

Abstract
Astrocytes comprise approximately half of the volume of the adult mammalian brain and are the primary neuronal structural and trophic supportive elements. Astrocytes are organized into distinct nonoverlapping domains and extend elaborate and dense fine processes that interact intimately with synapses and cerebrovasculature. The recognition in the mid 1990s that astrocytes undergo elevations in intracellular calcium concentration following activation of G protein-coupled receptors by synaptically released neurotransmitters demonstrated not only that astrocytes display a form of excitability but also that astrocytes may be active participants in brain information processing. The roles that astrocytic calcium elevations play in neurophysiology and especially in modulation of neuronal activity have been intensely researched in recent years. This review will summarize the current understanding of the function of astrocytic calcium signaling in neurophysiological processes and discuss areas where the role of astrocytes remains controversial and will therefore benefit from further study.
Glia: From Passive Glue to Excitable Cells
When neuroglia were first described, there was considerable debate as to whether neuroglia were a connective tissue or a true population of cells (Somjen, 1988). While this issue was resolved in the late 1800s, little attention was paid to the role of glia in neurophysiology for nearly a century. During this period, neuroscientists generally considered glia as chemical and physical insulators that enabled neurons to carry out the diverse functions of the brain. This view was reinforced by the findings of early neurophysiologists who impaled glial cells with sharp electrodes and found only passive membrane currents. Given the large fraction of brain contributed by glia, the prevailing view until the early 1970s was that over half of the mammalian brain was, in effect, silent. This view began to change as investigators found that glial cells in culture exhibited a large number of G protein-coupled receptors (GPCRs) linked to a diverse array of intracellular signaling cascades (McCarthy and de Vellis, 1978; van Calker and Hamprecht, 1981; van Calker et al., 1978). Concerns about the expression of GPCRs by glia being a “culture” phenomenon were put to rest as it was demonstrated that glia in situ and in vivo also express GPCRs (Porter and McCarthy, 1997). Today, it is generally accepted that glia throughout the brain and spinal cord as well as peripheral glia residing within ganglia and aligning axons express members of most of the different families of GPCRs known to be expressed by neurons (Porter and McCarthy, 1997). Stimulation of these GPCRs evokes a variety of glial cell responses, the most studied of which is elevation of intracellular calcium (Ca2+) concentration that is widely considered a form of glial excitability. The question is no longer whether glia exhibit GPCRs, but under what conditions are these GPCRs activated and what is the role of glial GPCR-mediated signaling in neurophysiology?
Astrocytes Are the Predominant Glial Cell Type in the Central Nervous System
Like the term “neuron,” glia refers to a diverse set of cell types that are likely to carry out distinct functions in neurophysiology. There are four major groups of glial cells in the nervous system: (1) Schwann cells and oligodendrocytes, which produce and wrap layers of myelin around axons in the peripheral and central nervous systems, respectively; (2) microglia, the immune cell type of the nervous system, which participate in inflammatory responses; (3) nerve/glial antigen 2 (NG2)-positive glia, which include oligodendrocyte and astrocyte progenitor cells as well as NG2+ cells that persist in the mature brain; and (4) astrocytes. Astrocytes are found throughout the brain and spinal cord and, on the basis of number, surface area, and volume, are the predominant glial cell type. There are many distinct subsets of astrocytes that can be distinguished on the basis of their morphology and biochemical characteristics. For example, Mueller glia in the retina and Bergmann glia in the cerebellum are generally grouped with astrocytes because of their expression of glial fibrillary acidic protein (GFAP) but exhibit striking differences in morphology, pharmacology, and physiology (Grosche et al., 1999, 2002; Metea and Newman, 2006; Pinto and Gotz, 2007). It is likely that, even within a localized brain region, adjacent astrocytes that appear identical morphologically and immunocytochemically may vary in their expression of GPCRs and their response to activation of GPCRs. While such diversity is generally accepted when considering neurons, it is rarely taken into account when interpreting data derived from astrocytes.
Protoplasmic astrocytes are the most common type of astrocytes. These cells exhibit a very complex morphology and contact most, if not all, other cell types in the brain and spinal cord. The morphology of an astrocyte resembles a bush with processes radiating out from a central cell body (Figure 1A). Within the CA1 stratum radiatum of the hippocampus, an individual astrocyte has a soma diameter of 7-9 μm and, with its fine processes, occupies a volume of ∼66,000 μm3 (Bushong et al., 2002). Interestingly, individual astrocytes tend to occupy distinct, nonoverlapping domains (Figure 1A) (Bushong et al., 2002). The fine processes of an individual astrocyte are connected to one another through reflexive gap junctions and to other astrocytes via gap junctions at their boundaries. Patch-clamping a single astrocyte with an electrode filled with a gap-junction-permeable dye rapidly leads to the filling of hundreds, if not thousands, of astrocytes (Konietzko and Muller, 1994). Astrocytes likely function as a syncytium contacting essentially all other cellular elements in brain, including neurons, oligodendrocytes, NG2+ cells, microglia, and vasculature. A striking feature of astrocytes is that processes from a single astrocyte can envelop approximately 140,000 synapses (Figure 1B) (Bushong et al., 2002), while >99% of the cerebrovascular surface is ensheathed by astrocyte processes (Kacem et al., 1998; Rama Rao et al., 2003; Simard et al., 2003; Haydon and Carmignoto, 2006; Takano et al., 2006). In addition to the diversity among astrocytes, there may be substantial diversity within individual astrocytes with respect to interactions with the local environment. For instance, it is possible that, within a single astrocyte, a subset of processes interacts autonomously with a neighborhood of neuronal synapses, while other regions of that astrocyte interact with different groups of synapses or with other cellular elements, such as the cerebrovasculature. Further, under physiological conditions, these local regions of interaction (microdomains) of an astrocyte may not always communicate with one another. Understanding how the different microdomains of astrocytes interact with neighboring cellular elements will be critical to determining their role in neurophysiology and neuropathology.
Figure 1. View of the Organization of Astrocytes in Nonoverlapping Anatomical Territories and Their Close Association to Neuronal Dendrites.

(A) View of two neighboring astrocytes in the CA1 area of hippocampus labeled with different colored fluorescent dyes (Alexa 488, green; Alexa 568, red). The elaborate and dense processes of each astrocyte do not overlap, and peripheral fine terminal processes interdigitate with one another (yellow). Pyramidal CA1 neurons appear in blue.
(B) View of an astrocyte (green) extending its highly ramified processes in close proximity to a CA1 neuronal dendrite (red). These processes can cover most synapses in the astrocyte domain. Panel (A), courtesy of M.H. Ellisman; panel (B), modified from Fiacco and McCarthy (2004).
Neuron-Astrocyte Interactions
The morphology of astrocytes places them in a unique situation to be able to listen to and respond to most cellular elements. Astrocytes exhibit a large number of GPCRs linked to Ca2+ mobilization from internal stores, most of them being Gq-coupled GPCRs (Gq GPCRs). While these receptors can be experimentally activated in situ by exogenous application of agonists (Porter and McCarthy, 1995a, 1995b), they are also activated by neurotransmitters released from presynaptic terminals (Araque et al., 2002; Kang et al., 1998; Navarrete and Araque, 2008; Pasti et al., 1997; Perea and Araque, 2005; Porter and McCarthy, 1996). This finding is relevant because it demonstrates the existence of neuron-to-astrocyte communication and that astrocytic Gq GPCRs appear to be a primary link between neuronal activity and astrocytic Ca2+ elevations. Evidence for a reciprocal effect of astrocytes on synaptic transmission through the Gq GPCR-mediated Ca2+-dependent release of neuroactive molecules (called gliotransmitters) was reported in vitro and in situ when Gq GPCR agonist application elicited Ca2+ increases in astrocytes, which correlated to changes in neuronal ionotropic glutamate receptor (iGluR) activity (Parpura et al., 1994; Pasti et al., 1997). Since these reports, several laboratories have reported that Ca2+ elevations in a small fraction of astrocytes and under certain conditions in situ can result in the release of gliotransmitters, including glutamate, ATP, and D-serine, that bind to pre- and/or postsynaptic neuronal receptors to modulate synaptic transmission and activity (Bezzi et al., 1998; Fiacco and McCarthy, 2004; Kang et al., 1998; Lee et al., 2007; Mothet et al., 2005; Navarrete and Araque, 2008; Pascual et al., 2005; Serrano et al., 2006; Yang et al., 2003). Thus, it appears that astrocytes in situ not only listen and react to ongoing neuronal activity but also have the ability to modulate this activity via the release of gliotransmitters. The recognition of the bidirectional communication between neurons and astrocytes at the synapse led to the concept of the “tripartite synapse” (Figure 2), in which the astrocyte, in addition to preand postsynaptic compartments, is a functional component of the synapse. A primary focus of this review is to discuss recent findings that have shaped our current understanding of the role of astrocyte Gq GPCR-mediated Ca2+ elevations on neuronal-astrocyte communication, focusing on the concept of “gliotransmission” and discussing both the issues that are well accepted and the ones that are currently in dispute.
Figure 2. Schematic Depicting the Tripartite Synapse.
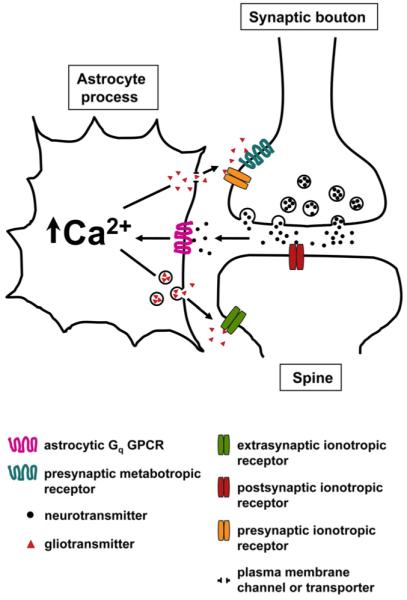
The presynaptic and postsynaptic compartments together with the enveloping astrocytic process form the tripartite synapse. Neurotransmitters released from presynaptic terminals act on postsynaptic receptors and astrocytic Gq GPCRs following spillover from the synaptic cleft. Astrocytic Gq GPCR-mediated Ca2+ elevations may trigger the release of gliotransmitters through unresolved pathways. Astrocyte-released gliotransmitters may then signal back to neurons by activating presynaptic or postsynaptic (extrasynaptic) neuronal receptors to modulate synaptic transmission.
Mechanisms of Ca2+ Increases in Astrocytes
The most widely accepted mechanism for astrocytic Ca2+ increases is the canonical phospholipase C (PLC)/inositol 1,4,5-trisphosphate (IP3) pathway. Upon Gq GPCR activation, PLC hydrolyzes the membrane lipid phosphatidylinositol 4,5-bisphosphate to generate diacylglycerol (DAG) and IP3, leading to IP3 receptor (IP3R) activation and Ca2+ release from the endoplasmic reticulum (ER). This is based on an exhaustive amount of data from both cultured astroglia and in situ astrocytes and has been the subject of numerous reviews (Fiacco and McCarthy, 2006; Parri and Crunelli, 2003; Scemes, 2000; Scemes and Giaume, 2006; Volterra and Steinhauser, 2004). Using a knockout (KO) of the IP3R type 2 (IP3R2), it has been recently demonstrated that, within the hippocampus, IP3R2 is the primary functional IP3R subtype in astrocytes in situ (Petravicz et al., 2008). These data are consistent with data in cultured astroglia showing that Ca2+ release sites correlate with IP3R2 immunostaining and that IP3R2 associates with other members of the PLC/IP3 pathway in lipid rafts (Sheppard et al., 1997; Weerth et al., 2007). In addition, immunostaining for IP3Rs in rodent brain sections indicates that astrocytes express primarily IP3R2, while type 1 and 3 IP3Rs are preferentially found in neurons (Hertle and Yeckel, 2007; Holtzclaw et al., 2002; Sharp et al., 1999).
