

Abstract
Rationale
Gender differences in cardiovascular disease have long been recognized and attributed to beneficial cardiovascular actions of estrogen. Class II histone deacetylases (HDACs) act as key modulators of heart disease by repressing the activity of the myocyte enhancer factor (MEF)2 transcription factor, which promotes pathological cardiac remodeling in response to stress. Although it is proposed that HDACs additionally influence nuclear receptor signaling, the effect of class II HDACs on gender differences in cardiovascular disease remains unstudied.
Objective
We aimed to examine the effect of class II HDACs on post–myocardial infarction remodeling in male and female mice.
Methods and Results
Here we show that the absence of HDAC5 or -9 in female mice protects against maladaptive remodeling following myocardial infarction, during which there is an upregulation of estrogen-responsive genes in the heart. This genetic reprogramming coincides with a pronounced increase in expression of the estrogen receptor (ER)α gene, which we show to be a direct MEF2 target gene. ERα also directly interacts with class II HDACs. Cardioprotection resulting from the absence of HDAC5 or -9 in female mice can be attributed, at least in part, to enhanced neoangiogenesis in the infarcted region via upregulation of the ER target gene vascular endothelial growth factor-a.
Conclusions
Our results reveal a novel gender-specific pathway of cardioprotection mediated by ERα and its regulation by MEF2 and class II HDACs.
Keywords: estrogen receptor, cardiac remodeling, myocardial infarction, class II HDACs
Heart failure secondary to myocardial infarction (MI) and ischemic cardiomyopathy is the primary cause of cardiovascular mortality. Cardiac remodeling following MI induces expansion and alterations in the infarcted and noninfarcted regions of the heart and increases internal load, which promotes further stress, dilation, and hypertrophy of the noninfarcted area.1 Although neoangiogenesis within the infarcted tissue is also an integral component of the remodeling process, the microvascular network that develops following MI is generally unable to support the increased demands of the hypertrophied myocardium, resulting in progressive loss of viable tissue, infarct extension and fibrous replacement.2 For reasons that remain incompletely understood, premenopausal women appear to be partially protected against remodeling of the infarct area and remote myocardium during the course of ischemic cardiomyopathy because of the presence of estrogen.3
Estrogen receptors (ERs) are ligand-dependent transcription factors that mediate cardioprotective processes such as vasodilation and angiogenesis, which limit myocardial remodeling after infarction and attenuate cardiac hypertrophy.4 After estrogen binding, the ER undergoes a conformational change, which allows the receptor to dissociate from chaperone proteins and translocate to the nucleus where it binds DNA and activates estrogen-responsive genes.5 Like other nuclear hormone receptors, the activity of the ER is also modulated by association with histone acetyltransferases and histone deacetylases (HDACs).6
There are 2 general classes of HDACs that have been shown to have profound influences on the response of the heart to stress.7 Class I HDACs (HDACs 1, 2, and 3) are relatively simple in structure, containing only a catalytic domain, which efficiently deacetylates histones. HDACs 1 and 2 function redundantly to control myocardial growth.8,9 HDAC2 also functions as a positive regulator of myocyte hypertrophy in response to stress and appears to be a key target for HDAC inhibitors, which prevent pathological cardiac remodeling.10 HDAC3 has been implicated in the control of cardiac lipid metabolism.11 In contrast, the 4 class IIa HDACs (HDACs 4, 5, 7, and 9) contain a large N-terminal regulatory domain and a catalytic domain with weak activity.7 These HDACs exert their functions through recruitment of class I HDACs and other corepressors to transcription factors, such as myocyte enhancer factor (MEF)2.
The response of the heart to biomechanical stress and other forms of pathological signaling is governed by the class II HDACs, HDAC5 and -9, which serve as signal-responsive repressors of the MEF2 transcription factor.6 In the unstimulated state, these HDACs interact with MEF2 and repress the transcription of MEF2 target genes, whereas on activation of intracellular signaling pathways involving calcium, calmodulin-dependent protein kinase or protein kinase D, these HDACs become phosphorylated, triggering their nuclear export and the consequent activation of MEF2 target genes. Consistent with the roles of HDAC5 and -9 as repressors of pathological MEF2 activity, male mice lacking these HDACs display enhanced MEF2 activity and heightened sensitivity to cardiac stress.12,13
In the present study, we investigated the potential involvement of the MEF2/class II HDAC pathway in post-MI remodeling in male and female mice. Whereas male mice lacking HDAC5 or -9 were hypersensitive to the pathological effects of MI, we found, unexpectedly, that female mice lacking HDAC5 and -9 were protected against pathological remodeling of the heart following MI. In the absence of HDAC5 or -9, there is a pronounced increase in expression of the ERα gene, which contains essential MEF2 binding sites in its promoter. In addition, HDAC5 and -9 directly interact with α to repress transcriptional activation of the receptor by estrogen. Upregulation of ERα signaling in female mice mutant for either HDAC5 or -9 dramatically diminishes cardiac dysfunction and deleterious left ventricular remodeling following MI. This protection appears to be due, at least in part, to neoangiogenesis in the infarcted region via upregulation of the ER target gene vascular endothelial growth factor (VEGF)a. These findings reveal a key role for MEF2 and class II HDACs in the regulation of cardiac ER signaling and the mechanisms underlying the cardioprotective effects of estrogen.
Methods
An expanded Methods section is available in the Online Data Supplement at http://circres.ahajournals.org.
Surgical Procedures and Echocardiography
All animal protocols were approved by the Institutional Animal Care and Use Committee of the University of Texas Southwestern Medical Center. Adult age matched HDAC9 knockout (KO), HDAC5 KO mice, and wild-type (WT) mice of either sex received a MI as described before.14 Sham-operated animals underwent the same procedure without occlusion of the left coronary artery. At 4 weeks of age, mice were either sham-operated or ovariectomized and either left untreated or treated with 17β-estradiol (0.16 μg per day) for 4 weeks as described.15 Three weeks following MI, echocardiography was performed in conscious mice using the fully digital Vingmed System (GE Vingmed Ultrasound, Horten, Norway) and a 11.5-MHz linear array transducer as previously described.16 All surgeries and subsequent analyses were performed in a blinded fashion for genotype.
Infarct Size
To measure the infarct size 3 weeks post-MI, the heart was excised, and the left ventricles (LVs) were cut from apex to base into 3 transverse sections. Five-micron sections were cut and stained with Masson’s trichrome. Infarct length was measured along the endocardial and epicardial surfaces from each of the cardiac sections, and the values from all specimens were summed. Infarct size (in percentage) was calculated as percentage of infarcted LV versus total LV circumference. Values represent the average of 3 slides per animal.17
Histology and Histochemistry
After collection, heart tissue was either cryoembedded or incubated for 30 minutes in Krebs buffer (118 mmol/L NaCl, 4.7 mmol/L KCL, 1.2 mmol/L KH2PO4, 1.2 MgSO4, 25 mmol/L NaHCO3, 11 mmol/L glucose) to arrest the heart in diastole, fixed in 3.7% paraformaldehyde, and embedded in paraffin. Sections were stained with hematoxylin/eosin to visualize infarcted area (n=3 to 6 in each group). To examine the capillary density, all sections were incubated with biotinylated Griffiona simplicifolia lectin (Vector Laboratories, United Kingdom) for 2 hours at room temperature, as described previously.18 The number of capillaries was counted under microscopy for 5 random fields in the remote, border zone, or infarcted region of each longitudinal slices in both WT and HDAC9 KO female animals post-MI.
