

Abstract
Background
Plasmodium falciparum dihydrofolate reductase-thymidylate synthase (pfDHFR-TS) is a well-defined target of anti-malarial drug, such as pyrimethamine and cycloguanil. Emergence of malaria parasites resistant to these drugs has been shown to be associated with point mutations of the gene coding for the target enzymes. Although the 3D-structure of P. falciparum bifunctional pfDHFR-TS has been reported previously, relatively little is known about the interactions between the pfDHFR and pfTS domains and the roles of the junctional region that links the two domains together. Therefore, a thorough understanding of the interaction of the two domains and the role of the junctional region of this target is important as the knowledge could assist the development of new effective anti-malarial drugs aimed at overcoming drug-resistant malaria.
Methods
A system was developed to investigate the interaction between pfDHFR and pfTS domains and the role of the junctional region on the activity of the recombinant pfTS. Based on the ability of co-transformed plasmids coding for pfDHFR and pfTS with truncated junctional region to complement the growth of TS-deficient Escherichia coli strain χ2913recA(DE3) on minimum media without thymidine supplementation, active pfTS mutants with minimal length of junctional region were identified. Interactions between active pfDHFR and the pfTS domains were demonstrated by using a bacterial two-hybrid system.
Results
Using TS-deficient E. coli strain χ2913recA(DE3), the authors have shown for the first time that in P. falciparum a junctional region of at least 44 amino acids or longer was necessary for the pfTS domain to be active for the synthesis of thymidylate for the cells. Truncation of the junctional region of the bifunctional pfDHFR-TS further confirmed the above results, and suggested that a critical length of the junctional peptide of pfDHFR-TS would be essential for the activity of TS to catalyze the synthesis of thymidylate.
Conclusion
The present study demonstrated the interactions between the pfDHFR and pfTS domains of the bifunctional pfDHFR-TS, and revealed that the junctional region linking the two protein domains is essential for the expression of catalytically active pfTS domain. The findings could be useful since inhibition of the pfDHFR-TS domain-domain interaction could form a basis for the development of new anti-malarial drugs based on targeting the non-active site region of this important enzyme.
Keywords: Malaria, Plasmodium falciparum, DHFR-TS, Junctional region
Background
Malaria remains an important disease in many tropical and sub-tropical countries [1]. The disease has become a global health threat, with over one million deaths annually, mostly children in sub-Saharan Africa [2]. The emergence of malaria resistance to almost all the currently available anti-malarial drugs has highlighted an urgent need to identify new malarial targets, and develop new effective drugs to combat the drug-resistant parasites [3-8]. Plasmodium falciparum dihydrofolate reductase-thymidylate synthase (pfDHFR-TS) is a well-defined target of antifolate drugs such as pyrimethamine and cycloquanil. The enzyme is responsible for the production of folates as well as thymidylate (dTMP) required for DNA synthesis [9]. Unfortunately, the emergence of anti-folate resistance has compromised the utility of the drugs and presented an urgent need to discover new drug targets and to develop novel effective drugs to combat drug-resistant parasites.
Structural studies of P. falciparum DHFR-TS revealed that the native enzyme is a homodimeric protein comprising 231 residues of DHFR domain (~27 kDa) at the N-terminus, followed by a short junctional region of 89 residues (~11 kDa) and 288 residues (~34 kDa) of the TS domain at the C-terminus of the protein [10,11]. It has been postulated that such a bifunctional arrangement could have evolved as a mechanism for the tight coupling generation of reduced folates required for the synthesis of amino acids, purine, pyrimidine, and dTMP, a phenomenon called “substrate channelling”. Support for this hypothesis comes from the evidence of metabolic channelling of the H2folate produced in the TS-catalyzed reaction, which was found to proceed at a faster rate than the diffusion rate [12]. Data from the bifunctional DHFR-TS of Leishmania major and Toxoplasma gondii also supported the substrate channelling hypothesis [12,13]. Nevertheless, the mechanism of substrate channelling for pfDHFR-TS remains unclear since, based on the structure of pfDHFR-TS, such a mechanism could not explain the delivery of H2folate from the pfTS domain to the active site of pfDHFR, and an electrostatic channelling mechanism was proposed as a possible alternative method [14-16].
The junctional region (JR) linked between the DHFR and TS domains in parasitic protozoa reported thus far varies significantly in length depending upon the source [9,10,16,17]. The long JR found in P. falciparum (89 amino acids) provides a number of interactions which facilitate contacts with the DHFR domain of the opposite half of the DHFR-TS dimer, and brings the two DHFR domains closer together. Structural alignments of DHFR-TS enzymes from Cryptosporidium hominis and P. falciparum revealed that the JR played an important role in the orientation of the DHFR domain relative to TS [17]. Therefore, inhibition of the interaction between the JR, the DHFR and TS domains could be a possible approach for the development of novel effective anti-malarial drugs [18]. The present study therefore describes an approach towards understanding the interactions between DHFR and TS domains of the bifunctional pfDHFR-TS, and the important role of JR on the activity of the TS domain. This is the first successful expression of the catalytically active pfTS domain that has JR attached at its N-terminus, as all attempts in the past to express pfTS domain failed. Through deletion of the JR, it is demonstrated that a critical length of JR is required for proper folding of the pfTS domain leading to an active molecule of pfTS. The findings highlight the importance of the JR which links DHFR and TS domains and suggest a possible alternative in exploiting the non-active site region of this important enzyme in developing new anti-malarial drugs to overcome drug-resistant parasites.
Methods
Materials
Restriction endonucleases and T4 DNA ligase were obtained from New England Biolabs, Life Technology, Inc. 2′-deoxyuridylate, 5-fluoro-2′- deoxyuridylate, and NADPH were from Sigma. MTX-sepharose CL-6B (~ 1 μmole/ml) [12], H2folate [19], and CH2H4folate [20] were prepared as described. Custom primer syntheses and DNA sequencing were from BioDesign Co Ltd and Genome Institute. TS-deficient Escherichia coli strain χ2913recA(DE3) was used for the genetic complementation studies to monitor the function of TS.
Construction and transformation of recombinant plasmids
The gene coding for P. falciparum DHFR (amino acids 1–228) was amplified using a template DNA from pETpfDHFR-TS [21]. The pfJRTS mutants were constructed by the PCR mutagenesis method. The PCR reaction (100 μl) is composed of 50 ng pETpfDHFR-TS as a DNA template, 25 ρmole of each primer (Table 1), 200 μM of dNTPs, 1.5 mM MgCl2, and 2.5 units of Taq DNA polymerase in 1 x reaction buffer. The PCR conditions were as follows: 1 cycle of 94°C for 3 min, then 25 cycles of 94°C for 45 sec, annealing at 45°C for 30 sec, and extension at 72°C for 1 min. This was followed by a final extension at 72°C for 5 min. Deletion mutants of bifunctional pfDHFR-TS were constructed using the whole plasmid amplification PCR approach [22] employing recombinant plasmid pET15b carrying the P. falciparum DHFR-TS(3D7) gene as a template and sets of primers as shown in Table 2. The PCR products were analysed by agarose electrophoresis, and were further purified using a Qiaquick Gel Extraction kit.
Table 1.
