

Four-dimensional live-cell imaging is combined with single-particle tracking to identify key steps in polarized epithelium cell entry by the prototype enteric virus reovirus.
Abstract
Polarized epithelial cells that line the digestive, respiratory, and genitourinary tracts form a barrier that many viruses must breach to infect their hosts. Current understanding of cell entry by mammalian reovirus (MRV) virions and infectious subvirion particles (ISVPs), generated from MRV virions by extracellular proteolysis in the digestive tract, are mostly derived from in vitro studies with nonpolarized cells. Recent live-cell imaging advances allow us for the first time to visualize events at the apical surface of polarized cells. In this study, we used spinning-disk confocal fluorescence microscopy with high temporal and spatial resolution to follow the uptake and trafficking dynamics of single MRV virions and ISVPs at the apical surface of live polarized Madin–Darby canine kidney cells. Both types of particles were internalized by clathrin-mediated endocytosis, but virions and ISVPs exhibited strikingly different trafficking after uptake. While virions reached early and late endosomes, ISVPs did not and instead escaped the endocytic pathway from an earlier location. This study highlights the broad advantages of using live-cell imaging combined with single-particle tracking for identifying key steps in cell entry by viruses.
INTRODUCTION
During natural infections by many viruses, polarized epithelial cells that line the digestive, respiratory, and genitourinary tracts form a barrier that the viruses must breach to infect their hosts. In addition, viruses encounter similarly polarized cells in other settings, including endothelial cells in the circulatory system and ependymal cells in the CNS. Knowledge of the routes and mechanisms used by viruses to enter such polarized cells is of general interest, given the broad implications for understanding pathogenesis of viral diseases and for design of novel therapeutics and vaccines.
The nonfusogenic mammalian reoviruses (MRVs) constitute one of five approved species in genus Orthoreovirus, family Reoviridae. They are large (diameter ≈ 85 nm, excluding their surface fibers) nonenveloped viruses, with a double-stranded RNA genome comprising 10 segments, and provide useful models for investigating the detailed routes and mechanisms of cell entry (Chandran and Nibert, 2003; Danthi et al., 2010) and host pathogenesis (Fields, 1992). Current understanding of the stepwise process of cell entry by MRV virions is mostly derived from in vitro studies with nonpolarized cells, as summarized in the next paragraphs.
To initiate cell entry, MRV virions attach to cell surface sugar (sialyl) moieties (Gentsch and Pacitti, 1985; Paul et al., 1989; Barton et al., 2001a; Helander et al., 2003), as well as to cell surface protein junctional adhesion molecule A (JAM-A; Barton et al., 2001b; Campbell et al., 2005). Attachment to each of these factors is mediated through a separate region of the MRV outer-fiber protein, σ1: sialyl binding through a portion of the σ1 fiber and JAM-A binding through the σ1 head (Kirchner et al., 2008; Reiter et al., 2011). Binding to sialyl moieties is thought to favor subsequent binding to JAM-A through a process of multistep adhesion strengthening (Barton et al., 2001a).
Following attachment to cells, MRV virions are internalized via the well-known, dynamin-dependent pathway of clathrin-mediated endocytosis (Sturzenbecker et al., 1987; Rubin et al., 1992; Ehrlich et al., 2004; Maginnis et al., 2008; Schulz et al., 2012). Several aspects of the data, however, suggest that other uptake pathways may be used as well (Ehrlich et al., 2004; Maginnis et al., 2008; Schulz et al., 2012). In at least some cells, β1 integrins are important cofactors for productive uptake of MRV virions (Maginnis et al., 2006, 2008), possibly through a direct interaction with the MRV core-turret protein, λ2, and/or through a role in inducing JAM-A clustering and internalization (Cera et al., 2009) or in endocytic trafficking after uptake (Maginnis et al., 2006, 2008; Mainou and Dermody, 2012).
After internalization, the MRV virion is ultimately trafficked into endosomes and lysosomes (Silverstein and Dales, 1968; Borsa et al., 1979; Sturzenbecker et al., 1987; Chandran et al., 2003; Mainou and Dermody, 2012), where it is attacked by cellular proteases (Sturzenbecker et al., 1987; Baer and Dermody, 1997; Kothandaraman et al., 1998; Ebert et al., 2002). The virion is thereby converted to an infectious subvirion particle (ISVP; Shatkin and LaFiandra, 1972; Borsa et al., 1973) via proteolytic degradation of the outer-clamp protein, σ3, and nondegradative cleavage of the T = 13 outer-shell protein, μ1 (Nibert and Fields, 1992; Dryden et al., 1993). The ISVP is an essential intermediate, because it is competent to undergo further structural changes associated with escape to the cytosol (Borsa et al., 1981; Chandran et al., 2002, 2003; Agosto et al., 2008). The structural changes in μ1 are coupled with release of its myristoylated N-terminal peptide, μ1N, which plays a direct role in membrane penetration (Odegard et al., 2004; Agosto et al., 2006; Ivanovic et al., 2008; Zhang et al., 2009).
On top of the preceding summary, during naturally occurring enteric infections, MRV virions encounter host proteases in the lumen of the digestive tract that mediate their extracellular conversion to ISVPs (Bodkin et al., 1989). As a consequence, ISVPs, not virions, are considered to initiate the first round of attachment, uptake, and entry of cells in the small intestine (Bass et al., 1990; Amerongen et al., 1994; Nibert et al., 1995). Despite this key role of ISVPs, however, their routes and mechanisms for entering cells remain a matter of debate, and indeed, little if anything is known about the strategies used by either virions or ISVPs to infect polarized epithelial cells per se.
Inhibiting clathrin-mediated endocytosis has been shown to have little or no effect on infection by ISVPs in nonpolarized cells (Maginnis et al., 2008; Schulz et al., 2012), and a recent report suggests that ISVPs might enter some nonpolarized cells via caveolar endocytosis (Schulz et al., 2012). It has also been suggested that ISVPs can penetrate the cytosol directly through the plasma membrane of nonpolarized cells (Borsa et al., 1979; Lucia-Jandris et al., 1993), without the need for any form of endocytosis. Biochemical and genetic experiments have demonstrated that JAM-A constitutes the primary receptor for productive infection of nonpolarized cells by ISVPs, as well as by virions (Barton et al., 2001b; Campbell et al., 2005), while β1 integrin appears to be important only for infection by virions (Maginnis et al., 2006). Together these observations suggest that ISVPs, as for virions described above, may exploit alternative pathways for productive entry, although those pathways have not been directly imaged.
Until recently, it was almost impossible to visualize entry of viruses from the apical surface of polarized cells, but a four-dimensional (4D) visualization approach with high temporal and spatial resolution is now available (Boulant et al., 2011; Kural et al., 2012), allowing us to follow the uptake and trafficking dynamics of single virus particles in live cells. In the current study, we took advantage of this new approach to compare the entry routes of MRV virions and ISVPs at the apical surface of polarized Madin–Darby canine kidney (MDCK) cells and found that both were internalized by clathrin-mediated endocytosis. We also found that, despite their similar uptake mechanism, virions and ISVPs exhibited strikingly different trafficking after internalization, with virions reaching early and late endosomes, while ISVPs escaped the endocytic pathway from a location reached before early endosomes.
RESULTS
Fluorescent MRV virions and ISVPs used for imaging experiments
Purified virions of MRV strain type 1 Lang (T1L) were covalently labeled with fluorescent dye Alexa Fluor (AF) 647, using a labeling density per particle that did not reduce infectivity as measured by plaque assay (3.7 ± 0.9 × 1011 plaque-forming units [PFU]/ml and 4.0 ± 1.1 × 1011 PFU/ml for AF647-labeled and nonlabeled virions, respectively). SDS–PAGE revealed that all three major surface-exposed proteins in the virions—outer-clamp protein σ3, T = 13 outer-shell protein μ1/μ1C, and core-turret protein λ2—were covalently labeled with the dye (Figure 1A, left). Fluorescent ISVPs were generated by standard chymotrypsin treatment of the AF647-labeled virions (Figure 1A), and plaque assays confirmed no reduction in infectivity (6.2 ± 1.3 × 1011 and 6.5 ± 1.5 × 1011 PFU/ml for AF647-labeled and unlabeled ISVPs, respectively). SDS–PAGE confirmed the expected removal of dye-labeled σ3 from the ISVPs (Dryden et al., 1993), but the retention of labeled proteins μ1/μ1C (in the form of cleavage products μ1/μ1C and φ, as expected [ Nibert and Fields, 1992]) and λ2 (Figure 1A, left). Imaging of the fluorescent particles by spinning-disk confocal microscopy revealed that they each belong to a single class of small, diffraction-limited objects (Figure 1A, right), consistent with presence of individual particles and absence of aggregates. Owing to the proteolytic removal of σ3, the average fluorescence intensity of AF647-labeled ISVPs was lower than that of AF647-labeled virions (Figure 1B). Comparable results were obtained for virions and ISVPs labeled with fluorescent dye AF568.
