

Abstract
The critical role of polyamines in cell growth has led to the development of a number of agents that interfere with polyamine metabolism including a novel class of polyamine analogues, oligoamines. Here we demonstrate that oligoamines specifically suppress the mRNA and protein expression of estrogen receptor α (ERα) and ERα target genes in ER-positive human breast cancer cell lines, whereas neither ERβ nor other steroid hormonal receptors are affected by oligoamines. The constitutive expression of a cytomegalovirus promoter-driven exogenous ERα in ER-negative MDA-MB-231 human breast cancer cells was not altered by oligoamines, suggesting that oligoamines specifically suppress ERα transcription rather than affect mRNA or protein stability. Further analysis demonstrated that oligoamines disrupted the DNA binding activity of Sp1 transcription factor family members to an ERα minimal promoter element containing GC/CA-rich boxes. Treatment of MDA-MB-231 cells with the JNK-specific inhibitor SP600125 or expression of the c-Jun dominant negative inhibitor TAM67 blocked the oligoamine-activated JNK/c-Jun pathway and enhanced oligoamine-inhibited ERα expression, suggesting that AP-1 is a positive regulator of ERα expression and that oligoamine-activated JNK/AP-1 activity may antagonize the down-regulation of ERα induced by oligoamines. Taken together, these results suggest a novel antiestrogenic mechanism for specific polyamine analogues in human breast cancer cells.
Polyamines are naturally occurring polycationic alkylamines that are absolutely required for cell growth. Because of their positively charged amine groups, polyamines interact with negatively charged molecules like DNA, RNA, proteins, and phospholipids to modify their structure and conformation. Rapid tumor growth is associated with significantly increased polyamine biosynthesis (1, 2). In human breast cancer, studies have demonstrated the important roles of polyamine biosynthesis and action in tumor development and metastasis, and increased polyamine levels are often associated with aggressive forms of breast tumors (3–7). Because of the requirement for polyamines in breast cell growth and the demonstration of dysregulated polyamine metabolism in breast tumor cells, polyamine metabolism has become a rational target for breast cancer therapy. Polyamine analogues were designed based upon the theory that the analogues can mimic some of the self-regulatory functions of natural polyamines but are unable to substitute for natural polyamines in their growth promoting roles (8). Recently, a novel class of polyamine analogues has been developed that includes conformationally restricted, cyclic, and long chain oligoamine analogues (9). Our recent studies showed that oligoamines effectively inhibit growth of human breast cancer cell lines in culture and mouse xenografts (10). We also demonstrated that specific oligoamines reduced ornithine decarboxylase activity and induced the activity of the polyamine catabolic enzyme, spermidine/spermine N1-acetyltransferase, thereby significantly decreasing the intracellular polyamine pools in several human breast cancer cell lines (10, 11).
Estrogens are thought to play a major role in breast cancer development. Because of the pivotal role of the estrogen-estrogen receptor (ER)3 axis in breast cancer progression, targeting ER or its ligands is a major strategy for breast cancer treatment. Estrogen effects are exerted through the activation of specific estrogen receptors. Unfortunately, many breast tumors develop estrogen independence, perhaps as a consequence of multiple mechanisms including altered balance of co-regulatory proteins or the emergence of other growth signaling pathways. These molecular mechanisms are still poorly understood, and this lack of understanding has posed a major clinical challenge in breast cancer therapy. Therefore, elucidation of ER regulatory pathways is critical. A potential interaction between polyamines and ER is suggested by several lines of evidence. For instance, in ER-positive breast cancer cells, estradiol up-regulates ornithine decarboxylase and increases polyamine levels, which accompanies cell proliferation (12). Tamoxifen-induced growth inhibition is often associated with the down-regulation of polyamine biosynthesis (13). It also has been demonstrated that spermine facilitates the binding of ERα to estrogen response elements (ERE) and that increased polyamine concentrations may induce a DNA sequence containing EREs to undergo conformational transition and enhance the binding of ER to EREs, suggesting a possible mechanism for polyamine-mediated alterations in the interaction of ER with ERE (14).
In this study, we demonstrate that selective oligoamines specifically suppress the expression and activity of ERα in human breast cancer cells. Oligoamines disrupt the DNA binding activity of Sp1 transcription factor family members at the GC-and CA-rich boxes of the ERα proximal promoter. Additionally, a c-Jun NH2-terminal kinase (JNK) specific inhibitor or expression of a dominant negative c-Jun inhibited ERα expression and enhanced inhibition of ERα expression by the oligoamines, suggesting that the JNK/AP-1 signaling pathway is a positive regulator of ERα expression and that oligoamine activated c-Jun activity may antagonize the down-regulation of ERα by oligoamines.
EXPERIMENTAL PROCEDURES
Compounds and Culture Conditions
Oligoamines were synthesized as previously reported (9, 15). Stock solutions (10 mM in double distilled H2O) of each analogue were diluted with medium to the desired concentrations for specific experiments. Spermine was purchased from Sigma and was prepared as a stock solution of 10 mM in H2O. 17β-Estradiol was purchased from Sigma. SP600125 (anthra[1,9-cd]pyrazol-6(2H)-one) from BIOMOL Research Laboratories, Inc. (Plymouth Meeting, PA) was prepared as a stock solution of 20 mM in 100% Me2SO. The human breast cancer MCF-7, T47D, and MDA-MB-231 cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 5% fetal bovine serum and 2 mM glutamine at 37 °C in a 5% CO2 atmosphere.
Plasmid Construction and Stable Transfection
The entire coding region of ERα was PCR-amplified using MCF7 cDNA as template and cloned into the expression vector, pIRESNEO (Clontech, Palo Alto, CA). The cloned cDNA was verified by sequencing (16). Empty vector or pIRES-ER construct was then transfected into MDA-MB-231 cells using Gene Jammer as recommended by the manufacturer (Invitrogen). Stable colonies were selected and maintained using G418 at a concentration of 500 μg/ml in Dulbecco’s modified Eagle’s medium. The cDNA of mutant c-Jun (TAM67), which lacks amino acids 3–122 of the transactivation domain, was a generous gift from Drs. Steve Georas (Johns Hopkins) and Michael Birrer (National Institutes of Health) and was subcloned into the pcDNA3.1 mammalian expression vector (Invitrogen) (11). TAM67 constructs were transfected into MCF-7 cells using the same method described above, and stable transfectants were selected and maintained as above.