RNA Extraction and RT-PCR Analysis
Total RNA from the infarcted area, including the border zone region, was isolated using TRIzol (Invitrogen). A 10-μg aliquot representative of 3 animals per sample group was then analyzed on Affymetrix U74Av2 microarrays. A subset of differentially expressed RNAs was further characterized by either semiquantitative PCR or quantitative real-time PCR (see Online Table I for primer sequences).
In Situ Hybridization
Section in situ hybridization was performed on fresh-frozen tissue 4 days after either sham surgery or MI as described,19 with some minor modifications because of the use of cryosections. The coding regions of VEGFa and VEGFr2 were subcloned into pCDNA, linearized, and transcribed as follows: antisense VEGFa, BamHI, and T3; antisense VEGFr2, NotI, and T7 (generously provided by Dr T. Sato, University of Texas Southwestern Medical Center).
Electrophoretic Mobility-Shift Assays
Oligonucleotides corresponding to the conserved MEF2-binding site in the ERα regulatory region and the mutated MEF2-binding sites were used to detect MEF2 binding (see Online Table II for primer sequences). Annealed oligonucleotides were radiolabeled with [32P]dCTP using the Klenow fragment of DNA polymerase and purified using G50 spin columns (Roche). Nuclear cell extracts were isolated from COS-1 cells that were transfected with pcDNAMYC-MEF2C. Reaction conditions of the gel mobility-shift assays were previously described.20
Generation of ERα Reporter Constructs
A mouse genomic DNA fragment covering either the region from −3990 to +1 relative to the ERα transcription initiation site was isolated from genomic DNA C57Bl6. These ERα promoter fragments were cloned into pGL2 luciferase vector as a KpN/Nhe fragment. Mutations of the MEF2 sites were introduced into the sequence by PCR-based site-directed mutagenesis. All constructs were verified by DNA sequence analysis.
Cell Culture, Transfection, and Luciferase Assays
MYC- and FLAG-tagged derivatives of MEF2C, asHDAC9, and asHDAC5 and the signal-resistant counterparts (S259/498A, adH-DAC5 S>A) have been described.6,21 Primary rat cardiomyocytes were prepared as described.22 Eighteen hours after plating, cells were infected with adenovirus for 2 hours and subsequently cultured in serum-free medium for 48 hours before collection. Both COS-1 and HeLa cells were maintained in DMEM with FBS (10%), L-glutamine (2 mmol/L), and penicillin-streptomycin, and transfections were performed as described previously.6 COS-1 cells were transfected with pcDNAMYC-MEF2C to obtain nuclear cell extracts for electrophoretic mobility-shift assays (EMSAs). HeLa cells were transfected with a reporter construct containing 3 estrogen response elements (3ERE-Luc), full-length ERα, and increasing concentrations of either HDAC5 and -9 and β-galactosidase (internal control) reporter plasmids using Fugene 6 (Stratagene) according to the instructions of the manufacturer. Luciferase activity is reported as the fold induction compared with that of empty 3ERE-Luc alone. Relative promoter activities are expressed as luminescence relative units normalized for β-galactosidase expression in the cell extracts.
Glutathione S-Transferase Pull Down
Glutathione S-transferase (GST)-ERα was generated by subcloning PCR amplified fragment into EcoRI and XhoI sites of the pGEX-KG vector, whereas GST-dHDAC9 (aa131–586) was subcloned in the pGEX-KG vector as a XbaI-XhoI fragment. The GST pull down was performed as described previously.6
Statistical Analysis
Data are shown as means±SD. Comparison between 2 groups were analyzed by the 2-tailed Student’s t test. Values of P<0.05 were considered statistically significant.
Results
The Absence of HDAC5 and -9 Protects Female Mice Against Post-MI Cardiac Remodeling
To explore the potential influence of class II HDACs on gender-specific responses of the heart to injury, we compared the cardiac remodeling responses of male and female WT and HDAC5 and -9 KO mice following MI. Survival up to 3 weeks after MI was comparable in WT males and females (63% versus 65%, respectively). Whereas a dramatic decrease in survival was evident in HDAC5 and -9 mutant males (36% and 40%, respectively), female mice deficient for HDAC5 and -9 displayed remarkably improved postinfarction survival (85% and 83%, respectively) especially in the first week after the insult (Figure 1A).
Figure 1.

Female class II HDAC KO mice are protected against post-MI cardiac remodeling. A, Post-MI mortality was comparable between male and female WT mice. Whereas male HDAC5 and -9 mutant animals had an elevated mortality compared to WT, females lacking HDAC5 or -9 showed an increase in survival after MI (WT males, n=13; WT females, n=17; HDAC5 KO males, n=15; HDAC5 KO females, n=16; HDAC9 KO males, n=16; HDAC9 KO females, n=16). B, Cardiac function was measured by echocardiography 3 weeks after permanent left coronary artery ligation. The data demonstrate progressive dilation and loss of contractile function in WT mice as indicated by left ventricular systolic dimension (LVIDs) and fractional shortening, whereas the loss of cardiac function was substantially attenuated in female HDAC5 and -9 KO mice (n=5 in sham groups; n=9 to 10 in MI groups). *P<0.05 vs corresponding WT group. C, The infarct size 3 weeks post-MI expressed as a fraction of the total cross-sectional circumference of the LV indicates that the infarct size in HDAC5 and -9 KO females is significantly smaller than the infarct size in WT females (34% and 37% vs 55% in WT, n=4). *P<0.05 vs corresponding WT group. D, Hematoxylin/eosin staining on transverse section 3 weeks post-MI shows the infarct of HDAC9 KO females to be nontransmural because of the presence of an additional layer of surviving cells at the epicardial surface of the infarcted area. The region in the box is enlarged in the lower panel. Scale bars: top, 2 mm; bottom, 20 μm.
Echocardiography 3 weeks after MI surgery indicated that left ventricular diastolic dimension, left ventricular systolic dimension, and fractional shortening worsened severely in both male and female WT mice, whereas there were no differences in the sham-operated animals. The loss of cardiac function was even more dramatic for male HDAC mutant mice (data not shown). However, both parameters were significantly preserved in HDAC5 and HDAC9 mutant females compared to WT females, indicative of better-maintained cardiac function (Figure 1B, Online Table III, and Online Figure I). The fact that genetic deletion of both class II HDACs leads to the protection in females points to a threshold of class II HDACs below which there is a protective effect.
Three weeks after MI, morphometric analysis indicated the presence of cardiac hypertrophy and concomitant signs of cardiac failure, such as an increase in lung weight, in WT females, whereas this pathological remodeling response was lacking in the HDAC5 and -9 mutant females (Online IV, only shown for HDAC9 KO females). Infarct size measurements as percentage of LV circumference indicated 55% of the LV to be affected by the infarct in WT females, whereas the infarct accounted for 37% and 34% of the LV in HDAC5 and -9 KO females, respectively (Figure 1C). Histological examination of the infarcted area clearly indicated the presence of a distinct layer of surviving cardiomyocytes at the epicardial surface of the infarcted region in the HDAC9 KO females (Figure 1D). Because of the presence of this additional layer of muscle cells, the integrity of the infarcted area was better maintained after MI and likely prevented the heart from dilating to the same extent as in the WT mice, which results in a better maintenance of cardiac function post-MI.