While the mechanisms of astrocytic Ca2+ increases in response to Gq GPCR activation are relatively well understood, less is known about (1) how astrocytic second messenger pathways may be regulating the spatiotemporal dynamics of Gq GPCR-mediated Ca2+ transients and (2) how the interactions among the Gq GPCR signaling molecules contribute to a cellular response. Several factors can influence the activity of IP3Rs, including the cytoplasmic Ca2+ elevations themselves that activate IP3Rs due to the coagonistic action of Ca2+ on these receptors, the additional generation of IP through the Ca2+-dependent activation of PLC, and the phosphorylation and consequent potentiation of IP3Rs following ATP binding to the receptor (Foskett et al., 2007). Furthermore, studies of the DAG/protein kinase C (PKC) pathway indicate that PKC is involved in the termination of astrocytic Ca2+ transients (Codazzi et al., 2001; Parri and Crunelli, 2003). While the molecular target of PKC phosphorylation remains unknown, the two most likely candidates are either Gq GPCRs or the IP3R itself. Further research into second messenger regulation will provide a better understanding of the signaling mechanisms governing astrocyte PLC-dependent Ca2+ regulation.
Astrocytic Ca2+ elevations following stimulation of neuronal afferents are attributed to activation of astrocytic Gq GPCRs and not to activation of voltage-gated Ca2+ channels (VGCCs) (Beck et al., 2004; Carmignoto et al., 1998; Duffy and MacVicar, 1994; Jabs et al., 1994; Nett et al., 2002; Parri and Crunelli, 2003; Porter and McCarthy, 1995b; Straub et al., 2006). However, while astrocytic VGCCs do not seem to play a role in the initiation of evoked astrocytic Ca2+ increases, they may be important for initiating Ca2+ oscillations that occur independent of neuronal input, generally referred to as spontaneous or intrinsic astrocytic Ca2+ oscillations (Aguado et al., 2002; Parri et al., 2001; Parri and Crunelli, 2003). To date, there is little convincing evidence that astrocytes in situ exhibit ryanodine receptor-mediated increases in Ca2+ (Beck et al., 2004; Carmignoto et al., 1998; Nett et al., 2002; Parri and Crunelli, 2003; Porter and McCarthy, 1995b; Straub et al., 2006). However, it remains possible that this alternate Ca2+ source could be important in the fine processes of astrocytes where it is difficult to study Ca2+ regulation. Overall, the field as a whole is only beginning to appreciate the complexity of signaling molecules activated when astrocytic Gq GPCRs are stimulated and how these molecules, together with Ca2+, shape the cellular response.
Tools for Studying Astrocytic Ca2+ Responses
In spite of the fact that astrocytic signaling has been studied for nearly three decades, very little is known concerning the role of astrocytic Gq GPCRs in neurophysiology. Our lack of progress in this area stems, in large part, from the difficulty in selectively activating or blocking astrocytic Gq GPCRs in situ or in vivo. Astroglial Ca2+ signaling can easily be assessed in vitro using purified cultured astroglia. However, most investigators in the field acknowledge that cultured astroglia present a very poor model for studying the functions of astrocytic Gq GPCRs in situ or in vivo. For example, the morphological characteristics of astrocytes in situ are lost in cultured astroglia. In addition, cultured astroglia express genes that are not necessarily expressed in vivo, favoring the concept of a “glial” cell class, while the gene profiles of different subpopulations of astrocytes are dissimilar in situ (Cahoy et al., 2008; Lovatt et al., 2007; Wilhelm et al., 2004). Consequently, acutely isolated brain slices have been utilized to study astrocytic Gq GPCR-mediated Ca2+ dynamics. In this situation, it is possible to load astrocytes with Ca2+ indicator dyes by either bulk loading with membrane-permeable Ca2+ indicator dyes or patch-clamp pipette loading with membrane-impermeable dyes. Bulk loading in situ preferentially loads astrocytes relative to neurons (Porter and McCarthy, 1995b). However, until the recent development of two-photon microscopy, bulk loading has not been useful for studying Ca2+ signaling in the processes of astrocytes due to the high fluorescent background within the neuropil. This is a serious technical limitation when using conventional fluorescence microscopy, given that the cell body constitutes a very small proportion of an astrocyte, and most neuronal-astrocyte interactions involving Gq GPCR signaling are likely to initiate at neurotransmitter release sites within the neuropil. Additionally, while there is evidence that most Ca2+ indicator dyes preferentially bulk load into astrocytic cell bodies, this may not be the case in the neuropil, where the small-diameter processes of many cell types may load with dye. There are two advantages to using patch-clamp pipettes to load astrocytes with Ca2+ indicator dye. First, patch-clamp electrode recordings of electrophysiological properties are the best way to definitively distinguish between the two primary subtypes of GFAP-positive cells observed in immature (postnatal day 6-20) rat and mouse brain slices: the passive astrocytes, which are the subject of this review, and the so-called “complex” glia, which exhibit voltage- and time-dependent electrical currents in situ (Jabs et al., 2005; Lin and Bergles, 2004; Matthias et al., 2003). Second, because of the decrease in neuropil loading, it is possible to examine Ca2+ signaling in small astrocytic compartments.
A difficulty in studying astrocytic signaling in situ or in vivo is the fact that astrocytes and other neural cells (e.g., neurons, microglia, NG2+ glia) exhibit a similar array of Gq GPCRs linked to Ca2+ mobilization. As a consequence, it is very difficult to determine the effect of selectively stimulating astrocytic Ca2+ signaling cascades on physiological processes such as synaptic transmission. Approaches used to selectively increase astrocytic Ca2+ include mechanical stimulation with glass electrodes (Kozlov et al., 2006); strong, nonphysiological depolarization of astrocytes (Jourdain et al., 2007; Kang et al., 1998); use of agonists to endogenous Gq GPCRs (Lee et al., 2007; Navarrete and Araque, 2008; Pasti et al., 1997); uncaging caged IP3 or Ca2+ that was loaded via patch-clamp pipettes into astrocytes (Fellin et al., 2004; Fiacco and McCarthy, 2004; Liu et al., 2004; Perea and Araque, 2007); or stimulation of a transgenic Gq GPCR known to be restricted to astrocytes and whose ligand fails to activate endogenous Gq GPCRs (Fiacco et al., 2007) (Figure 3). There are caveats associated with each of these approaches. However, there is particular concern about using mechanical stimulation where responses are likely to reflect cellular damage. The use of agonists to endogenous Gq GPCRs to evoke Ca2+ increases from internal stores in astrocytes is also problematic given that it is nearly impossible to rule out the presence of the same Gq GPCRs on alternate cells. Consequently, application of a Gq GPCR agonist directly stimulates these receptors on surrounding cells (e.g., neurons), making interpretation of the findings difficult. Uncaging Ca2+ or IP3 that was selectively loaded into astrocytes at first appears to be an excellent approach for assessing the role of Ca2+ signaling in astrocytes. Many laboratories have used this approach to demonstrate that increases in astrocytic Ca2+ can lead to gliotransmitter release that affects the activity of neighboring neurons (Fellin et al., 2004; Fiacco et al., 2004; Liu et al., 2004; Perea and Araque, 2007). However, there are also caveats associated with this approach. First, uncaging either Ca2+ or IP3 within astrocytic cell bodies leads to an intracellular Ca2+ wave that moves rapidly throughout the entire astrocyte from the soma into the processes. This fails to mimic the spatiotemporal dynamics and magnitude of Ca2+ increases normally observed when activating endogenous astrocytic Gq GPCRs with bath application of agonists or following stimulation of neuronal fibers (Fiacco et al., 2007; Grosche et al., 1999; Wang et al., 2006). During endogenous astrocytic G1 GPCR activation, Ca2+ elevates at discrete initiation points within the astrocyte processes before propagating as an intracellular Ca2+ wave to other parts of the cell. The second caveat associated with uncaging Ca2+ or IP3 is that this approach does not activate the fabric of endogenous regulators (e.g., Gbγ, PLC, DAG) normally associated with stimulation of Gq GPCRs. Consequently, alternate signaling cascades participating in astrocytic Gq GPCR responses, which could markedly modify astrocytic responses, are absent when uncaging Ca2+ or IP3. The use of caged IP3 seems to be a better choice over caged Ca2+ because it engages some of the endogenous Ca2+-release mechanisms (i.e., activation of IP3 R-dependent Ca2+ release from the ER, rather than solely increasing cytosolic Ca2+). Despite such drastic uncaging methods to elevate astrocytic Ca2+, only a small fraction of the stimulated astrocytes have been reported to significantly affect neuronal activity.
Figure 3. Tools for Evoking Astrocytic Ca2+ Elevations in Brain Slices In Situ.

Schematic depicting the main approaches used to increase astrocytic Ca2+ in situ. Abbreviations: α, Gq α subunit; βγ, Gq βγ subunits; Gq GPCR, Gq-protein coupled receptor; ER, endoplasmic reticulum; IP3, inositol 1,4,5-trisphosphate; PIP2, phosphatidylinositol 4,5-bisphosphate; IP3R, IP3 receptor; PLC, phospholipase C.
In order to circumvent a number of these caveats, genetically modified mice that enable either selective activation or inactivation of Gq GPCR-mediated Ca2+ signaling in astrocytes were developed. The first of these lines was made using the MrgA1 receptor (MrgA1R) (Fiacco et al., 2007) that is normally expressed in a subset of dorsal root ganglion nociceptive sensory neurons but is not expressed in brain (Dong et al., 2001). The synthetic ligand that activates the MrgA1R has no apparent effect on endogenous brain Gq GPCRs, and MrgA1Rs do not appear to respond to endogenous ligands released in the brain. The advantage of this approach is that it is possible to specifically activate the fabric of Gq GPCR second messenger signaling molecules in a large population of astrocytes. A potential disadvantage of the transgenic approach is that Gq GPCR transgenes are driven by a heterologous promoter (i.e., a fragment of the human GFAP promoter), which could affect the level and/or the spatiotemporal characteristics of GPCR transgene expression. With respect to the spatial expression of the MrgA1R transgene, this does not appear to be a significant problem. Activation of MrgA1Rs and endogenous metabotropic glutamate receptors (mGluRs) leads to increases in Ca2+ that originate from the same spatial location within astrocyte processes and move as an intracellular Ca2+ wave with the same speed from this location (Fiacco et al., 2007). These results suggest that the same clustered signaling molecules required for Ca2+ release from internal stores are activated by both endogenous astrocytic Gq GPCRs and transgenic astrocytic MrgA1Rs. The fact that MrgA1R responses behave similarly to endogenous mGluRs suggests that MrgA1Rs are likely functional in astrocytes. However, the possibility of loss of cellular regulatory mechanisms or changes in the downstream signaling molecules necessary for initiating astrocyte responses cannot be entirely ruled out.
Another genetically modified mouse line was made by knocking out the IP3R2 to selectively block Gq GPCR-dependent changes in astrocytic Ca2+. Hippocampal astrocytes derived from these mice do not respond to agonists that activate Gq GPCRs and do not exhibit spontaneous or intrinsic Ca2+ oscillations. CA1 pyramidal neuron Ca2+ responses to Gq GPCR agonists are unaffected in these mice, reflecting earlier observations that IP3Rs 1 and 3 are the predominant neuronal IP3R isoforms (Petravicz et al., 2008). These results indicate that IP3R2 is the primary functional IP3R isoform in astrocytes and that IP3R2 is not required for neuronal Gq GPCR-mediated Ca2+ elevations. The absence of evoked and spontaneous astrocytic Gq GPCR-mediated Ca2+ increases suggests that other IP3Rs are not upregulated in astrocytes to compensate for the loss of IP3R2 and astrocytic Ca2+ signaling.
Astrocytes Listen to Neuronal Conversations In Situ
The number of different types of GPCRs expressed by astrocytes in situ is striking. For instance, hippocampal CA1 stratum radiatum astrocytes respond to application of ligands that activate purinergic, adrenergic, muscarinic, glutamatergic, GABAergic, histaminergic, endothelin, endocannabinoid, bradykinin, thrombin, opiate, and substance P receptors with an increase in Ca2+ (Bowser and Khakh, 2007; Duffy and MacVicar, 1995; Fiacco et al., 2007; Lee et al., 2007; Navarrete and Araque, 2008; Pasti et al., 1997; Porter and McCarthy, 1997; Serrano et al., 2006; Shelton and McCarthy, 1999, 2000; Verkhratsky et al., 1998). An individual astrocyte within the hippocampus can respond to at least four different neuroligands (Shelton and McCarthy, 2000). It remains unclear whether different astrocytic GPCRs on a single cell regulate different physiological processes.