FIGURE 1:
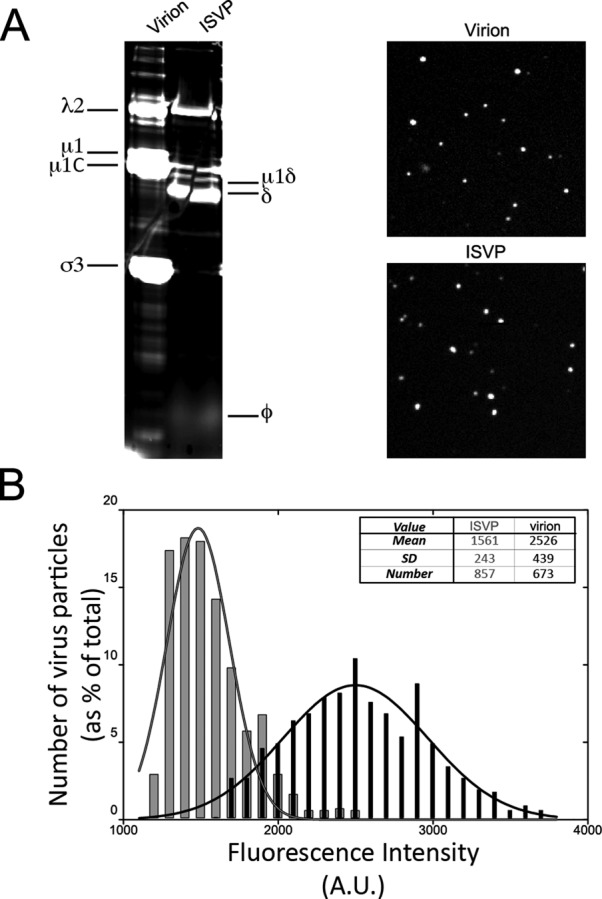
Fluorescence labeling of MRV particles. (A) Left, AF647-labeled virions and ISVPs were analyzed by SDS–PAGE, and the gel was visualized using a TYPHOON imager to identify which MRV proteins were labeled with the fluorescent dye. Right, AF647-labeled virions (top) and ISVPs (bottom) were deposited on a glass coverslip and imaged by spinning-disk confocal microscopy. (B) The fluorescence intensities of the labeled virions (black bars) and ISVPs (gray bars) imaged on coverslips were measured by use of Slidebook5 to identify particles by intensity thresholding and to record their intensities. Fluorescence intensity per pixel (in arbitrary units) is shown in bins on the x-axis, and percentage of total virions or ISVPs falling within each bin is shown on the y-axis. A Gaussian fit of the data for each type of particle is also shown. The legend shows the mean, SD, and total number of particles analyzed for each type.
Virion and ISVP uptake from the apical surface of polarized MDCK cells
To examine the uptake and entry routes of MRV virions and ISVPs in polarized cells, we used MDCK cells, which are known to support MRV infection when nonpolarized (Kothandaraman et al., 1998). Polarization of these cells was confirmed by establishment of tight junctions between cells as determined by 1) immunostaining of ZO-1 in a typical “belt” pattern surrounding the cells (Figure 2, A and C); 2) accumulation of actin, reflecting development of apical microvilli (Figure 2A); and 3) increase of transepithelial resistance from ∼ 300 to ∼ 600 ohm/cm2 after growth on Transwell supports for 3 d or longer (Figure 2B). Given the role of JAM-A as an MRV cell surface receptor (Barton et al., 2001b), we also immunostained the polarized MDCK cells for JAM-A and found it concentrated near the tight junctions, as expected, but also distributed in lower amounts throughout the apical surface, in contrast to ZO-1, which was more strongly restricted to the tight junctions (Figure 2C). Fluorescent MRV particles were added to the polarized MDCK cells and allowed to internalize for up to 30 min. Internalized MRV particles were then identified by their inaccessibility to immunostaining with anti-virion antibodies in nonpermeabilized cells.
FIGURE 2:

Evidence for MDCK cell polarization and associated localization of JAM-A. (A) MDCK cells expressing AP2-GFP were plated on coverslips 1 d (top: nonpolarized) or 3–5 d (bottom: polarized) before being fixed and then immunostained for actin (blue) and tight-junction protein ZO-1 (red). Images were obtained by spinning-disk confocal microscopy, as described in Materials and Methods. A representative transversal (side) view is also shown for each panel at top right. (B) MDCK cells were seeded on Transwell supports, and the trans-epithelial resistance (TER) was measured at 1, 2, 3, and 4 d after plating. Results are shown as the mean value ± SD from three independent experiments. (C) MDCK cells were grown on Transwell supports for 4 d, and after polarization was confirmed by TER measurement, cells were fixed and then immunostained for tight-junction protein ZO-1 (left: red) and JAM-A (right: red). Nuclei were stained using 4′,6-diamidino-2-phenylindole (blue). Z-series were obtained by scanning confocal microscopy using a Zeiss LSM-780. Representative transversal (side) views are shown for each panel and correspond to the YZ view (left strip) and the XZ view (top strip). Black arrows correspond to the focal-plane display in the XY view toward the top of the cells.
Because clathrin-mediated endocytosis is dynamin dependent, we probed the importance of this route for uptake of MRV particles by using dynasore, which inhibits dynamin-mediated scission of clathrin-coated vesicles from the plasma membrane (Macia et al., 2006; Kural et al., 2012). Clathrin-mediated endocytosis at the apical surface of polarized cells is actin dependent as well (Gottlieb et al. 1993; Jackman et al., 1994; Shurety et al., 1996; Boulant et al., 2011), and we therefore additionally probed the importance of this route by using jasplakinolide (jasp), which inhibits actin dynamics by inducing actin polymerization and stabilization (Holzinger, 2009). To probe the importance of macropinocytosis by comparison, we used amiloride, which inhibits submembranous Na+/H+ exchange essential to that process (Meier et al., 2002; Mercer and Helenius, 2009). In control experiments, we confirmed the expected activities of these inhibitors in polarized MDCK cells by showing that both dynasore and jasp blocked the apical uptake of fluorescently labeled transferrin, whereas amiloride blocked the apical uptake of fluorescently labeled dextran (Figure 3A). Dynasore and jasp also inhibited uptake of MRV virions from the apical surface of polarized MDCK cells, by ∼ 80%, whereas amiloride alone or in combination with either dynasore or jasp had little if any effect (Figure 3B). Effects on ISVP uptake by these inhibitors were very similar to those on virions (Figure 3B). These findings suggested to us that clathrin-mediated endocytosis is a major uptake route for both virions and ISVPs at the apical surface of polarized MDCK cells, whereas macropinocytosis makes little or no contribution. It is possible that jasp disrupted tight junctions, allowing for some MRV particles to be internalized at the basolateral surface by the mostly actin-independent, clathrin-mediated pathway (Boulant et al., 2011), but we consider this unlikely, because the control experiments with jasp showed no evidence for basolateral uptake of transferrin added to the apical surface (Figure 3A). Moreover, a level of residual MRV uptake (and infection; see Figure 4B) similar to that observed with jasp was also observed with dynasore treatment (Figure 3B) at conditions that fully block coated-pit formation from both surfaces (Boulant et al., 2011). The ∼20% residual uptake of MRV particles observed in cells treated with either dynasore or jasp (Figure 3B) therefore appears to be mediated by a distinct dynamin- and actin-independent pathway.
FIGURE 3:

Internalization of MRV particles from the apical surface of polarized MDCK cells. (A) Polarized MDCK cells, which had been plated on coverslips 3 d previously, were pretreated with or without jasp or amiloride. Fluorescently labeled transferrin or dextran was added to the cells in the presence or absence of the inhibitors and allowed to internalize for 7 min at 37°C; this was followed by an acid wash to remove membrane-bound transferrin or dextran. Cells were then fixed, and images were obtained by laser-scanning confocal microscopy. The panels correspond to representative maximum-intensity projections. (B) Polarized MDCK cells, which had been plated on coverslips 3 d previously, were pretreated or not with the inhibitors. AF568-labeled virions or ISVPs were then allowed to attach to the cells, after which unbound particles were removed, and internalization of bound particles was measured in the presence or absence of inhibitor(s). Results are expressed as the percentage of internalized particles among total particles counted in each cell, and are shown as the mean value ± SD from at least 10 cells for each condition.