Upregulation of VEGFa in the Infarcted Region of HDAC9 KO Females
Gene expression analysis to further explore the mechanistic basis of gender-specific cardioprotection in HDAC mutant females indicated that a collection of genes implicated in ER signaling was expressed at significantly higher levels in the infarcted areas of the HDAC9 KO females compared to their gender-matched WT littermates (Online Table V). Because of the obvious restriction of the cardioprotective effect of class II HDAC deletion to females, the upregulation of genes involved in ER signaling, and previous reports on the involvement of HDACs in the ER signaling pathway,23–27 we speculated that class II HDAC removal led to an increase in expression of ER target genes.
One of the beneficial effects of estrogen on the cardiovascular system is to improve myocardial perfusion by the formation of new capillaries and by the enlargement of preexisting collateral vessels, when the myocardium is deprived of blood.28–31 One key regulator of this proangiogenic response is the ER target gene VEGFa, which encodes an extracellular ligand for its corresponding endothelial receptors flt-1 (Fms-related tyrosine kinase 1, or VEGF receptor 1) and flk-1 (fetal liver kinase 1, or VEGF receptor 2).2,32–35 Real-time PCR analysis indicated numerous angiogenic factors to be more highly expressed in the infarcted area of the HDAC9 KO females compared to WT females. VEGFa and its receptors (VEGFr1 and VEGFr2) were upregulated most dramatically (68-, 12-, and 20-fold versus WT, respectively) (Figure 2A). This observation is consistent with the fact that VEGFa induces expression of its receptors to create a positive feed-forward loop allowing endothelial cells to become responsive to and activated by VEGF.33 The expression levels of the angiogenic factors tested did not differ in the sham-operated animals between either sex of WT and HDAC9 KO animals, nor did they differ between male and female HDAC9 KO mice (data not shown).
Figure 2.

Enhanced angiogenic response in infarcted region of HDAC9 mutant females 3 weeks post-MI. A, Real-time PCR for angiogenic factors indicates an increased angiogenic response in the infarcted region of the HDAC9 KO females vs the WT females. Data are represented as fold change compared to WT animals. B, In situ hybridization analysis revealed an increase in both VEGFa and its endothelial cell surface receptor VEGFr2 at the border zone of the infarcted region 4 days post-MI, which is more pronounced in the HDAC9 KO females. Red square indicates the enlarged area shown on the right. Scale bars: 40 μm. C, Representative sections stained for lectin show an equal distribution of capillaries surrounding cardiomyocytes in the noninfarcted, remote areas, whereas the ischemic region shows irregular patterning of vasculature with additional and enlarged vessels in the border zone and the infarcted region of the HDAC9 mutant females compared to the WT females. Scale bars: 20 μm. D, Semi-quantitative analysis of capillary density in the myocardium indicates there to be no difference in number of vessels in the remote region between WT and HDAC9 KO females. However, HDAC9 KO females show an increase in vessel density in the border zone compared to WT females. The capillary count of these sections is expressed as number of vessels per microscopic field (n=4 to 6; means±SD). *P<0.05 vs WT group.
During the first few days following MI there is a neoangiogenic response within the infarcted tissue. In situ hybridization 4 days after MI indicated that VEGFa and VEGFr2 expression was significantly induced in the border zone, which was even more pronounced in the HDAC9 KO females (Figure 2B). This indicates that removal of class II HDACs in females increases the expression of VEGFa and its downstream receptors, which likely promotes cardioprotection after ischemic damage.
To visualize the vasculature, we used Griffonia Simplicifolia lectin I (GS-I) as an endothelial surface marker.18 There was a regular distribution of capillaries around cardiomyocytes in both sham-operated groups, with no detectable differences is vessel density. Three weeks after MI, the border zone of the infarcted area contained regions of low vascularity in the WT animals, which was even more decreased in the infarct. However, both the border zone and the infarcted region of the HDAC9 KO females appeared to be highly vascular, with enlarged, thin-walled vessels (Figure 2C). The number of capillaries that was counted in the remote, border zone, or infarcted region of longitudinal slices in both WT and HDAC9 KO female animals post-MI indicated there to be no difference in vessel density in the remote region, whereas this number was significantly increased in the border zone of the HDAC9 KO females (Figure 2D). These findings suggest that the cardioprotection observed in HDAC5 and -9 mutant females is attributable, at least in part, to an increase in vessel formation post-MI, which might involve VEGFa, because prolonged VEGFa expression in the myocardium results in a high density of vessels with enlarged lumens.32
Cardioprotective Effect Dependent on Estrogen
Based on the fact that the cardioprotective effect after MI in the absence of class II HDACs appeared to be restricted to females, and the known interaction between HDACs and ERα, we speculated this gender-dependent response in the class II HDAC-null background to be attributable to an enhancement of cardiac ER signaling. To explore this hypothesis, we subjected HDAC9 mutant females at 4 weeks of age to ovariectomy, followed by supplementation of a physiological dose of estrogen or a placebo pellet before we induced MI. Ovariectomy in these females before MI resulted in a thin dilated infarcted region, whereas additional estrogen supplementation again inhibited the infarct to become transmural (Figure 3A). In line with the functional data, we observed an increase in post-MI mortality in the mutant ovariectomized group, although this increase was abolished in the presence of estrogen supplementation (Figure 3B). Although ovariectomy at baseline had no overt effect on cardiac function in the mutant females, MI phenocopied the decrease in cardiac function present in WT females after MI, whereas supplementation of a physiological dose of estrogen in these animals resulted in a comparable resistance to cardiac dysfunction post-MI, as seen in the HDAC9 KO females (Figure 3C and Online Table VI). However, short-term estrogen supplementation in male HDAC9 KO animals was unable to reverse the remodeling and induce the protective effect seen in females (data not shown). It is likely that this is because of the ongoing adverse cardiac remodeling that took place because of the extended absence of HDAC9 in these males.12,13 The levels of circulating estrogen did not differ at baseline between WT and HDAC9 mutant females (data not shown).
Figure 3.

Cardioprotection regulated by the effect of estrogen. A, Histological sections show that ovariectomy in the HDAC9 females ablates the protective effect on cardiac remodeling, whereas supplementation of estrogen restores the protective effect 3 weeks post-MI. Scale bars: 40 μm. B, Post-MI mortality in ovariectomized (ovex) females was comparable to the post-MI mortality seen in WT females. Supplementation of estrogen in ovariectomized HDAC9 KO females was able to largely restore the protective effect (n=5 to 10). C, Cardiac function was measured by echocardiography 3 weeks after MI. The data demonstrate more dilation and loss of contractile function in HDAC9 KO females after ovariectomy as indicated by left ventricular systolic dimension (LVIDs) and fractional shortening, whereas these effects were substantially attenuated after estrogen supplementation (n=5 in sham group; n=7 to 10 in MI groups). *P<0.05 vs corresponding HDAC9 KO post-MI group.