Immunoblotting
Whole cellular proteins were extracted as previously described (10). Cytoplasmic and nuclear fractions were prepared using the NE-PER™ nuclear and cytoplasmic extraction kit (Pierce). The protein concentrations were determined, and Western blotting was performed using the methods described previously (10). Primary antibodies against ERα, ERβ, vitamin D receptor, progesterone receptor (PR), retinoic acid receptor β, cyclin D1, Sp1, Sp3, c-Jun, and JNK1 were from Santa Cruz Biotechnology Company (Santa Cruz, CA). Proliferating cell nuclear antigen monoclonal antibody was purchased from Oncogene Research Products (Cambridge, MA). Actin was used to normalize for protein loading. All of the experiments were performed at least twice with similar results.
Luciferase Reporter Activity Assays
Deletion constructs of the ERα 5′-flanking region in luciferase reporter vector pGL3-Basic were provided by Dr. Suzanne Fuqua (Baylor College of Medicine). ERE-tk-luc vectors contain four consecutive ERE and were used to measure ER-mediated transcription activity. The cells were seeded at 2 × 105 cells/well in 24-well plates 24 h prior to transfection. 3 μl of Gene Jammer transfection reagent (Stratagene, La Jolla, CA) was used to transiently transfect 0.5 μg of luciferase reporter gene constructs. One μg of β-galactosidase expression vector was co-transfected with all plasmids to determine transfection efficiency. Luciferase activity was measured on a Monolight luminometer using the BrightGlo luciferase assay kit (Promega, Madison, WI), and β-galactosidase activity was determined using the β-galactosidase activity kit (Promega). The experiments were completed three times in triplicate. The Student’s t test was used to determine the statistical differences between various experimental and control groups. p ≤ 0.05 was considered significant. Shown are means ± S.D.
Reverse Transcription PCR
RNA was harvested using TRIzol reagent (Invitrogen) as previously described (16). cDNA was synthesized from 3 μg of total RNA using Moloney murine leukemia virus reverse transcriptase (Invitrogen) and oligo(dT) primers (Invitrogen) at 37 °C for 1 h. PCR was performed using the following primers: ERα sense, GCA CCC TGA AGT CTC TGG AA; ERα antisense, TGG CTA AAG TGG TGC ATG AT (55 °C for 35 cycles); actin sense, ACC ATG GAT GAT GAT ATC GC; and actin antisense, ACA TGG CTG GGG TGT TGA AG (60 °C for 30 cycles).
Electrophoretic Mobility Shift Assay (EMSA)
Nuclear and cytoplasmic proteins were extracted using the NE-PER™ nuclear and cytoplasmic extraction kit (Pierce). EMSA was performed as described previously (16, 17). Briefly, 10 μg of nuclear extracts were incubated with γ-32P-labeled oligonucleotides (2 × 104 counts/min/lane), which span the ERα promoter from −245 to −182 bp. For supershift assays, the cell extracts were preincubated with 2 μg of antibody for 20 min on ice. A 100-fold excess of unlabeled 21-bp oligonucleotide probe, which contains the minimal consensus binding site for Sp1 transcription factor (Santa Cruz Biotechnology Company) was used as a competitor. The reactions were then separated on 5% polyacrylamide gels at 150 V in 0.5 × Tris borate-EDTA buffer followed by autoradiography.
Chromatin Immunoprecipitation
Chromatin immunoprecipitation assays were performed as described previously (18). After treatment, the cells were exposed to formaldehyde to cross-link proteins, and chromatin samples were sonicated on ice three times for 10 s each (to give an average length of 1–1.5 kb sheered genomic DNA) followed by centrifugation for 10 min. The antibodies for Sp1 family members were used for immunoprecipitation of protein-DNA complexes. PCR primers flanking the Sp1-binding sites of the ERα promoter were designed as follows: sense, 5′-ACC TTA GCA GAT CCT CGT; and antisense, 5′-GCT GCT GGA TAG AGG CTG A. Genomic DNA was used as a positive control (input). Chromatin eluted from immunoprecipitations lacking antibody was used as a “no antibody” control. PCR products were run on 1% agarose gel and analyzed by ethidium bromide staining.
Immunoprecipitation and Kinase Assays
The immunoprecipitation of JNK kinase and kinase activity assays were performed as described previously (19). Briefly, JNK complexes were immunoprecipitated by anti-JNK rabbit polyclonal antibodies (Santa Cruz Biotechnology) bound to protein A-Sepharose (Amersham Biosciences). The kinase assays were performed by the addition of 2 μg of substrate protein, 20 μM cold ATP, and 10 μCi of [γ-32P] ATP for 30 min at 30 °C and stopped by the addition of 2× SDS-PAGE sample buffer. The product was analyzed by SDS-polyacrylamide gel electrophoresis followed by autoradiography. The experiments were performed at least twice with similar results.
RESULTS
Inhibition of ERα Expression by Oligoamines
Three representative oligoamine analogues, CGC-11144, CGC-11159, and CGC-11172, were selected for this study (Fig. 1A). To investigate whether oligoamines affect hormonal signaling pathways in breast cancer cells, the protein expression of several breast cell growth-related hormone receptors including the ERα, ERβ, vitamin D receptor, and retinoic acid receptor β were examined. All three oligoamines specifically down-regulated the protein level of ERα in two ER-positive human breast cancer cell lines MCF-7 and T47D, whereas the protein expression of ERβ, vitamin D receptor, and retinoic acid receptor β was not affected by oligoamines (Fig. 1B). Further, Western blot analysis showed that the expression of two ERα-regulated genes, progesterone receptors (PRA and PRB), and cyclin D1 was also inhibited by oligoamines (Fig. 1B). These data suggest that oligoamines specifically target ERα and its downstream genes in breast cancer cells. Down-regulation of ERα by oligoamines displays clear time-, concentration-, and cell type-dependent features in that ERα expression was inhibited by oligoamines after a 72-h exposure in MCF-7 cells and a 16-h exposure in T47D cells (Fig. 1C). A minimum CGC-11144 concentration of 5 μM is required to decrease ERα protein in MCF-7 cells treated for 96 h and T47D cells treated for 24 h (Fig. 1, C and D).