In situ, communication from neurons to astrocytes is generally thought to be mediated by spillover of neurotransmitter from the synaptic cleft to activate astrocytic GPCRs or from neurotransmitter released from ectopic sites along axons. Neuronally mediated astrocyte Ca2+ elevations have been shown in different brain areas, including the hippocampus, cerebellum, and retina. In hippocampal brain slices, electrical stimulation of neurons elicits Ca2+ rises in neighboring astrocytes (Araque et al., 2002; Kang et al., 1998; Navarrete and Araque, 2008; Pasti et al., 1997; Perea and Araque, 2005; Porter and McCarthy, 1997). Astrocytic Ca2+ responses can be evoked by the release of a number of different transmitters from neurons, activating glial mGluRs (Fellin et al., 2004; Pasti et al., 1997; Perea and Araque, 2005; Porter and McCarthy, 1996), γ-amino-butyric-acid B (GABAB) receptors (Kang et al., 1998), muscarinic acetylcholine receptors (Araque et al., 2002; Perea and Araque, 2005), and endocannabinoid receptors (Navarrete and Araque, 2008). In the cerebellum, electrical stimulation of cerebellar parallel fibers or neurons in the granule cell layer elicits Ca2+ increases in Bergmann glial cells; these responses appear to be mediated by nitric oxide or activation of α-adrenergic, purinergic, mGluR1, and/or AMPA receptors (Beierlein and Regehr, 2006; Grosche et al., 2002; Kulik et al., 1999; Matsui and Jahr, 2004; Matsui et al., 2005; Matyash et al., 2001; Piet and Jahr, 2007). In the isolated rat retina, neuron-to-glial signaling occurs in response to physiological stimuli (light flashes) and is mediated by neuronal release of adenosine triphosphate (ATP) and activation of glial purinergic receptors (Newman, 2005). Interestingly, in hippocampus, cortex, and thalamus, analyses reveal that most astrocytes also exhibit spontaneous Ca2+ oscillations (Aguado et al., 2002; Nett et al., 2002; Parri et al., 2001). Spontaneous astrocyte Ca2+ activity persists when neuronal miniature and action-potential-mediated synaptic release is blocked, suggesting that astrocytes might be able to initiate dialog in the absence of neuronal input (Nett et al., 2002). This astrocyte Ca2+ activity is considered intrinsic to the astrocyte and may be driven by constitutive Gq GPCR activity. In the presence of neuronal activity, spontaneous astrocytic Ca2+ oscillations are synchronized by active neuronal networks (Aguado et al., 2002).
Neuronal-Mediated Increases in Astrocytic Ca2+ In Vivo
Recent studies in vivo demonstrate that astrocytes exhibit both spontaneous Ca2+ increases (Hirase et al., 2004; Nimmerjahn et al., 2004; Wang et al., 2006) and Ca2+ responses to neuronal activity (Bekar et al., 2008; Dombeck et al., 2007; Gobel et al., 2007; Hirase et al., 2004; Schummers et al., 2008; Wang et al., 2006). It is worthwhile noting that, in the cerebral cortex in vivo, frequent spontaneous Ca2+ elevations have been detected in the soma of astrocytes under basal conditions (without evoked synaptic transmission), suggesting that Ca2+ elevations initiating in astrocyte processes are able to propagate to the soma without any apparent need for elevated levels of neuronal activity (Hirase et al., 2004; Nimmerjahn et al., 2004). However, one study demonstrated that spontaneous Ca2+ increases in astrocytic cell bodies were rare, while in contrast, Ca2+ activity was much more frequent and dynamic in astrocyte processes or vascular endfeet (Wang et al., 2006). These discrepancies in the literature may be explained in part by the use of different ranges of (1) Ca2+ dyes and techniques used to load astrocytes, (2) confocal/two-photon laser power, and/or (3) anesthetic concentration. Increased two-photon laser power has been reported to increase the frequency of Ca2+ oscillations (Wang et al., 2006), while small increases in anesthetic concentration could produce a dramatic reduction in astrocyte responses (Hirase et al., 2004; Schummers et al., 2008). Use of anesthetics has also been shown to greatly reduce overall brain activity and markedly affect several forms of neural dynamics (Dombeck et al., 2007). It is also likely that different subpopulations of astrocytes, or different microdomains within astrocyte processes, will exhibit different levels of Ca2+ activity. Overall, the magnitude, frequency, and pattern of spontaneous Ca2+ dynamics observed in astrocytes in vivo are qualitatively similar to those described in acute slice preparations (Hirase et al., 2004; Nimmerjahn et al., 2004; Wang et al., 2006). In addition, coordination of cortical astrocyte spontaneous Ca2+ events is observed, suggesting that networks of coactive astrocytes occur in the cerebral cortex in vivo under basal neuronal activity (Hirase et al., 2004), similar to what was observed previously in situ (Aguado et al., 2002).
Several studies have investigated whether astrocytes respond to physiological stimuli in vivo. Astrocytes in the barrel cortex of anesthetized adult mice promptly respond to whisker stimulation with increases in Ca2+ in both processes and cell bodies (Wang et al., 2006). Increases in astrocytic somatic Ca2+ were delayed by ∼3 s after the onset of whisker stimulation. Calcium increases in the processes occurred ∼1 s earlier than those in the soma, suggesting that the Ca2+ increases likely initiated in the fine processes that wrap presynaptic boutons. The number of astrocytes showing increases in Ca2+ was dependent on the frequency of whisker stimulation. Inhibition of mGluRs reduced astrocytic Ca2+ responses to sensory stimulation, indicating that synaptic release of glutamate is a key mediator of neuron-to-astrocyte signaling in barrel cortex. Another study used two-photon imaging of Ca2+ signals in the anesthetized adult ferret visual cortex in vivo to discover that astrocytes exhibit robust somatic Ca2+ responses to visual stimuli that are dependent on stimulus features and have many of the receptive-field characteristics of neurons (Schummers et al., 2008). The astrocyte responses were delayed roughly 3-4 s from stimulus onset, suggesting the occurrence of a neuron-to-astrocyte communication. A third study performed on anesthetized adult mice demonstrated that astrocytes throughout the somatosensory cortex respond to robust foot stimulation via norepinephrine release from the locus coeruleus, with Ca2+ responses peaking within 6 s of stimulation (Bekar et al., 2008). Finally, in a fourth study using awake, mobile, adolescent mice, somatic Ca2+ transients from large neuronal and astrocytic populations in sensory cortex were routinely measured and correlated with running behavior (Dombeck et al., 2007). The running-correlated astrocyte Ca2+ elevations were delayed by ∼1-2 s from the onset of running. The astrocyte Ca2+ signals were qualitatively similar to that seen in anesthetized animals in vivo in response to physiological whisker stimulation, visual stimuli, or foot stimulation (Schummers et al., 2008; Wang et al., 2006). In conclusion, in vivo studies have led to a wealth of information regarding astrocyte activity and, most importantly, demonstrate that neuronal-to-astrocyte signaling occurs spontaneously or during elevated levels of neuronal activity in vivo.
Localized Signaling Domains and Intracellular Ca2+ Waves
Application of a Gq GPCR agonist leads to increases in astrocytic Ca2+ that initiate at specific sites within the processes and then spread into other regions of the astrocyte (Fiacco et al., 2007; Sheppard et al., 1997; Yagodin et al., 1994, 1995). Intracellular Ca2+ waves have been observed in many different cell types and are thought to propagate along organized clusters of IP3Rs on the membrane of the ER (Simpson et al., 1997; Thomas et al., 2000; Sheppard et al., 1997; Shuai and Jung, 2003; Roth et al., 1995; Weerth et al., 2007). Studies in vitro and in situ indicate that stimulation of different Gq GPCRs evokes Ca2+ elevations initiating within the same region of astrocyte processes and traveling in the same pattern through the astrocytic volume (Fiacco et al., 2007; Yagodin et al., 1994). Similar data have also been reported in other cell types, including oligodendrocytes (Simpson et al., 1997; Thomas et al., 2000). Although it has not been investigated for all Gq GPCRs, these findings suggest that individual astrocytes exhibit a unique Ca2+ “fingerprint” following Gq GPCR activation. Why would a discrete site within an astrocyte process initiate intracellular Ca2+ waves? One possibility is that, like neurons, a threshold of activation (e.g., IP3 levels) needs to be obtained to activate a sensitive Ca2+ initiation site to evoke a rise in Ca2+. An intracellular Ca2+ wave then travels for some distance from its point of origin depending upon the level of IP3 and IP3Rs in the local area. In support of this, in situ studies have demonstrated that astrocytes are generally activated by intense electrical stimulation of neuronal afferents mimicking high synaptic activity (Araque et al., 2002; Pasti et al., 1997; Porter and McCarthy, 1996). Astrocytes respond to high levels of neuronal activation with a global intracellular Ca2+ increase, which is likely initiated within the processes enwrapping active synapses and then migrates along the processes to reach the soma. In contrast, low-frequency stimulation of neuronal afferents may evoke Ca2+ transients that stay restricted to microdomains within astrocyte processes but fail to induce detectable Ca2+ transients in the soma (Matyash et al., 2001). Recent in vivo studies demonstrate that robust physiological stimuli can evoke astrocytic Ca2+ responses that initiate in cortical astrocyte processes and then propagate rapidly to the soma (Bekar et al., 2008; Wang et al., 2006). In addition, astrocytes in the visual cortex in vivo respond to visual stimuli with cytosolic increases in Ca2+ that are sharply tuned for stimulus orientation and spatial frequency. Such a sharp tuning of astrocytes suggests the possibility that a high level of local neuronal activity is necessary to elicit astrocyte responses (Schummers et al., 2008). If astrocytes had a higher proportion of processes and/or a higher density of Gq GPCRs clustered in microdomains adjacent to the strongest synapses, this could effectively increase (1) astrocyte response selectivity and (2) the chance for an intracellular Ca2+ wave to initiate and propagate to reach the cell body. However, considering astrocyte diversity within the same or different brain regions, it is possible that some subsets of astrocytes respond differently to neuronal inputs, either preferentially undergoing localized Ca2+ transients or being more susceptible to exhibiting intracellular Ca2+ waves.
Astrocyte Intercellular Ca2+ Waves
In the early 1990s, a series of papers from Steven Smith’s laboratory demonstrated intra- and intercellular Ca2+ waves propagating through an astrocytic syncytium in cultured astroglia (Cornell-Bell et al., 1990; Dani et al., 1992). These papers set in motion the idea that intercellular Ca2+ waves could be a mechanism whereby astrocytes convey information over long distances in the brain. While the idea that astrocytes signal over long distances via Ca2+ waves is attractive, there is little evidence to suggest that this actually occurs in situ or in vivo during physiological levels of activity. Assessing intercellular Ca2+ waves in situ or in vivo requires being able to demonstrate that a rise in Ca2+ in one astrocyte directly leads to the rise in Ca2+ in secondary astrocytes. The difficulty is determining how to selectively increase Ca2+ in a single astrocyte while recording Ca2+ responses from surrounding astrocytes. One approach is to monitor intrinsic Ca2+ oscillations (i.e., in absence of neuronal activity) occurring in a single astrocyte and look for correlations in Ca2+ increases among surrounding astrocytes; correlations in small clusters of two to five astrocyte neighbors in hippocampal and thalamic slices have been observed (Aguado et al., 2002; Parri et al., 2001). Another approach to study intercellular Ca2+ waves, although less physiological, is to uncage IP3 or Ca2+ in single astrocytes while monitoring Ca2+ transients in neighboring astrocytes. Two comparable studies reported that (1) uncaging glutamate over individual hippocampal astrocytes in situ results in global Ca2+ increases that propagate into four neighboring astrocytes (Sul et al., 2004) and (2) uncaging Ca2+ in single cortical astrocytes in vivo triggers intercellular waves recruiting ∼30 astrocytes (Tian et al., 2006). However, when IP3 or Ca2+ is uncaged in single astrocytes of hippocampal slices, a robust and global intracellular Ca2+ response is observed that does not propagate into surrounding astrocytes (Fiacco and McCarthy, 2004; Liu et al., 2004). In vivo studies have reported that physiological stimuli do not induce intercellular Ca2+ waves propagating through large networks of astrocytes, demonstrating that astrocytes behave relatively independently of each other and do not generally function as a broadly interconnected network ((Wang et al., 2006; Schummers et al., 2008). Overall, findings in this area suggest that, while intercellular Ca2+ waves might occur under certain experimental conditions in situ and in vivo, there is little evidence for long-distance signaling between astrocytes under physiological conditions.