Virion and ISVP infection from the apical surface of polarized MDCK cells
To determine which uptake pathways are specifically important for MRV infection of polarized MDCK cells, we took advantage of the neutralizing activity of monoclonal antibody 5C6 (Virgin et al., 1991), directed against the MRV receptor-binding outer-fiber protein σ1, to assay for infection (protocol diagrammed in Figure 4A; see also Materials and Methods). We thereby found that dynasore or jasp reduced apical infection by virions by ∼70%, whereas amiloride alone or in combination with either dynasore or jasp had little or no effect (Figure 4B). The inhibitors of vacuolar acidification NH4Cl and bafilomycin A1, included as positive controls, each blocked infection by virions by > 90% (Sturzenbecker et al., 1987; Martínez et al., 1996; Figure 4B). Effects on ISVP infection by the uptake inhibitors were only slightly less than those on virions (Figure 4B). These findings suggested to us that clathrin-mediated endocytosis is a major productive route of uptake for both virions and ISVPs at the apical surface of polarized MDCK cells and parallel the preceding findings obtained by imaging of internalized particles (see Figure 3B).
FIGURE 4:
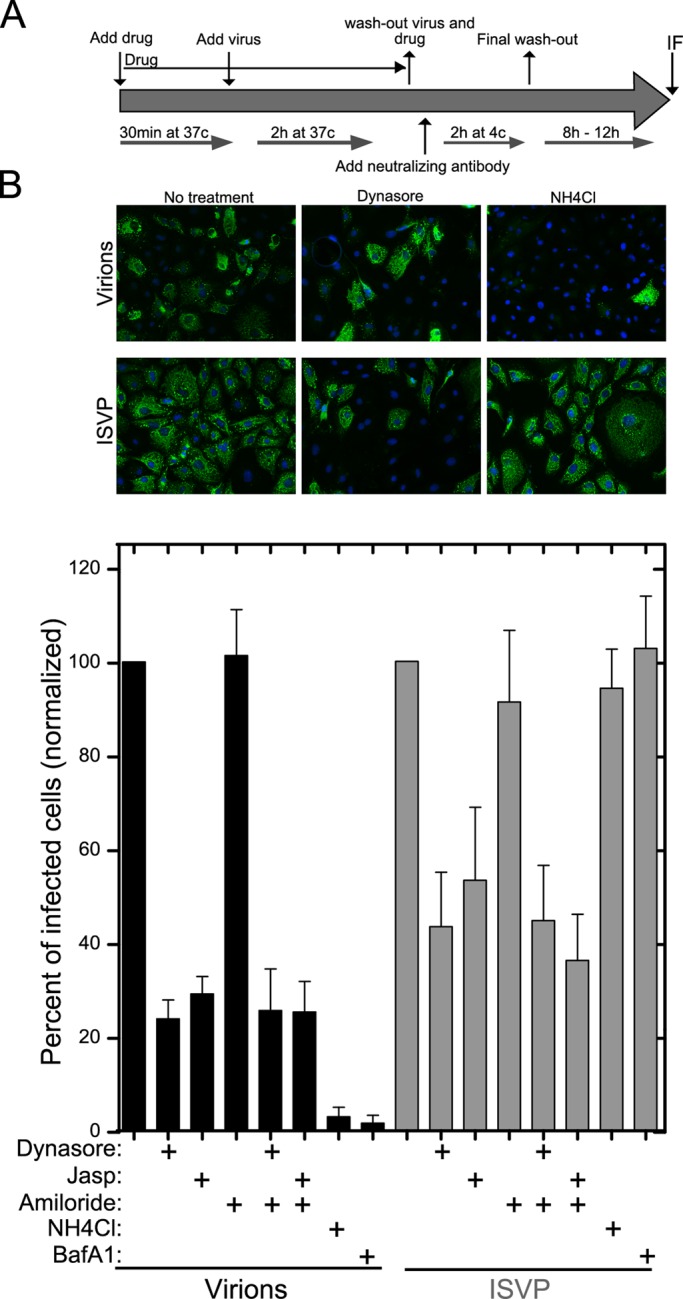
Productive infection by MRV particles following uptake at the apical surface of polarized MDCK cells. (A) The assay procedure, as detailed in Materials and Methods, is depicted schematically. MDCK cells were seeded 5 d before infection to allow complete polarization. (B) Top, representative fields of polarized MDCK cells subjected to the infection assay with virions or ISVPs in the absence or presence of inhibitors as indicated. Immunostained μNS protein, indicative of productive infection, appears green. Bottom, the number of infected cells in the presence of each inhibitor was assessed by μNS immunostaining and normalized to the number of infected cells in mock-treated samples. Data are shown as the mean value ± SD from at least 10 fields in each of three independent experiments.
Live-cell imaging of clathrin-mediated endocytosis of virions and ISVPs at the apical surface of polarized MDCK cells
We directly visualized the clathrin-mediated uptake of MRV particles from the apical surface of polarized MDCK cells using our newly implemented approach for live-cell 4D imaging by spinning-disk confocal microscopy (Boulant et al., 2011). This imaging approach allows efficient visualization with high temporal resolution (1–2 s) of diffraction-limited fluorescently tagged objects (virions, ISVPs, and clathrin/AP2-coated pits and vesicles) located on or near the dome-shaped apical surface of polarized cells. The imaging experiments were carried out using a recently developed MDCK line expressing the σ2 subunit of adaptor protein 2 fused to green fluorescent protein (AP2-GFP) (Boulant et al., 2011), which allows tracking of all AP2-containing endocytic carriers. Some experiments included transient expression of clathrin light chain A fused to fluorescent tomato protein (CLCa-TOM), to confirm that AP2 and clathrin always colocalized (Ehrlich et al., 2004; Cureton et al., 2009, 2010, 2012).
By using this approach, we obtained the first, real-time visualizations of capture and uptake of single MRV virions and ISVPs by the formation of clathrin/AP2-coated structures at the apical surface of polarized MDCK cells. Both virions and ISVPs colocalized extensively with clathrin/AP2 structures; this was followed by efficient internalization. We observed multiple such uptake events for single virions or ISVPs in all of the 4D series, as illustrated by the examples for a single virion tracked over time in Figure 5A and for virions and ISVPs in Supplemental Movies S1–S4. Typically, 30–40% of virions or ISVPs attached to the apical surface were internalized by clathrin-mediated endocytosis during the 3- to 5-min interval of the time series (Figure 5B and Movies S1–S4). Uptake by this route was divided into five phases: 1) apical cell surface attachment of an MRV virion or ISVP, and slow lateral diffusion; 2) cessation of lateral diffusion that always coincided with the site at which a clathrin/AP2 structure was forming; 3) growth of the coated pit (increase in AP2-GFP and CLCa-TOM fluorescence intensity); 4) separation of the virion- or ISVP-containing coated vesicular carrier from the apical membrane, followed by rapid release of the clathrin/AP2 coat (abrupt disappearance of AP2-GFP and CLCa-TOM fluorescence); and 5) rapid and directed intracellular movement of the virion- or ISVP-containing vesicular carrier (Figure 5A and Movies S1–S4). Also noteworthy was the distribution of clathrin-mediated uptake events across the entire apical surface of the polarized MDCK cells, and not concentrated near the edges where tight junctions are found (Movies S3 and S4), consistent with the apical distribution of some MRV receptor JAM-A (Figure 2C). In addition, the observed efficiency of virion and ISVP uptake by clathrin-mediated endocytosis in these cells was not specifically due to their polarization, as nonpolarized MDCK cells also exhibited efficient uptake of both particle types through this pathway (Figure 5B).
FIGURE 5:

Live-cell imaging and single-particle tracking of single MRV virion and ISVP particles during their association with endocytic clathrin-coated pits and vesicles at the apical surface of polarized MDCK cells. Polarized MDCK cells stably expressing σ2-GFP and transiently expressing CLCa-TOM were inoculated with fluorescent virus particles, and images were acquired by 4D spinning-disk confocal microscopy. (A) A series of 50 images acquired at 3-s intervals trace a representative example of the clathrin/AP2-associated uptake of a single MRV virion: green, AP2-GFP; red, CLCa-TOM; and blue, virion. Phases of the uptake process as described in the text are labeled. (B) Left, representative images depicting virions (top) or ISVPs (bottom) colocalizing with clathrin/AP2-coated pits on the apical surface of polarized MDCK cells. Selected examples of colocalization are highlighted by arrowheads. Right, fractions of virus particles undergoing uptake by clathrin-mediated endocytosis in polarized (plated 5 d before infection) or nonpolarized (plated 1 d before infection) MDCK cells quantified by counting. Data are shown as the mean value ± SD from at least five independent experiments.