Primers used for the construction of truncated pfJRTS mutants
| Name | Length (bases) | Sequence (5′ ⟶ 3′) | Utilities and description |
|---|---|---|---|
| JRTSΔ232-235 |
28 |
AAAGAATCCCATGGAACAAAATTGTATA |
Sense strand PCR primer for the construction of pET-pfJRTSΔ232-235. Bold-face letters represent Met introduced in front of Glu236. Underlined sequence is the restriction site for NcoI. |
| JRTSΔ232-251 |
28 |
AAAGAATCCCATGGAAAAGAATGATGAC |
Sense strand PCR primer for the construction of pET-pfJRTSΔ232-251. Bold-face letters represent Met-Glu introduced infront of Lys252. Underlined sequence is the restriction site for NcoI. |
| JRTSΔ232-265 |
28 |
AAAGAATCCCATGGAATTTTACAAAAAT |
Sense strand PCR primer for the construction of pET-pfJRTSΔ232-265. Bold-face letters represent Met introduced in front of Glu266. Underlined sequence is the restriction site for NcoI. |
| JRTSΔ232-271 |
28 |
AAAGAATCCCATGGACAAATATAAAATT |
Sense strand PCR primer for the construction of pET-pfJRTSΔ232-271. Bold-face letters represent Met introduced in front of Asp272. Underlined sequence is the restriction site for NcoI |
| JRTSΔ232-274 |
57 |
CCCCTCTAGAAATAATTTTGTTTAACTTTAAGAAGGAGATATACCATGAAAATTAAT |
Sense strand PCR primer for theconstruction of pET-pfJRTSΔ232-274. Bold-face letters represent Met introduced in front of Lys275. Underlined sequence is the restriction site for XbaI |
| JRTSΔ232-276 |
57 |
CCCCTCTAGAAATAATTTTGTTTAACTTTAAGAAGGAGATATACCATGAATTATGAA |
Sense strand PCR primer for the construction of pET-pfJRTSΔ232-276. Bold-face letters represent Met introduced in front of Asn277. Underlined sequence is the restriction site for XbaI |
| JRTSΔ232-277 |
57 |
CCCCTCTAGAAATAATTTTGTTTAACTTTAAGAAGGAGATATACCATGTATGAAAAT |
Sense strand PCR primer to prepare construct pET-pfJRTSΔ232-277. Bold-face letters represent Met introduced in front of Tyr278. Underlined sequence is the restriction site for XbaI |
| JRTSΔ232-299 |
28 |
AAAGAATCCCATGGAAGAGAAAAATAAA |
Sense strand PCR primer to prepare construct pET-pfJRTSΔ232-299. Bold-face letters represent Met introduced in front of Glu300. Underlined sequence is the restriction site for NcoI |
| N3TS | 22 | ACTCATGGATCCTTAAGCAGCC | Antisense strand PCR primer for the construction of all pET-pfJRTS mutants. Underlined sequence is the restriction site for BamHI |
Table 2.
Primers used for construction of truncated bifunctional pfDHFR-TS
| Name | Length (bases) | Sequence (5′ ⟶ 3′) | Utilities and description |
|---|---|---|---|
| Δ229-277 Sense |
60 |
ACAACATTGGATTTTATCATTTATAAGAAATATGAAAATGATGATGATGATGAAGAAGAA |
Sense strand PCR primer for the construction of pfDHFR-TSΔ229-277. Lys228 (italic) was followed by Tyr278(bold) |
| Δ229-277 Antisense |
60 |
TTCTTCTTCATCATCATCATCATTTTCATATTTCTTATAAATGATAAAATCCAATGTTGT |
Antisense strand PCR primer for the construction of pfDHFR-TSΔ229-277 |
| Δ229-276 Sense |
60 |
ACAACATTGGATTTTATCATTTATAAGAAAAATTATGAAAATGATGATGATGATGAAGAA |
Sense strand PCR primer for the construction of pfDHFR-TSΔ229-276. Lys228 (italic) was followed by Asn277(bold) |
| Δ229-276 Antisense |
60 |
TTCTTCATCATCATCATCATTTTCATAATTTTTCTTATAAATGATAAAATCCAATGTTGT |
Antisense strand PCR primer to amplify pfDHFR-TSΔ229-276 |
| Δ229-275 Sense |
60 |
ACAACATTGGATTTTATCATTTATAAGAAAATTAATTATGAAAATGATGATGATGATGAA |
Sense strand PCR primer for the construction of pfDHFR-TSΔ229-275. Lys228 (italic) was followed by Ile276(bold) |
| Δ229-275 Antisense |
60 |
TTCATCATCATCATCATTTTCATAATTAATTTTCTTATAAATGATAAAATCCAATGTTGT |
Antisense strand PCR primer to amplify pfDHFR-TSΔ229-275 |
| Δ229-274 Sense |
60 |
ACAACATTGGATTTTATCATTTATAAGAAAAAAATTAATTATGAAAATGATGATGATGAT |
Sense strand PCR primer for the construction of pfDHFR-TSΔ229-274. Lys228 (italic) was followed by Lys275(bold) |
| Δ229-274 Antisense |
60 |
ATCATCATCATCATTTTCATAATTAATTTTTTTCTTATAAATGATAAAATCCAATGTTGT |
Antisense strand PCR primer to amplify pfDHFR-TSΔ229-274 |
| Δ229-271 Sense |
60 |
ACAACATTGGATTTTATCATTTATAAGAAAGACAAATATAAAATTAATTATGAAAATGAT |
Sense strand PCR primer for the construction of pfDHFR-TSΔ229-271. Lys228 (italic) was followed by Asp272(bold) |
| Δ229-271 Antisense |
60 |
ATCATTTTCATAATTAATTTTATATTTGTCTTTCTTATAAATGATAAAATCCAATGTTGT |
Antisense strand PCR primer to amplify pfDHFR-TSΔ229-271 |
| Δ229-265 Sense |
60 |
ACAACATTGGATTTTATCATTTATAAGAAAGAATTTTACAAAAATGTAGACAAATATAAA |
Sense strand PCR primer for the construction of pfDHFR-TSΔ229-265. Lys228 (italic) was followed by Glu266(bold) |
| Δ229-265 Antisense | 60 | TTTATATTTGTCTACATTTTTGTAAAATTCTTTCTTATAAATGATAAAATCCAATGTTGT | Antisense strand PCR primer to amplify pfDHFR-TSΔ229-265 |
The amplified product was cloned into pAC28 expression plasmid [23] to express catalytically active pACpfDHFR. Likewise, sequences coding for truncated JR with the pfTS domain attached were amplified using the primers as listed in Table 1, and cloned into pET-15b to yield plasmids pET-pfJRTS△232-235, pET-pfJRTS△232-251, pET-pfJRTS△232-265, pET-pfJRTS△232-271, pET-pfJRTS△232-274, pET-pfJRTS△232-276, pET-pfJRTS△232-277, and pET-pfJRTS△232-299. These mutant pET-pfJRTS plasmids were transformed into electro-competent E. coli strain χ2913 cells by electroporation using pulses set at 1.8 kV, 400Ω, 25 μF and a pulse length of ~8-10 min. After centrifugation at 6,500 g at 4°C for 10 min and resuspending the cell pellets in 2 ml of 10% glycerol, a second electroporation was performed to transform 100 μl of the first transformed cells with ~50 ng of pAC-pfDHFR. After recovering the cells by addition of 0.9 ml of LB broth followed by vigorous shaking for 1 hour at 37°C, the cells were plated onto LB agar containing 100 μg/ml ampicillin and 30 μg/ml kanamycin. Colonies appeared on the plates after overnight incubation at 37°C were individually picked and grown overnight in 1 ml LB broth containing 100 μg/ml ampicillin and 30 μg/ml kanamycin at 37°C.
Truncation of JR of the bifunctional pfDHFR-TS was performed by PCR amplification using the whole recombinant plasmid pET15b carrying gene coding for the bifunctional P. falciparum DHFR-TS (3D7) as a template, and the primers as listed in Table 2. The resulting truncated mutants, i.e., pfDHFR-TSΔ229-265, pfDHFR-TSΔ229-271, pfDHFR-TSΔ229-274, pfDHFR-TSΔ229-275, pfDHFR-TSΔ229-276, and pfDHFR-TSΔ229-277, were used to co-transform pAC-28 plasmid in the complementation study.