These data indicate that the reduction of the maladaptive alterations of both the remote myocardium and the infarcted area in the postinfarcted heart of HDAC9 KO females is likely attributable to an enhancement of estrogen signaling because ovariectomy in HDAC9 KO females results in a post-MI response comparable to WT females and subsequent addition of estrogen can recapitulate the cardioprotective effect before ovariectomy.
Regulation of ER Expression by MEF2 and Class II HDACs
Class II HDACs associate with MEF2 and inhibit MEF2-dependent transcription.36,37 To determine whether loss of HDAC5 and -9 might enhance cardiac ER signaling through upregulation of the receptor isoforms via MEF2, we scanned the 5′ flanking regions of both the ERα and ERβ genes and identified 2 conserved putative MEF2 binding sites located between −3990 and +1 bp relative to the translation initiation codon of the ERα gene (Figure 4A). EMSA was used to check for binding of MEF2C to the putative MEF2 sites of the ERα promoter using nuclear extracts from COS-1 cells transfected with a MYC-MEF2C expression plasmid and the radiolabeled MEF2 site. Both MEF2 consensus sequences bound MYC-tagged MEF2C avidly, with the highest binding seen for the sole perfect MEF2 binding sequence (site 2). The MEF2-DNA complex was supershifted by anti-MYC antibody and was abolished in the presence of an excess of the unlabeled cognate DNA sequence or the prototypical MEF2 site from the muscle creatine kinase enhancer,38 which was used as a competitor, whereas a mutant sequence that did not bind MEF2C failed to compete for MEF2C binding (Figure 4B).
Figure 4.

Cardiac expression of ERα is regulated by MEF2 and class II HDACs. A, Cross-species conservation of genomic sequences upstream of the ERα gene is shown at the top. Sequence of putative MEF2 transcription factor-binding sites (in boxes) in the promoter of the mouse ERα gene. Numbers in brackets refer to location of the MEF2 site relative to the ATG start codon for ERα. B, EMSA for binding of MEF2C to the putative MEF2 sites of the ERα promoter using nuclear extracts from COS-1 cells transfected with a MYC-MEF2C expression plasmid and the radiolabeled MEF2 site. The MEF2-DNA complex was supershifted by anti-MYC antibody and was abolished in the presence of an excess of the unlabeled cognate DNA sequence or the prototypical MEF2 site from the muscle creatine kinase enhancer as a competitor, whereas a mutant sequence that did not bind MEF2C failed to compete for MEF2C binding. C, Luciferase reporter experiments using an upstream promoter fragment of the ERα gene indicates that MEF2 can induce transcriptional activity, which is enhanced in the presence of myocardin. Mutational analysis indicates that the MEF2 site 770 bp upstream of the transcriptional start is responsible for the MEF2 responsiveness of the ERα gene. D, Real-time PCR analysis of ERα expression in noninfected neonatal rat cardiomyocytes compared with cells infected with AdCMV-β gal or AdCMV-asHDAC5 or asHDAC9 indicates that both asHDAC5 and asHDAC9 increases ERα expression in myocytes in a dose-dependent manner. MOI indicates multiplicity of infection. Additionally, adenoviral overexpression of MEF2C (AdMEF2C) is capable of inducing ERα expression, whereas titrating in a signal resistant form of HDAC5 (AdHDAC5 S>A) reduces this induction. E, Real-time PCR analysis of ERα expression in cardiac samples of WT, HDAC5 and HDAC9 KO animals shows an in vivo upregulation of ERα expression in the absence of HDAC5 and -9 compared to WT. *P<0.05 compared to corresponding WT male or female.
Reporter assays transfecting the 4-kb enhancer fragment fused to luciferase into COS-1 cells indicated this region to be responsive to MEF2, which was further enhanced by the MEF2 coactivator myocardin. Mutating MEF2 site 1 was unable to abrogate MEF2 transcriptional activation, whereas a comparable mutation of site 2 completely abolished transcription, indicating that MEF2 responsiveness is dependent on the MEF2 site located 770 bp upstream of the transcriptional start of the ERα gene (Figure 4C).
To determine whether the expression of ERα in cardiomyocytes is regulated by MEF2 and its interaction with class II HDACs, we infected neonatal rat ventricular myocytes with increasing amounts of an adenovirus expressing anti-sense HDAC5 or -9. Real-time PCR analysis indicated a dose-dependent increase in ERα expression with loss of these class II HDACs (Figure 4D). Additionally, adenoviral over-expression of MEF2C induced the expression of ERα, and this effect was blunted in the presence of an increasing amount of a signal-resistant HDAC5 mutant, referred to as HDAC5S/A, which functions as a MEF2 super-repressor (Figure 4D).6 Consistent with the conclusion that HDAC5 and -9 negatively regulate the expression of ERα in vivo, ERα expression was significantly upregulated in cardiac samples of both HDAC5 and -9 KO animals, whereas ERβ expression levels were unchanged (Figure 4E and data not shown). We conclude that MEF2 binds directly to the regulatory region of the ERα gene and is responsible for the increased expression of ERα in the absence of either class II HDAC5 or -9.
Class II HDACs Regulate ER Signaling by Direct Interaction With ERα
Because HDAC1 and 4 have been shown to downregulate the transcriptional activity of ERα by directly interacting with the receptor,27,39 we aimed to determine whether HDAC5 and -9 could also interact with the receptor. For this purpose, full-length ERα was fused to GST, expressed in Escherichia coli, and tested for the ability to associate with in vitro-translated HDAC4, -5, -9, and -1. Whereas the class II HDACs 4, 5, and 9 interacted with ERα, we were unable to detect an interaction with HDAC1, a class I HDAC (Figure 5A).
Figure 5.

The MEF2 binding domain of class II HDACs directly interact binding with ERα. A, Either GST alone or GST-ERα was expressed in E coli, conjugated to glutathione-agarose beads, and incubated with the indicated [35S]methionine-labeled HDACs. Although the class II HDACs 4, 5, and 9 all bind, HDAC1 failed to interact with ERα in this assay. B, To map the domain of interaction, GST alone or GST-ERα was incubated with the indicated [35S]methionine-labeled HDAC5 deletion mutants. The HDAC mutants lacking the MEF2 binding domain failed to interact with ERα in this assay.
To map the region of class II HDACs that mediates the association with ERα, we assessed the capacities of a series of HDAC5 deletion mutants to associate with GST-ERα (Figure 5B). Deletion of residues from amino acid 260 to the carboxyl terminus of HDAC5 had no effect on ERα binding. Similarly, an N-terminal deletion mutant lacking the CtBP binding domain (mutant 51-C) interacted with ERα, whereas deletion mutants lacking the MEF2 binding domain failed to bind ERα.
To determine which region on ERα was able to interact with class II HDACs, we used GST-HDAC5 (aa131–586) and generated a series of ERα deletion mutants. ERα contains the following functional domains: a hormone-independent transcription function domain (AF-1); a DNA-binding domain; a hinge region containing 3 nuclear localization sequences that mediate the translocation of the receptor from cytosol to nucleus; and a conserved ligand-binding domain, possessing dimerization and hormone-dependent activation functions (activation function [AF]-2).40 Whereas an N-terminal deletion mutant lacking the ligand-binding domain was able to interact with HDAC5, this interaction was absent using N- and C-terminal deletion mutants lacking the nuclear localization sequence domain. We therefore conclude that the MEF2 binding domain in class II HDACs directly interacts with ERα by binding to the nuclear localization sequence domain (Figure 6A).