We compared the formation characteristics of clathrin/AP2 structures alone or in association with MRV particles by determining the lifetime of coated pits. Pit lifetime is the elapsed time between the appearance and sudden disappearance of the coat constituents (Figure 6A). The lifetimes of coated pits containing or lacking MRV particles were measured in the same polarized MDCK cells (Figure 6A). Coated pits containing virions had significantly longer lifetimes (78 ± 21 s) than those lacking virions (49 ± 8 s) (Figure 6B). Similarly, coated pits containing ISVPs had significantly longer lifetimes (61 ± 13 s) than those without, but significantly shorter lifetimes than those with virions (Figure 6B). These increased coated-pit lifetimes with virions or ISVPs are not easily explained by the time it would take to assemble a larger coat, however, because pits containing or lacking MRV particles recruited similar amounts of AP2, an indicator of coat size (Ehrlich et al., 2004; Figure 6C). Thus, though not increasing coat size in polarized MDCK cells, both particle types (virions to a greater extent than ISVPs) slowed the progression of pit formation.
FIGURE 6:
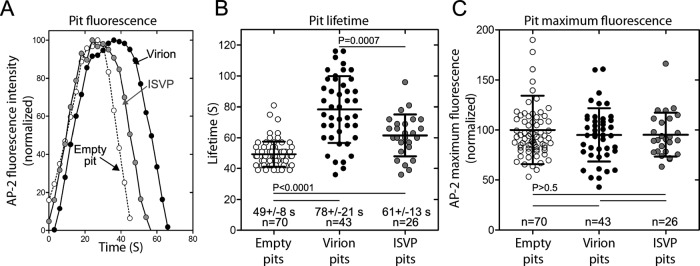
Characteristics of clathrin-coated pits associated with entry of MRV virion and ISVP particles at the apical surface of polarized MDCK cells. Data were acquired by 4D spinning-disk confocal microscopy, as indicated for Figure 5. (A) Kinetic intensity profiles of single, representative coated pits: one empty, that is, not containing a fluorescent virus particle; one containing a fluorescent virion; and one containing a fluorescent ISVP. The AP2-GFP fluorescence intensity for each time point has been normalized to the maximum AP2-GFP fluorescence intensity reached during formation of the clathrin-coated pit in each example. (B) Scatter plot of the lifetimes of coated pits lacking or containing an MRV particle. Data are shown as the mean value ± SD from three cells for pits with each type of cargo; n = number of pits analyzed. Statistical significance values for the observed differences in pit lifetimes are shown. (C) Scatter plot of the maximum AP2-GFP fluorescence intensities of coated pits lacking or containing an MRV particle. The maximum fluorescence intensity of each pit during the course of uptake has been normalized to the average maximum fluorescence intensity of the empty pits. Data are shown as the mean value ± SD from three cells for pits with each type of cargo; n = number of pits analyzed. No statistically significant differences were found.
Our live-cell imaging approach further allowed us to track MRV-containing clathrin-coated vesicles immediately after they budded from the plasma membrane. Soon after recruitment of AP2-GFP reached its peak (Figure 7A, green circles), we observed a small displacement of the MRV particle away from the plasma membrane into the cell interior (Figure 7A, red circles). This displacement corresponds to inward movement of the virion-containing coated vesicle just after budding, but before complete release of the clathrin/AP2 coat, as depicted in the schematic (Figure 7A, top panel). At about the same time that uncoating was completed, we observed a sudden but brief, high-velocity displacement of the MRV particle (Figure 7A, blue line), presumably still within its vesicular carrier. This movement is similar to one previously described during the clathrin-dependent uptake of vesicular stomatitis virus (Cureton et al., 2010). The average Z-displacement from the apical membrane of virion-containing coated vesicles before completion of uncoating (loss of AP2-GFP signal) was 472 ± 83 nm, similar to the value obtained for coated vesicles not containing virions (451 ± 124 nm) (Figure 7B). The value for ISVP-containing coated vesicles was also similar (388 ± 83 nm). Thus the presence of an enclosed MRV particle, either virion or ISVP, did not significantly affect the distance traveled from the plasma membrane before release of the clathrin/AP2 coat.
FIGURE 7:

Displacement of clathrin-coated vesicles mediating uptake of MRV virion and ISVP particles at the apical surface of polarized MDCK cells. Fluorescent virions or ISVPs were added to polarized MDCK cells stably expressing AP2-GFP, and their uptake was imaged by 4D live-cell spinning-disk confocal microscopy, as described for Figure 5. (A) Kinetic data for a single, representative virion-uptake event. The fluorescence intensity of AP2-GFP associated with the clathrin-coated pit is tracked in green, the Z-displacement of the virion is tracked in red, and the velocity of X/Y-displacement of the virion is tracked in cyan. (B) Kinetic data for single, representative uptake events involving an empty pit (open circles), a virion-containing pit (black circles), or an ISVP-containing pit (gray circles). The Z-displacement of each pit is tracked relative to its original position on the cell surface. Inset shows scatter plot of the total Z-displacement of pits with each different cargo. In all cases, the Z-position was defined by the AP2-GFP signal. No statistically significant differences were found.
Trafficking of virions and ISVPs after uptake from the apical surface of polarized MDCK cells
We next used our live-cell 4D imaging approach to examine the trafficking of virions and ISVPs following their uptake by clathrin-mediated endocytosis. To mark endosomal compartments, we used polarized MDCK cells transiently expressing low levels of Rab5-GFP (marking early endosomes) or Rab7-GFP (marking late endosomes). Additionally, in order to track the two types of MRV particles simultaneously, we coinfected the cells with AF568-labeled virions and AF647-labeled ISVPs (red and blue, respectively, in Figure 8A). This tracking strategy corrected for potential cell-to-cell variability in intracellular trafficking kinetics. Virions were readily found associated with both early and late endosomes (Figure 8A), in numbers that increased with time and reached a plateau at ∼20–25 min postinfection (Figure 8B). In striking contrast, substantially fewer ISVPs were found in either early or late endosomes (Figure 8A), and their numbers in these compartments did not increase with time (Figure 8A and B). Furthermore, we observed numerous, intracellular ISVPs that failed to colocalize with either Rab5 or Rab7 (Figure 8A).
FIGURE 8:

Endosomal association of MRV virion and ISVP particles after cell entry. Cells transiently expressing Rab5-GFP or Rab7-GFP for 16 h were inoculated with fluorescent virus particles, and Z-series images were acquired from individual cells at different times postinoculation by 4D spinning-disk confocal microscopy, as described in Materials and Methods. (A) Maximum-intensity projection images of MDCK cells expressing early endosome marker Rab5-GFP (left panels) or late endosome marker Rab7-GFP (right panels) at 30 min postinoculation with both AF563-labeled virions (red) and AF647-labeled ISVPs (blue). White boxes correspond to the magnified insets. (B) Quantification of the fraction of virions and ISVPs (same particles as shown in A) colocalizing with Rab5-GFP (left panel) and Rab7-GFP (right panel). The number of virions or ISVPs in these endosomal compartments at different times postinoculation was normalized to the total number of virions or ISVPs in those cells at each respective time. The histogram represents ± SD for 10 cells analyzed. (C) Similar experiment as described in (B), but this time inoculating the cells with AF647-labeled ISVPs (blue) and AF563-labeled μ1(N42A)-ISVPs (magenta). The histogram represents the mean ± SD for 10 cells analyzed.
Failure to detect substantial numbers of ISVPs in early or late endosomes of polarized MDCK cells could be the result of ISVPs 1) having escaped the endocytic pathway from a location reached before early endosomes or 2) having been redirected such that they were unable to reach those endosomes. To try to distinguish between these two main possibilities, we coinfected polarized MDCK cells with AF568-labeled ISVPs and AF647-labeled μ1(N42A)-ISVPs, the latter of which are unable to escape to the cytosol, because the mutant μ1 protein cannot mediate membrane penetration (Odegard et al., 2004; Agosto et al., 2006). The μ1(N42A)-ISVPs were found to associate with AP2-GFP and undergo uptake by clathrin-mediated endocytosis at the apical surface of polarized MDCK cells with similar efficiency to native ISVPs (Movie S5). However, while native ISVPs failed to associate with early or late endosomes in the coinfected cells (Figure 8C), μ1(N42A)-ISVPs were readily detected in both these compartments, marked by Rab5-GFP and Rab7-GFP, respectively (Figure 8C). Given that the penetration-incompetent μ1(N42A)-ISVPs, but not the penetration-competent native ISVPs, reached early and late endosomes after efficient uptake by clathrin-mediated endocytosis, we conclude that native ISVPs escaped the endocytic pathway from a location reached before early endosomes.