FIGURE 1. Effects of oligoamines on ERα expression.
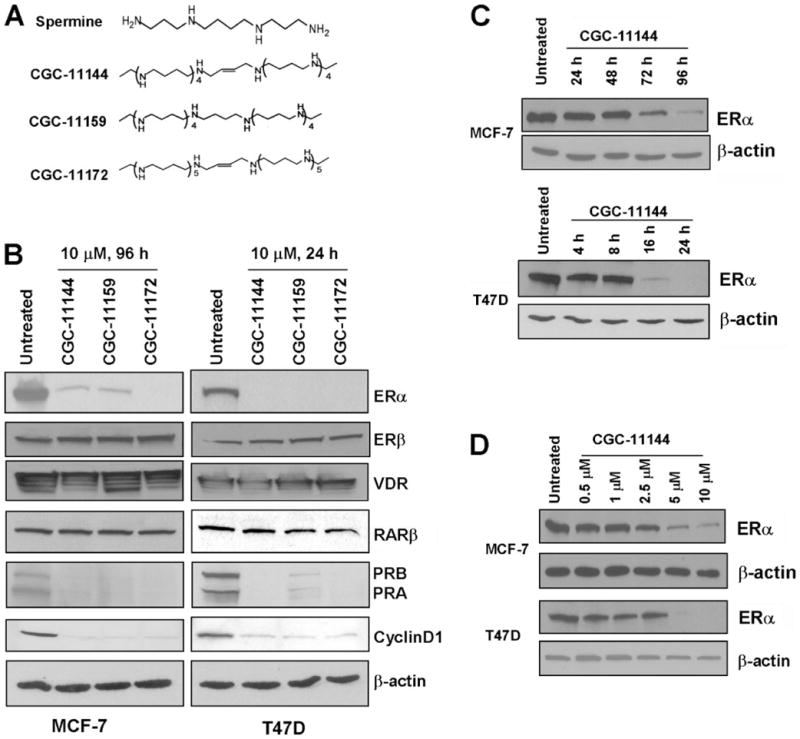
A, structures of natural spermine and oligoamines. B, MCF-7 and T47D cells were treated with 10 μM of the indicated oligoamines for 96 and 24 h, respectively. Western blots were performed to detect the expression of ERα, ERβ, vitamin D receptor, PRA, PRB, and cyclin D1. C, MCF-7 and T47D cells were treated with 10 μM CGC-11144 for the indicated times. Western blots were performed to detect the expression of ERα. D, MCF-7 and T47D cells were treated with increasing concentrations of CGC-11144 for 96 h in MCF-7 and 24 h in T47D, respectively. Western blots were performed to detect the expression of ERα. Actin protein was blotted as a control. All of the experiments were performed at least twice with similar results.
Oligoamines Down-regulate ER-mediated Transcription Activity
Ligand-activated ER dimers can bind to the estrogen response element (ERE) of target genes and regulate their transcription. To determine whether oligoamine down-regulation of ERα affects ER-mediated transcriptional activity, MCF-7 or T47D cells were treated with various oligoamines for different times with 10 nM 17β-estradiol (E2), and the subcellular localization of ERα and ERβ was examined by immunoblotting using nuclear and cytoplasmic extracts. Treatment with E2 alone (control) led to nuclear localization of ERα, whereas the nuclear ERα protein was significantly decreased by treatment with oligoamines. By contrast, most of the ERβ protein remained in the cytosol after the stimulation of E2, and oligoamine treatment did not affect either the level of protein expression or the cellular localization of ERβ(Fig. 2A). Polyamine analogues specifically down-regulate ERα expression without affecting the expression of ERβ. To verify that decreased ERα expression is associated with decreased ERα-mediated transcription activities, MCF-7 and T47D cells were transiently transfected with the ERE-tk-luciferase constructs. Reporter gene activities were measured after 24 h of oligoamine treatment in T47D cells and after 96 h of treatment in MCF-7 cells. E2 significantly increased ERE reporter gene activities in both ER-positive human breast cancer cell lines. Treatment with all oligoamines significantly suppressed the level of reporter gene activity in both cell lines, suggesting that down-regulation of ERα protein expression by oligoamines leads to the subsequent suppression of ER-mediated transcriptional activities (Fig. 2B).
FIGURE 2. Effects of oligoamines on transactivation of ERα.
