL
control
- ECL
enhanced chemoluminescence
- EGFP
enhanced green fluorescent protein
- ER
endoplasmic reticulum
- ERAD
ER-associated protein degradation
- EV
empty vector
- Flag-gp78
Flag-tagged Gp78
- Flag-RINGmut
Flag-Gp78 containing a C356S mutation in the Ring finger domain
- Gp78
glycoprotein 78
- IRES
internal ribosome entry site
- LC3
microtubule-associated protein light chain 3
- MFN
mitofusin
- Mfn1
mitofusin1
- Mfn2
mitofusin2
- MG132
carbobenzoxy-Leu-Leu-leucinal
- OxPhosV
oxidative phosphorylation complex subunit V
- PFA
paraformaldehyde
- RFP
red fluorescent protein
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-08-0607) on February 20, 2013.
*These authors contributed equally.
REFERENCES
- Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, Ktistakis NT. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. 2008;182:685–701. doi: 10.1083/jcb.200803137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benlimame N, Le PU, Nabi IR. Localization of autocrine motility factor receptor to caveolae and clathrin-independent internalization of its ligand to smooth endoplasmic reticulum. Mol Biol Cell. 1998;9:1773–1786. doi: 10.1091/mbc.9.7.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benlimame N, Simard D, Nabi IR. Autocrine motility factor receptor is a marker for a distinct tubular membrane organelle. J Cell Biol. 1995;129:459–471. doi: 10.1083/jcb.129.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci USA. 2004;101:15927–15932. doi: 10.1073/pnas.0407043101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- Dagda RK, Cherra SJ 3rd, Kulich SM, Tandon A, Park D, Chu CT. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J Biol Chem. 2009;284:13843–13855. doi: 10.1074/jbc.M808515200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H, Dodson MW, Huang H, Guo M. The Parkinson's disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc Natl Acad Sci USA. 2008;105:14503–14508. doi: 10.1073/pnas.0803998105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn WAJ. Studies on the mechanism of autophagy: formation of the autophagic vacuole. J Cell Biol. 1990;110:1923–1935. doi: 10.1083/jcb.110.6.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairbank M, St-Pierre P, Nabi IR. The complex biology of autocrine motility factor/phosphoglucose isomerase (AMF/PGI) and its receptor, the gp78/AMFR E3 ubiquitin ligase. Mol Biosyst. 2009;5:793–801. doi: 10.1039/b820820b. [DOI] [PubMed] [Google Scholar]
- Fang S, Ferrone M, Yang C, Jensen JP, Tiwari S, Weissman AM. The tumor autocrine motility factor receptor, gp78, is a ubiquitin protein ligase implicated in degradation from the endoplasmic reticulum. Proc Natl Acad Sci USA. 2001;98:14422–14427. doi: 10.1073/pnas.251401598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang S, Lorick KL, Jensen JP, Weissman AM. RING finger ubiquitin protein ligases: implications for tumorigenesis, metastasis and for molecular targets in cancer. Semin Cancer Biol. 2003;13:5–14. doi: 10.1016/s1044-579x(02)00095-0. [DOI] [PubMed] [Google Scholar]
- Fu M, Li L, Albrecht T, Johnson JD, Kojic LD, Nabi IR. Autocrine motility factor/phosphoglucose isomerase regulates ER stress and cell death through control of ER calcium release. Cell Death Differ. 2011;18:1057–1070. doi: 10.1038/cdd.2010.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gegg ME, Cooper JM, Chau K-Y, Rojo M, Schapira AHV, Taanman J-W. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet. 2010;19:4861–4870. doi: 10.1093/hmg/ddq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- Goetz JG, Genty H, St. Pierre P, Dang T, Joshi B, Sauvé R, Vogl W, Nabi IR. Reversible interactions between smooth domains of the endoplasmic reticulum and mitochondria are regulated by physiological cytosolic calcium levels. J Cell Sci. 2007;120:3553–3564. doi: 10.1242/jcs.03486. [DOI] [PubMed] [Google Scholar]
- Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol. 2009;11:1433–1437. doi: 10.1038/ncb1991. [DOI] [PubMed] [Google Scholar]
- Joshi B, Li L, Nabi IR. A role for KAI1 in promotion of cell proliferation and mammary gland hyperplasia by the gp78 ubiquitin ligase. J Biol Chem. 2010;285:8830–8839. doi: 10.1074/jbc.M109.074344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanki T, Klionsky DJ. Mitochondrial abnormalities drive cell death in Wolfram syndrome 2. Cell Res. 2009;19:922–923. doi: 10.1038/cr.2009.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott AB, Perkins G, Schwarzenbacher R, Bossy-Wetzel E. Mitochondrial fragmentation in neurodegeneration. Nat Rev. 2008;9:505–518. doi: 10.1038/nrn2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, aging. Rejuvenation Res. 2005;8:3–5. doi: 10.1089/rej.2005.8.3. [DOI] [PubMed] [Google Scholar]
- Liang J-S, Kim T, Fang S, Yamaguchi J, Weissman AM, Fisher EA, Ginsberg HN. Overexpression of the tumor autocrine motility factor receptor Gp78, a ubiquitin protein ligase, results in increased ubiquitinylation and decreased secretion of apolipoprotein B100 in HepG2 cells. J Biol Chem. 2003;278:23984–23988. doi: 10.1074/jbc.M302683200. [DOI] [PubMed] [Google Scholar]