DISCUSSION
The above results lead us to the following main conclusions. 1) MRV virions and ISVPs underwent efficient uptake at the apical surface of polarized MDCK cells by clathrin-mediated endocytosis, with little or no evidence for uptake by macropinocytosis. 2) Clathrin-mediated endocytosis at the apical surface of these cells led efficiently in turn to productive infection by both virions and ISVPs. 3) Clathrin-mediated endocytosis of virions and ISVPs was widely distributed at the apical surface of these cells and not concentrated at regions near tight junctions, consistent with our evidence that the MRV receptor JAM-A was found throughout the apical surface in these cells. Additional conclusions pertain to real-time studies by live-cell 4D imaging of individual MRV particles undergoing clathrin-mediated endocytosis at the apical surface of polarized MDCK cells. 4) Virions and ISVPs both lengthened the lifetime of coated-pit formation, and virions did so to a greater extent than ISVPs. 5) Virions and ISVPs did not affect coat size or intracellular displacement before uncoating. Finally, 5) following internalization, virions underwent efficient trafficking to early and late endosomes, whereas ISVPs did not and instead escaped the endocytic pathway from a location reached before early endosomes. Important implications of these conclusions are discussed below.
Details of clathrin-mediated endocytosis of MRV revealed by live-cell 4D imaging
Incorporation of MRV virions into clathrin-coated pits at the apical surface of polarized MDCK cells significantly prolonged the pits’ mean lifetime (78 vs. 49 s for pits lacking an MRV virion in the same cells) but did not alter the number of clathrin and adaptor molecules that ultimately assembled into the endocytic coat. Virion incorporation therefore altered the rate of one or more steps in the pit-maturation process, but not the eventual size of the endocytic vesicle. Analyses of individual traces revealed that coated pits containing or lacking an MRV virion recruited clathrin and adaptor at similar rates during the earlier stages of pit formation, but virion-containing pits took longer to complete the later stages of pit maturation. These findings suggest that assembly of the coat around the first one-half to two-thirds of the virion does not constitute a limitation, but that completion of coat assembly and pit constriction into a vesicle likely take longer due to steric hindrance created by the virion. Some perturbation to coated-pit formation is in fact expected, because the diameter of the MRV virion (85 nm, excluding the σ1 fibers, which would nearly double the particle diameter if fully extended [ Furlong et al., 1988; Dryden et al., 1993]) approaches the upper limit for the internal diameter of a canonical coated vesicle (Fotin et al., 2006; Kirchhausen, 2009). Because the σ1 fiber can bend at several points along its length, including near its base (Furlong et al., 1988; Fraser et al., 1990), we suspect it may contribute little to the effective diameter of virions and ISVPs as far as coated-pit formation is concerned. Completion of coated-pit formation may therefore have been slowed less by ISVPs (mean pit lifetime: 61 s) than by virions, because the effective diameter of the ISVP is smaller, due to the absence of outer-clamp protein σ3 (Dryden et al., 1993).
We previously reported that the lifetime of coated pits engaged in the capture of MRV virions averaged 400 s in nonpolarized BSC-1 cells (Ehrlich et al., 2004), much longer than the 78-s mean lifetime in polarized MDCK cells reported here. Our previous study indicated, however, that only a small fraction of MRV virions was internalized by clathrin-mediated endocytosis. In contrast, we now find evidence for very efficient clathrin-mediated capture of virions at the apical surface of polarized MDCK cells. We believe that this difference may have been due in part to a relative deficiency of one or more MRV cell surface receptors in the BSC-1 cells. Indeed, BSC-1 cells are greatly deficient in JAM-A (the MRV protein receptor [ Barton et al., 2001a; Campbell et al., 2005]) relative to MDCK cells, and transient expression of JAM-A in BSC-1 cells substantially increases the capture of virions by clathrin/AP2 carriers (S. Boulant, unpublished data).
Alternative uptake pathways for MRV virions
Several studies preceding this one have provided evidence that MRV virions can enter nonpolarized cells by clathrin-mediated endocytosis (Sturzenbecker et al., 1987; Rubin et al., 1992; Ehrlich et al., 2004; Maginnis et al., 2008; Schulz et al., 2012); however, in some of those same studies, inhibitors of clathrin-mediated endocytosis did not completely block virion uptake and infection, suggesting that virions can productively enter cells via one or more alternative routes as well. In our current study, even though we obtained evidence that clathrin-mediated endocytosis is a major uptake route for productive infection by MRV virions at the apical surface of polarized MDCK cells, 20–30% of virion uptake and infection occurred, despite inhibition of the clathrin-dependent route. Thus one or more clathrin-independent routes for productive uptake of MRV virions must have also been available for use, though those routes remain to be elucidated in polarized MDCK cells.
One question that naturally arises from the current results is whether different, productive uptake routes may be used by MRV virions to infect nonpolarized versus polarized cells, and to infect polarized cells from their apical versus basolateral surfaces. This study was not designed to address those questions, but during the course of our experiments, we found that MDCK cells before polarization appeared to be as efficient at internalizing MRV virions by clathrin-mediated endocytosis as after polarization. Thus the large difference in high efficiency of clathrin-mediated endocytosis identified here with polarized MDCK cells compared with the lower efficiency described earlier in nonpolarized BSC-1 cells (Ehrlich et al., 2004) is likely to reflect a difference between these two cell types unrelated to polarization, perhaps due to the lower level of JAM-A expression in BSC-1 cells. We suspect that clathrin-mediated endocytosis is the main productive uptake route for MRV virions in most types of cells, unless a deficiency in JAM-A, and/or perhaps β1 integrins (Maginnis et al., 2006, 2008), allows alternative uptake routes, such as macropinocytosis, to gain greater prominence.
Uptake, trafficking, and escape of ISVPs
We propose here that clathrin-mediated endocytosis constitutes an efficient and productive uptake route for ISVPs, as well as virions. To our knowledge, this is the first such finding, and given that it was obtained in polarized cells, we propose that clathrin-mediated endocytosis may be an important uptake route for ISVPs in polarized epithelial cells during natural infections. Nonetheless, since inhibition of clathrin-mediated endocytosis did not completely block ISVP uptake and infection in this study, ISVPs must also exploit one or more alternative uptake routes in these cells, as also recently suggested for other, nonpolarized cells (Schulz et al., 2012).
Another key conclusion from our study is that ISVPs escaped the endocytic pathway of polarized MDCK cells after clathrin/AP2-vesicle budding and uncoating but before reaching early endosomes. These findings likely explain why previous studies in nonpolarized cells have detected few infecting ISVPs in endosomes and have furthermore detected ISVPs free in the cytosol near the plasma membrane (Borsa et al., 1979). They are also consistent with our “sequestration” model, whereby the ISVP undergoes structural changes associated with membrane penetration (Chandran et al., 2002, 2003; Odegard et al., 2004) only after its surrounding volume is restricted inside an endocytic vesicle or similar structure (Agosto et al., 2008). Whether infecting ISVPs escaped directly from uncoated vesicles in this study remains to be demonstrated. Another question of interest concerns ISVPs generated from virions only after proteolysis inside late endosomes or lysosomes (Silverstein and Dales, 1968; Borsa et al., 1979; Sturzenbecker et al., 1987; Baer and Dermody, 1997; Mainou and Dermody, 2012; Kothandaraman et al., 1998; Ebert et al., 2002; Chandran et al., 2003). Can ISVPs escape directly from either of those compartments, or must they first be trafficked elsewhere?
MATERIAL AND METHODS
Virus particles
Nonlabeled virions of MRV strain T1L, derived from stocks originally obtained from Bernard N. Fields (Harvard Medical School), were grown and purified by standard protocols (Furlong et al., 1988). Nonlabeled ISVPs were generated from nonlabeled virions (1 × 1013 particles/ml) by treatment with chymotrypsin (200 μg/ml) in virion buffer (150 mM NaCl, 10 mM MgCl2, 10 mM Tris, pH 7.5) at 32°C for 10 min, after which the protease was inactivated with 1 mM phenylmethylsulfonyl fluoride. Before being fluorescently labeled in vitro, virions were dialyzed overnight against phosphate-buffered saline (PBS). Amine-reactive dyes AF647 or AF568 (Molecular Probes) were solubilized in dimethyl sulfoxide at 10 mg/ml and diluted to a final concentration of 62.5 μg/ml in PBS, along with purified virions at a final concentration of 1 mg/ml. After incubation at room temperature for 90 min, labeled virions were separated from free dye using a PD-10 desalting column (GE Healthcare, Waukesha, WI) and dialyzed against virion buffer. Fluorescent ISVPs were obtained by treatment of labeled virions, as described above for nonlabeled particles. Recoated μ1(N42A)-virions (Odegard et al., 2004) were generated from T1L cores and proteins and purified using standard protocols (Chandran et al., 1999). They were then fluorescently labeled with AF647 or AF568, as described above for native virions. Fluorescent μ1(N42A)-ISVPs were prepared from μ1(N42A)-virions, as described above. All particles were titrated by plaque assay, as described previously (Middleton et al., 2002).