The Concept of Gliotransmission
In general, gliotransmission refers to the process whereby astrocytes release gliotransmitters (e.g., glutamate, ATP, D-serine) to affect synaptic activity (Halassa et al., 2007). Astrocytic release of glutamate and ATP has been the most extensively studied. Early findings derived from cultured astroglia demonstrated that these cells release a number of gliotransmitters in response to brief exposure to hypotonic solutions (Kimelberg et al., 1990). At the time, these findings were thought to reflect processes that occurred following brain trauma. More recent studies suggest that cultured astroglia release gliotransmitters via a number of different mechanisms, including swelling-induced activation of volume-regulated anion channels (Kimelberg et al., 2006), connexin hemichannels (Kang et al., 2008; Stout et al., 2002; Ye et al., 2003), pore-forming P2X7 purinergic receptors (Duan et al., 2003; Fellin et al., 2006), and reversal of glutamate transporters (Anderson and Swanson, 2000). In each of these situations, it is likely that the release mechanism is Ca2+ independent and active primarily under pathological conditions. However, recent data now suggest that a fraction of astrocytes in situ release neuroactive molecules in a Ca2+-dependent manner to affect synaptic transmission via preand postsynaptic mechanisms (Bezzi et al., 1998; Fiacco and McCarthy, 2004; Kang et al., 1998; Lee et al., 2007; Mothet et al., 2005; Navarrete and Araque, 2008; Pascual et al., 2005; Pasti et al., 1997; Serrano et al., 2006; Yang et al., 2003). The mechanisms underlying Ca2+-dependent gliotransmitter release are currently under investigation and remain controversial (Kang et al., 2008; Parpura et al., 2004). However, exocytotic vesicular release of gliotransmitters has received considerable attention due to its Ca2+ dependency and potential to occur under physiological conditions (Parpura et al., 2004). There are many excellent reviews in this area describing the mechanisms and outcomes of gliotransmission in vitro and in situ (Halassa et al., 2007; Haydon and Carmignoto, 2006; Iadecola and Nedergaard, 2007; Ransom et al., 2003; Volterra and Steinhauser, 2004). The implications of these findings are profound, with astrocytes representing a potential feed-forward excitatory or inhibitory influence on neuronal activity. However, the physiological relevance of these results remains unclear, since Ca2+-dependent gliotransmission and subsequent modulation of neuronal synaptic activity is both controversial and has not yet been demonstrated to occur in vivo.
The hippocampal circuit, specifically the Schaffer collateral (SC)-CA1 pyramidal neuron synapse (SC-CA1 synapse), has been used extensively to study gliotransmission and therefore provides for facilitated comparison among research groups. Although results obtained at other synapses or in other brain areas should not be diminished, the goal of the following sections is to highlight the key findings concerning gliotransmission at this synapse (i.e., findings showing a causal link between Ca2+ increases in astrocytes and effect on neuronal activity). With the aim of providing the reader with the most accurate picture of the findings to date, indirect evidence with no clear causality/directionality established will not be presented. In addition, given that astrocytes in vitro are becoming recognized as a poor model of astrocyte-neuron interaction, only data from intact tissue preparations will be presented.
Gliotransmission and the Regulation of Glutamate Release from Presynaptic Terminals
A number of laboratories have reported that increases in astrocytic Ca2+ lead to either an increase or decrease in glutamate release from Schaffer collateral terminals innervating CA1 pyramidal neurons. Uncaging IP3 to selectively increase Ca2+ in single astrocytes evokes a global intracellular astrocytic Ca2+ increase that triggers an increase in the frequency of spontaneous excitatory postsynaptic AMPA currents (sEPSCs) of CA1 neurons (Fiacco and McCarthy, 2004). Incubation with group I mGluR antagonists blocked the astrocyte-induced increase in AMPA sEPSC frequency, suggesting that astrocytically released glutamate activates mGluRs on neighboring SC presynaptic terminals to elevate presynaptic Ca2+ and increase the probability of neurotransmitter release (Fiacco and McCarthy, 2004). More recently, it has also been reported that uncaging Ca2+ in astrocytes elicits the release of glutamate, causing increased glutamate release from 47% of the SC presynaptic terminals studied (Perea and Araque, 2007). These investigators further demonstrated that, when increases in astrocytic Ca2+ are coincident with postsynaptic depolarization, glutamate release from astrocytes can result in persistent potentiation at 41% of the SC-CA1 synapses. This effect was blocked when tetanus toxin was loaded via patch-clamp along with caged Ca2+ to cleave synaptobrevin 2 and interfere with vesicular gliotransmitter release. Overall, these findings suggest that the use of IP3 or Ca2+ uncaging in a single astrocyte leads to a global intracellular Ca2+ elevation and glutamate release that activates presynaptic mGluRs on a fraction of neighboring SC terminals to enhance presynaptic vesicular release of glutamate.
Several groups have reported at this same synapse in situ that electrical SC stimulation evokes an increase in astrocytic Ca2+, triggering the release of ATP that is converted extracellularly into adenosine, which then suppresses glutamate release from presynaptic terminals via the activation of inhibitory presynaptic adenosine receptors (A1Rs) (Pascual et al., 2005; Serrano et al., 2006). The expression of a dominant-negative mutation of synaptobrevin 2 (dnSNARE) to interfere with the vesicular release of ATP/adenosine from astrocytes resulted in a decrease of the tonic suppression of synaptic transmission by A1Rs (Pascual et al., 2005). As a consequence, the dynamic range of synaptic transmission was decreased, leading to an inhibition of LTP in situ. This was a surprising finding given that glutamate, not ATP, has been shown to be released by an exocytotic synaptobrevin 2-dependent process in cultured astroglia (Araque et al., 2000; Zhang et al., 2004) and in situ (Perea and Araque, 2007). These results suggest that, when astrocytic Ca2+ elevations are evoked by neurotransmitter release from SC terminals in situ, synaptobrevin 2-dependent exocytotic release might apply to ATP release but not glutamate release. In contrast, glutamate might be preferentially released from astrocytes when astrocytic Ca2+ increases are evoked by pharmacological stimuli such as Ca2+ and IP3 uncaging (Fiacco and McCarthy, 2004; Perea and Araque, 2007). Work from Richard Robitaille’s laboratory also suggested that increases in astrocytic Ca2+ trigger ATP/adenosine release to suppress synaptic transmission at the SC-CA1 synapse (Serrano et al., 2006). In this situation, electrical stimulation of the SC pathway led to activation of GABAergic interneurons, resulting in release of GABA onto astrocytes to activate GABAB receptors to increase astrocytic Ca2+. A rise in astrocytic Ca2+ was reported to stimulate ATP/adenosine release that activated presynaptic A1Rs, suppressing SC presynaptic glutamate release to enhance heterosynaptic depression (Serrano et al., 2006).
In contrast to the above reports, studies using genetic models to either selectively activate astrocytic Gq GPCR signaling (MrgA1 transgenic mice) or abolish astrocyte Ca2+ activity (IP3R2 KO mice) failed to find any evidence that astrocytic Ca2+ affects glutamate release at the SC-CA1 synapse in situ (Fiacco et al., 2007; Petravicz et al., 2008). Slice perfusion with the ligand that selectively activates MrgA1Rs led to increases in Ca2+ in 80%-90% of astrocytes but did not result in the modulation of the frequency or amplitude of miniature EPSCs (mEPSCs), indicating that the presynaptic and postsynaptic activity of SC-CA1 synapses were not affected. Experiments performed to determine whether the expression of transgenic MrgA1Rs interfered with gliotransmitter release revealed that this was not the case, since uncaging IP3 in MrgA1R+ astrocytes led to an increase in sEPSCs, similar to findings in wild-type mice. These results demonstrated that glutamate is released from astrocytes following Ca2+ increases due to IP3 uncaging, but not when stimulating Gq GPCRs to increase astrocytic Ca2+. In addition to data obtained using the transgenic MrgA1 astrocytic receptors, activation of endogenous astrocytic Gq GPCRs including endothelin receptors and mGluRs was also found to produce no effect in either the amplitude or frequency of sEPSCs or mEPSCs (Fiacco et al., 2007). It has been recently reported that certain Gq GPCRs, such as the purinergic P2Y1 receptors, are not coupled to the release machinery in astrocytes (Shigetomi et al., 2008). Therefore, the possibility that activation of transgenic MrgA1Rs and native endothelin and mGlu receptors produces Ca2+ elevations, which may also be uncoupled from release machinery. However, this hypothesis seems difficult to reconcile with data showing that (1) stimulation of endothelin receptors evokes glutamate release from cultured astroglia (Sasaki et al., 1997) and (2) mGluR-mediated Ca2+ increases have been reported to produce glutamate release from astrocytes in situ to activate postsynaptic receptors (Angulo et al., 2004; Fellin et al., 2004; Pasti et al., 1997; see following paragraph). Although in the case of these latter studies (Angulo et al., 2004; Fellin et al., 2004; Pasti et al., 1997) the authors did not report any presynaptic effect, which is consistent with the data mentioned above (Fiacco et al., 2007), it remains unclear as to why mGluR-mediated Ca2+ increases would affect only the postsynaptic compartment at the SC-CA1 synapse in these studies, while no postsynaptic effect was found by Fiacco et al. (2007). In addition, it is unclear as to why astrocytic glutamate release that targets presynaptic receptors would be evoked by IP3 or Ca2+ uncaging (Fiacco and McCarthy, 2004; Perea and Araque, 2007) while this modulation would not be evoked by stimulation of Gq GPCRs (Fiacco et al., 2007). A rational explanation for these discrepancies will require further study.
If astrocytic Ca2+ elevations regulate glutamate release from astrocytes, it is likely that locking astrocytic cytosolic Ca2+ concentrations to basal levels would affect neuronal sEPSCs. Knockout of astrocytic IP3R2 not only eliminates the ability of astrocytes to respond to agonist application with Ca2+ elevations but, in addition, these astrocytes exhibit no spontaneous Ca2+ activity. The frequency and amplitude of sEPSCs measured in CA1 pyramidal neurons derived from wild-type and IP3R2 KO mice were compared, and no significant differences were detected (Petravicz et al., 2008), suggesting that astrocytic Ca2+ increases are not involved in modulation of neuronal activity. It remains a possibility that astrocytic Ca2+ does not modulate basal neuronal activity but instead higher levels of neuronal activity so this should be borne in mind. However, it is striking that the absence of astrocytic Ca2+ activity in the IP3R2 KO mice has no apparent effect on brain development, mouse behavior, and spontaneous neuronal synaptic activity, which is in agreement with the idea that astrocytic Ca2+ increases might not play a central role in astrocyte-to-neuronal interactions (Petravicz et al., 2008).
Overall, these findings indicate that astrocytic gliotransmitter release that affects presynaptic activity can be induced under certain conditions, such as when electrically stimulating SCs (Pascual et al., 2005; Serrano et al., 2006) or using certain pharmacological treatments (e.g., uncaging IP3 or Ca2+ in astrocytes;Fiacco and McCarthy, 2004; Perea and Araque, 2007), but may not be induced when stimulating astrocytic Gq GPCRs (Fiacco et al., 2007; Petravicz et al., 2008).