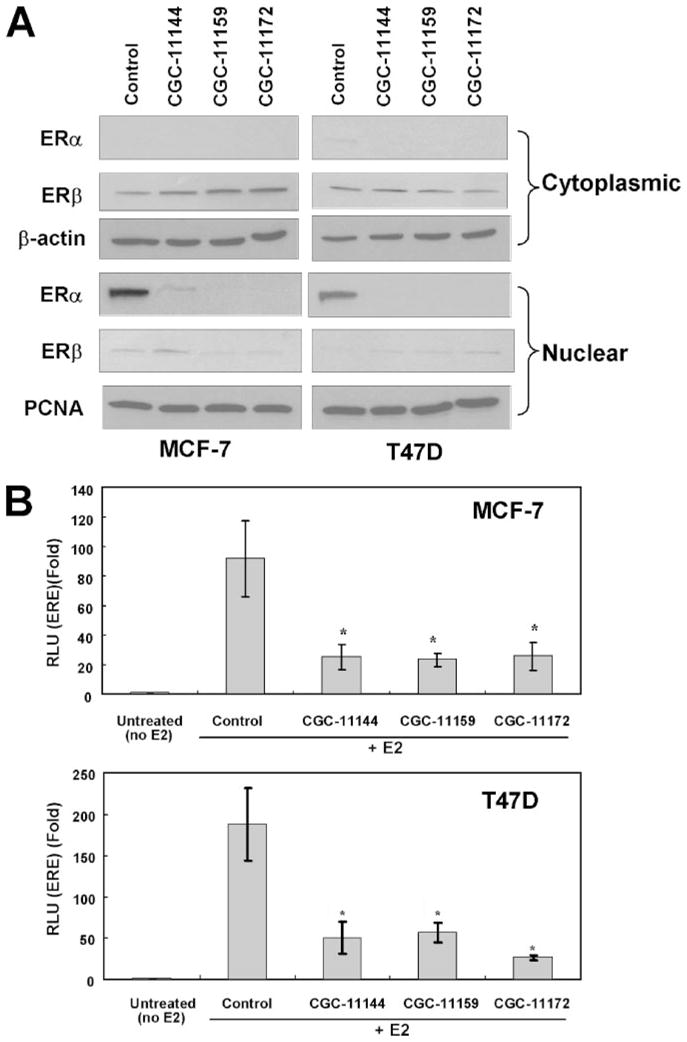
The cells were cultured in phenol red-free Dulbecco’s modified Eagle’s medium supplemented with 5% charcoal dextran-stripped fetal bovine serum and treated with 10 nM 17 β-estradiol (E2). A, MCF-7 and T47D cells were treated with 10 μM of the specific oligoamine for 96 h in MCF-7 and 24 h in T47D cells. Cytoplasmic or nuclear protein was extracted for immunoblotting with anti-ERα and ERβ antibodies. Actin protein was blotted as a loading control for cytoplasmic extracts and proliferating cell nuclear antigen (PCNA) was used as a loading control for nuclear extracts. B, MCF-7and T47D cells were co-transfected with ERE-tk-luciferase and CMV-β-galactosidase. Reporter gene activities were measured after 24 h of oligoamine treatment for T47D cells and 96 h treatment for MCF-7 cells. The experiments were completed three times, and each measurement was taken in triplicate. *, p < 0.05, statistically significant differences using Student’s t test between control cells (treated with E2 only) and cells treated with E2 and oligoamines.
Down-regulation of ERα by Oligoamines Is Caused by Transcriptional Repression
To elucidate the possible mechanism by which oligoamines down-regulate ERα, reverse transcription-PCR was performed to determine the ERα mRNA level in MCF-7 and T47D cells after treatment with oligoamines. Reverse transcription-PCR results showed that ERα mRNA was markedly decreased by oligoamines in both cell lines (Fig. 3A). To investigate whether the down-regulation of ERα by oligoamines occurs through the regulation of the promoter activity of the ERα gene, 4.1 kb of the ERα 5′-flanking region linked to a luciferase reporter pGL3-Basic vector was transiently transfected into T47D cells followed by treatment with CGC-11144 as a representative oligoamine. The results showed that endogenous ERE-luciferase activity is significantly suppressed by CGC-11144 (Fig. 3B).
FIGURE 3. Effects of oligoamines on ERα mRNA expression and promoter activity.
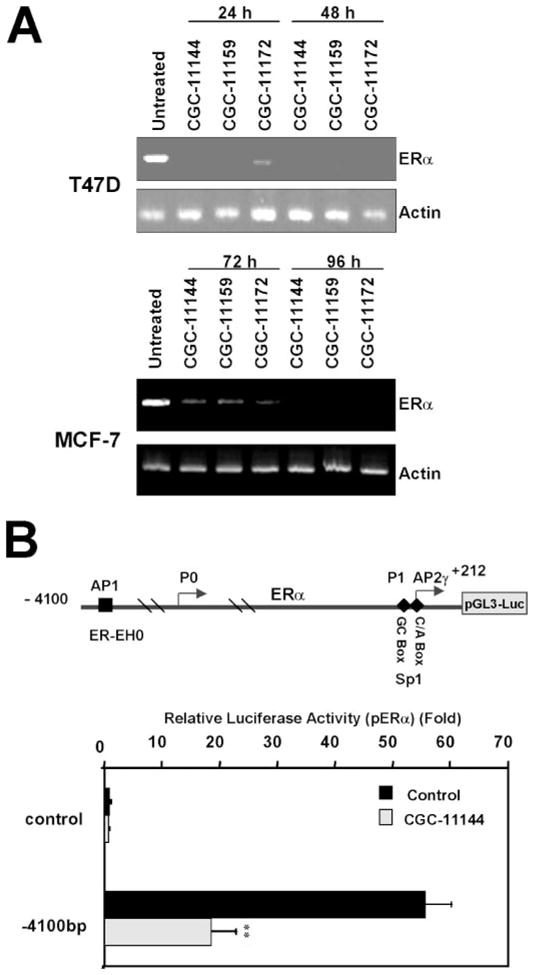
A, MCF-7 and T47D cells were treated with the specific oligoamine for the time indicated. ERα mRNA expression was determined by reverse transcription-PCR. B, luciferase reporter constructs containing the ERα promoter region from −4100 to +212 bp were transfected with CMV-β-galactosidase into T47D cells followed by treatment of 10 μM CGC-11144 for 24 h. Reporter gene activities were then measured. The experiments were completed three times, and each measurement was taken in triplicate. **, p < 0.01.
To further validate that oligoamine-induced down-regulation of ERα is caused by the repression of ER transcription rather than by enhanced protein degradation, the ER-negative MDA-MB-231 cell line was stably transfected with an ERα cDNA construct that lacks any 5′ ERα promoter sequence and that is driven by the CMV promoter in a pIRES vector (MDA-MB-231-ERα). Two single clones (ER19 and ER28) that express ERα at a level comparable with that observed in MCF-7 cells were identified and selected for a series of functional assays (Fig. 4A). To test whether constitutively expressed ERα is functional in MDA-MB-231 cells, an ERE-tk-luciferase reporter gene activity assay was used to demonstrate that E2 induced ERE reporter gene expression (luciferase) in both ERα transfected clones, but not in vector controls (Fig. 4B). These results indicate that exogenously introduced ERα in ERα-negative breast cancer cells has intact ER signaling in response to estradiol. Further, Western blotting demonstrated that the ERα protein level was not affected by oligoamine treatment in either clone ER19 (Fig. 4C) or ER28 (not shown), thereby providing evidence that oligoamine-induced down-regulation of ERα indeed occurs through repression of ERα promoter activity rather than enhanced protein degradation.