Genetic complementation studies
Overnight culture of the co-transformants was streaked on a minimal agar plate [24] containing 100 μg/ml ampicillin, 30 μg/ml kanamycin, and 0.025 mM IPTG without thymidine supplementation. The plates were incubated at 37°C for 48 hours. Colonies appearing on this plate were considered to have positive genetic complementation, and were selected for subsequent characterization and verification of the expressed enzymes.
Expression and purification of the expressed enzymes
An overnight culture of E. coli χ2913 harbouring two plasmids, i.e., pAC-pfDHFR and truncated pET-pfJRTS with different lengths of truncated JR, was inoculated at 1% inoculum in LB containing 100 μg/ml of ampicillin, 30 μg/ml kanamycin. The bacteria were grown at 37°C until the OD600 of the cell suspension reached ~0.5-0.6, and isopropyl β-D thiogalactopyranoside (IPTG) was added at a final concentration of 0.025 mM. The culture was allowed to grow with shaking at 20°C for 20 hours. The cells were harvested by centrifugation at 6,500 g for 10 min at 4°C, washed once with 250 ml cold phosphate buffered saline, pH 7.4, resuspended in buffer A (20 mM potassium phosphate buffer, pH 7.0, 0.1 mM EDTA, 10 mM DTT, 20% glycerol) containing 0.2 M KCl, and passed through a French Pressure Cell (American Instruments Co Inc, USA) at 12,000 psi three times. After centrifugation at 20,000 g for 1 hour at 4°C, the clear supernatant of the crude sample was circulated at a flow rate of ~0.5 ml/min in a methotrexate-sepharose CL-6B column (1.5 × 5.0 cm) pre-equilibrated with buffer A containing 0.2 M KCl. After overnight circulation, the column was washed with 30 ml of buffer A containing 0.75 M KCl, followed by 20 ml of buffer A containing 0.2 M KCl. The column was then washed with 30 ml of elution buffer (50 mM TES pH 7.8, 0.1 mM EDTA, 10 mM DTT, 20% glycerol, 50 mM KCl) containing 4 mM H2folate to elute DHFR. Fractions of 1 ml were collected. Active fractions with DHFR activity were pooled, concentrated, and H2folate in the pooled fraction was removed by passing the pooled fraction through a pre-packed NAP-25 column (Pharmacia) pre-equilibrated with buffer A.
Enzyme assays and protein analysis
The activity of DHFR was determined spectrophotometrically by monitoring the rate of decrease in absorbance at 340 nm [12,25]. The standard DHFR assay (1 ml) in a 1-cm path-length cuvette was composed of 100 μM H2folate, 100 μM NADPH, 50 mM TES, pH 7.0, 75 mM β-mercaptoethanol, 1 mg/ml bovine serum albumin, and ~0.01 units of enzyme. The reaction was initiated with H2folate. One unit of DHFR activity is defined as the amount of enzyme that produces 1 μmole of product per minute at 25°C.
The activity of TS was determined by monitoring the increase of absorbance at 340 nm due to the formation of H2folate at 25°C [26]. The reaction (1 ml) in 1-cm path-length cuvette was composed of 50 mM TES, pH 7.4, 25 mM MgCl2, 1 mM EDTA, 6.5 mM HCHO, 75 mM β-mercaptoethanol, 100 μM (6R) CH2H4folate, 125 μM dUMP and the enzyme. The reaction components, except for dUMP, were incubated at 25°C for at least 5 min to obtain the baseline prior to initiation the reaction with dUMP. One unit of TS activity is defined as the amount of enzyme that produces 1 nmole of product per minute at 25°C. The activities of the expressed enzymes were reported as mean ± standard deviation.
[3H]-FdUMP binding assay
The ability of the pfJRTS mutants with truncated JR to form covalent complex with [3H]-FdUMP and CH2H4folate was investigated by incubating the mutant proteins with 0.5 μM [3H]-FdUMP (19.3 Ci/mmol), 0.1 mM CH2H4folate, and 6.5 mM formaldehyde in 50 mM TES pH 7.4, 25 mM MgCl2, 1 mM EDTA, and 75 mM β-mercaptoethanol for 15 min at room temperature. The reaction was then electrophoresed on 12.5% SDS-PAGE. After Coomassie-Blue staining and destaining, the gel was soaked with autoradiography enhancer (EN3HANCE™, New England Nuclear) with gentle shaking for 30 min at room temperature. This was followed by washing the gel with cold water and drying the gel on a piece of filter paper under vacuum at 80°C for 30 min. The dried gel was then exposed on AGFA X-ray film at -80°C with intensifying screens for three to five days before development of the X-ray film.
Bacterial two-hybrid system
Interactions between pfDHFR domain and pfTS domain including those pfTS mutants with truncated JR were investigated using an E. coli two-hybrid system (Stratagene Inc) [27,28]. The sequence coding for pfDHFR domain was cloned into pBT (bait) vector to yield pBT-pfDHFR, whereas the pfJRTS was cloned into pTRG (target) vector to yield the pTRG-pfJRTS plasmid. The two plasmids were co-transformed into BacterioMatch two-hybrid system reporter strain E. coli XL1-blue MRF’. Positive interaction is indicated by the ability of the bacteria to grow on LB agar plate supplemented with 250 μg/ml carbenicillin, 15 μg/ml tetracycline, 34 μg/ml chloramphenical, and 50 μg/ml kanamycin (LB-CTCK).
Results
Construction of pfJRTS and pfDHFR-TS mutants and growth complementation studies
Unlike the DHFR domain of the bifunctional pfDHFR-TS, which can be heterologously expressed to yield the catalytically active form of enzyme [29-32], all attempts to express the catalytically active domain of pfTS have failed. Evidence from deletion of pfDHFR-TS suggested that the amino terminus of the pfDHFR domain is important for the function of the pfTS domain, and interactions between the pfDHFR and pfTS domains are important [18]. To address the important function of JR, mutant constructs of the pfJRTS domain with various lengths of JR sequences were constructed and co-transformed with plasmid expressing the catalytically active pfDHFR domain (pAC-pfDHFR) in TS-deficient E. coli strain χ2913recA(DE3). Successful expression of catalytically active pfTS was monitored by the ability of the co-transformed bacteria to complement growth upon plating on minimum agar plate and the detectable TS activity in the crude extract of the cells.
Figure 1 illustrates the constructs of pfJRTS mutants containing varying lengths of JR attached to the C-terminus of the pfDHFR domain (Asn231). The mutant with the longest JR (pET-pfJRTS△232-235) had only four amino acids of the JR (Lys232-Asn235) deleted, while mutants with the shortest JR (pET-pfJRTS△232-299) had 68 residues (Lys232-Lys299) removed. A similar approach was undertaken to construct bifunctional pfDHFR-TS deletion mutants of which the JR sequence was shortened to compare the effects of JR deletion with the truncated mutants of pfJRTS constructs. The bifunctional pfDHFR-TS deletion mutants being constructed include pET-pfDHFR-TS△229-265, pET-pfDHFR-TS△229-271, pET-pfDHFR-TS△229-274, pET-pfDHFR-TS△229-275, pET-pfDHFR-TS△229-276, and pET-pfDHFR-TS△229-277 (Figure 2). Figure 3 shows the 89 amino acids of P. falciparum JR sequence and indicates the positions of amino acids that were chosen to prepare truncated and deletion mutants. Results from growth complementation monitored after incubation of the plates overnight at 37°C revealed that pET-pfJRTS△232-235, pET-pfJRTS△232-251, pET-pfJRTS△232-265, pET-pfJRTS△232-271, pET-pfJRTS△232-274, and pET-pfJRTS△232-276 with corresponding JR length of 85, 69, 55, 49, 46, 44 amino acid residues, respectively, could grow on minimum media (Figure 4A, lanes 2–7), while pET-pfJRTS△232-277 and pET-pfJRTS△232-299 which had the JR length of 43 and 21 residues, respectively, did not show growth complementation (Figure 4A, lanes 8–9). The results suggest that the length of JR was important for the proper folding of the TS domain and hence affected its function. The data revealed that at least 44 residues (the mutant pET-pfJRTS△232-276) were required in the case of JR of P. falciparum in order for the pfTS domain to function properly.