Gliotransmission and the Direct Activation of Neuronal Postsynaptic Glutamatergic Receptors
Several laboratories have reported that increases in Ca2+ in a subpopulation of astrocytes leads to the release of glutamate that activates postsynaptic neuronal glutamatergic receptors (Angulo et al., 2004;Fellin et al., 2004; Jourdain et al., 2007; Kozlov et al., 2006; Lee et al., 2007; Navarrete and Araque, 2008; Pasti et al., 1997; Perea and Araque, 2007; Shigetomi et al., 2008). At the SC-CA1 synapse, Gq GPCR agonist application in the presence of inhibitors of synaptic neurotransmitter exocytosis has been reported to elicit Ca2+ elevations in a large population of hippocampal astrocytes in situ that immediately precede increases in neuronal Ca2+. Neuronal Ca2+ increases were blocked in 20% of the neurons by iGluR antagonists, suggesting that glutamate derived from astrocytes was activating neuronal postsynaptic iGluRs in a small population of neurons (Pasti et al., 1997). Two independent groups using either Gq GPCR agonist application, Ca2+ uncaging and/or mechanical stimulation to increase astrocytic Ca2+ reported that Ca2+-evoked glutamate release from astrocytes induced extrasynaptic NR2B subtype NMDAR-mediated slow inward currents (SICs) in a population of hippocampal CA1 pyramidal neurons to synchronize their activity (Angulo et al., 2004; Fellin et al., 2004). To facilitate activation of NMDARs, these experiments were performed in the absence of extracellular magnesium. Approximately 60% of single astrocytes that were mechanically stimulated evoked SICs in an adjacent neuron (Angulo et al., 2004). In addition, when a large population of astrocytes was stimulated by bath application of Gq GPCR agonists, ∼30% of neurons gave rise to SICs (Fellin et al., 2004). Under physiological conditions (i.e., 1 mM extracellular magnesium), activation of astrocytes by Gq GPCR agonists evoked SICs in a significantly lower percentage of neurons (∼10%). The occurrence of SICs was unaffected by treatments that block the vesicular release of glutamate from SC terminals, suggesting that the source of glutamate driving neuronal SICs had an astrocytic and not a neuronal origin. The slow kinetics of SICs were attributed to both the longer diffusion distance of glutamate in the extrasynaptic space as opposed to the synaptic cleft and the slower kinetics inherent to NR2B subtype NMDARs. Increases in astrocytic Ca2+ were thought to be necessary and sufficient to induce glutamate release from astrocytes that directly evoked SICs in CA1 pyramidal neurons (Fellin and Carmignoto, 2004). Since these reports, other laboratories have reported that astrocytic Ca2+ evoked by application of Gq GPCR agonists, neuronal depolarization, or astrocyte depolarization leads to glutamate release that activates extrasynaptic NR2B subunit subtype NMDARs on CA1 pyramidal neurons to generate SICs (Navarrete and Araque, 2008; Perea and Araque, 2005; Shigetomi et al., 2008). Control experiments showed that blocking astrocytic Ca2+ elevations (by loading astrocytes with Ca2+ chelators) blocked neuronal SICs, suggesting that astrocytic Ca2+ increases were necessary to evoke SICs (Navarrete and Araque, 2008; Perea and Araque, 2005). In these studies, however, the source of glutamate was not directly shown to have a nonneuronal origin (experiments were performed in the presence of presynaptic neurotransmitter release), and therefore, the possibility that glutamate released from neurons contributed to the effect cannot be ruled out (Navarrete and Araque, 2008; Perea and Araque, 2005; Shigetomi et al., 2008). It should be noted here that SICs were reported to happen spontaneously, in the absence of astrocyte stimulation. In addition, in four of the studies cited above, an increase in SIC frequency associated with astrocytic Ca2+ elevations was found only in a small fraction (∼10%-60%) of cells tested (Angulo et al., 2004; Fellin et al., 2004; Pasti et al., 1997; Shigetomi et al., 2008). These observations suggest that averaging data together rather than analyzing only the cells showing an effect may have led to a different conclusions (i.e., that astrocytic Ca2+ elevations would not have correlated significantly to changes in the frequency of SICS). However, two other studies did not mention such a selection criteria (Navarrete and Araque, 2008; Perea and Araque, 2005) and therefore provide a better demonstration for the occurrence of astrocyte-to-neuron signaling through postsynaptic iGluRs.
In contrast to the studies described above, a number of other groups have provided conflicting data about the occurrence of SICs in CA1 neurons or have failed to observe any SICs following astrocytic Gq GPCR-mediated Ca2+ increases. For example, the activation of protease-activated receptor 1 (PAR1) on hippocampal astrocytes was shown to produce astrocytic Ca2+ increases that caused CA1 neuronal depolarization and enhanced the NMDAR-mediated component of synaptic mEPSCs, evoked EPSCs, and evoked EPSPs, but no evidence of SICs was observed in these recordings (Lee et al., 2007). Interestingly, a different study showed that activation of astrocytic PAR1 evoked SICs in 60% of CA1 neurons, whereas activation of astrocytic P2Y1 purinergic receptors did nothing (Shigetomi et al., 2008). This led the authors to conclude that Gq GPCR-mediated increases in astrocytic Ca2+ and consequent release of glutamate depends on the Gq GPCR activated (PAR1 versus P2Y1). Although these results (Lee et al., 2007; Shigetomi et al., 2008) suggest that PAR1 may uniquely tap into processes important in gliotransmitter release, it remains a concern that some investigators observe PAR1-mediated SICs without neuronal depolarizations (Shigetomi et al., 2008), while others are unable to see SICs but evoke PAR1-mediated neuronal depolarizations (Lee et al., 2007). It is also a concern that Shigetomi et al. reported no effect of stimulation of astrocytic P2Y1 receptors, when several studies have reported that ATP stimulation of astrocytes leads to SICs (Angulo et al., 2004; Perea and Araque, 2005) and other effects in neurons (Jourdain et al., 2007; Domercq et al., 2006), presumably due to activation of P2Y1 receptors on astrocytes (Jourdain et al., 2007; Domercq et al., 2006). These discrepancies suggest that there may be some technical variable(s) affecting the ability of astrocytes to release gliotransmitters while also providing clear examples as to why Ca2+-dependent glutamate release by astrocytes remains controversial. For example, the possibility that PAR1 agonists might directly activate neurons cannot be ruled out, as there are several reports supporting the expression of PAR1 receptors in both hippocampal neurons and astrocytes in rats and humans (Gingrich et al., 2000; Niclou et al., 1998; Junge et al., 2004; Striggow et al., 2001). In addition, the criteria (kinetics and amplitude) used for defining SICs by Shigetomi et al. (2008) are different from those previously defined by other investigators. The SICs described by Shigetomi et al. are approximately half the amplitude and twice as fast when compared to previously reported SICs, raising the possibility that they recorded NMDAR-mediated currents that other investigators such as Lee et al. (2007) might not have qualified as SICs but instead as synaptic NMDAR currents. Similarly, most NMDA receptor-dependent EPSCs described by Perea and Araque (2005) (with the exception of the ATP-evoked SICs) are of much smaller amplitude and faster kinetics than SICs described by other groups, making it unclear whether these events are synaptic NMDA mEPSCs or SICs. Overall, due to the difficulty comparing kinetics between groups, it is unclear how many investigators differentiate between synaptic NMDA mEPSCs (which are significantly slower than synaptic AMPA mEPSCs) and SICs. Researchers in the field would benefit from collective agreement on a nomenclature to describe SICs, to facilitate the exchange and the comparison of information and to build a foundation for future progress in the field.
Over the course of several years of experiments attempting to replicate these findings, our group has never observed any evidence for Ca2+-induced glutamate release from astrocytes activating postsynaptic neuronal receptors. In earlier studies relying primarily on uncaging IP3 in single astrocytes to stimulate gliotransmitter release, findings indicated that glutamate could indeed be released from astrocytes to activate presynaptic mGluRs but not postsynaptic iGluRs (Fiacco and McCarthy, 2004). Then, using the MrgA1 mouse model to selectively increase Ca2+ in a large population of astrocytes, not only were no changes in SC presynaptic transmission detected, but also no changes in postsynaptic CA1 pyramidal neuron activity were observed (Fiacco et al., 2007). Additionally, stimulating endogenous Gq GPCRs such as endothelin receptors or mGluRs failed to induce any detectable SICs or changes in the ambient postsynaptic NMDAR current noise (Fiacco et al., 2007). Overall, these findings argue that an increase in astrocytic Ca2+ is not sufficient to induce glutamate release from astrocytes to affect neuronal postsynaptic glutamatergic receptors in situ. These data are conflicting with comparable data from three other groups (discussed in detail above) showing that Gq GPCR agonist application results in SICs (Angulo et al., 2004; Fellin et al., 2004; Shigetomi et al., 2008). A critical evaluation of the differences among all these results has to be done in order to help resolve what appear to be persistent discrepancies among the primary research groups working on this phenomenon. One notable difference should be pointed out here. Investigators reporting that stimulation of astrocytic Ca2+ increases triggers SICs (regardless of the stimuli used, including Gq GPCR agonist application; Angulo et al., 2004; Fellin et al., 2004; Navarrete and Araque, 2008; Perea and Araque, 2005; Shigetomi et al., 2008), have all reported the occurrence of spontaneous SICs in basal conditions independent of astrocytic activity. It is striking to note that the investigators that have not reproduced such data using two kinds of stimuli (IP3 uncaging or agonist application to stimulate several Gq GPCRs) have never recorded any SICs in basal conditions (Fiacco and McCarthy, 2004; Fiacco et al., 2007; Lee et al., 2007). This raises the possibility that there are other variable(s) in the acute slice preparations that are primarily responsible for the occurrence of SICs. What could be this difference? There may be a combination of several variables from tissue preparation to the content of internal and external solutions. Interestingly, currents similar to the NR2B subtype NMDAR-mediated SICs have been reported to be induced 100% of the time when the osmolarity of the recording buffer is reduced. The SICs induced by a hypo-osmotic shift in buffer are (1) indistinguishable from the large-amplitude, very slow SICs reported by other groups following treatments that increase astrocytic Ca2+ and (2) occur independent of IP3R-dependent astrocytic Ca2+ increases or presynaptic vesicular release of glutamate (Fiacco et al., 2007). These results suggest that SIC-like currents are driven predominantly by glutamate release via a pathway that does not involve astrocytic Ca2+ elevations but involves alterations in cell volume, which are more typical in pathological conditions. In agreement with this hypothesis, another report indicates that perfusion with hypotonic buffer leads to a significant increase in SIC frequency in olfactory bulb neurons (Kozlov et al., 2006). This latter set of data from different groups (Fiacco et al., 2007; Kozlov et al., 2006) might reconcile the discrepancies found in the literature with the possibility that spontaneous and/or evoked glutamate release from astrocytes might be preferentially associated with certain nonphysiological states of the tissue and might not be necessarily related to Ca2+ elevations in astrocytes. Finally, investigators in the field would certainly benefit from discussion with other laboratories to determine whether the occurrence of spontaneous SICs is a common phenomenon or not among neurophysiologists outside of the field.
Gliotransmission: Neurophysiology, Neuropathology, or Pharmacology?
There is no doubt that under certain experimental conditions astrocytes can release gliotransmitters, such as glutamate or ATP, that activate presynaptic and postsynaptic neuronal metabotropic or ionotropic receptors. As discussed above, there are conflicting data concerning whether or not gliotransmission occurs in situ. Data from several laboratories suggest that gliotransmission occurs and might play an important role in physiology. However, there are also data arguing that gliotransmission does not occur when activating Gq GPCRs in situ (Table 1); therefore, the significance of gliotransmission in neurophysiology or neuropathology remains unclear (Figure 4). Three distinct possibilities emerge from these discrepancies with respect to gliotransmission. First, gliotransmission could occur under physiological conditions, and subtle differences in experimental conditions among laboratories may explain the qualitatively different findings reported. Second, gliotransmission may only occur under pathological conditions, and the variability in the health of acutely isolated brain slices or other experimental parameters could account for the different findings reported in this field. Third, we can make gliotransmission occur using nonphysiological stimulations (e.g., mechanical stimulation, Ca2+ or IP3 uncaging, depolarization of astrocytes, electrical stimulation of nerve fibers), but this phenomenon does not occur in the absence of such stimulations. There are credible arguments for and against each of these three possibilities. As such, how do we make progress in sorting out these possibilities? All elements of experimental protocols need to be carefully examined in an attempt to define the conditions affecting gliotransmitter release. Reagents should be compared and shared, and reciprocal laboratory visits between investigators obtaining conflicting findings should be carried out. Study of gliotransmission in vivo rather that in situ using imaging and electrophysiological methods may partially circumvent the pathology associated with acutely isolated brain slices. Genetic models are likely to be critically important for elucidating the role of gliotransmission in synaptic transmission. However, multiple gene manipulations will be required to ensure that the genetic manipulation is truly affecting the targeted process. Innovative tools (pharmacological and/or genetic) to activate Gq GPCR-mediated Ca2+ elevations selectively in astrocytes need to be generated in order to overcome the caveats associated with the current tools used. A deeper appreciation of the heterogeneity among astrocytes also needs to be considered in the interpretation of findings in this field. It remains possible that subsets of astrocytes, and/or different domains within astrocytes, participate in gliotransmission. As additional investigators begin to address the question of gliotransmission and as new methods are applied, we can expect to attain a better understanding of this process.