FIGURE 4. Effects of oligoamines on exogenous ERα expression in MDA-MB-231 cells.
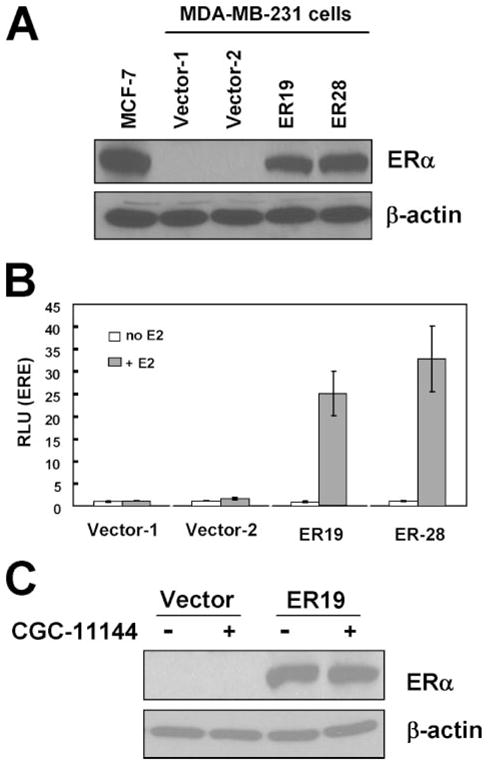
A, MDA-MB-231 cells were stably transfected with pIRES-ERα. Proteins isolated from MCF-7 cells, empty vector MDA-MB-231 transfectants, and ERα transfected MDA-MB-231 single colonies were subjected to immunoblot with an ERα antibody. Two G418-resistant clones (ER19 and ER28) were shown to express high levels of ERα. B, vector control and ERα transfected MDA-MB-231 cells were co-transfected with ERE-tk-luciferase and CMV-β-galactosidase and cultured in medium with 5% charcoal dextran-stripped fetal bovine serum with or without 10 nM E2. The bars represent the means ± S.D. of at least three independent experiments. C, vector control and ERα transfected ER19 cells were treated with oligoamines for 72 h, and ERα expression was determined by Western blot.
Oligoamines Disrupt ERα Promoter Activity within a Proximal Region Containing an Sp1-binding Site
The exact molecular mechanisms for regulation of ERα expression in breast cancer cells are unclear, but studies suggest that the regulation is at least partially transcriptional (20). Using deletion luciferase reporter constructs of the ERα promoter, we found that CGC-11144 inhibited the reporter gene activity of the proximal ERα minimal promoter region −245 to +212 bp (Fig. 5A). This fragment contains the GC- and CA-rich boxes that are binding sites for Sp1 transcription factor family members and other zinc finger transcription factors. To ascertain whether oligoamines alter the recruitment of Sp1 family members to this element, a radiolabeled oligonucleotide spanning the ERα promoter region from −245 to −182 bp was used as a probe with T47D nuclear extracts in EMSA analysis. Shifted protein-DNA complexes were clearly observed in untreated cells, and oligoamines markedly reduced protein binding to this element (Fig. 5B). The binding of Sp1 to this element was confirmed by supershift analysis (Fig. 5B). Competition with an unlabeled probe consisting of the minimal Sp1 consensus sequence clearly inhibits the binding of transcription factor complex to the ERα minimal promoter region (Fig. 5C). This result suggests that the EMSA is generated by a protein contacting the Sp1-binding site at the ERα minimal promoter and not other sequences, such as the E box. In addition, the labeled probe was incubated with Sp1 antibody only, and no shift was observed. This result rules out the possibility of a direct interaction between labeled probe and antibody (Fig. 5C).
FIGURE 5. Effects of oligoamines on ERα proximal promoter activity.
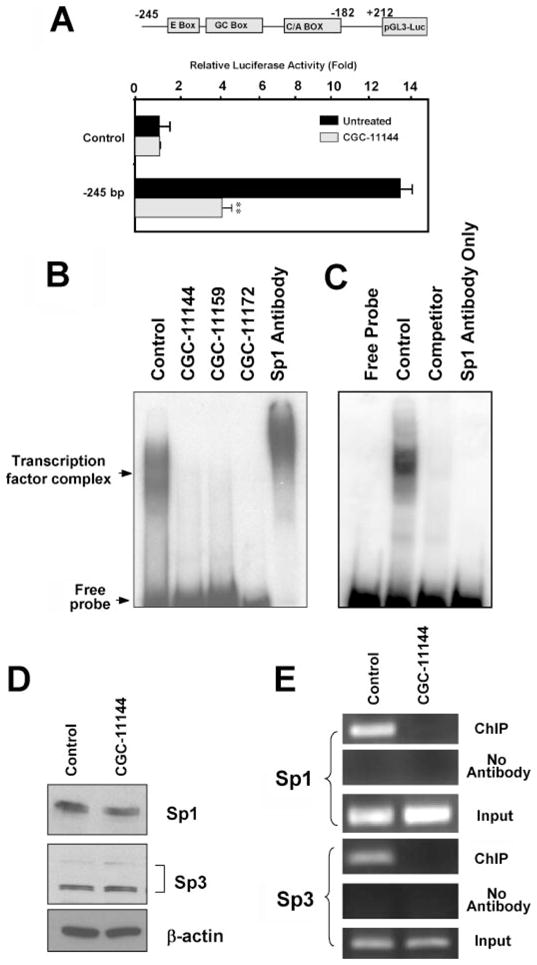
A, a luciferase reporter construct containing the ERα promoter fragment (−245 to +212 bp) was transfected into T47D cells with CMV-β-galactosidase vectors followed by treatment with 10 μM CGC-11144 for 24 h. Reporter gene activities were then measured. **, p < 0.01. B, T47D cells were treated with 10 μM oligoamine for 24 h. EMSA was performed using 10 μg of T47D extracts incubated with γ-32P-labeled oligonucleotides that spanned the ERα promoter from −245 to −182 bp. Super gel shift was performed by using anti-Sp1 antibody. C, EMSA was performed using γ-32P-labeled oligonucleotides of the ERα minimal promoter (−245 to −182 bp) with control T47D nuclear extracts with or without 100-fold excess of nonlabeled Sp1 consensus DNA as competitor and the Sp1 antibodies only. D, T47D cells were treated with 10 μM CGC-11144 for 24 h. Western blots were performed to detect the expression of Sp1 and Sp3. E, cross-linked chromatin prepared from untreated and CGC-11144-treated T47D cells was immunoprecipitated with Sp1 or Sp3 antibody. The immunoprecipitates were subjected to PCR analysis using primer pairs spanning the ERα promoter from −245 to −182 bp. Aliquots of chromatin taken before immunoprecipitation were used as Input controls, whereas chromatin eluted from immunoprecipitations lacking antibody were used as No Antibody controls.