Figure 6.

Class II HDACs repress the transcriptional binding activity of ERα through direct binding of the MEF2 domain with the N terminus of ERα. A, To map the domain of interaction, GST alone or GST-HDAC5 was incubated with the indicated [35S]methionine-labeled ERα deletion mutants. The ERα mutant lacking the nuclear localization sequences failed to interact with class II HDAC5 in this assay. B, HeLa cells were transfected with 3ERE-Luc, full-length ERα, and increasing concentrations of either HDAC5 and -9 and β-galactosidase (internal control) reporter plasmids. Luciferase activity is reported as the fold induction relative to empty 3ERE-Luc alone. ERα induces luciferase activity which is dose-dependently decreased by the presence of either HDAC5 or -9. C, Titrating in either HDAC5 or -9 both severely reduces the transcriptional activity, whereas HDAC inhibition by TSA, a pharmacological HDAC inhibitor, strongly enhances the transcriptional activity of ERα.
Because class II HDACs are known to interact with the nuclear receptor corepressors SMRT and N-CoR, we predicted that the association of these HDACs with the ER would inhibit ER function by repressing its transcriptional activity. Indeed, transfection experiments in HeLa cells showed significant suppression of the ER-dependent reporter 3ERE-Luc reporter with both HDAC5 and -9 in a concentration-dependent manner (Figure 6B). Combined treatment of cells with ERα and the HDAC inhibitor trichostatin A (TSA) resulted in robust synergistic activation of ERα transcriptional activity (Figure 6C). Together, these findings suggest that class II HDACs are capable of direct interaction with ERα and repress the transcriptional activity of the receptor.
Discussion
Estrogens protect the female heart against cardiovascular disease.3 Through its nuclear receptor, estrogen can attenuate left ventricular hypertrophy, enhance angiogenesis, improve contractility, and prevent pathological cardiac remodeling after ischemic insult. Our results show that class II HDACs play a key role in the regulation of cardiac ER signaling and that removal of either HDAC5 or -9 elicits a cardioprotective mechanism in female mice that is dependent on signaling through ERα (Figure 7). Class II HDACs associate with MEF2 and inhibit MEF2 dependent transcription.13,36 This phenomenon applies not only to post-MI remodeling, because the age dependent cardiac remodeling seen in male HDAC5 and -9 mutant animals is also largely absent in female littermates (Online Figure III). Our data demonstrate the ERα gene to be under control of MEF2 and to be upregulated in the absence of either HDAC5 or -9, providing a mechanism to account for the cardioprotection observed in HDAC5 and -9 mutant females.
Figure 7.
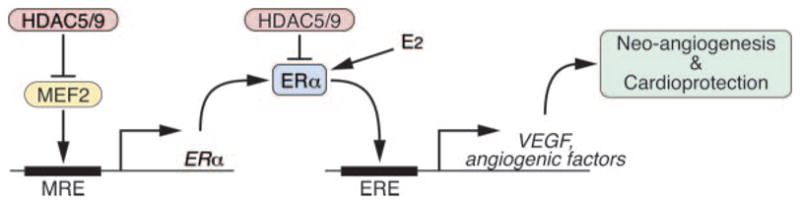
Model for the control of ERα expression and cardioprotection by MEF2 and class II HDACs. Class II HDACs repress ERα expression through a MEF2 response element (MRE) upstream of the ERα gene. Additionally, class II HDACs directly interact with ERα and repress the transcriptional activity of the receptor in the presence of its ligand, estrogen (E2). Class II HDAC inhibition increases the cardioprotective effect of estrogen by both increasing the level of ERα and the transcriptional activity of the receptor. ERα acts through an ERE in the promoter of the VEGF gene and other genes encoding angiogenic factors.
Modulation of ER Activity by HDACs
In response to pathological signals, class II HDACs are shuttled from the nucleus to the cytoplasm, allowing for activation of MEF2-dependent gene expression (reviewed elsewhere41). Thus, our results predict that cardiac stress should induce ERα expression. In line with our predictions, ERα expression has been shown to be increased in both male and female human hypertrophic and end-stage failing hearts.42,43
In addition to regulating gene expression through MEF2, class II HDACs interact with other transcriptional regulators. SMRT and N-CoR interact with nuclear receptors and repress the activity of the receptors through the recruitment of either class I or class II HDACs.44 Transcriptional repression of N-CoR and SMRT is mediated by multiple repression domains, which engage distinct HDAC complexes. Whereas class I HDACs interact with SMRT and N-CoR indirectly through mSin3, class II HDACs directly interact with SMRT and N-CoR through a different repressor domain.45 Kraus and colleagues have shown that ERα, but not ERβ, is a target for acetylation by p300 and have identified acetylation as modulator of the ligand-dependent gene regulatory activity of ERα.46 Besides the fact that the recruitment of HDACs to the ER would promote chromatin condensation of ER target genes, the transcriptional activity and estrogen sensitivity of ERα is enhanced by p300-induced acetylation of the receptor itself, which can be increased by TSA.46 In this study, we show that, in addition to this distinct manner of interaction between class I and II HDACs with the SMRT/N-CoR corepressor complex, and in contrast to class I HDACs, class II HDACs directly interact with ERα. These findings reveal a substantial difference between nuclear receptor corepressor–HDAC complexes involving either class I or II HDACs and provide a rationale for a distinct, tissue-specific recruitment of HDACs by the SMRT/N-CoR complex to regulate the transcriptional activity of ERα.
Modulation of Angiogenesis by Estrogens
Irrespective of whether class II HDAC removal enhances cardiac ER signaling through upregulation of the receptor, through an increase in transcriptional activity of the receptor, or a combination of both, our findings have important implications because of the beneficial effects of ER signaling on the heart. Estrogens are capable of inducing angiogenesis both in vitro and in vivo47 and have been postulated to induce myocardial revascularization post-MI. Although the mechanisms involved in the proangiogenic effects of estrogen are probably multifactorial, upregulation of VEGFa, its receptors, and additional angiogenic proteins is clearly involved.48–52 Estrogen is able to regulate VEGFa expression by increasing the recruitment of ERα and p300 to Sp1 bound to the promoter of the VEGFa gene.53 Recruitment of hypoxia-inducible factor 1α, another key regulator of the transcriptional response during oxygen deprivation, is also required for the increase in VEGFa expression in response to estrogen.53 The requirement of both hypoxia-inducible factor 1α and ERα to induce VEGFa expression would be in line with our data, because we were unable to detect a significant difference at baseline in expression level of angiogenic factors in HDAC mutant mice, whereas post-MI, the absence of class II HDACs caused an exaggerated expression of angiogenic factors in females, with VEGFa being the most dominant. We speculate that the exaggerated VEGFa expression in the border zone of the infarcted area on removal of class II HDACs enables the endothelial cells of the existing coronary network to branch and thereby improve the perfusion of the ischemic myocardial tissue, preventing infarcts from becoming transmural. However, the protective effects of estrogen include many facets of heart disease. Estrogen inhibits the development of pathological hypertrophy and changes the expression of a number of cardioprotective genes such as nitric oxide synthase and heat shock proteins54–56 and alters a number of genes involved in metabolism such as those encoding lipoprotein lipase, prostaglandin D2 synthase, and PGC-1α (peroxisome proliferator-activated receptor γ coactivator 1α).55 The most highly upregulated gene in the HDAC9 mutant females compared to WT females was PPARBP (peroxisome proliferator-activated receptor binding protein) (TRAP220), a transcriptional coactivator that directly enhances the activity of the ER.57 How this cofactor might contribute to cardioprotection remains to be determined. Conceivably, the protective effect seen in the mutant class II HDACs females reflects the net effect of a combination of these responses.