Figure 1.
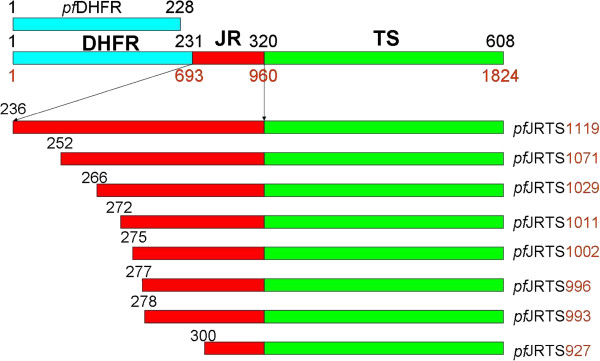
Schematic diagram representing full-length pfDHFR-TS, pfDHFR domain, and pfTS domains with truncated junction region (JR). Gene encoding amino acid residues 1–228 of pfDHFR domain was cloned into pAC28 expression vector, whereas gene fragments encoding the pfTS domain with different lengths of JR that are attached to the N-terminus (amino acid residue 320) of pfTS were cloned into the pET15b expression vector. The numbers of amino acid are shown above the gene and the names of the mutants are indicated at the right.
Figure 2.
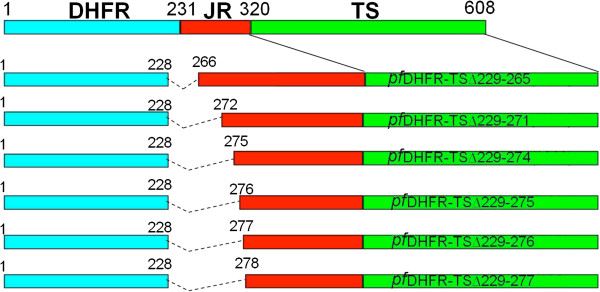
Schematic diagram representing full-length and truncated pfDHFR-TS. Constructs of pfDHFR domain containing amino acids 1–228 followed by truncated JR sequences of different lengths linked to the N-terminus of the pfTS domain were cloned into the pET15b expression vector. The numbers of amino acid are shown above the gene and the names of the mutants are indicated.
Figure 3.
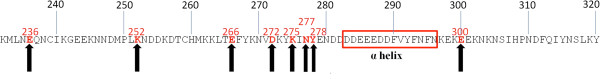
Full-length sequence of Plasmodium falciparum JR and the sequences of truncated pfJRTS mutants. The 89 amino acids of the full-length sequence of P. falciparum JR are shown, with amino acid numbers in relation to the full-length bifunctional pfDHFR-TS indicated above the sequence. Arrows indicate the amino acids corresponding to the first amino acid of the truncated pfJRTS constructs. The region where the α-helix is located is marked in the box.
Figure 4.

Growth complementation of TS-deficient Escherichia coli cells harbouring. (A) pAC-pfDHFR co-transformed with pET-pfJRTS containing truncated JR. (B) pAC28 co-transformed with pET-pfDHFR-TS with truncated JR. Cells from overnight culture were streaked on Luria-Bertani (LB), minimum media (MM), and minimum media supplemented with 0.025 mM IPTG (MM + IPTG). pET-pfDHFR-TS (3D7) was used as a controlled plasmid.
As with the result for the construct pfJRTS TSΔ229-277, TS-deficient E. coli strain χ2913 recA(DE3) transformed with pET-pfDHFR-TS△229-277and pAC-28 could not grow on a minimum agar plate (Figure 4B, lane 7). Therefore, pfDHFR-TS mutants with a longer JR sequence, i.e. pET-pfDHFR-TS△229-265, pET-pfDHFR-TS△229-271, pET-pfDHFR-TS△229-274, pET-pfDHFR-TS△229-275, and pET-pfDHFR-TS△229-276, were constructed and tested for growth complementation. Complementation studies revealed that the constructs pET-pfDHFR-TS△229-265, pET-pfDHFR-TS△229-271, and pET-pfDHFR-TS△229-274 were able to grow on both LB agar and minimum agar supplemented with 0.025 mM IPTG (Figure 4B, lanes 2–4), whereas the construct pET-pfDHFR-TS△229-275 and pET-pfDHFR-TS△229-276 failed to show complementation (Figure 4B, lanes 5–6). The results using deletion mutants of bifunctional pfDHFR-TS are in good agreement with those from co-transformation of plasmids encoding pfJRTS with varying lengths of JR and plasmid expressing catalytically active pfDHFR.
[3H]-FdUMP binding studies of pfJRTS and pfDHFR-TS mutant constructs expressing pfTS
[3H]-FdUMP binding studies were carried out to monitor the expression of catalytically active pfTS from truncated pfJRTS mutants and the pfDHFR-TS deletion mutants found to show growth complementation on minimum media. Crude extracts from E. coli χ2913 co-transformed with plasmid expressing pfDHFR and mutant pfJRTSs with varying length of JR were subjected to SDS-PAGE (Figure 5A(I) and B(I)) and the gel was exposed to X-ray film (Figure 5A(II) and B(II)). In agreement with the results from genetic complementation results, co-transformation of pAC-pfDHFR and pET-pfJRTS△232-235, pET-pfJRTS△232-251, pET-pfJRTS△232-265, pET-pfJRTS△232-271, pET-pfJRTS△232-274 and pET-pfJRTS△232-276 formed [3H]-FdUMP-enzyme complexes which could be visualized on X-ray film as a band according to the size predicted from that of pfJRTS (Figure 5B, lanes 2–7). Co-transformation of pAC-pfDHFR and pfJRTS△232-277 and pfJRTS△232-299, however, showed relatively weak signal from [3H]-FdUMP-enzyme complexes (Figure 5B, lanes 8–9).
Figure 5.

[3H]-FdUMP binding of crude extracts and partially purified proteins. Crude extracts and partially purified proteins from plasmids encoding pfDHFR co-transformed with pfJRTS mutants with truncated JR (panels A and B, respectively), and plasmids encoding pAC28 co-transformed with pfDHFR-TS with truncated JR (panel C and D, respectively) were labeled with [3H]-FdUMP. For panels A and B: lane 1, pfDHFR-TS (3D7); lane 2, pfJRTS△232-235 co-expressed with pAC28-pfDHFR; lane 3, pfJRTS△232-251 co-expressed with pAC28-pfDHFR; lane 4, pfJRTS△232-265 co-expressed with pAC28-pfDHFR; lane 5, pfJRTS△232-271 co-expressed with pAC28-pfDHFR; lane 6, pfJRTS△232-274co-expressed with pAC28-pfDHFR; lane 7, pfJRTS△232-276 co-expressed with pAC28-pfDHFR; lane 8, pfJRTS△232-277 co-expressed with pAC28-pfDHFR; lane 9, pfJRTS△232-299 co-expressed with pAC28-pfDHFR. For panels C and D: lane 1, pfDHFR-TS (3D7); lane 2, pfDHFR-TS△229-265; lane 3, pfDHFR-TS△229-271; lane 4, pfDHFR-TS△229-274; lane 5, pfDHFR-TS△229-275; lane 6, pfDHFR-TS△229-276; lane 7, pfDHFR-TS△229-277993. M is the molecular weight standard markers. (I) Coomassie blue stained SDS-PAGE, (II) autoradiogram of the SDS gel.