To further investigate the mechanisms underlying the interruption of the DNA binding activity of the Sp1 family of transcription factors by oligoamines, Western analysis was used to examine whether oligoamines affected the protein expression of Sp1 and/or Sp3, two transcription factors that directly bind to GC boxes and transactivate the ER minimal promoter (17). CGC-11144 did not change the protein level of Sp1 or Sp3 (Fig. 5D), indicating that its interruption of Sp1 transcription factor family activity is not through diminished protein expression. Using the chromatin immunoprecipitation assay, the binding of Sp1 and Sp3 to the −245 to −182 element of the ERα promoter was confirmed, and CGC-11144 treatment led to a significant decrease of Sp1 and Sp3 recruitment to the ERα promoter (Fig. 5E).
Effects of JNK/AP-1 on Oligoamine-regulated ERα Expression
The cross-talk between the estrogen signaling and mitogen-activated protein kinase pathways has been intensively studied. To date, one enhancer region termed ER-EH0 (−3778 to −3744 bp; Fig. 3B) containing an AP-1-binding site has been identified as a potential regulatory region in the ERα promoter (21). Our previous study showed that activation of the JNK/AP-1 signaling cascade protects breast cancer cells from oligoamine-induced cell death in breast cancer cells (11). To further investigate whether the JNK/AP-1 pathway is involved in regulation of oligoamine-induced ERα down-regulation, an in vitro JNK kinase assay was used to examine whether oligoamines affect JNK kinase activity in MCF-7 cells. c-Jun fusion protein containing the JNK phosphorylation sites was used as substrate for the kinase assay, and the JNK complex was immunoprecipitated from MCF-7 cells by using a JNK1 monoclonal antibody. The results indicate that the phosphorylation of substrate c-Jun protein (p-c-Jun) is stimulated by CGC-11144. However, simultaneous treatment with SP600125, a selective JNK inhibitor, significantly blocked both the basal JNK and CGC-11144-induced activity (Fig. 6A). SP600125 alone inhibited ERα expression, but combined treatment with SP600126 and CGC-11144 reduced ERα expression below detectable levels (Fig. 6A). These results indicate that JNK/AP-1 is a positive regulator of ERα expression and that oligoamine enhanced JNK/AP-1 activity may antagonize the down-regulation of ERα by oligoamines.
FIGURE 6. Effect of JNK/c-Jun activation on oligoamine-regulated ERα expression.
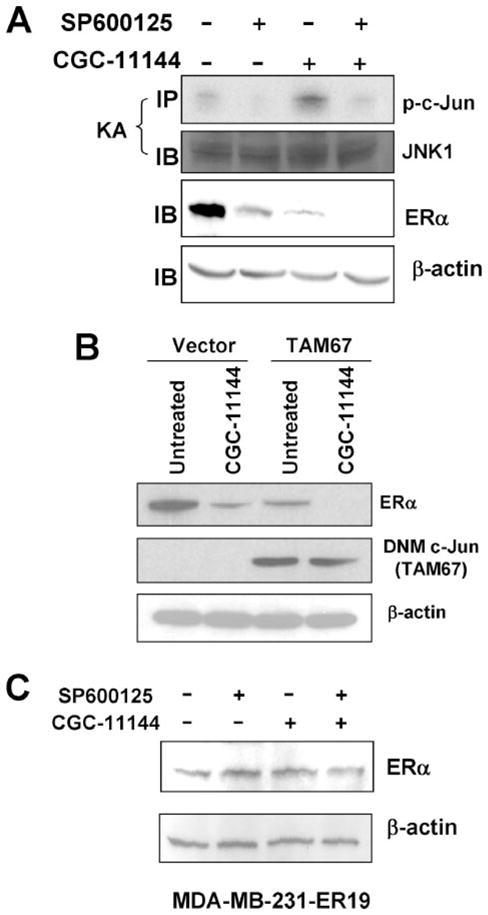
A, MCF-7 cells were treated with 10 μM CGC-11144 or SP600125 alone or both for 96 h. JNK complex was immunoprecipitated (IP) with an anti-JNK1 antibody and then was subjected to the in vitro kinase assay (KA) by using c-Jun fusion protein as the substrate. Total JNK protein was analyzed by Western blotting (IB) for anti-JNK1 protein as a control. Immunoblotting with anti-ERα was performed with actin protein as a control. B, empty and dominant negative mutant (DNM) c-Jun (TAM67) transfected MCF-7 cells were treated with 10 μM CGC-11144 for 96 h, and Western blot with anti-ERα or anti-c-Jun antibodies was performed. Actin protein was blotted as a loading control. C, MDA-MB-231-ER19 cells were treated with 10 μM CGC-11144 or SP600125 or both for 72 h. Western blots were performed by using anti-ERα and anti-actin antibodies.