Antibodies and inhibitors
The following primary antibodies were used: rabbit polyclonal antibodies against virions of MRV strain T1L (Tyler et al., 1993), used at 1/1000 dilution for immunofluorescence and immunoblotting; rabbit polyclonal antibodies against MRV nonstructural protein μNS (Broering et al., 2000), used at 1/1000 dilution for immunofluorescence and immunoblotting; mouse monoclonal antibodies against ZO-1 (Invitrogen, Carlsbad, CA) and JAM-A (J10.4; Santa Cruz Biotechnology, Santa Cruz, CA) used at 1/1000 and 1/200 dilutions for immunofluorescence; and mouse monoclonal antibody 5C6 against MRV attachment protein σ1 (Virgin et al., 1991), used at 1–2 μg/ml in PBS for neutralization. Secondary antibodies were conjugated with AF488, AF-568 or AF-647 and directed against the corresponding animal source of primary antibodies (Molecular Probes). The following pharmacological inhibitors were used: 1 μM jasplakinolide (Alexis Biochemical, Farmingdale, NY), 80 μM dynasore (Macia et al., 2006), 20 mM 5-(N-ethyl-N-isopropyl) amiloride (Sigma-Aldrich, St. Louis, MO), 20 mM NH4Cl (Sigma-Aldrich), and 1 μM bafilomycin A (Sigma-Aldrich).
Cell culture and transfection
MDCK-2 cells stably expressing AP2-EGFP (Boulant et al., 2011) were grown in DMEM containing 10% fetal calf serum (FCS), penicillin, and streptomycin (Invitrogen). Cells were seeded on coverslips at 5 d before infection to allow formation of polarized monolayers. Transfected plasmids were ones encoding 1) CLCa-TOM (Cureton et al., 2009, 2010), 2) EGFP fused to early endosome marker Rab5 (Rab5-GFP; Addgene, Cambridge, MA), and 3) GFP fused to late endosome marker Rab7 (Rab7-GFP; Addgene). Transfection was carried out using TransIT-LT1 (Mirus Bio, Madison, WI), and cells were analyzed 12–16 h after transfection unless otherwise indicated.
Spinning-disk confocal microscopy
Imaging of MRV entry was performed as described for vesicular stomatitis virus and canine parvovirus (Cureton et al., 2009, 2010, 2012), and the microscopy setup was identical to that described for another recent study of polarized MDCK cells (Boulant et al., 2011). Briefly, imaging was performed in a temperature-controlled, 5% CO2, humidified chamber (20/20 Technologies, Wilmington, NC) mounted on a Marianas system platform (Intelligent Imaging Innovations, Denver, CO) based on a Zeiss AxioVert 200M inverted microscope equipped with a CSU-X1 spinning-disk confocal unit (Yokogawa Electric, Tokyo, Japan) and a spherical-aberration correction device (Infinity Photo-Optical, Boulder, CO). The correction device was needed because of the severe spherical aberration on the apical surface of polarized MDCK cells located 8–10 μm away from the coverslip.
Immunostaining
Cells grown on coverslips were fixed at room temperature for 15 min with 4% wt/vol paraformaldehyde dissolved in PBS and then processed by sequential incubation with the appropriate primary and secondary antibodies dissolved in PBS containing 0.05% wt/vol Triton X-100 and 2% FCS. Sequential optical planes spaced 200 nm in the z-axis were acquired using spinning-disk confocal microscopy, and representative maximum-intensity projection images are shown.
Internalization assay
Polarized monolayers of MDCK cells on coverslips were pretreated with respective pharmacological inhibitors for 15 min at 37°C in serum-free DMEM. Fluorescent virions or ISVPs (MOI = 100) were then added on top of the cells in the presence of the same respective inhibitors and incubated at 37°C for indicated times up to 30 min. Cells were then washed with ice-cold PBS and fixed with 0.5–1% paraformaldehyde for 5 min on ice. Cells were washed three times with ice-cold PBS and immunostained, without being permeabilized, using anti-virion polyclonal antibodies. The fraction of internalized particles determined by spinning-disk confocal microscopy was obtained using the intensity-thresholding routine in Slidebook 5 (Intelligent Imaging Innovations) to determine the number of fluorescent virus particles colocalizing (“outside”) or not (“inside”) with differently fluorescent secondary antibodies specific for the anti-virion primary antibodies. The virus particles and secondary antibodies were respectively labeled with AF568 and AF647, or with AF647 and AF568. AF647-labeled human transferrin and fluorescein-labeled dextran (Molecular Probes) were used at 50 and 200 μg/ml, respectively.
Infection assay
Polarized monolayers of MDCK cells were washed with PBS, and serum-free DMEM with respective pharmacological inhibitors was added for 30 min at 37°C. Virions or ISVPs were then added on top of the cells (MOI = 3) and incubated for 2 h at 37°C to allow attachment and uptake in the presence of the same respective inhibitors. Cells were washed three times with ice-cold PBS, and anti-σ1 monoclonal antibody 5C6 was then added for 2 h at 4°C to neutralize any MRV particles that had not yet been internalized. Cells were washed three times with ice-cold PBS, and fresh complete medium was added for 8–12h at 37°C to allow MRV gene expression and replication. Cells were then fixed with 4% paraformaldehyde and immunostained using anti-μNS polyclonal antibodies and fluorescent secondary antibodies. The fraction of infected cells (i.e., cells expressing μNS) was determined by fluorescence microscopy. The neutralizing activity of 5C6 was validated beforehand in comparable experiments in nonpolarized cells.
Live-cell imaging of virus uptake
MRV particles (MOI = 100) were added atop polarized monolayers of MDCK cells at 2–3 min before imaging. Virus uptake was then monitored by 4D spinning-disk confocal microscopy. Three-dimensional (3D) movies comprising Z-stacks of three to five consecutive optical planes spaced 0.5 μm apart were acquired at a frequency of 0.3 Hz using a 100-ms exposure time for a total of 50–100 frames. Two-dimensional movies were generated from these 3D movies by merging the maximum-intensity Z-projection for a particular virus particle from each frame. AP2-coated fluorescent structures were identified and tracked by using a 3D-tracking MATLAB routine (Kural et al., 2012).
Live-cell imaging of virus trafficking
At 16 h before infection, MDCK cells were transfected according to the manufacturer's instructions with plasmids encoding Rab5-GFP or Rab7-GFP. Fluorescent virions, ISVPs, or μ1(N42A)-ISVPs (MOI = 100) were then added on top of the transfected cells, and at different times postinfection, a Z-series encompassing the full volume of at least 10 cells was acquired by 4D spinning-disk confocal microscopy. Virus particles were identified using an intensity-thresholding routine in Slidebook 5 (Intelligent Imaging Innovations), and their colocalization with the endosomal markers was ascertained. The fraction of virus particles colocalizing with either marker was normalized to the total number of particles detected in the respective compartments at 30 min postinfection. A minimum of 75 particles was counted for each histogram value shown in Figure 8.
Supplementary Material
Acknowledgments
We thank E. Marino (supported by National Institutes of Health [NIH] grant U54-AI057159, New England Regional Center of Excellence in Biodefense and Emerging Infectious Diseases) for maintaining the imaging resource used in this study. This work was additionally supported by NIH grants R01-GM075252 (T.K.) and R56-AI067445 (M.L.N). S.B. was also supported by a Harvard Digestive Disease Consortium Feasibility Award, a GlaxoSmithKline fellowship, and the C.H.S. Foundation.
Abbreviations used:
- 3D
three-dimensional
- 4D
four-dimensional
- AF
Alexa Fluor
- AP2-GFP
adaptor protein 2 fused to green fluorescent protein
- CLCa-TOM
clathrin light chain A fused to fluorescent tomato protein
- ISVP
infectious subvirion particle
- JAM-A
junctional adhesion molecule A
- jasp
jasplakinolide
- MDCK
Madin–Darby canine kidney
- MRV
mammalian reovirus
- PBS
phosphate-buffered saline
- PFU
plaque-forming units
- T1L
type 1 Lang
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-12-0852) on February 20, 2013.
Present addresses: *Schaller Research Group at University of Heidelberg and DKFZ and Department of Infectious Diseases/Virology, University of Heidelberg, 69120 Heidelberg, Germany
†Department of Physics, Ohio State University, Columbus, OH 43210
‡Influenza Division, Centers for Disease Control and Prevention, Atlanta, GA 30333.
§Co–senior authors.