To investigate the role of JR in bifunctional pfDHFR-TS, formation of [3H]-FdUMP-enzyme complexes were monitored from the extracts of E. coli cells co-transformed with pAC28 plasmid and pET-pfDHFR-TS△229-265, pET-pfDHFR-TS△229-271, pET-pfDHFR-TS△229-274, pET-pfDHFR-TS△229-275, and pET-pfDHFR-TS△229-276 (Figure 5C and 5D). The results showed that both crude extracts and partially purified proteins of these constructs could form complexes with [3H]-FdUMP, though growth complementation could not be observed from pET-pfDHFR-TS△229-275, pET-pfDHFR-TS△229-276, and pET-pfDHFR-TS△229-277 (Figure 5D, lanes 5–7).
Purification and characterization of the expressed enzymes
Crude extracts of the transformed TS-deficient E. coli that were both positive and negative from the growth complementation studies were assayed for DHFR and TS activities. Extracts that were positive were passed through a MTX-affinity column for purification of the pfDHFR domain [32]. The results from enzyme assays revealed that the specific activity of pfDHFR from all the co-transformants were not significantly different among the crude extracts (average 36.2 ± 3.1 nmole/min/mg), but the activity was only about half of that observed from the crude extract of the bifunctional pfDHFR-TS (Table 3). The specific activities of the partially purified pfDHFR upon affinity purification were 1,371 ± 208 nmole/min/mg (Table 3), about 3 times lower than that obtained from the purified pfDHFR-TS. Interestingly, the TS activities of the purified enzymes from the truncated pfJRTS mutants are in most cases 1–2 times higher than that from the purified pfDHFR-TS. The activity of pfTS was found to co-elute with that of pfDHFR, suggesting that the expressed pfTS domain was somehow associated with the pfDHFR domain. As indicated in Table 3, the TS activity of the pfJRTS mutants was found to decrease upon shortening the length of JR in the construct.
Table 3.
DHFR and TS activities in crude extracts and purified enzymes from TS-deficient Escherichia coli χ2913 harbouring pAC- pf DHFR and pET- pf JRTS with different lengths of JR
|
Specific activity | ||||
|---|---|---|---|---|
|
Plasmid |
Crude extract* |
Purified enzyme** |
||
| |
(nmole/min/mg) |
(nmole/min/mg) |
||
| DHFR | TS | DHFR | TS | |
| 1. pET-pfDHFRTS (3D7) |
70.3 ± 3.6 |
3.7 ± 0.4 |
5331.9 ± 113.8 |
99.0 ± 1.0 |
| 2. pAC-pfDHFR + pET-pfJRTSΔ232-235 |
39.5 ± 5.8 |
2.6 ± 0.2 |
1511.1 ± 227.5 |
164.1 ± 22.6 |
| 3. pAC-pfDHFR + pET-pfJRTSΔ232-251 |
34.2 ± 5.8 |
2.1 ± 0.4 |
1394.5 ± 275.7 |
134.4 ± 8.9 |
| 4. pAC-pfDHFR + pET-pfJRTSΔ232-265 |
37.8 ± 5.3 |
3.8 ± 0.5 |
1668.4 ± 234.7 |
132.0 ± 7.4 |
| 5. pAC-pfDHFR + pET-pfJRTSΔ232-271 |
39.9 ± 7.3 |
3.9 ± 0.7 |
1601.7 ± 206.1 |
107.9 ± 23.3 |
| 6. pAC-pfDHFR + pET-pfJRTSΔ232-274 |
38.3 ± 5.6 |
1.5 ± 0.3 |
1328.8 ± 231.7 |
70.1 ± 0.2 |
| 7. pAC-pfDHFR + pET-pfJRTSΔ232-276 |
34.2 ± 9.1 |
1.1 ± 0.1 |
1140.1 ± 137.8 |
44.2 ± 7.5 |
| 8. pAC-pfDHFR + pET-pfJRTSΔ232-277 |
31.0 ± 8.1 |
ND |
1136.5 ± 132.0 |
ND |
| 9. pAC-pfDHFR + pETpfJRTSΔ232-279 |
34.9 ± 7.2 |
ND |
1193.6 ± 111.8 |
ND |
| 10. pAC-28 + pET-15b | 7.7 ± 0.4 | ND | - | - |
* Average values from three independent experiments.
**Average values from two independent experiments.
ND: not detectable.
The importance of the length of the JR sequence on the activity of pfTS was also investigated using the bifunctional pfDHFR-TS. Site-directed mutagenesis of the bifunctional pfDHFR-TS was performed to yield mutants containing the same lengths of JR as for the pfJRTS mutant constructs described above. These mutants include pET-pfDHFR-TS△229-265, pET-pfDHFR-TS△229-271, pET-pfDHFR-TS△229-274, pET-pfDHFR-TS△229-275, pET-pfDHFR-TS△229-276, and pET-pfDHFR-TS△229-277. Table 4 summarizes the pfDHFR and pfTS activities of the bifunctional DHFR-TS and the bifunctional mutants from the crude extracts and upon partial purification. In agreement with the growth complementation experiments, no pfTS activity was detected from the crude extracts and partial purification of pET-pfDHFR-TS△229-275, pET-pfDHFR-TS△229-276, and pET-pfDHFR-TS△229-277 mutants. However, the activities of the pfDHFR domain of these mutants remained active, though they were found to gradually decrease upon shortening the length of the JR sequence.
Table 4.
DHFR and TS activities in crude extracts and purified enzymes from TS-deficient Escherichia coli χ2913 harbouring pET- pf DHFR-TS and pET- pf DHFR-TS constructs with different lengths of JR
|
Specific activity | ||||
|---|---|---|---|---|
|
Plasmid |
Crude extract* |
Purified enzyme** |
||
| |
(nmole/min/mg) |
(nmole/min/mg) |
||
| DHFR | TS | DHFR | TS | |
| 1. pET-pfDHFR-TS (3D7) |
70.3 ± 3.6 |
3.7 ± 0.4 |
5331.9 ± 113.8 |
99.0 ± 1.0 |
| 2. pET-pfDHFR-TSΔ229-265 |
64.7 ± 5.8 |
1.1 ± 0.3 |
4583.2 ± 228.9 |
44.8 ± 1.9 |
| 3. pET-pfDHFR-TSΔ229-271 |
75.5 ± 12.4 |
1.0 ± 0.3 |
3844.6 ± 130.6 |
40.9 ± 5.9 |
| 4. pET-pfDHFR-TSΔ229-274 |
74.4 ± 18.5 |
0.9 ± 0.2 |
3487.8 ± 213.2 |
35.8 ± 2.4 |
| 5. pET-pfDHFR-TSΔ229-275 |
66.7 ± 7.3 |
ND |
4719.3 ± 391.3 |
ND |
| 6. pET-pfDHFR-TSΔ229-276 |
83.9 ± 1.0 |
ND |
3357.8 ± 443.3 |
ND |
| 7. pET-pfDHFR-TSΔ229-277 |
82.1 ± 3.0 |
ND |
3137.8 ± 210.3 |
ND |
| 8. pET-15b | 3.1 ± 0.1 | ND | - | - |
* Average values from three independent experiments.
** Average values from two independent experiments.
ND: not detectable.
Demonstration of pfDHFR-TS domain-domain interaction by Escherichia coli two-hybrid system
The interaction between the pfDHFR domain and pfJRTS was demonstrated using E. coli BacterioMatch™ two-hybrid system (Strategene). The pfDHFR domain was cloned into NotI-BamHI sites of a pBT bait plasmid, resulting in a pBT-pfDHFR plasmid, which contains the pfdhfr gene fused at the end of the bacteriophage λcI gene. The pfJRTS domain was cloned into BamHI-XhoI sites of the pTRG target plasmid, resulting in constructs that express truncated pfJRTS domains fused with the α-subunit of RNA polymerase. If the pfJRTS domain could interact with the pfDHFR domain, then this would stabilize the binding of RNA polymerase located close to the promoter and activate the transcription of the AmpR reporter gene. As a consequence, the transformed reporter strain (E. coli XL1-blue MRF’) showed growth on Luria-Bertani agar containing carbenicillin, tetracycline, chloramphenical and kanamycin (LB-CTCK).