To define more precisely the effect of the JNK/AP-1 pathway on oligoamine-induced ERα down-regulation, a previously described vector expressing the c-Jun dominant negative mutant, TAM67, was stably transfected into MCF-7 cells (11). As shown in Fig. 6B, MCF-7 cells transfected with the pcDNA3.1-TAM67 vector expressed a ~29-kDa dominant negative mutant c-Jun protein. Overexpression of TAM67 inhibited ERα expression, and treatment with CGC-11144 further reduced ERα expression in TAM67-transfected cells. These results provide further evidence that the JNK/AP-1 pathway is important for ERα expression and that inhibition of this pathway may enhance down-regulation of ERα expression by oligoamines.
To further confirm that the regulation of ERα by c-Jun occurs through an effect on the promoter activity of the ERα gene, we investigated ERα protein expression in CGC-11144-and/or SP600125-treated MDA-MB-231-ER19 cells stably expressing exogenous ERα. Treatment with either SP600125 or CGC11144 or the combination of the two agents did not alter ERα protein expression driven by the CMV promoter (Fig. 6C). This result clearly indicates that the effect of AP-1 on ERα expression occurs through transcriptional regulation on the natural promoter of ERα rather than through an effect on ERα protein itself.
DISCUSSION
Natural polyamine levels in breast tumors are usually higher than in adjacent normal tissues, and dysregulated polyamine metabolism has been well demonstrated in breast cancer (1). Our previous studies demonstrated that a novel class of polyamine analogues, oligoamines, effectively inhibits growth of human breast cancer cells in culture and nude mouse xenografts (10). However, the molecular events underlying oligoamine cytotoxicity in breast cancer cells are currently unknown. A number of recent studies have identified a wide range of important molecular targets for polyamine analogues in human breast cancer cells and one of these targets is ERα (1). Our current studies demonstrate that oligoamines specifically inhibit ERα protein expression but do not affect the expression of ERβ or other specific steroid hormonal receptors. Oligoamine inhibition of ERα expression also leads to the down-regulation of ERα-mediated transcriptional activity and the loss of expression of several important ERα target or partner genes, including PR and cyclin D1. Based on these observations, we envision that ERα is an important molecular target of oligoamines in breast cancer cells and that down-regulation of ERα may contribute to oligoamine cytotoxicity in ER-positive human breast cancer cells. There is a significant difference in the timing of the decrease in ERα expression between T-47D and MCF-7 cells by oligoamines. The possible mechanisms include variable polyamine transport, intracellular polyamine metabolism regulation, drug resistance mechanisms, etc., which may affect the accumulation of polyamines and/or analogues in tumor cells. In addition, it will be important to further elucidate whether the regulation of oligoamines on ERα target genes occurs through down-regulation of ERα or a more global effect of the analogues.
Our analyses using a series of ERα transcription activity assays demonstrate that oligoamine-inhibited ERα expression results from the suppression of ERα transcription rather than changes in protein stability. Furthermore, oligoamine treatment affects ERα transcription apparently by altering the binding of Sp1 transcription factor family members to the −245 to −182 bp proximal promoter element, which contains the GC-rich box of binding sites. It has been known for some time that polyamine-mediated conformational changes in DNA can alter the transcriptional activity of genes. Although it is still unclear whether polyamines interact with DNA in a sequence-specific manner, several studies have suggested that the GC-rich region in the major groove of DNA is one of the preferred sites for spermine binding (22–24). Polyamine-modulated protein-DNA interactions may also regulate the binding activity of regulatory proteins to specific DNA sequences in the gene promoter (25). The Sp1 transcription factor family is an important class of transcription factors whose activity may be regulated by natural polyamines. For example, a recent study found that the binding of Sp1 to the GC box of the spermidine/spermine N1-acetyltransferase promoter is essential for transcription of this key polyamine catabolism enzyme, and a polyamine-responsive element seemed to be responsible for the elevated transcription (26). Because of structural similarity, oligoamines may strongly compete with the binding of natural polyamines to specific DNA sequences like the GC-rich regions, thus leading to the interruption of normal transcription machinery. Our recent studies indicate that co-treatment with spermine prevents the decreased expression of ERα by oligoamines, and luciferase activity assay shows that oligoamine-suppressed ERE reporter gene expression was prevented by simultaneous treatment with spermine (data not shown). However, there are multiple mechanisms by which these findings can result. Because spermine competes with the oligoamines for uptake, one likely interpretation is that spermine prevents down-regulation of ERα expression by competing with oligoamines for transport into the cells. The net effect is that the combination treatment leads to maintenance of the natural polyamine level that prevents the oligoamine-induced ERα down-regulation.
The JNK/AP-1 signaling pathway is a key component of many signal transduction pathways and plays a critical role in the control of growth in breast cancer cells. A 35-bp element called ER-EH0 mapped from −3778 to −3744 bp upstream of the ERα mRNA start site forms multiple DNA-protein complexes, including an AP-1-containing complex with strong promoter activity in ER-positive breast cancer cells (21). Our data show that oligoamines activate the JNK/AP-1 signaling pathway. Use of the JNK-specific inhibitor (SP600125) or a dominant negative mutant c-Jun (TAM67) markedly inhibits ERα expression and potentiates oligoamine down-regulation of ERα expression. These findings provide evidence that activation of JNK signaling stabilizes ERα transcription and may play a role in modulating oligoamine down-regulation of ERα expression. Further elucidation of the mechanisms of the effect of the JNK/AP-1 pathway on oligoamine down-regulation of ERα gene expression may be helpful in designing treatment strategies combining oligoamines and JNK/AP-1 inhibitors.
In summary, we have demonstrated that oligoamines specifically suppress the expression and activity of ERα, a principal determinant of growth and differentiation in human breast cancer cells. Several lines of evidence suggest that oligoamines may interact with other critical regulatory proteins like Sp1 and AP-1 to mediate ERα expression. These data indicate a new approach to modulating estrogen signaling utilizing these novel analogues. These findings also suggest a relationship between polyamines and ERα expression in breast cancer cells and underscore the rationale of targeting polyamine metabolism as a potential approach to breast cancer therapy and/or prevention.
Footnotes
This work was supported by National Institutes of Health Grants P50CA88843, CA98454, and CA51085; Department of Defense Grants DAMD 17-03-1-0376 and W81XWH-04-1-0457; and funds from the Breast Cancer Research Foundation, the Flight Attendant Medical Research Institute (FAMRI), and the Avon Foundation.