Table 1. Astrocytic Ca2+-Mediated Neuromodulatory Effects Obtained at the SC-CA1 Synapse Using Varied Approaches to Activate Astrocytes In Situ.
| Agonists/Stimuli | Astrocytic Gq GPCRs (activated by agonists) | Gliotransmitters released | Presynaptic modulation | Postsynaptic modulation |
|---|---|---|---|---|
| DHPG | Group I mGluRs | Glutamate | NR | SICs 1, 2 |
| No release | No effect | No SICs 3 | ||
| tACPD | Group I and II mGluRs | Glutamate | NR | Neuronal Ca2+ increase 4 |
| TFLLR | PAR1 | Glutamate | NR | SICs 5 |
| Glutamate | NR | Potention of NMDA currents/no SICs 6 | ||
| ATP | P2Y1 | No release | NR | No SICs 5 |
| ATP | Glutamate | NR | SICs 1, 12 | |
| Endothelin (1 and 3) | Endotheline receptors (types 1 and 3) | No release | No effect | No SICs 3 |
| FLRFaMrgA1 mouse | MrgA1 | No release | No effect | No SICs 3 |
| Ca2+ ungaging | Glutamate | NR | SICs 2 | |
| Glutamate | Induction of a form of presynapic LTP 8 | NR 8 | ||
| IP3 uncaging | ||||
| Wild type mouse | Glutamate 3 |
|
No SICs 3, 9 | |
| MrgA1 mouse | Glutamate 9 | |||
| Electrical SC stimulation | ||||
| Wild type mouse | ATP 11 |

|
NR 10, 11 | |
| dn SNARE mouse | ATP blocked 10 | |||
| Mechanical stimulation | Glutamate | NR | SICs 1 | |
| Neuronal depolarization | Endocannabinoid receptors (CB1R) | Glutamate | NR | SICs 7 |
Abbreviations: 2AG, AEA, and MAEA, agonist for endocannabinoid receptors (CB1Rs); DHPG [(S)-3,5-dihydroxyphenylglycine], specific group I mGluR agonist; dnSNARE mouse, transgenic mouse expressing a dominant-negative synaptobrevin 2 protein selectively in astrocytes to block SNARE-dependent exocytosis of gliotransmitters; FLRFa, selective MrgA1R agonist; mGluR, metabotropic glutamate receptor; MrgA1R, transgenic Gq GPCR specifically expressed in astrocytes; PAR1, proteinase-activated receptor-1; P2Y1, purinergic Gq GPCR; tACPD [trans-(+)-1-amino-1,3-ciclopentanedicarboxylic acid], specific group I and II mGluR agonist; TFLLR, selective PAR1 agonist. Notes: (1) mouse translines (wild-type versus transgenic) are listed for experiments performed in transgenic mice. When not listed, experiments were carried out using wild-type rats or mice; (2) CB1Rs are mainly coupled to Gi/o proteins that regulate cAMP levels, but these receptors can also be coupled to Gq proteins (Lauckner et al., 2005); (3) when presynaptic or postsynaptic modulations were not reported, it is indicated as NR in the table.
Angulo et al., 2004;
Fellin et al., 2004;
Fiacco et al., 2007;
Pasti et al., 1997;
Shigetomi et al., 2008;
Lee et al., 2007;
Navarrete and Araque, 2008;
Perea and Araque, 2007;
Fiacco and McCarthy, 2004;
Pascual et al., 2005;
Serrano et al., 2006;
Perea and Araque, 2005.
Figure 4. Schematic Showing the Current Understanding of Astrocytic Ca2+ Signaling Involvement at the Synapse.

(Left panel) Both in situ and in vivo studies strongly support the conclusion that synaptic release of neurotransmitters, under basal and heightened levels of stimulation, elicits Ca2+ increases in astrocytes mostly via the activation of Gq GPCRs. These astrocytic Ca2+ elevations can remain localized within small territories (microdomains) within the cell or propagate as intracellular waves into more distant compartments, depending on the level of neuronal activity. (Right panel) Whether or not astrocytic Ca2+ increases evoke the release of gliotransmitters to modulate pre- or postsynaptic metabotropic or ionotropic neuronal receptors is still under debate. To date, there are no in vivo data available, and data in situ argue both for and against the concept of gliotransmission. The potential significance of gliotransmission in neurophysiology and neuropathophysiology remains an open issue.
Conclusions
In situ and in vivo studies clearly demonstrate that astrocytes express a wide variety of GPCRs that affect a diverse set of signaling cascades, including many different Gq GPCRs, which are activated by neurotransmitters released from presynaptic terminals to produce astrocytic Ca2+ increases (Figure 4). However, for the most part, the physiological outcome of astrocytic Gq GPCR activation remains unclear. Prevailing evidence indicates that spontaneous astrocytic Ca2+ activity generally remains localized and is unlikely to activate distant astrocytes via intercellular Ca2+ waves. It is also probable that the level of stimulation of astrocytic Gq GPCRs determines the volume of the astrocyte that will participate in the signaling cascade.
Whether or not gliotransmission plays a role in neurophysiology or neuropathology or is an artifact of the experimental preparations used in this field is one of the most important questions facing researchers in this area (Figure 4). To a large extent, the only tool for monitoring astrocyte Gq GPCR activation is Ca2+ imaging. Currently, Ca2+ changes within single astrocyte processes wrapping individual synapses where Ca2+ signaling originates and probably carries out its primary effects cannot be measured accurately. Further, most techniques for selectively manipulating Ca2+ in astrocytes rely on pharmacological tools of questionable selectivity or of questionable ability to evoke Ca2+ increases that mimic endogenous Ca2+ signaling dynamics. Advances in this field will be dependent on the development of novel physiological tools for selectively altering astrocyte function.
ACKNOWLEDGMENTS
This work was supported by grants from the US National Institutes of Health (NS033938 and NS020212).
REFERENCES
- Aguado F, Espinosa-Parrilla JF, Carmona MA, Soriano E. Neuronal activity regulates correlated network properties of spontaneous calcium transients in astrocytes in situ. J. Neurosci. 2002;22:9430–9444. doi: 10.1523/JNEUROSCI.22-21-09430.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson CM, Swanson RA. Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia. 2000;32:1–14. [PubMed] [Google Scholar]
- Angulo MC, Kozlov AS, Charpak S, Audinat E. Glutamate released from glial cells synchronizes neuronal activity in the hippocampus. J. Neurosci. 2004;24:6920–6927. doi: 10.1523/JNEUROSCI.0473-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araque A, Li N, Doyle RT, Haydon PG. SNARE protein-dependent glutamate elease from astrocytes. J. Neurosci. 2000;20:666–673. doi: 10.1523/JNEUROSCI.20-02-00666.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araque A, Martin ED, Perea G, Arellano JI, Buno W. Synaptically released acetylcholine evokes Ca2+ elevations in astrocytes in hippocampal slices. J. Neurosci. 2002;22:2443–2450. doi: 10.1523/JNEUROSCI.22-07-02443.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck A, Nieden RZ, Schneider HP, Deitmer JW. Calcium release from intracellular stores in rodent astrocytes and neurons in situ. Cell Calcium. 2004;35:47–58. doi: 10.1016/s0143-4160(03)00171-4. [DOI] [PubMed] [Google Scholar]
- Beierlein M, Regehr WG. Brief bursts of parallel fiber activity trigger calcium signals in bergmann glia. J. Neurosci. 2006;26:6958–6967. doi: 10.1523/JNEUROSCI.0613-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekar LK, He W, Nedergaard M. Locus coeruleus {alpha}-adrenergic-mediated activation of cortical astrocytes in vivo. Cereb. Cortex. 2008 doi: 10.1093/cercor/bhn040. in press. Published online March 27, 2008. 10.1093/cercor/bhn040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezzi P, Carmignoto G, Pasti L, Vesce S, Rossi D, Rizzini BL, Pozzan T, Volterra A. Prostaglandins stimulate calcium-dependent glutamate release in astrocytes. Nature. 1998;391:281–285. doi: 10.1038/34651. [DOI] [PubMed] [Google Scholar]
- Bowser DN, Khakh BS. Vesicular ATP is the predominant cause of intercellular calcium waves in astrocytes. J. Gen. Physiol. 2007;129:485–491. doi: 10.1085/jgp.200709780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushong EA, Martone ME, Jones YZ, Ellisman MH. Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J. Neurosci. 2002;22:183–192. doi: 10.1523/JNEUROSCI.22-01-00183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J. Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmignoto G, Pasti L, Pozzan T. On the role of voltage-dependent calcium channels in calcium signaling of astrocytes in situ. J. Neurosci. 1998;18:4637–4645. doi: 10.1523/JNEUROSCI.18-12-04637.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codazzi F, Teruel MN, Meyer T. Control of astrocyte Ca(2+) oscillations and waves by oscillating translocation and activation of protein kinase C. Curr. Biol. 2001;11:1089–1097. doi: 10.1016/s0960-9822(01)00326-8. [DOI] [PubMed] [Google Scholar]
- Cornell-Bell AH, Finkbeiner SM, Cooper MS, Smith SJ. Glutamate induces calcium waves in cultured astrocytes: long-range glial signaling. Science. 1990;247:470–473. doi: 10.1126/science.1967852. [DOI] [PubMed] [Google Scholar]
- Dani JW, Chernjavsky A, Smith SJ. Neuronal activity triggers calcium waves in hippocampal astrocyte networks. Neuron. 1992;8:429–440. doi: 10.1016/0896-6273(92)90271-e. [DOI] [PubMed] [Google Scholar]
- Dombeck DA, Khabbaz AN, Collman F, Adelman TL, Tank DW. Imaging large-scale neural activity with cellular resolution in awake, mobile mice. Neuron. 2007;56:43–57. doi: 10.1016/j.neuron.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domercq M, Brambilla L, Pilati E, Marchaland J, Volterra A, Bezzi P. P2Y1 receptor-evoked glutamate exocytosis from astrocytes: control by tumor necrosis factor-alpha and prostaglandins. J. Biol. Chem. 2006;281:30684–30696. doi: 10.1074/jbc.M606429200. [DOI] [PubMed] [Google Scholar]
- Dong X, Han S, Zylka MJ, Simon MI, Anderson DJ. A diverse family of GPCRs expressed in specific subsets of nociceptive sensory neurons. Cell. 2001;106:619–632. doi: 10.1016/s0092-8674(01)00483-4. [DOI] [PubMed] [Google Scholar]
- Duan S, Anderson CM, Keung EC, Chen Y, Swanson RA. P2X7 receptor-mediated release of excitatory amino acids from astrocytes. J. Neurosci. 2003;23:1320–1328. doi: 10.1523/JNEUROSCI.23-04-01320.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy S, MacVicar BA. Potassium-dependent calcium influx in acutely isolated hippocampal astrocytes. Neuroscience. 1994;61:51–61. doi: 10.1016/0306-4522(94)90059-0. [DOI] [PubMed] [Google Scholar]
- Duffy S, MacVicar BA. Adrenergic calcium signaling in astrocyte networks within the hippocampal slice. J. Neurosci. 1995;15:5535–5550. doi: 10.1523/JNEUROSCI.15-08-05535.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellin T, Carmignoto G. Neurone-to-astrocyte signalling in the brain represents a distinct multifunctional unit. J. Physiol. 2004;559:3–15. doi: 10.1113/jphysiol.2004.063214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellin T, Pascual O, Gobbo S, Pozzan T, Haydon PG, Carmignoto G. Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron. 2004;43:729–743. doi: 10.1016/j.neuron.2004.08.011. [DOI] [PubMed] [Google Scholar]
- Fellin T, Pozzan T, Carmignoto G. Purinergic receptors mediate two distinct glutamate release pathways in hippocampal astrocytes. J. Biol. Chem. 2006;281:4274–4284. doi: 10.1074/jbc.M510679200. [DOI] [PubMed] [Google Scholar]