The interaction between the DHFR monomer and the donated helix within the JR sequence has earlier been noted from the structures of the bifunctional DHFR-TS enzymes of C. hominis and P. falciparum[17,33]. An interaction of the pfDHFR and pfJRTS domains was demonstrated using the E. coli two-hybrid system [27,28,34]. The E. coli reporter strain co-transformed with plasmids pBT-pfDHFR and pTRG-pfJRTS (with full-length JR sequence) can grow on a LB-CTCK agar plate (Figure 6). The data is in agreement with the complementation results and results from [3H]-FdUMP binding studies supporting the importance of JR on the folding of the pfTS domain.
Figure 6.
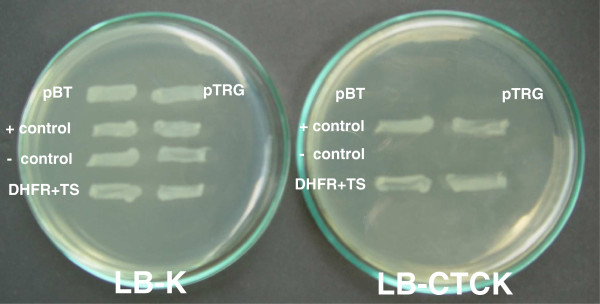
Escherichia coli two-hybrid showing interaction between pfDHFR and pfTS domains.Escherichia coli BacterioMatch™ two-hybrid system (Strategene) was employed for the study of the domain-domain interaction between P. falciparum DHFR and TS domains. The gene coding for pfDHFR was cloned into the bait plasmid pBT, whereas the gene for pfJRTS was cloned into the target plasmid pTRG. The two recombinant plasmids were transformed into the E. coli XL1-blue MRF’ reporter strain. Positive control is E. coli XL1-blue MRF’ co-transformed with pBT-LDF2 and pTRG-Gal11p provided by the manufacturer. Negative control is E. coli XL1-blue MRF’. Agar plate on the left is LB-kanamycin (LB-K) and that on the right is LB- carbenicillin, tetracycline, chloramphenical and kanamycin (LB-CTCK).
Discussion
The JR represents a junctional region linked between the DHFR and the TS domain of parasitic protozoa. This region has been proposed as potential target for drug development in many parasitic protozoa. Indeed, this region in C. hominis from one monomer was reported to make extension contacts with the DHFR active site of the other monomer [17]. In P. falciparum, the amino acid residues Asp283-Asn296 of JR are strongly predicted to be involved in domain-domain interaction. Unfortunately, the major portions of JR (Lys232-Asn280) were not seen in the crystal structure previously reported [33]. Therefore, it remains unclear whether each DHFR domain of P. falciparum is linked to the TS domain as seen in C. hominis or there is a domain swapping assembly. The role of JR was characterized with respect to interdomain interaction.
A series of truncated mutants of pfJRTS were constructed containing varying lengths of JR and their interactions with the active pfDHFR domain were examined. By employing TS-deficient E. coli χ2913 and monitoring its growth complementation in minimum media without thymidine supplementation, the study showed that the pfJRTS construct with the shortest length of JR that could still show growth complementation in TS-deficient E. coli χ2913 was pfJRTS△232-276. The specific activity of DHFR determined for the crude extract of the monofunctional pfDHFR upon co-expression with pfJRTS△232-276 was 34.2 ± 9.1 nmole/min/mg, a value which was about half that obtained from the bifunctional pfDHFR-TS (70.3 ± 3.6 nmole/min/mg), but was about the same level for all truncated pfJRTS mutants. However, the specificity of TS of this truncated construct was only 1.1 ± 0.1 nmole/min/mg (Table 3), which is about 29% of the wild-type enzyme (3.7 ± 0.4 nmole/min/mg). It is noteworthy that the DHFR specific activities among the co-transformants investigated were comparable whereas the TS specific activities were dramatically reduced upon shortening of the JR sequence. The pfDHFR and pfJRTS domains could be co-purified by using a MTX-affinity column, suggesting that the pfDHFR and pfTS domains interacted with each other because methotrexate binds only to the pfDHFR domain. For the co-transformants that lose the pfTS activity including pfJRTS△232-277 and pfJRTS△232-299, the constructs still expressed active pfDHFR (Table 3). The expression of catalytically active pfTS upon cotransformation with pfDHFR plasmid was further confirmed by showing a positive autoradiogram of covalent complex formed as a result of [3H]-FdUMP bound to the expressed pfTS from the pfJRTS△232-276 mutant construct.
These results agree well with the previous studies, suggesting that the pfDHFR domain is essential for pfTS to be active [18,35]. However, one interesting piece of data from this laboratory showed that co-expression of the death mutant pfDHFR (Ser108 to Trp108 mutation) [36] and pfJRTS△232-235 or P. vivax DHFR with pfJRTS△232-235 could not confer expression of catalytically active pfTS (data not shown). This finding suggests that not only minimal length of JR is required but active pfDHFR domain is also important for the pfTS domain to be active. Although endogenous DHFR from an E. coli host exists, the enzyme could not promote the growth of TS-deficient E. coli. It is possible that amino acids within the pfDHFR domain are critically necessary to stabilize the DHFR tertiary structure which could indirectly influence the correct folding and hence the activity of the pfTS domain.
The domain-domain interaction of the bifunctional pfDHFR-TS was further demonstrated using an E. coli two hybrid system and the results obtained were consistent with other experiments. The pBT-pfDHFR and pTRG-pfJRTS co-transformant showed interaction and turned on the AmpR gene, allowing the growth of E. coli XL1-blue MRF’. The results suggest that the minimal length of JR which could allow expression of the catalytically active pfTS was 44 amino acids, approximately half the full length of JR of the P. falciparum DHFR-TS.
The importance of the length of JR was investigated by the ability to complement the growth of TS-deficient E. coli in the absence of thymine of co-transformed plasmids harbouring the genes for pfDHFR and truncated pfJRTS compared to plasmid harbouring the gene for the bifunctional pfDHFR-TS. Complementation experiments support the conclusion that the length of JR sequence is crucial. The finding that positive complementation was still observed from the construct of truncated bifunctional pfDHFR-TS△229-274 which possesses two more amino acid residues than the case of monofunctional pfJRTS△232-276 could be due to the more complex structure of the bifunctional protein as compared to the monofunctional enzymes resulting from the interaction, or associated with the sensitivity of the complementation assay system. To further address this discrepancy, a more sensitive [3H]-FdUMP binding assay method was used to confirm that pfDHFR-TS△229-276 is the construct with the shortest length of JR that could still express active pfTS. The result was in good agreement with the results from complementation studies using monofunctional pfJRTS△229-276. The fact that the bifunctional mutant failed to show complemention in TS-deficient E. coli but could not detect the activity of pfTS by in vitro enzymatic assay could be due to the insufficient expression of protein in combination with the sensitivity of the complemention method, since a minimum TS activity of ~1-5 nmole/min/mg is necessary for complementation to be observed in E. coli χ2913 [37]. It should also be noted that the low expression observed in the truncated bifunctional pfDHFR-TS was consistent with a previous mutation study which proposed that the significant intracellular proteolysis activity associated with the α-helix structure in this region could be attributed to the low expression of the mutant proteins [38].