HDAC Inhibitors and the Heart
Reduced post-MI remodeling through enhanced angiogenesis is only one of the many cardioprotective responses induced by ER signaling. Our data imply that removal of class II HDACs likely constitutes a more general effect of cardioprotection through enhancement of this signaling cascade. Interestingly, HDAC inhibitors have shown efficacy in humans and animal studies in suppressing tumor cell growth and hypertrophy.58,59 Although to date, 1 in vitro study reported on selective inhibition of class II HDACs,60 the in vivo efficacy and utility of such inhibitors needs to be tested. Future studies will provide insight into whether it is possible to enhance ER signaling through the use of HDAC inhibitors, and, if so, whether this will not only increase estrogen-dependent cardioprotection by the ER but also the neuro- and musculo-protective effects of ER signaling. Finally, increasing the transcriptional activity of the ER by relieving it from the repressive effects of class II HDACs may prove beneficial in numerous cardiac pathologies because of the broad spectrum of beneficial cardiovascular effects of ER signaling.
Supplementary Material
Acknowledgments
Sources of Funding
Work in the laboratory of E.N.O. was supported by grants from the NIH, the Donald W. Reynolds Cardiovascular Clinical Research Center, the Robert A. Welch Foundation, and the Leducq Foundation. Additional support was received by European Union Contract no. LSHM-CT-2005-018833/EUGeneHeart and a VIDI award 917-863-72 from The Netherlands Organization for Health Research and Development (to L.J.D.W.). E.v.R. received funding from a Scientist Development Grant from the American Heart Association.
Non-standard Abbreviations and Acronyms
- AF
activation function
- EMSA
electrophoretic mobility-shift assay
- ER
estrogen receptor
- ER
Eestrogen response element
- GST
glutathione S-transferase
- HDAC
histone deacetylase
- KO
knockout
- LV
left ventricle
- MEF2
myocyte enhancer factor 2
- MI
myocardial infarction
- TSA
trichostatin A
- VEGF
vascular endothelial growth factor
- WT
wild type
Footnotes
This manuscript was sent to Steven Houser, Consulting Editor, for review by expert referees, editorial decision, and final disposition.
Disclosures
None.
References
- 1.Roenicke V, Leclair S, Hoffmann A, Becker M, Szado T, Kirsch T, Stoss O, Nave BT, Henkel T. Emerging therapeutic targets in chronic heart failure: part II. Expert Opin Ther Targets. 2003;7:49–59. doi: 10.1517/14728222.7.1.49. [DOI] [PubMed] [Google Scholar]
- 2.Kocher AA, Schuster MD, Szabolcs MJ, Takuma S, Burkhoff D, Wang J, Homma S, Edwards NM, Itescu S. Neovascularization of ischemic myocardium by human bone-marrow-derived angioblasts prevents cardiomyocyte apoptosis, reduces remodeling and improves cardiac function. Nat Med. 2001;7:430–436. doi: 10.1038/86498. [DOI] [PubMed] [Google Scholar]
- 3.Mendelsohn ME, Karas RH. Molecular and cellular basis of cardiovascular gender differences. Science. 2005;308:1583–1587. doi: 10.1126/science.1112062. [DOI] [PubMed] [Google Scholar]
- 4.Mendelsohn ME, Karas RH. The protective effects of estrogen on the cardiovascular system. N Engl J Med. 1999;340:1801–1811. doi: 10.1056/NEJM199906103402306. [DOI] [PubMed] [Google Scholar]
- 5.Schultz JR, Petz LN, Nardulli AM. Cell- and ligand-specific regulation of promoters containing activator protein-1 and Sp1 sites by estrogen receptors alpha and beta. J Biol Chem. 2005;280:347–354. doi: 10.1074/jbc.M407879200. [DOI] [PubMed] [Google Scholar]
- 6.McKinsey TA, Zhang CL, Lu J, Olson EN. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature. 2000;408:106–111. doi: 10.1038/35040593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Montgomery RL, Davis CA, Potthoff MJ, Haberland M, Fielitz J, Qi X, Hill JA, Richardson JA, Olson EN. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 2007;21:1790–1802. doi: 10.1101/gad.1563807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Antos CL, McKinsey TA, Dreitz M, Hollingsworth LM, Zhang CL, Schreiber K, Rindt H, Gorczynski RJ, Olson EN. Dose-dependent blockade to cardiomyocyte hypertrophy by histone deacetylase inhibitors. J Biol Chem. 2003;278:28930–28937. doi: 10.1074/jbc.M303113200. [DOI] [PubMed] [Google Scholar]
- 10.Trivedi CM, Luo Y, Yin Z, Zhang M, Zhu W, Wang T, Floss T, Goettlicher M, Noppinger PR, Wurst W, Ferrari VA, Abrams CS, Gruber PJ, Epstein JA. Hdac2 regulates the cardiac hypertrophic response by modulating Gsk3 beta activity. Nat Med. 2007;13:324–331. doi: 10.1038/nm1552. [DOI] [PubMed] [Google Scholar]
- 11.Montgomery RL, Potthoff MJ, Haberland M, Qi X, Matsuzaki S, Humphries KM, Richardson JA, Bassel-Duby R, Olson EN. Maintenance of cardiac energy metabolism by histone deacetylase 3 in mice. J Clin Invest. 2008;118:3588–3597. doi: 10.1172/JCI35847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang S, McKinsey TA, Zhang CL, Richardson JA, Hill JA, Olson EN. Histone deacetylases 5 and 9 govern responsiveness of the heart to a subset of stress signals and play redundant roles in heart development. Mol Cell Biol. 2004;24:8467–8476. doi: 10.1128/MCB.24.19.8467-8476.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA, Olson EN. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell. 2002;110:479–488. doi: 10.1016/s0092-8674(02)00861-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Rooij E, Doevendans PA, Crijns HJ, Heeneman S, Lips DJ, van Bilsen M, Williams RS, Olson EN, Bassel-Duby R, Rothermel BA, De Windt LJ. MCIP1 overexpression suppresses left ventricular remodeling and sustains cardiac function after myocardial infarction. Circ Res. 2004;94:18–26. doi: 10.1161/01.RES.0000118597.54416.00. [DOI] [PubMed] [Google Scholar]