- Fiacco TA, McCarthy KD. Intracellular astrocyte calcium waves in situ increase the frequency of spontaneous AMPA receptor currents in CA1 pyramidal neurons. J. Neurosci. 2004;24:722–732. doi: 10.1523/JNEUROSCI.2859-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiacco TA, McCarthy KD. Astrocyte calcium elevations: properties, propagation, and effects on brain signaling. Glia. 2006;54:676–690. doi: 10.1002/glia.20396. [DOI] [PubMed] [Google Scholar]
- Fiacco TA, Agulhon C, Taves SR, Petravicz J, Casper KB, Dong X, Chen J, McCarthy KD. Selective stimulation of astrocyte calcium in situ does not affect neuronal excitatory synaptic activity. Neuron. 2007;54:611–626. doi: 10.1016/j.neuron.2007.04.032. [DOI] [PubMed] [Google Scholar]
- Foskett JK, White C, Cheung KH, Mak DO. Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev. 2007;87:593–658. doi: 10.1152/physrev.00035.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingrich MB, Junge CE, Lyuboslavsky P, Traynelis SF. Potentiation of NMDA receptor function by the serine protease thrombin. J. Neurosci. 2000;20:4582–4595. doi: 10.1523/JNEUROSCI.20-12-04582.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobel W, Kampa BM, Helmchen F. Imaging cellular network dynamics in three dimensions using fast 3D laser scanning. Nat. Methods. 2007;4:73–79. doi: 10.1038/nmeth989. [DOI] [PubMed] [Google Scholar]
- Grosche J, Matyash V, Moller T, Verkhratsky A, Reichenbach A, Kettenmann H. Microdomains for neuron-glia interaction: parallel fiber signaling to Bergmann glial cells. Nat. Neurosci. 1999;2:139–143. doi: 10.1038/5692. [DOI] [PubMed] [Google Scholar]
- Grosche J, Kettenmann H, Reichenbach A. Bergmann glial cells form distinct morphological structures to interact with cerebellar neurons. J. Neurosci. Res. 2002;68:138–149. doi: 10.1002/jnr.10197. [DOI] [PubMed] [Google Scholar]
- Halassa MM, Fellin T, Haydon PG. The tripartite synapse: roles for gliotransmission in health and disease. Trends Mol. Med. 2007;13:54–63. doi: 10.1016/j.molmed.2006.12.005. [DOI] [PubMed] [Google Scholar]
- Haydon PG, Carmignoto G. Astrocyte control of synaptic transmission and neurovascular coupling. Physiol. Rev. 2006;86:1009–1031. doi: 10.1152/physrev.00049.2005. [DOI] [PubMed] [Google Scholar]
- Hertle DN, Yeckel MF. Distribution of inositol-1,4,5-trisphosphate receptor isotypes and ryanodine receptor isotypes during maturation of the rat hippocampus. Neuroscience. 2007;150:625–638. doi: 10.1016/j.neuroscience.2007.09.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirase H, Qian L, Bartho P, Buzsaki G. Calcium dynamics of cortical astrocytic networks in vivo. PLoS Biol. 2004;2:E96. doi: 10.1371/journal.pbio.0020096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzclaw LA, Pandhit S, Bare DJ, Mignery GA, Russell JT. Astrocytes in adult rat brain express type 2 inositol 1,4,5-trisphosphate receptors. Glia. 2002;39:69–84. doi: 10.1002/glia.10085. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Nedergaard M. Glial regulation of the cerebral microvasculature. Nat. Neurosci. 2007;10:1369–1376. doi: 10.1038/nn2003. [DOI] [PubMed] [Google Scholar]
- Jabs R, Kirchhoff F, Kettenmann H, Steinhauser C. Kainate activates Ca(2+)-permeable glutamate receptors and blocks voltage-gated K+ currents in glial cells of mouse hippocampal slices. Pflugers Arch. 1994;426:310–319. doi: 10.1007/BF00374787. [DOI] [PubMed] [Google Scholar]
- Jabs R, Pivneva T, Huttmann K, Wyczynski A, Nolte C, Kettenmann H, Steinhauser C. Synaptic transmission onto hippocampal glial cells with hGFAP promoter activity. J. Cell Sci. 2005;118:3791–3803. doi: 10.1242/jcs.02515. [DOI] [PubMed] [Google Scholar]
- Jourdain P, Bergersen LH, Bhaukaurally K, Bezzi P, Santello M, Domercq M, Matute C, Tonello F, Gundersen V, Volterra A. Glutamate exocytosis from astrocytes controls synaptic strength. Nat. Neurosci. 2007;10:331–339. doi: 10.1038/nn1849. [DOI] [PubMed] [Google Scholar]
- Junge CE, Lee CJ, Hubbard KB, Zhang Z, Olson JJ, Hepler JR, Brat DJ, Traynelis SF. Protease-activated receptor-1 in human brain: localization and functional expression in astrocytes. Exp. Neurol. 2004;188:94–103. doi: 10.1016/j.expneurol.2004.02.018. [DOI] [PubMed] [Google Scholar]
- Kacem K, Lacombe P, Seylaz J, Bonvento G. Structural organization of the perivascular astrocyte endfeet and their relationship with the endothelial glucose transporter: a confocal microscopy study. Glia. 1998;23:1–10. [PubMed] [Google Scholar]
- Kang J, Jiang L, Goldman SA, Nedergaard M. Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nat. Neurosci. 1998;1:683–692. doi: 10.1038/3684. [DOI] [PubMed] [Google Scholar]
- Kang J, Kang N, Lovatt D, Torres A, Zhao Z, Lin J, Nedergaard M. Connexin 43 hemichannels are permeable to ATP. J. Neurosci. 2008;28:4702–4711. doi: 10.1523/JNEUROSCI.5048-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimelberg HK, Anderson E, Kettenmann H. Swelling-induced changes in electrophysiological properties of cultured astrocytes and oligodendrocytes. II. Whole-cell currents. Brain Res. 1990;529:262–268. doi: 10.1016/0006-8993(90)90836-z. [DOI] [PubMed] [Google Scholar]
- Kimelberg HK, Macvicar BA, Sontheimer H. Anion channels in astrocytes: biophysics, pharmacology, and function. Glia. 2006;54:747–757. doi: 10.1002/glia.20423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konietzko U, Muller CM. Astrocytic dye coupling in rat hippocampus: topography, developmental onset, and modulation by protein kinase C. Hippocampus. 1994;4:297–306. doi: 10.1002/hipo.450040313. [DOI] [PubMed] [Google Scholar]
- Kozlov AS, Angulo MC, Audinat E, Charpak S. Target cell-specific modulation of neuronal activity by astrocytes. Proc. Natl. Acad. Sci. USA. 2006;103:10058–10063. doi: 10.1073/pnas.0603741103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulik A, Haentzsch A, Luckermann M, Reichelt W, Ballanyi K. Neuron-glia signaling via alpha(1) adrenoceptor-mediated Ca(2+) release in Bergmann glial cells in situ. J. Neurosci. 1999;19:8401–8408. doi: 10.1523/JNEUROSCI.19-19-08401.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauckner JE, Hille B, Mackie K. The cannabinoid agonist WIN55,212-2 increases intracellular calcium via CB1 receptor coupling to Gq/11 G proteins. Proc. Natl. Acad. Sci. USA. 2005;102:19144–19149. doi: 10.1073/pnas.0509588102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CJ, Mannaioni G, Yuan H, Woo DH, Gingrich MB, Traynelis SF. Astrocytic control of synaptic NMDA receptors. J. Physiol. 2007;581:1057–1081. doi: 10.1113/jphysiol.2007.130377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SC, Bergles DE. Synaptic signaling between GABAergic interneurons and oligodendrocyte precursor cells in the hippocampus. Nat. Neurosci. 2004;7:24–32. doi: 10.1038/nn1162. [DOI] [PubMed] [Google Scholar]
- Liu QS, Xu Q, Kang J, Nedergaard M. Astrocyte activation of presynaptic metabotropic glutamate receptors modulates hippocampal inhibitory synaptic transmission. Neuron Glia Biol. 2004;1:307–316. doi: 10.1017/S1740925X05000190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovatt D, Sonnewald U, Waagepetersen HS, Schousboe A, He W, Lin JH, Han X, Takano T, Wang S, Sim FJ, et al. The transcriptome and metabolic gene signature of protoplasmic astrocytes in the adult murine cortex. J. Neurosci. 2007;27:12255–12266. doi: 10.1523/JNEUROSCI.3404-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui K, Jahr CE. Differential control of synaptic and ectopic vesicular release of glutamate. J. Neurosci. 2004;24:8932–8939. doi: 10.1523/JNEUROSCI.2650-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui K, Jahr CE, Rubio ME. High-concentration rapid transients of glutamate mediate neural-glial communication via ectopic release. J. Neurosci. 2005;25:7538–7547. doi: 10.1523/JNEUROSCI.1927-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthias K, Kirchhoff F, Seifert G, Huttmann K, Matyash M, Kettenmann H, Steinhauser C. Segregated expression of AMPA-type glutamate receptors and glutamate transporters defines distinct astrocyte populations in the mouse hippocampus. J. Neurosci. 2003;23:1750–1758. doi: 10.1523/JNEUROSCI.23-05-01750.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matyash V, Filippov V, Mohrhagen K, Kettenmann H. Nitric oxide signals parallel fiber activity to Bergmann glial cells in the mouse cerebellar slice. Mol. Cell. Neurosci. 2001;18:664–670. doi: 10.1006/mcne.2001.1047. [DOI] [PubMed] [Google Scholar]
- McCarthy KD, de Vellis J. Alpha-adrenergic receptor modulation of beta-adrenergic, adenosine and prostaglandin E1 increased adenosine 3′:5′-cyclic monophosphate levels in primary cultures of glia. J. Cyclic Nucleotide Res. 1978;4:15–26. [PubMed] [Google Scholar]
- Metea MR, Newman EA. Calcium signaling in specialized glial cells. Glia. 2006;54:650–655. doi: 10.1002/glia.20352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mothet JP, Pollegioni L, Ouanounou G, Martineau M, Fossier P, Baux G. Glutamate receptor activation triggers a calcium-dependent and SNARE protein-dependent release of the gliotransmitter D-serine. Proc. Natl. Acad. Sci. USA. 2005;102:5606–5611. doi: 10.1073/pnas.0408483102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarrete M, Araque A. Endocannabinoids mediate neuronastrocyte communication. Neuron. 2008;57:883–893. doi: 10.1016/j.neuron.2008.01.029. [DOI] [PubMed] [Google Scholar]
- Nett WJ, Oloff SH, McCarthy KD. Hippocampal astrocytes in situ exhibit calcium oscillations that occur independent of neuronal activity. J. Neurophysiol. 2002;87:528–537. doi: 10.1152/jn.00268.2001. [DOI] [PubMed] [Google Scholar]
- Newman EA. Calcium increases in retinal glial cells evoked by light-induced neuronal activity. J. Neurosci. 2005;25:5502–5510. doi: 10.1523/JNEUROSCI.1354-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niclou SP, Suidan HS, Pavlik A, Vejsada R, Monard D. Changes in the expression of protease-activated receptor 1 and protease nexin-1 mRNA during rat nervous system development and after nerve lesion. Eur. J. Neurosci. 1998;10:1590–1607. doi: 10.1046/j.1460-9568.1998.00183.x. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Kerr JN, Helmchen F. Sulforhodamine 101 as a specific marker of astroglia in the neocortex in vivo. Nat. Methods. 2004;1:31–37. doi: 10.1038/nmeth706. [DOI] [PubMed] [Google Scholar]
- Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S, Haydon PG. Glutamate-mediated astrocyte-neuron signalling. 1994;369:744–774. doi: 10.1038/369744a0. [DOI] [PubMed] [Google Scholar]
- Parpura V, Scemes E, Spray DC. Mechanisms of glutamate release from astrocytes: gap junction “hemichannels”, purinergic receptors and exocytotic release. Neurochem. Int. 2004;45:259–264. doi: 10.1016/j.neuint.2003.12.011. [DOI] [PubMed] [Google Scholar]
- Parri HR, Gould TM, Crunelli V. Spontaneous astrocytic Ca2+ oscillations in situ drive NMDAR-mediated neuronal excitation. Nat. Neurosci. 2001;4:803–812. doi: 10.1038/90507. [DOI] [PubMed] [Google Scholar]
- Parri HR, Crunelli V. The role of Ca2+ in the generation of spontaneous astrocytic Ca2+ oscillations. Neuroscience. 2003;120:979–992. doi: 10.1016/s0306-4522(03)00379-8. [DOI] [PubMed] [Google Scholar]