The present results clearly demonstrated that shortening the JR sequence in the pfDHFR-TS could affect the activity of both pfTS and pfDHFR. Contrary to that observed in T. gondii and C. hominis which have longer JR sequences, mutations of the JR sequence were reported to affect only the activity of DHFR but not the activity of TS [39,40]. This and other data support the conclusion that the length of JR sequence is necessary for the activity of both pfDHFR and pfTS domains, and hence highlights the importance of the JR sequence in P. falciparum bifunctional DHFR-TS.
It was not so surprising that the constructs pfJRTS△232-277 and pfDHFR-TS△229-277 showed no expression of catalytically active pfTS, despite the fact that the constructs contain a sequence that encompasses Asp283-Asn296, the region previously predicted to form α-helix and responsible for electrostatic interaction between the negatively charge residues and positive charge of the pfDHFR and pfTS from different domains [33]. A number of possible explanations could be as follows: (i) the length of JR in the constructs is too short, and this could affect the formation of a stable helix and consequently disturb the interaction with pfDHFR and interfere with the proper formation of pfTS conformation; (ii) too short a JR sequence could affect the structure which could result in a steric constraint or functional infringement of one domain upon the other; (iii) the existence of translational autoregulation could block translation of mRNA coding for bifunctional enzyme from binding to the DHFR domain of the protein [41]; and, (iv) the mutation could cause exposure of some parts of the enzyme which are sensitive to proteolysis [38]. Therefore, too short a JR sequence could neither support interaction between pfDHFR and pfTS domains nor contribute to proper folding of the pfDHFR-TS bifunctional protein.
Conclusions
The data presented in this study show that: a) the presence of an active pfDHFR domain and the appropriate length of JR are critical for pfTS to fold correctly to be catalytically active; and, b) deletion of JR affects both the activities of the pfTS and the pfDHFR domain of the bifunctional pfDHFR-TS. From this study, the JR of P. falciparum has a unique role which is different from other long linker parasitic protozoa. The data described here could be useful for the development of compounds that bind to the JR and interfere with the dimerization of the pfTS subdomains. These studies could potentially lead to novel means of development of inhibitors targeting the JR sequence of the parasite.
Abbreviations
DHFR-TS: Dihydrofolate reductase-thymidylate synthase; SHMT: Serine hydroxymethyltransferase; dTMP: 2′-deoxythymidine 5′-monophosphate; [3H]-FdUMP: Tritiated-5-fluoro-2′-deoxyuridine 5′-monophosphate; JR: Junctional region; MM: Minimal media; MTX: Methotrexate
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
NC participated in performing experiments and drafted the manuscript. RS helped to perform the experiments and discussion. WS conceived the study, participated in its design, data analysis and critical revision of the manuscript. All authors read and approved the final manuscript.
Contributor Information
Natpasit Chaianantakul, Email: natpasit.c@gmail.com.
Rachada Sirawaraporn, Email: grrsr@mahidol.ac.th.
Worachart Sirawaraporn, Email: worachart.sir@mahidol.ac.th.
Acknowledgements
The authors are grateful to Drs. Michael A. Allen, Alfredo Villarroel and Alejandro Sáiz for critical review of the manuscript. This work was supported in part by the UNICEF/UNDP/World Bank/WHO Special Programme for Research and Training in Tropical Diseases (TDR) and the Office of the Higher Education Commission and Mahidol University under the National Research Universities Initiative to WS. NC is a recipient of a grant under the programme Strategic Scholarships for Frontier Research Network for the Joint PhD Program Thai Doctoral degree from the Commission on Higher Education, Thailand.
References
- Hay SI, Guerra CA, Tatem AJ, Noor AM, Snow RW. The global distribution and population at risk of malaria: past, present, and future. Lancet Infect Dis. 2004;4:327–336. doi: 10.1016/S1473-3099(04)01043-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouma MJ, Sondorp HE, van der Kaay HJ. Climate change and periodic epidemic malaria. Lancet. 1994;343:1440. doi: 10.1016/s0140-6736(94)92569-0. [DOI] [PubMed] [Google Scholar]
- Cowman AF, Morry MJ, Biggs BA, Cross GA, Foote SJ. Amino acid changes linked to pyrimethamine resistance in the dihydrofolate reductase-thymidylate synthase gene of Plasmodium falciparum. Proc Natl Acad Sci USA. 1988;85:9109–9113. doi: 10.1073/pnas.85.23.9109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foote SJ, Galatis D, Cowman AF. Amino acids in the dihydrofolate reductase-thymidylate synthase gene of Plasmodium falciparum involved in cycloguanil resistance differ from those involved in pyrimethamine resistance. Proc Natl Acad Sci USA. 1990;87:3014–3017. doi: 10.1073/pnas.87.8.3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson DS, Walliker D, Wellems TE. Evidence that a point mutation in dihydrofolate reductase-thymidylate synthase confers resistance to pyrimethamine in falciparum malaria. Proc Natl Acad Sci USA. 1988;85:9114–9118. doi: 10.1073/pnas.85.23.9114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson DS, Milhous WK, Wellems TE. Molecular basis of differential resistance to cycloguanil and pyrimethamine in Plasmodium falciparum malaria. Proc Natl Acad Sci USA. 1990;87:3018–3022. doi: 10.1073/pnas.87.8.3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyde JE. Point mutations and pyrimethamine resistance in Plasmodium falciparum. Parasitol Today. 1989;5:252–255. doi: 10.1016/0169-4758(89)90257-3. [DOI] [PubMed] [Google Scholar]
- Zolg JW, Plitt JR, Chen GX, Palmer S. Point mutations in the dihydrofolate reductase-thymidylate synthase gene as the molecular basis for pyrimethamine resistance in Plasmodium falciparum. Mol Biochem Parasitol. 1989;36:253–262. doi: 10.1016/0166-6851(89)90173-4. [DOI] [PubMed] [Google Scholar]
- Ivanetich KM, Santi DV. Bifunctional thymidylate synthase-dihydrofolate reductase in protozoa. FASEB J. 1990;4:1591–1597. doi: 10.1096/fasebj.4.6.2180768. [DOI] [PubMed] [Google Scholar]
- Bzik DJ, Li WB, Horii T, Inselburg J. Molecular cloning and sequence analysis of the Plasmodium falciparum dihydrofolate reductase-thymidylate synthase gene. Proc Natl Acad Sci USA. 1987;84:8360–8364. doi: 10.1073/pnas.84.23.8360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyde JE. The dihydrofolate reductase-thymidylate synthetase gene in the drug resistance of malaria parasites. Pharmacol Ther. 1990;48:45–59. doi: 10.1016/0163-7258(90)90017-V. [DOI] [PubMed] [Google Scholar]
- Meek TD, Garvey EP, Santi DV. Purification and characterization of the bifunctional thymidylate synthetase-dihydrofolate reductase from methotrexate-resistant Leishmania tropica. Biochemistry. 1985;24:678–686. doi: 10.1021/bi00324a021. [DOI] [PubMed] [Google Scholar]