REFERENCES
- Agosto MA, Ivanovic T, Nibert ML. Mammalian reovirus, a nonfusogenic nonenveloped virus, forms size-selective pores in a model membrane. Proc Natl Acad Sci USA. 2006;103:16496–16501. doi: 10.1073/pnas.0605835103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agosto MA, Myers KS, Ivanovic T, Nibert ML. A positive-feedback mechanism promotes reovirus particle conversion to the intermediate associated with membrane penetration. Proc Natl Acad Sci USA. 2008;105:10571–10576. doi: 10.1073/pnas.0802039105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amerongen HM, Wilson GA, Fields BN, Neutra MR. Proteolytic processing of reovirus is required for adherence to intestinal M cells. J Virol. 1994;68:8428–8432. doi: 10.1128/jvi.68.12.8428-8432.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baer GS, Dermody TS. Mutations in reovirus outer-capsid protein sigma3 selected during persistent infections of L cells confer resistance to protease inhibitor E64. J Virol. 1997;71:4921–4928. doi: 10.1128/jvi.71.7.4921-4928.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton ES, Connolly JL, Forrest JC, Chappell JD, Dermody TS. Utilization of sialic acid as a coreceptor enhances reovirus attachment by multistep adhesion strengthening. J Biol Chem. 2001a;276:2200–2211. doi: 10.1074/jbc.M004680200. [DOI] [PubMed] [Google Scholar]
- Barton ES, Forrest JC, Connolly JL, Chappell JD, Liu Y, Schnell FJ, Nusrat A, Parkos CA, Dermody TS. Junction adhesion molecule is a receptor for reovirus. Cell. 2001b;104:441–451. doi: 10.1016/s0092-8674(01)00231-8. [DOI] [PubMed] [Google Scholar]
- Bass DM, Bodkin D, Dambrauskas R, Trier JS, Fields BN, Wolf JL. Intraluminal proteolytic activation plays an important role in replication of type 1 reovirus in the intestines of neonatal mice. J Virol. 1990;64:1830–1833. doi: 10.1128/jvi.64.4.1830-1833.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodkin DK, Nibert ML, Fields BN. Proteolytic digestion of reovirus in the intestinal lumens of neonatal mice. J Virol. 1989;63:4676–4681. doi: 10.1128/jvi.63.11.4676-4681.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borsa J, Copps TP, Sargent MD, Long DG, Chapman JD. New intermediate subviral particles in the in vitro uncoating of reovirus virions by chymotrypsin. J Virol. 1973;11:552–564. doi: 10.1128/jvi.11.4.552-564.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borsa J, Morash BD, Sargent MD, Copps TP, Lievaart PA, Szekely JG. Two modes of entry of reovirus particles into L cells. J Gen Virol. 1979;45:161–170. doi: 10.1099/0022-1317-45-1-161. [DOI] [PubMed] [Google Scholar]
- Borsa J, Sargent MD, Lievaart PA, Copps TP. Reovirus: evidence for a second step in the intracellular uncoating and transcriptase activation process. Virology. 1981;111:191–200. doi: 10.1016/0042-6822(81)90664-4. [DOI] [PubMed] [Google Scholar]
- Boulant S, Kural C, Zeeh JC, Ubelmann F, Kirchhausen T. Actin dynamics counteract membrane tension during clathrin-mediated endocytosis. Nat Cell Biol. 2011;13:1124–1131. doi: 10.1038/ncb2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broering TJ, McCutcheon AM, Centonze VE, Nibert ML. Reovirus nonstructural protein μNS binds to core particles but does not inhibit their transcription and capping activities. J Virol. 2000;74:5516–5524. doi: 10.1128/jvi.74.12.5516-5524.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JA, Schelling P, Wetzel JD, Johnson EM, Forrest JC, Wilson GA, Aurrand-Lions M, Imhof BA, Stehle T, Dermody TS. Junctional adhesion molecule A serves as a receptor for prototype and field-isolate strains of mammalian reovirus. J Virol. 2005;79:7967–7978. doi: 10.1128/JVI.79.13.7967-7978.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cera MR, Fabbri M, Molendini C, Corada M, Orsenigo F, Rehberg M, Reichel CA, Krombach F, Pardi R, Dejana E. JAM-A promotes neutrophil chemotaxis by controlling integrin internalization and recycling. J Cell Sci. 2009;122:268–277. doi: 10.1242/jcs.037127. [DOI] [PubMed] [Google Scholar]
- Chandran K, Farsetta DL, Nibert ML. Strategy for nonenveloped virus entry: a hydrophobic conformer of the reovirus membrane penetration protein μ1 mediates membrane disruption. J Virol. 2002;76:9920–9933. doi: 10.1128/JVI.76.19.9920-9933.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandran K, Nibert ML. Animal cell invasion by a large nonenveloped virus: reovirus delivers the goods. Trends Microbiol. 2003;11:374–382. doi: 10.1016/s0966-842x(03)00178-1. [DOI] [PubMed] [Google Scholar]
- Chandran K, Parker JSL, Ehrlich M, Kirchhausen T, Nibert ML. The δ region of outer-capsid protein μ1 undergoes conformational change and release from reovirus particles during cell entry. J Virol. 2003;77:13361–13375. doi: 10.1128/JVI.77.24.13361-13375.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandran K, Walker SB, Chen Y, Contreras CM, Schiff LA, Baker TS, Nibert ML. In vitro recoating of reovirus cores with baculovirus-expressed outer-capsid proteins μ1 and σ3. J Virol. 1999;73:3941–3950. doi: 10.1128/jvi.73.5.3941-3950.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cureton DK, Harbison CE, Cocucci E, Parrish CR, Kirchhausen T. Limited transferrin receptor clustering allows rapid diffusion of canine parvovirus into clathrin endocytic structures. J Virol. 2012;86:5330–5340. doi: 10.1128/JVI.07194-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cureton DK, Massol RH, Saffarian S, Kirchhausen TL, Whelan SPJ. Vesicular stomatitis virus enters cells through vesicles incompletely coated with clathrin that depend upon actin for internalization. PLoS Pathog. 2009;5:e1000394. doi: 10.1371/journal.ppat.1000394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cureton DK, Massol RH, Whelan SPJ, Kirchhausen T. The length of vesicular stomatitis virus particles dictates a need for actin assembly during clathrin-dependent endocytosis. PLoS Pathog. 2010;6:e1001127. doi: 10.1371/journal.ppat.1001127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danthi P, Guglielmi KM, Kirchner E, Mainou B, Stehle T, Dermody TS. From touchdown to transcription: the reovirus cell entry pathway. Curr Top Microbiol Immunol. 2010;343:91–119. doi: 10.1007/82_2010_32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dryden KA, Wang G, Yeager M, Nibert ML, Coombs KM, Furlong DB, Fields BN, Baker TS. Early steps in reovirus infection are associated with dramatic changes in supramolecular structure and protein conformation: analysis of virions and subviral particles by cryoelectron microscopy and image reconstruction. J Cell Biol. 1993;122:1023–1041. doi: 10.1083/jcb.122.5.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert DH, Deussing J, Peters C, Dermody TS. Cathepsin L and cathepsin B mediate reovirus disassembly in murine fibroblast cells. J Biol Chem. 2002;277:24609–24617. doi: 10.1074/jbc.M201107200. [DOI] [PubMed] [Google Scholar]
- Ehrlich M, Boll W, Van Oijen A, Hariharan R, Chandran K, Nibert ML, Kirchhausen T. Endocytosis by random initiation and stabilization of clathrin-coated pits. Cell. 2004;118:591–605. doi: 10.1016/j.cell.2004.08.017. [DOI] [PubMed] [Google Scholar]
- Fields BN. Studies of reovirus pathogenesis reveal potential sites for antiviral intervention. Adv Exp Med Biol. 1992;312:1–14. doi: 10.1007/978-1-4615-3462-4_1. [DOI] [PubMed] [Google Scholar]