- 15.Srivastava S, Weitzmann MN, Kimble RB, Rizzo M, Zahner M, Milbrandt J, Ross FP, Pacifici R. Estrogen blocks M-CSF gene expression and osteoclast formation by regulating phosphorylation of Egr-1 and its interaction with Sp-1. J Clin Invest. 1998;102:1850–1859. doi: 10.1172/JCI4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harrison BC, Kim MS, van Rooij E, Plato CF, Papst PJ, Vega RB, McAnally JA, Richardson JA, Bassel-Duby R, Olson EN, McKinsey TA. Regulation of cardiac stress signaling by protein kinase d1. Mol Cell Biol. 2006;26:3875–3888. doi: 10.1128/MCB.26.10.3875-3888.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shiomi T, Tsutsui H, Hayashidani S, Suematsu N, Ikeuchi M, Wen J, Ishibashi M, Kubota T, Egashira K, Takeshita A. Pioglitazone, a per-oxisome proliferator-activated receptor-gamma agonist, attenuates left ventricular remodeling and failure after experimental myocardial infarction. Circulation. 2002;106:3126–3132. doi: 10.1161/01.cir.0000039346.31538.2c. [DOI] [PubMed] [Google Scholar]
- 18.Ren G, Michael LH, Entman ML, Frangogiannis NG. Morphological characteristics of the microvasculature in healing myocardial infarcts. J Histochem Cytochem. 2002;50:71–79. doi: 10.1177/002215540205000108. [DOI] [PubMed] [Google Scholar]
- 19.Shelton JM, Lee MH, Richardson JA, Patel SB. Microsomal triglyceride transfer protein expression during mouse development. J Lipid Res. 2000;41:532–537. [PubMed] [Google Scholar]
- 20.McFadden DG, Charite J, Richardson JA, Srivastava D, Firulli AB, Olson EN. A GATA-dependent right ventricular enhancer controls dHAND transcription in the developing heart. Development. 2000;127:5331–5341. doi: 10.1242/dev.127.24.5331. [DOI] [PubMed] [Google Scholar]
- 21.Zhang CL, McKinsey TA, Olson EN. The transcriptional corepressor MITR is a signal-responsive inhibitor of myogenesis. Proc Natl Acad Sci U S A. 2001;98:7354–7359. doi: 10.1073/pnas.131198498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J, Grant SR, Olson EN. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 1998;93:215–228. doi: 10.1016/s0092-8674(00)81573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M. Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell. 2000;103:843–852. doi: 10.1016/s0092-8674(00)00188-4. [DOI] [PubMed] [Google Scholar]
- 24.Margueron R, Duong V, Bonnet S, Escande A, Vignon F, Balaguer P, Cavailles V. Histone deacetylase inhibition and estrogen receptor alpha levels modulate the transcriptional activity of partial antiestrogens. J Mol Endocrinol. 2004;32:583–594. doi: 10.1677/jme.0.0320583. [DOI] [PubMed] [Google Scholar]
- 25.Liu XF, Bagchi MK. Recruitment of distinct chromatin-modifying complexes by tamoxifen-complexed estrogen receptor at natural target gene promoters in vivo. J Biol Chem. 2004;279:15050–15058. doi: 10.1074/jbc.M311932200. [DOI] [PubMed] [Google Scholar]
- 26.Kurtev V, Margueron R, Kroboth K, Ogris E, Cavailles V, Seiser C. Transcriptional regulation by the repressor of estrogen receptor activity via recruitment of histone deacetylases. J Biol Chem. 2004;279:24834–24843. doi: 10.1074/jbc.M312300200. [DOI] [PubMed] [Google Scholar]
- 27.Kawai H, Li H, Avraham S, Jiang S, Avraham HK. Overexpression of histone deacetylase HDAC1 modulates breast cancer progression by negative regulation of estrogen receptor alpha. Int J Cancer. 2003;107:353–358. doi: 10.1002/ijc.11403. [DOI] [PubMed] [Google Scholar]
- 28.Arany Z, Foo SY, Ma Y, Ruas JL, Bommi-Reddy A, Girnun G, Cooper M, Laznik D, Chinsomboon J, Rangwala SM, Baek KH, Rosenzweig A, Spiegelman BM. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1alpha. Nature. 2008;451:1008–1012. doi: 10.1038/nature06613. [DOI] [PubMed] [Google Scholar]
- 29.Lamping KG, Christensen LP, Tomanek RJ. Estrogen therapy induces collateral and microvascular remodeling. Am J Physiol Heart Circ Physiol. 2003;285:H2039–H2044. doi: 10.1152/ajpheart.00405.2003. [DOI] [PubMed] [Google Scholar]
- 30.Battegay E. Angiogenesis–mechanisms and therapeutic approaches. Schweiz Rundsch Med Prax. 1995;84:118–121. [PubMed] [Google Scholar]
- 31.Lee SH, Wolf PL, Escudero R, Deutsch R, Jamieson SW, Thistlethwaite PA. Early expression of angiogenesis factors in acute myocardial ischemia and infarction. N Engl J Med. 2000;342:626–633. doi: 10.1056/NEJM200003023420904. [DOI] [PubMed] [Google Scholar]
- 32.Dor Y, Djonov V, Abramovitch R, Itin A, Fishman GI, Carmeliet P, Goelman G, Keshet E. Conditional switching of VEGF provides new insights into adult neovascularization and pro-angiogenic therapy. EMBO J. 2002;21:1939–1947. doi: 10.1093/emboj/21.8.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Herve MA, Meduri G, Petit FG, Domet TS, Lazennec G, Mourah S, Perrot-Applanat M. Regulation of the vascular endothelial growth factor (VEGF) receptor Flk-1/KDR by estradiol through VEGF in uterus. J Endocrinol. 2006;188:91–99. doi: 10.1677/joe.1.06184. [DOI] [PubMed] [Google Scholar]
- 34.Toyota E, Matsunaga T, Chilian WM. Myocardial angiogenesis. Mol Cell Biochem. 2004;264:35–44. doi: 10.1023/b:mcbi.0000044372.65864.18. [DOI] [PubMed] [Google Scholar]
- 35.Wurzel J, Goldman BI. Angiogenesis factors in acute myocardial ischemia and infarction. N Engl J Med. 2000;343:148–149. doi: 10.1056/NEJM200007133430216. [DOI] [PubMed] [Google Scholar]
- 36.Lu J, McKinsey TA, Nicol RL, Olson EN. Signal-dependent activation of the MEF2 transcription factor by dissociation from histone deacetylases. Proc Natl Acad Sci U S A. 2000;97:4070–4075. doi: 10.1073/pnas.080064097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu J, McKinsey TA, Zhang CL, Olson EN. Regulation of skeletal myogenesis by association of the MEF2 transcription factor with class II histone deacetylases. Mol Cell. 2000;6:233–244. doi: 10.1016/s1097-2765(00)00025-3. [DOI] [PubMed] [Google Scholar]
- 38.Gossett LA, Kelvin DJ, Sternberg EA, Olson EN. A new myocyte-specific enhancer-binding factor that recognizes a conserved element associated with multiple muscle-specific genes. Mol Cell Biol. 1989;9:5022–5033. doi: 10.1128/mcb.9.11.5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leong H, Sloan JR, Nash PD, Greene GL. Recruitment of histone deacetylase 4 to the N-terminal region of estrogen receptor alpha. Mol Endocrinol. 2005;19:2930–2942. doi: 10.1210/me.2005-0178. [DOI] [PubMed] [Google Scholar]