The abbreviations used are: ER, estrogen receptor; ERE, estrogen response element; PR, progesterone receptor; JNK, c-Jun NH2-terminal kinase; CMV, cytomegalovirus; EMSA, electrophoretic mobility shift assay.
References
- 1.Huang Y, Pledgie AM, Casero RA, Davidson NE. Anti-cancer Drugs. 2005;16:229–241. doi: 10.1097/00001813-200503000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Russell DH. Crit Rev Clin Lab Sci. 1983;18:261–311. doi: 10.3109/10408368209085073. [DOI] [PubMed] [Google Scholar]
- 3.LaMuraglia GM, Lacaine F, Malt RA. Ann Surg. 1986;204:89–93. doi: 10.1097/00000658-198607000-00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Porter CW, Herrera-Ornelas L, Pera P, Petrelli NF, Mittelman A. Cancer. 1987;60:1275–1281. doi: 10.1002/1097-0142(19870915)60:6<1275::aid-cncr2820600619>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 5.Manni A. In Vivo. 2002;16:493–500. [PubMed] [Google Scholar]
- 6.Manni A. Cancer Treat Res. 1994;71:209–225. doi: 10.1007/978-1-4615-2592-9_11. [DOI] [PubMed] [Google Scholar]
- 7.Leveque J, Foucher F, Bansard JY, Havouis R, Grall JY, Moulinoux JP. Breast Cancer Res Treat. 2000;60:99–105. doi: 10.1023/a:1006319818530. [DOI] [PubMed] [Google Scholar]
- 8.Marton LJ, Pegg AE. Annu Rev Pharmacol Toxicol. 1995;35:55–91. doi: 10.1146/annurev.pa.35.040195.000415. [DOI] [PubMed] [Google Scholar]
- 9.Valasinas A, Reddy VK, Blokhin AV, Basu HS, Bhattacharya S, Sarkar A, Marton LJ, Frydman B. Bioorg Med Chem. 2003;11:4121–4131. doi: 10.1016/s0968-0896(03)00453-x. [DOI] [PubMed] [Google Scholar]
- 10.Huang Y, Hager ER, Phillips DL, Dunn VR, Hacker A, Frydman B, Kink JA, Valasinas AL, Reddy VK, Marton LJ, Casero RA, Davidson NE. Clin Cancer Res. 2003;9:2769–2777. [PMC free article] [PubMed] [Google Scholar]
- 11.Huang Y, Keen JC, Hager E, Smith R, Hacker A, Frydman B, Valasinas AL, Reddy VK, Marton LJ, Casero RA, Jr, Davidson NE. Mol Cancer Res. 2004;2:81–88. [PubMed] [Google Scholar]
- 12.Thomas T, Thomas TJ. Breast Cancer Res Treat. 1993;29:189–201. doi: 10.1007/BF00665680. [DOI] [PubMed] [Google Scholar]
- 13.Cohen FJ, Manni A, Glikman P, Bartholomew M, Demers L. Cancer Res. 1988;48:6819–6825. [PubMed] [Google Scholar]
- 14.Shah N, Thomas TJ, Lewis JS, Klinge CM, Shirahata A, Gelinas C, Thomas T. Oncogene. 2001;20:1715–1729. doi: 10.1038/sj.onc.1204247. [DOI] [PubMed] [Google Scholar]
- 15.Bacchi CJ, Weiss LM, Lane S, Frydman B, Valasinas A, Reddy V, Sun JS, Marton LJ, Khan IA, Moretto M, Yarlett N, Wittner M. Antimicrob Agents Chemother. 2002;46:55–61. doi: 10.1128/AAC.46.1.55-61.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keen JC, Zhou Q, Park BH, Pettit C, Mack KM, Blair B, Brenner K, Davidson NE. J Biol Chem. 2005;280:29519–29524. doi: 10.1074/jbc.M505317200. [DOI] [PubMed] [Google Scholar]
- 17.deGraffenried LA, Hilsenbeck SG, Fuqua SA. J Steroid Biochem Mol Biol. 2002;82:7–18. doi: 10.1016/s0960-0760(02)00151-6. [DOI] [PubMed] [Google Scholar]
- 18.Sharma D, Blum J, Yang X, Beaulieu N, Macleod AR, Davidson NE. Mol Endocrinol. 2005;19:1740–1751. doi: 10.1210/me.2004-0011. [DOI] [PubMed] [Google Scholar]
- 19.Huang Y, Pledgie A, Rubin E, Marton LJ, Woster PM, Sukumar S, Casero RA, Davidson NE. Cancer Biol Ther. 2005;4:1006–1013. doi: 10.4161/cbt.4.9.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.deConinck EC, McPherson LA, Weigel RJ. Mol Cell Biol. 1995;15:2191–2196. doi: 10.1128/mcb.15.4.2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tang Z, Treilleux I, Brown M. Mol Cell Biol. 1997;17:1274–1280. doi: 10.1128/mcb.17.3.1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haworth IS, Rodger A, Richards WG. Proc Biol Sci. 1991;244:107–116. doi: 10.1098/rspb.1991.0058. [DOI] [PubMed] [Google Scholar]
- 23.Zakrzewska K, Pullman B. Biopolymers. 1986;25:375–392. doi: 10.1002/bip.360250302. [DOI] [PubMed] [Google Scholar]
- 24.Feuerstein BG, Williams LD, Basu HS, Marton LJ. J Cell Biochem. 1991;46:37–47. doi: 10.1002/jcb.240460107. [DOI] [PubMed] [Google Scholar]
- 25.Panagiotidis CA, Artandi S, Calame K, Silverstein SJ. Nucleic Acids Res. 1995;23:1800–1809. doi: 10.1093/nar/23.10.1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tomitori H, Nenoi M, Mita K, Daino K, Igarashi K, Ichimura S. Biochim Biophys Acta. 2002;1579:180–184. doi: 10.1016/s0167-4781(02)00545-6. [DOI] [PubMed] [Google Scholar]