- Fotin A, Kirchhausen T, Grigorieff N, Harrison SC, Walz T, Cheng Y. Structure determination of clathrin coats to subnanometer resolution by single particle cryo-electron microscopy. J Struct Biol. 2006;156:453–460. doi: 10.1016/j.jsb.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser RD, Furlong DB, Trus BL, Nibert ML, Fields BN, Steven AC. Molecular structure of the cell-attachment protein of reovirus: correlation of computer-processed electron micrographs with sequence-based predictions. J Virol. 1990;64:2990–3000. doi: 10.1128/jvi.64.6.2990-3000.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furlong DB, Nibert ML, Fields BN. Sigma 1 protein of mammalian reoviruses extends from the surfaces of viral particles. J Virol. 1988;62:246–256. doi: 10.1128/jvi.62.1.246-256.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentsch JR, Pacitti AF. Effect of neuraminidase treatment of cells and effect of soluble glycoproteins on type 3 reovirus attachment to murine L cells. J Virol. 1985;56:356–364. doi: 10.1128/jvi.56.2.356-364.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb TA, Ivanov IE, Adesnik M, Sabatini DD. Actin microfilaments play a critical role in endocytosis at the apical but not the basolateral surface of polarized epithelial cells. J Cell Biol. 1993;120:695–710. doi: 10.1083/jcb.120.3.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helander A, Silvey KJ, Mantis NJ, Hutchings AB, Chandran K, Lucas WT, Nibert ML, Neutra MR. The viral σ1 protein and glycoconjugates containing α2-3-linked sialic acid are involved in type 1 reovirus adherence to M cell apical surfaces. J Virol. 2003;77:7964–7977. doi: 10.1128/JVI.77.14.7964-7977.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzinger A. Jasplakinolide: an actin-specific reagent that promotes actin polymerization. Methods Mol Biol. 2009;586:71–87. doi: 10.1007/978-1-60761-376-3_4. [DOI] [PubMed] [Google Scholar]
- Ivanovic T, Agosto MA, Zhang L, Chandran K, Harrison SC, Nibert ML. Peptides released from reovirus outer capsid form membrane pores that recruit virus particles. EMBO J. 2008;27:1289–1298. doi: 10.1038/emboj.2008.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman MR, Shurety WW, Ellis JA, Luzio JP. Inhibition of apical but not basolateral endocytosis of ricin and folate in Caco-2 cells by cytochalasin D. J Cell Sci. 1994;107:2547–2556. doi: 10.1242/jcs.107.9.2547. [DOI] [PubMed] [Google Scholar]
- Kirchhausen T. Imaging endocytic clathrin structures in living cells. Trends Cell Biol. 2009;19:596–605. doi: 10.1016/j.tcb.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchner E, Guglielmi KM, Strauss HM, Dermody TS, Stehle T. Structure of reovirus σ1 in complex with its receptor junctional adhesion molecule-A. PLoS Pathog. 2008;4:e1000235. doi: 10.1371/journal.ppat.1000235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kothandaraman S, Hebert MC, Raines RT, Nibert ML. No role for pepstatin-A-sensitive acidic proteinases in reovirus infections of L or MDCK cells. Virology. 1998;251:264–272. doi: 10.1006/viro.1998.9434. [DOI] [PubMed] [Google Scholar]
- Kural C, Tacheva-Grigorova SK, Boulant S, Cocucci E, Baust T, Duarte D, Kirchhausen T. Dynamics of intracellular clathrin/AP1- and clathrin/AP3-containing carriers. Cell Rep. 2012;2:1111–1119. doi: 10.1016/j.celrep.2012.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucia-Jandris P, Hooper JW, Fields BN. Reovirus M2 gene is associated with chromium release from mouse L cells. J Virol. 1993;67:5339–5345. doi: 10.1128/jvi.67.9.5339-5345.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T. Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell. 2006;10:839–850. doi: 10.1016/j.devcel.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Maginnis MS, Forrest JC, Kopecky-Bromberg SA, Dickeson SK, Santoro SA, Zutter MM, Nemerow GR, Bergelson JM, Dermody TS. β1 integrin mediates internalization of mammalian reovirus. J Virol. 2006;80:2760–2770. doi: 10.1128/JVI.80.6.2760-2770.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maginnis MS, Mainou BA, Derdowski A, Johnson EM, Zent R, Dermody TS. NPXY motifs in the β1 integrin cytoplasmic tail are required for functional reovirus entry. J Virol. 2008;82:3181–3191. doi: 10.1128/JVI.01612-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mainou BA, Dermody TS. Transport to late endosomes is required for efficient reovirus infection. J Virol. 2012;86:8346–8358. doi: 10.1128/JVI.00100-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez CG, Guinea R, Benavente J, Carrasco L. The entry of reovirus into L cells is dependent on vacuolar proton-ATPase activity. J Virol. 1996;70:576–579. doi: 10.1128/jvi.70.1.576-579.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier O, Boucke K, Hammer SV, Keller S, Stidwill RP, Hemmi S, Greber UF. Adenovirus triggers macropinocytosis and endosomal leakage together with its clathrin-mediated uptake. J Cell Biol. 2002;158:1119–1131. doi: 10.1083/jcb.200112067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer J, Helenius A. Virus entry by macropinocytosis. Nat Cell Biol. 2009;11:510–520. doi: 10.1038/ncb0509-510. [DOI] [PubMed] [Google Scholar]
- Middleton JK, Severson TF, Chandran K, Gillian AL, Yin J, Nibert ML. Thermostability of reovirus disassembly intermediates (ISVPs) correlates with genetic, biochemical, and thermodynamic properties of major surface protein μ1. J Virol. 2002;76:1051–1061. doi: 10.1128/JVI.76.3.1051-1061.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nibert ML, Chappell JD, Dermody TS. Infectious subvirion particles of reovirus type 3 Dearing exhibit a loss in infectivity and contain a cleaved σ1 protein. J Virol. 1995;69:5057–5067. doi: 10.1128/jvi.69.8.5057-5067.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nibert ML, Fields BN. A carboxy-terminal fragment of protein μ1/μ1C is present in infectious subvirion particles of mammalian reoviruses and is proposed to have a role in penetration. J Virol. 1992;66:6408–6418. doi: 10.1128/jvi.66.11.6408-6418.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odegard AL, Chandran K, Zhang X, Parker JSL, Baker TS, Nibert ML. Putative autocleavage of outer capsid protein μ1, allowing release of myristoylated peptide μ1N during particle uncoating, is critical for cell entry by reovirus. J Virol. 2004;78:8732–8745. doi: 10.1128/JVI.78.16.8732-8745.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul RW, Choi AH, Lee PWK. The α-anomeric form of sialic acid is the minimal receptor determinant recognized by reovirus. Virology. 1989;172:382–385. doi: 10.1016/0042-6822(89)90146-3. [DOI] [PubMed] [Google Scholar]
- Reiter DM, Frierson JM, Halvorson EE, Kobayashi T, Dermody TS, Stehle T. Crystal structure of reovirus attachment protein σ1 in complex with sialylated oligosaccharides. PLoS Pathog. 2011;7:e1002166. doi: 10.1371/journal.ppat.1002166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin DH, Weiner DB, Dworkin C, Greene MI, Maul GG, Williams WV. Receptor utilization by reovirus type 3, distinct binding sites on thymoma and fibroblast cell lines result in differential compartmentalization of virions. Microb Pathog. 1992;12:351–365. doi: 10.1016/0882-4010(92)90098-9. [DOI] [PubMed] [Google Scholar]
- Schulz WL, Haj AK, Schiff LA. Reovirus uses multiple endocytic pathways for cell entry. J Virol. 2012;86:12665–12675. doi: 10.1128/JVI.01861-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shatkin AJ, LaFiandra AJ. Transcription by infectious subviral particles of reovirus. J Virol. 1972;10:698–706. doi: 10.1128/jvi.10.4.698-706.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shurety W, Bright NA, Luzio JP. The effects of cytochalasin D and phorbol myristate acetate on the apical endocytosis of ricin in polarised Caco-2 cells. J Cell Sci. 1996;109:2927–2935. doi: 10.1242/jcs.109.12.2927. [DOI] [PubMed] [Google Scholar]
- Silverstein SC, Dales S. The penetration of reovirus RNA and initiation of its genetic function in L-strain fibroblasts. J Cell Biol. 1968;36:197–230. [PubMed] [Google Scholar]
- Sturzenbecker LJ, Nibert M, Furlong D, Fields BN. Intracellular digestion of reovirus particles requires a low pH and is an essential step in the viral infectious cycle. J Virol. 1987;61:2351–2361. doi: 10.1128/jvi.61.8.2351-2361.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler KL, Mann MA, Fields BN, Virgin HW., IV Protective anti-reovirus monoclonal antibodies and their effects on viral pathogenesis. J Virol. 1993;67:3446–3453. doi: 10.1128/jvi.67.6.3446-3453.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virgin HW, IV, Mann MA, Fields BN, Tyler KL. Monoclonal antibodies to reovirus reveal structure/function relationships between capsid proteins and genetics of susceptibility to antibody action. J Virol. 1991;65:6772–6781. doi: 10.1128/jvi.65.12.6772-6781.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Agosto MA, Ivanovic T, King DS, Nibert ML, Harrison SC. Requirements for the formation of membrane pores by the reovirus myristoylated μ1N peptide. J Virol. 2009;83:7004–7014. doi: 10.1128/JVI.00377-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.