- 40.Song RX, Zhang Z, Santen RJ. Estrogen rapid action via protein complex formation involving ERalpha and Src. Trends Endocrinol Metab. 2005;16:347–353. doi: 10.1016/j.tem.2005.06.010. [DOI] [PubMed] [Google Scholar]
- 41.Backs J, Olson EN. Control of cardiac growth by histone acetylation/deacetylation. Circ Res. 2006;98:15–24. doi: 10.1161/01.RES.0000197782.21444.8f. [DOI] [PubMed] [Google Scholar]
- 42.Mahmoodzadeh S, Eder S, Nordmeyer J, Ehler E, Huber O, Martus P, Weiske J, Pregla R, Hetzer R, Regitz-Zagrosek V. Estrogen receptor alpha up-regulation and redistribution in human heart failure. FASEB J. 2006;20:926–934. doi: 10.1096/fj.05-5148com. [DOI] [PubMed] [Google Scholar]
- 43.Nordmeyer J, Eder S, Mahmoodzadeh S, Martus P, Fielitz J, Bass J, Bethke N, Zurbrugg HR, Pregla R, Hetzer R, Regitz-Zagrosek V. Upregulation of myocardial estrogen receptors in human aortic stenosis. Circulation. 2004;110:3270–3275. doi: 10.1161/01.CIR.0000147610.41984.E8. [DOI] [PubMed] [Google Scholar]
- 44.Kao HY, Downes M, Ordentlich P, Evans RM. Isolation of a novel histone deacetylase reveals that class I and class II deacetylases promote SMRT-mediated repression. Genes Dev. 2000;14:55–66. [PMC free article] [PubMed] [Google Scholar]
- 45.Huang EY, Zhang J, Miska EA, Guenther MG, Kouzarides T, Lazar MA. Nuclear receptor corepressors partner with class II histone deacetylases in a Sin3-independent repression pathway. Genes Dev. 2000;14:45–54. [PMC free article] [PubMed] [Google Scholar]
- 46.Kim MY, Woo EM, Chong YT, Homenko DR, Kraus WL. Acetylation of estrogen receptor alpha by p300 at lysines 266 and 268 enhances the DNA binding and transactivation activities of the receptor. Mol Endocrinol. 2006;20:1479–1493. doi: 10.1210/me.2005-0531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morales DE, McGowan KA, Grant DS, Maheshwari S, Bhartiya D, Cid MC, Kleinman HK, Schnaper HW. Estrogen promotes angiogenic activity in human umbilical vein endothelial cells in vitro and in a murine model. Circulation. 1995;91:755–763. doi: 10.1161/01.cir.91.3.755. [DOI] [PubMed] [Google Scholar]
- 48.Shifren JL, Tseng JF, Zaloudek CJ, Ryan IP, Meng YG, Ferrara N, Jaffe RB, Taylor RN. Ovarian steroid regulation of vascular endothelial growth factor in the human endometrium: implications for angiogenesis during the menstrual cycle and in the pathogenesis of endometriosis. J Clin Endocrinol Metab. 1996;81:3112–3118. doi: 10.1210/jcem.81.8.8768883. [DOI] [PubMed] [Google Scholar]
- 49.Greb RR, Heikinheimo O, Williams RF, Hodgen GD, Goodman AL. Vascular endothelial growth factor in primate endometrium is regulated by oestrogen-receptor and progesterone-receptor ligands in vivo. Hum Reprod. 1997;12:1280–1292. doi: 10.1093/humrep/12.6.1280. [DOI] [PubMed] [Google Scholar]
- 50.Krasinski K, Spyridopoulos I, Asahara T, van der Zee R, Isner JM, Losordo DW. Estradiol accelerates functional endothelial recovery after arterial injury. Circulation. 1997;95:1768–1772. doi: 10.1161/01.cir.95.7.1768. [DOI] [PubMed] [Google Scholar]
- 51.Ferrara N. VEGF: an update on biological and therapeutic aspects. Curr Opin Biotechnol. 2000;11:617–624. doi: 10.1016/s0958-1669(00)00153-1. [DOI] [PubMed] [Google Scholar]
- 52.Hyder SM, Stancel GM, Chiappetta C, Murthy L, Boettger-Tong HL, Makela S. Uterine expression of vascular endothelial growth factor is increased by estradiol and tamoxifen. Cancer Res. 1996;56:3954–3960. [PubMed] [Google Scholar]
- 53.Kazi AA, Jones JM, Koos RD. Chromatin immunoprecipitation analysis of gene expression in the rat uterus in vivo: estrogen-induced recruitment of both estrogen receptor alpha and hypoxia-inducible factor 1 to the vascular endothelial growth factor promoter. Mol Endocrinol. 2005;19:2006–2019. doi: 10.1210/me.2004-0388. [DOI] [PubMed] [Google Scholar]
- 54.Hamilton KL, Gupta S, Knowlton AA. Estrogen and regulation of heat shock protein expression in female cardiomyocytes: cross-talk with NF kappa B signaling. J Mol Cell Cardiol. 2004;36:577–584. doi: 10.1016/j.yjmcc.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 55.Murphy E, Steenbergen C. Gender-based differences in mechanisms of protection in myocardial ischemia-reperfusion injury. Cardiovasc Res. 2007;75:478–486. doi: 10.1016/j.cardiores.2007.03.025. [DOI] [PubMed] [Google Scholar]
- 56.van Eickels M, Grohe C, Cleutjens JP, Janssen BJ, Wellens HJ, Doevendans PA. 17beta-estradiol attenuates the development of pressure-overload hypertrophy. Circulation. 2001;104:1419–1423. doi: 10.1161/hc3601.095577. [DOI] [PubMed] [Google Scholar]
- 57.Kang YK, Guermah M, Yuan CX, Roeder RG. The TRAP/mediator coactivator complex interacts directly with estrogen receptors alpha and beta through the TRAP220 subunit and directly enhances estrogen receptor function in vitro. Proc Natl Acad Sci U S A. 2002;99:2642–2647. doi: 10.1073/pnas.261715899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kong Y, Tannous P, Lu G, Berenji K, Rothermel BA, Olson EN, Hill JA. Suppression of class I and II histone deacetylases blunts pressure-overload cardiac hypertrophy. Circulation. 2006;113:2579–2588. doi: 10.1161/CIRCULATIONAHA.106.625467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Marks PA, Dokmanovic M. Histone deacetylase inhibitors: discovery and development as anticancer agents. Expert Opin Investig Drugs. 2005;14:1497–1511. doi: 10.1517/13543784.14.12.1497. [DOI] [PubMed] [Google Scholar]
- 60.Mai A, Massa S, Pezzi R, Simeoni S, Rotili D, Nebbioso A, Scognamiglio A, Altucci L, Loidl P, Brosch G. Class II (IIa)-selective histone deacetylase inhibitors. 1. Synthesis and biological evaluation of novel (aryloxopropenyl)pyrrolyl hydroxyamides. J Med Chem. 2005;48:3344–3353. doi: 10.1021/jm049002a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.