- Trujillo M, Donald RG, Roos DS, Greene PJ, Santi DV. Heterologous expression and characterization of the bifunctional dihydrofolate reductase-thymidylate synthase enzyme of Toxoplasma gondii. Biochemistry. 1996;35:6366–6374. doi: 10.1021/bi952923q. [DOI] [PubMed] [Google Scholar]
- Elcock AH, Potter MJ, Matthews DA, Knighton DR, McCammon JA. Electrostatic channeling in the bifunctional enzyme dihydrofolate reductase-thymidylate synthase. J Mol Biol. 1996;262:370–374. doi: 10.1006/jmbi.1996.0520. [DOI] [PubMed] [Google Scholar]
- Stroud RM. An electrostatic highway. Nat Struct Biol. 1994;1:131–134. doi: 10.1038/nsb0394-131. [DOI] [PubMed] [Google Scholar]
- Knighton DR, Kan CC, Howland E, Janson CA, Hostomska Z, Welsh KM, Matthews DA. Structure of and kinetic channelling in bifunctional dihydrofolate reductase-thymidylate synthase. Nat Struct Biol. 1994;1:186–194. doi: 10.1038/nsb0394-186. [DOI] [PubMed] [Google Scholar]
- O’Neil RH, Lilien RH, Donald BR, Stroud RM, Anderson AC. Phylogenetic classification of protozoa based on the structure of the linker domain in the bifunctional enzyme, dihydrofolate reductase-thymidylate synthase. J Biol Chem. 2003;278:52980–52987. doi: 10.1074/jbc.M310328200. [DOI] [PubMed] [Google Scholar]
- Shallom S, Zhang K, Jiang L, Rathod PK. Essential protein-protein interactions between Plasmodium falciparum thymidylate synthase and dihydrofolate reductase domains. J Biol Chem. 1999;274:37781–37786. doi: 10.1074/jbc.274.53.37781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futterman S. Enzymatic reduction of folic acid and dihydrofolic acid to tetrahydrofolic acid. J Biol Chem. 1957;228:1031–1038. [PubMed] [Google Scholar]
- Blakley RL. Spectrophotometric studies on the combination of formaldehyde with tetrahydropteroylglutamic acid and other hydropteridines. Biochem J. 1960;74:71–82. doi: 10.1042/bj0740071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirawaraporn W, Sirawaraporn R, Cowman AF, Yuthavong Y, Santi DV. Heterologous expression of active thymidylate synthase-dihydrofolate reductase from Plasmodium falciparum. Biochemistry. 1990;29:10779–10785. doi: 10.1021/bi00500a009. [DOI] [PubMed] [Google Scholar]
- Miyazaki K, Takenouchi M. Creating random mutagenesis libraries using megaprimer PCR of whole plasmid. Biotechniques. 2002;35:1033–1034. doi: 10.2144/02335st03. [DOI] [PubMed] [Google Scholar]
- Kholod N, Mustelin T. Novel vectors for co-expression of two proteins in E. coli. Biotechniques. 2001;31:322–323. doi: 10.2144/01312st03. 326–328. [DOI] [PubMed] [Google Scholar]
- Belfort M, Moelleken A, Maley GF, Maley F. Purification and properties of T4 phage thymidylate synthetase produced by the cloned gene in an amplification vector. J Biol Chem. 1983;258:2045–2051. [PubMed] [Google Scholar]
- Hillcoat BL, Nixon PF, Blakley RL. Effect of substrate decomposition on the spectrophotometric assay of dihydrofolate reductase. Anal Biochem. 1967;21:178–189. doi: 10.1016/0003-2697(67)90179-0. [DOI] [PubMed] [Google Scholar]
- Wahba AJ, Friedkin M. Direct spectrophotometric evidence for the oxidation of tetrahydrofolate during the enzymatic synthesis of thymidylate. J Biol Chem. 1961;236:PC11–PC12. [PubMed] [Google Scholar]
- Dove SL, Joung JK, Hochschild A. Activation of prokaryotic transcription through arbitrary protein-protein contacts. Nature. 1997;386:627–630. doi: 10.1038/386627a0. [DOI] [PubMed] [Google Scholar]
- Dove SL, Hochschild A. Conversion of the omega subunit of Escherichia coli RNA polymerase into a transcriptional activator or an activation target. Genes Dev. 1998;12:745–754. doi: 10.1101/gad.12.5.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall SJ, Sims PF, Hyde JE. Functional expression of the dihydrofolate reductase and thymidylate synthetase activities of the human malaria parasite Plasmodium falciparum in Escherichia coli. Mol Biochem Parasitol. 1991;45:317–330. doi: 10.1016/0166-6851(91)90100-K. [DOI] [PubMed] [Google Scholar]
- Sirawaraporn W, Prapunwattana P, Sirawaraporn R, Yuthavong Y, Santi DV. The dihydrofolate reductase domain of Plasmodium falciparum thymidylate synthase-dihydrofolate reductase. Gene synthesis, expression, and anti-folate-resistant mutants. J Biol Chem. 1993;268:21637–21644. [PubMed] [Google Scholar]
- Sano G, Morimatsu K, Horii T. Purification and characterization of dihydrofolate reductase of Plasmodium falciparum expressed by a synthetic gene in Escherichia coli. Mol Biochem Parasitol. 1994;63:265–273. doi: 10.1016/0166-6851(94)90062-0. [DOI] [PubMed] [Google Scholar]
- Hekmat-Nejad M, Lee PC, Rathod PK. Plasmodium falciparum: direct cloning and expression of pyrimethamine-sensitive and pyrimethamine-resistant dihydrofolate reductase domains. Exp Parasitol. 1997;85:303–305. doi: 10.1006/expr.1997.4145. [DOI] [PubMed] [Google Scholar]
- Yuvaniyama J, Chitnumsub P, Kamchonwongpaisan S, Vanichtanankul J, Sirawaraporn W, Taylor P, Walkinshaw MD, Yuthavong Y. Insights into antifolate resistance from malarial DHFR-TS structures. Nat Struct Biol. 2003;10:357–365. doi: 10.1038/nsb921. [DOI] [PubMed] [Google Scholar]
- Joung JK, Ramm EI, Pabo CO. A bacterial two-hybrid selection system for studying protein-DNA and protein-protein interactions. Proc Natl Acad Sci USA. 2000;97:7382–7387. doi: 10.1073/pnas.110149297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wattanarangsan J, Chusacultanachai S, Yuvaniyama J, Kamchonwongpaisan S, Yuthavong Y. Effect of N-terminal truncation of Plasmodium falciparum dihydrofolate reductase on dihydrofolate reductase and thymidylate synthase activity. Mol Biochem Parasitol. 2003;126:97–102. doi: 10.1016/S0166-6851(02)00240-2. [DOI] [PubMed] [Google Scholar]
- Sirawaraporn W, Yongkiettrakul S, Sirawaraporn R, Yuthavong Y, Santi DV. Plasmodium falciparum: asparagine mutant at residue 108 of dihydrofolate reductase is an optimal antifolate-resistant single mutant. Exp Parasitol. 1997;87:245–252. doi: 10.1006/expr.1997.4221. [DOI] [PubMed] [Google Scholar]
- Climie S, Ruiz-Perez L, Gonzalez-Pacanowska D, Prapunwattana P, Cho SW, Stroud R, Santi DV. Saturation site-directed mutagenesis of thymidylate synthase. J Biol Chem. 1990;265:18776–18779. [PubMed] [Google Scholar]
- Dasgupta T, Anderson KS. Probing the role of parasite-specific, distant structural regions on communication and catalysis in the bifunctional thymidylate synthase-dihydrofolate reductase from Plasmodium falciparum. Biochemistry. 2008;47:1336–1345. doi: 10.1021/bi701624u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belperron AA, Fox BA, O’Neil RH, Peaslee KA, Horii T, Anderson AC, Bzik DJ. Toxoplasma gondii: generation of novel truncation mutations in the linker domain of dihydrofolate reductase-thymidylate synthase. Exp Parasitol. 2004;106:179–182. doi: 10.1016/j.exppara.2004.03.002. [DOI] [PubMed] [Google Scholar]
- Vargo MA, Martucci WE, Anderson KS. Disruption of the crossover helix impairs dihydrofolate reductase activity in the bifunctional enzyme TS-DHFR from Cryptosporidium hominis. Biochem J. 2009;417:757–764. doi: 10.1042/BJ20081247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Rathod PK. Divergent regulation of dihydrofolate reductase between malaria parasite and human host. Science. 2002;296:545–547. doi: 10.1126/science.1068274. [DOI] [PMC free article] [PubMed] [Google Scholar]