- Matsuda N, et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol. 2010;189:211–221. doi: 10.1083/jcb.200910140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morito D, Hirao K, Oda Y, Hosokawa N, Tokunaga F, Cyr DM, Tanaka K, Iwai K, Nagata K. Gp78 Cooperates with RMA1 in endoplasmic reticulum-associated degradation of CFTRdeltaF508. Mol Biol Cell. 2008;19:1328–1336. doi: 10.1091/mbc.E07-06-0601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabi IR, Watanabe H, Raz A. Identification of B16-F1 melanoma autocrine motility-like factor receptor. Cancer Res. 1990;50:409–414. [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowikovsky K, Reipert S, Devenish RJ, Schweyen RJ. Mdm38 protein depletion causes loss of mitochondrial K+/H+ exchange activity, osmotic swelling and mitophagy. Cell Death Differ. 2007;14:1647–1656. doi: 10.1038/sj.cdd.4402167. [DOI] [PubMed] [Google Scholar]
- Park J, et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- Park Y-Y, Lee S, Karbowski M, Neutzner A, Youle RJ, Cho H. Loss of MARCH5 mitochondrial E3 ubiquitin ligase induces cellular senescence through dynamin-related protein 1 and mitofusin 1. J Cell Sci. 2010;123:619–626. doi: 10.1242/jcs.061481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci USA. 2011;108:10190–10195. doi: 10.1073/pnas.1107402108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu SW, Choi K, Park JH, Park YM, Kim S, Choi C. Mitofusin 1 inhibits an apoptosis-associated amino-terminal conformational change in Bax, but not its mitochondrial translocation, in a GTPase-dependent manner. Cancer Lett. 2012;323:62–68. doi: 10.1016/j.canlet.2012.03.038. [DOI] [PubMed] [Google Scholar]
- Song BL, Sever N, DeBose-Boyd RA. Gp78, a membrane-anchored ubiquitin ligase, associates with Insig-1 and couples sterol-regulated ubiquitination to degradation of HMG CoA reductase. Mol Cell. 2005;19:829–840. doi: 10.1016/j.molcel.2005.08.009. [DOI] [PubMed] [Google Scholar]
- St-Pierre P, Dang T, Joshi B, Nabi IR. Peripheral endoplasmic reticulum localization of the Gp78 ubiquitin ligase activity. J Cell Sci. 2012;125:1727–1737. doi: 10.1242/jcs.096396. [DOI] [PubMed] [Google Scholar]
- Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol. 2010;191:1367–1380. doi: 10.1083/jcb.201007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tooze SA, Yoshimori T. The origin of the autophagosomal membrane. Nat Cell Biol. 2010;12:831–835. doi: 10.1038/ncb0910-831. [DOI] [PubMed] [Google Scholar]
- Tsai YC, et al. The ubiquitin ligase gp78 promotes sarcoma metastasis by targeting KAI1 for degradation. Nat Med. 2007;13:1504–1509. doi: 10.1038/nm1686. [DOI] [PubMed] [Google Scholar]
- Twig G, Hyde B, Shirihai OS. Mitochondrial fusion, fission and autophagy as a quality control axis: the bioenergetic view. Biochim Biophys Acta. 2008;1777:1092–1097. doi: 10.1016/j.bbabio.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unoki M, Nakamura Y. Growth-suppressive effects of BPOZ and EGR2, two genes involved in the PTEN signaling pathway. Oncogene. 2001;20:4457–4465. doi: 10.1038/sj.onc.1204608. [DOI] [PubMed] [Google Scholar]
- Vives-Bauza C, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci USA. 2010;107:378–383. doi: 10.1073/pnas.0911187107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H-J, Benlimame N, Nabi IR. The AMF-R tubule is a smooth ilimaquinone-sensitive subdomain of the endoplasmic reticulum. J Cell Sci. 1997;110:3043–3053. doi: 10.1242/jcs.110.24.3043. [DOI] [PubMed] [Google Scholar]
- Wang H-J, Guay G, Pogan L, Sauve R, Nabi IR. Calcium regulates the association between mitochondria and a smooth subdomain of the endoplasmic reticulum. J Cell Biol. 2000;150:1489–1498. doi: 10.1083/jcb.150.6.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidberg H, Shvets E, Elazar Z. Biogenesis and cargo selectivity of autophagosomes. Annu Rev Biochem. 2011;80:125–156. doi: 10.1146/annurev-biochem-052709-094552. [DOI] [PubMed] [Google Scholar]
- Yang Y, Ouyang Y, Yang L, Beal MF, McQuibban A, Vogel H, Lu B. Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc Natl Acad Sci USA. 2008;105:7070–7075. doi: 10.1073/pnas.0711845105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ylä-Anttila P, Vihinen H, Jokitalo E, Eskelinen E-L. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy. 2009;5:1180–1185. doi: 10.4161/auto.5.8.10274. [DOI] [PubMed] [Google Scholar]
- Zhong X, Shen Y, Ballar P, Apostolou A, Agami R, Fang S. AAA ATPase p97/valosin-containing protein interacts with gp78, a ubiquitin ligase for endoplasmic reticulum-associated degradation. J Biol Chem. 2004;279:45676–45684. doi: 10.1074/jbc.M409034200. [DOI] [PubMed] [Google Scholar]
- Ziviani E, Tao RN, Whitworth AJ. Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc Natl Acad Sci USA. 2010;107:5018–5023. doi: 10.1073/pnas.0913485107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.