- Pascual O, Casper KB, Kubera C, Zhang J, Revilla-Sanchez R, Sul JY, Takano H, Moss SJ, McCarthy K, Haydon PG. Astrocytic purinergic signaling coordinates synaptic networks. Science. 2005;310:113–116. doi: 10.1126/science.1116916. [DOI] [PubMed] [Google Scholar]
- Pasti L, Volterra A, Pozzan T, Carmignoto G. Intracellular calcium oscillations in astrocytes: A highly plastic, bidirectional form of communication between neurons and astrocytes in situ. J. Neurosci. 1997;17:7817–7830. doi: 10.1523/JNEUROSCI.17-20-07817.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perea G, Araque A. Properties of synaptically evoked astrocyte calcium signal reveal synaptic information processing by astrocytes. J. Neurosci. 2005;25:2192–2203. doi: 10.1523/JNEUROSCI.3965-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perea G, Araque A. Astrocytes potentiate transmitter release at single hippocampal synapses. Science. 2007;317:1083–1086. doi: 10.1126/science.1144640. [DOI] [PubMed] [Google Scholar]
- Petravicz J, Fiacco TA, McCarthy KD. Loss of IP3 receptor-dependent Ca2+ increases in hippocampal astrocytes does not affect baseline CA1 pyramidal neuron synaptic activity. J. Neurosci. 2008;28:4967–4973. doi: 10.1523/JNEUROSCI.5572-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piet R, Jahr CE. Glutamatergic and purinergic receptor-mediated calcium transients in Bergmann glial cells. J. Neurosci. 2007;27:4027–4035. doi: 10.1523/JNEUROSCI.0462-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto L, Gotz M. Radial glial cell heterogeneity-the source of diverse progeny in the CNS. Prog. Neurobiol. 2007;83:2–23. doi: 10.1016/j.pneurobio.2007.02.010. [DOI] [PubMed] [Google Scholar]
- Porter JT, McCarthy KD. Adenosine receptors modulate [Ca2+]i in hippocampal astrocytes in situ. J. Neurochem. 1995a;65:1515–1523. doi: 10.1046/j.1471-4159.1995.65041515.x. [DOI] [PubMed] [Google Scholar]
- Porter JT, McCarthy KD. GFAP-positive hippocampal astrocytes in situ respond to glutamatergic neuroligands with increases in [Ca2+]i. Glia. 1995b;13:101–112. doi: 10.1002/glia.440130204. [DOI] [PubMed] [Google Scholar]
- Porter JT, McCarthy KD. Hippocampal astrocytes in situ respond to glutamate released from synaptic terminals. J. Neurosci. 1996;16:5073–5081. doi: 10.1523/JNEUROSCI.16-16-05073.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter JT, McCarthy KD. Astrocytic neurotransmitter receptors in situ and in vivo. Prog. Neurobiol. 1997;51:439–455. doi: 10.1016/s0301-0082(96)00068-8. [DOI] [PubMed] [Google Scholar]
- Rama Rao KV, Chen M, Simard JM, Norenberg MD. Increased aquaporin-4 expression in ammonia-treated cultured astrocytes. Neuroreport. 2003;14:2379–2382. doi: 10.1097/00001756-200312190-00018. [DOI] [PubMed] [Google Scholar]
- Ransom B, Behar T, Nedergaard M. New roles for astrocytes (stars at last) Trends Neurosci. 2003;26:520–522. doi: 10.1016/j.tins.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Roth BJ, Yagodin SV, Holtzclaw L, Russell JT. A mathematical model of agonist-induced propagation of calcium waves in astrocytes. Cell Calcium. 1995;17:53–64. doi: 10.1016/0143-4160(95)90102-7. [DOI] [PubMed] [Google Scholar]
- Sasaki Y, Takimoto M, Oda K, FrÅh T, Takai M, Okada T, Hori S. Endothelin evokes efflux of glutamate in cultures of rat astrocytes. J. Neurochem. 1997;68:2194–2200. doi: 10.1046/j.1471-4159.1997.68052194.x. [DOI] [PubMed] [Google Scholar]
- Scemes E. Components of astrocytic intercellular calcium signaling. Mol. Neurobiol. 2000;22:167–179. doi: 10.1385/MN:22:1-3:167. [DOI] [PubMed] [Google Scholar]
- Scemes E, Giaume C. Astrocyte calcium waves: what they are and what they do. Glia. 2006;54:716–725. doi: 10.1002/glia.20374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schummers J, Yu H, Sur M. Tuned responses of astrocytes and their influence on hemodynamic signals in the visual cortex. Science. 2008;320:1638–1643. doi: 10.1126/science.1156120. [DOI] [PubMed] [Google Scholar]
- Serrano A, Haddjeri N, Lacaille JC, Robitaille R. GABAergic network activation of glial cells underlies hippocampal heterosynaptic depression. J. Neurosci. 2006;26:5370–5382. doi: 10.1523/JNEUROSCI.5255-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp AH, Nucifora FC, Jr., Blondel O, Sheppard CA, Zhang C, Snyder SH, Russell JT, Ryugo DK, Ross CA. Differential cellular expression of isoforms of inositol 1,4,5-triphosphate receptors in neurons and glia in brain. J. Comp. Neurol. 1999;406:207–220. [PubMed] [Google Scholar]
- Shelton MK, McCarthy KD. Mature hippocampal astrocytes exhibit functional metabotropic and ionotropic glutamate receptors in situ. Glia. 1999;26:1–11. doi: 10.1002/(sici)1098-1136(199903)26:1<1::aid-glia1>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Shelton MK, McCarthy KD. Hippocampal astrocytes exhibit Ca2+-elevating muscarinic cholinergic and histaminergic receptors in situ. J. Neurochem. 2000;74:555–563. doi: 10.1046/j.1471-4159.2000.740555.x. [DOI] [PubMed] [Google Scholar]
- Sheppard CA, Simpson PB, Sharp AH, Nucifora FC, Ross CA, Lange GD, Russell JT. Comparison of type 2 inositol 1,4,5-trisphosphate receptor distribution and subcellular Ca2+ release sites that support Ca2+ waves in cultured astrocytes. J. Neurochem. 1997;68:2317–2327. doi: 10.1046/j.1471-4159.1997.68062317.x. [DOI] [PubMed] [Google Scholar]
- Shigetomi E, Bowser DN, Sofroniew MV, Khakh BS. Two forms of astrocyte calcium excitability have distinct effects on NMDA receptor-mediated slow inward currents in pyramidal neurons. J. Neurosci. 2008;28:6659–6663. doi: 10.1523/JNEUROSCI.1717-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuai JW, Jung P. Optimal ion channel clustering for intracellular calcium signaling. Proc. Natl. Acad. Sci. USA. 2003;100:506–510. doi: 10.1073/pnas.0236032100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard M, Arcuino G, Takano T, Liu QS, Nedergaard M. Signaling at the gliovascular interface. J. Neurosci. 2003;23:9254–9262. doi: 10.1523/JNEUROSCI.23-27-09254.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson PB, Mehotra S, Lange GD, Russell JT. High density distribution of endoplasmic reticulum proteins and mitochondria at specialized Ca2+ release sites in oligodendrocyte processes. J. Biol. Chem. 1997;272:22654–22661. doi: 10.1074/jbc.272.36.22654. [DOI] [PubMed] [Google Scholar]
- Somjen GG. Nervenkitt: notes on the history of the concept of neuroglia. Glia. 1988;1:2–9. doi: 10.1002/glia.440010103. [DOI] [PubMed] [Google Scholar]
- Stout CE, Costantin JL, Naus CC, Charles AC. Intercellular calcium signaling in astrocytes via ATP release through connexin hemichannels. J. Biol. Chem. 2002;277:10482–10488. doi: 10.1074/jbc.M109902200. [DOI] [PubMed] [Google Scholar]
- Straub SV, Bonev AD, Wilkerson MK, Nelson MT. Dynamic inositol trisphosphate-mediated calcium signals within astrocytic endfeet underlie vasodilation of cerebral arterioles. J. Gen. Physiol. 2006;128:659–669. doi: 10.1085/jgp.200609650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Striggow F, Riek-Burchardt M, Kiesel A, Schmidt W, Henrich-Noack P, Breder J, Krug M, Reymann KG, Reiser G. Four different types of protease-activated receptors are widely expressed in the brain and are upregulated in hippocampus by severe ischemia. Eur. J. Neurosci. 2001;14:595–608. doi: 10.1046/j.0953-816x.2001.01676.x. [DOI] [PubMed] [Google Scholar]
- Sul JY, Orosz G, Givens RS, Haydon PG. Astrocytic connectivity in the hippocampus. Neuron Glia Biol. 2004;1:3–11. doi: 10.1017/s1740925x04000031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano T, Tian GF, Peng W, Lou N, Libionka W, Han X, Nedergaard M. Astrocyte-mediated control of cerebral blood flow. Nat. Neurosci. 2006;9:260–267. doi: 10.1038/nn1623. [DOI] [PubMed] [Google Scholar]
- Thomas D, Lipp P, Tovey SC, Berridge MJ, Li W, Tsien RY, Bootman MD. Microscopic properties of elementary Ca2+ release sites in non-excitable cells. Curr. Biol. 2000;10:8–15. doi: 10.1016/s0960-9822(99)00258-4. [DOI] [PubMed] [Google Scholar]
- Tian GF, Takano T, Lin JH, Wang X, Bekar L, Nedergaard M. Imaging of cortical astrocytes using 2-photon laser scanning microscopy in the intact mouse brain. Adv. Drug Deliv. Rev. 2006;58:773–787. doi: 10.1016/j.addr.2006.07.001. [DOI] [PubMed] [Google Scholar]
- van Calker D, Hamprecht B. Effects of neurohormones on glial cells. In: Federoff S, Hertz L, editors. Advances in Cellular Neuribiology. Academic Press; Orlando, FL: 1981. pp. 32–55. [Google Scholar]
- van Calker D, Muller M, Hamprecht B. Adrenergic alpha and beta-receptors expressed by the same cell type in primary culture of perinatal mouse brain. J. Neurochem. 1978;30:713–718. doi: 10.1111/j.1471-4159.1978.tb10776.x. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Orkand RK, Kettenmann H. Glial calcium: homeostasis and signaling function. Physiol. Rev. 1998;78:99–141. doi: 10.1152/physrev.1998.78.1.99. [DOI] [PubMed] [Google Scholar]
- Volterra A, Steinhauser C. Glial modulation of synaptic transmission in the hippocampus. Glia. 2004;47:249–257. doi: 10.1002/glia.20080. [DOI] [PubMed] [Google Scholar]
- Wang X, Lou N, Xu Q, Tian GF, Peng WG, Han X, Kang J, Takano T, Nedergaard M. Astrocytic Ca2+ signaling evoked by sensory stimulation in vivo. Nat. Neurosci. 2006;9:816–823. doi: 10.1038/nn1703. [DOI] [PubMed] [Google Scholar]
- Weerth SH, Holtzclaw LA, Russell JT. Signaling proteins in raft-like microdomains are essential for Ca2+ wave propagation in glial cells. Cell Calcium. 2007;41:155–167. doi: 10.1016/j.ceca.2006.06.006. [DOI] [PubMed] [Google Scholar]
- Wilhelm A, Volknandt W, Langer D, Nolte C, Kettenmann H, Zim-mermann H. Localization of SNARE proteins and secretory organelle proteins in astrocytes in vitro and in situ. Neurosci. Res. 2004;48:249–257. doi: 10.1016/j.neures.2003.11.002. [DOI] [PubMed] [Google Scholar]
- Yagodin SV, Holtzclaw L, Sheppard CA, Russell JT. Nonlinear propagation of agonist-induced cytoplasmic calcium waves in single astrocytes. J. Neurobiol. 1994;25:265–280. doi: 10.1002/neu.480250307. [DOI] [PubMed] [Google Scholar]
- Yagodin S, Holtzclaw LA, Russell JT. Subcellular calcium oscillators and calcium influx support agonist-induced calcium waves in cultured astrocytes. Mol. Cell. Biochem. 1995;149-150:137–144. doi: 10.1007/BF01076572. [DOI] [PubMed] [Google Scholar]
- Yang Y, Ge W, Chen Y, Zhang Z, Shen W, Wu C, Poo M, Duan S. Contribution of astrocytes to hippocampal long-term potentiation through release of D-serine. Proc. Natl. Acad. Sci. USA. 2003;100:15194–15199. doi: 10.1073/pnas.2431073100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye ZC, Wyeth MS, Baltan-Tekkok S, Ransom BR. Functional hemichannels in astrocytes: a novel mechanism of glutamate release. J. Neurosci. 2003;23:3588–3596. doi: 10.1523/JNEUROSCI.23-09-03588.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Pangrsic T, Kreft M, Krzan M, Li N, Sul JY, Halassa M, Van Bockstaele E, Zorec R, Haydon PG. Fusion-related release of glutamate from astrocytes. J. Biol. Chem. 2004;279:12724–12733. doi: 10.1074/jbc.M312845200. [DOI] [PubMed] [Google Scholar]