

Abstract
Amplification of human Her2 and its aberrant signaling in 20-30% of early breast cancer patients is responsible for highly aggressive tumors with poor outcome. Grb7 is reported to be co-amplified with Her2. We report a concurrent high expression of mRNA (from FFPE tumor samples; mRNA correlation, Pearson r2= 0.806), and high levels of GRB7 protein (immunoblot) in HER2+ breast cancer cell lines. We demonstrated the signaling mechanism of HER2 and downstream effectors that contributes to proliferation and migration. Using HER2+ and trastuzumab-resistant breast cancer cell lines, we identified the interaction between GRB7 and HER2 in the control of HER2+ cell proliferation. Our co-IP data show that GRB7 recruits SHC into the HER2-GRB7 signaling complex. This complex formation leads to activation of RAS-GTP. We also observed that following integrin engagement, GRB7 is phosphorylated at tyrosine in a p-FAK (Y397) dependent manner. This FAK-GRB7 complex leads to downstream activation of RAC1-GTP (responsible for migration) probably through the recruitment of VAV2. Our CO-IP data demonstrate that GRB7 directly binds with VAV2 following fibronectin engagement in HER2+ cells. To address whether GRB7 could serve as a pathway specific therapeutic target, we used siRNA to suppress GRB7 expression. Knockdown of GRB7 expression in the HER2+ breast cancer cell lines decreases RAS activation, cell proliferation, 2D and 3D colony formation and also blocked integrin-mediated RAC1 activation along with integrin-directed cell migration. These findings dissected the HER2-mediated signaling cascade into (1) HER2+ cell proliferation (HER2-GRB7-SHC-RAS) and (2) HER2+ cell migration (alpha5 beta1/alpha4 beta1-FAK-GRB7-VAV2-RAC1). Our data clearly demonstrate that a coupling of GRB7 with HER2 is required for the proliferative and migratory signals in HER2+ breast tumor cells.
Keywords: Her2/Neu, GRB7, adapter protein, RAS, RAC, proliferation and migration
Introduction
Disregulation of HER2 signaling, including receptor amplification and/or overexpression is a significant factor in the progression of breast cancer. The Her2/Neu proto-oncogene is amplified in 25%-30% of human primary breast cancer and this alteration is associated with disease behavior [1]. Functional blocking antibody and receptor tyrosine kinase inhibitors have proven somewhat effective in this particular subset of breast cancer. Despite the clinical benefit of interruption of HER2 function with humanized HER2 monoclonal antibody such as trastuzumab, the precise mechanism of HER2 signaling in breast cancer progression is not well understood. Again, several preclinical and clinical reports have demonstrated that only 30% of the patients with HER2 overexpressing breast cancer respond to Herceptin® as a single agent. The majority of patients initially responds positively to this drug and subsequently develops resistance within years [2-4]. Therefore, further characterization of the pathophysiological roles of this pathway and development of pathway specific novel therapeutics should provide new opportunities to provide better management of breast cancer patients.
Gene expression profile analysis can generate a considerable amount of information for characterizing the nature of individual cancers; such information should be applied for elucidation of potential molecular targets for improving clinical strategies to treat neoplastic diseases [5,6]. These molecules are considered to be strong candidates for development of new therapeutic modalities. Since cytotoxic anti-cancer drugs often cause severe toxic reactions, it is obvious that careful selection of novel target molecules on the basis of well characterized mechanisms of action should be very helpful in developing molecular pathway specific and effective anti-cancer drugs with minimum toxic events. Toward this end, we developed a DASL-array based on a custom panel breast cancer gene profile (512 genes) and evaluated formalin fixed paraffin embedded (FFPE) breast cancer samples from 97 patients [7,8]. From this study, we have identified a significant subset-specific molecule that is overexpressed along with HER2 in a HER2 amplified breast cancer subset. Among many amplified genes in HER2 amplified breast cancer, here we report, the identification and characterization of a novel gene, GRB7, an important cytosolic adapter protein, to be a key molecule regulating a HER2 receptor signaling pathway in breast cancer. The objective of this study was to identify the potential downstream effectors of GRB7 as well as to understand the functional specificity of those downstream effectors in the HER2-GRB7 signaling cascade.
Grb7 maps to the HER2 amplicon on chromosome 17q and it has been reported by others that Grb7 is amplified concurrently with Her2/Neu in most, if not all, HER2+ breast cancers [9-11]. GRB7 isoforms are members of a super family of signaling mediators that includes GRB10, GRB14 and Caenorhabditis elegans MIG10 [12,13]. Members of the GRB7 family were originally cloned by their interaction with the EGF receptor, using the CORT (cloning of receptor target) system [14-16]. All mammalian members of this family share a domain structure which is represented by N- terminal proline-rich sequences, a homology domain of MIG10 (GM) which includes a RAS-associating (RA)-like domain, a pleckstrin homology region (PH), a C-terminal Src homology 2 (SH2) domain and a receptor binding domain located between the PH and SH2 domains termed BPS. The SH2 domain is responsible for GRB7’s adapter function (interaction of GRB7 with its binding partners). GRB7 is differentially overexpressed in a variety of tissues, namely breast, oesophageal and gastric cancers [17]. It has been also reported that GRB7 is a physiological factor during kidney [18] and liver development [19]. The function(s) of GRB7 in HER2+ breast cancers is not fully understood.
In this study, we report for the first time that HER2-GRB7 signaling complex facilitates the activation of both RAS and RAC1 GTPases in a cellular context (controlling proliferation and integrin-directed cell migration) with HER2 protein overexpression in breast cancer cell lines. Strikingly, we also observe that knockdown of GRB7, either by siRNA or by a GRB7 inhibitor peptide, is not capable of blocking either RAS or RAC1 activation in trastumab resistant cells. Based on GRB7’s interactions with a number of signaling mediators including upstream receptor kinases (HER2), non-receptor protein tyrosine kinase (FAK), cytosolic adapter protein (SHC), and further downstream small GTPases (activation of RAS-GTP following interaction with HER2 or RAC1-GTP following integrin-engagement), GRB7 may act as a signaling hub to integrate incoming signals. As well, it may act as a molecular scaffold to help assemble signaling complexes, ultimately controlling HER2 overexpressing breast cancer progression via tumor cell proliferation and migration (Figure 11). These findings indicate that expression of GRB7 in the HER2 overexpressed breast cancer subtype may be a surrogate marker of aberrant HER2 signaling that could contribute to the aggressive nature of the tumor.
Figure 11.
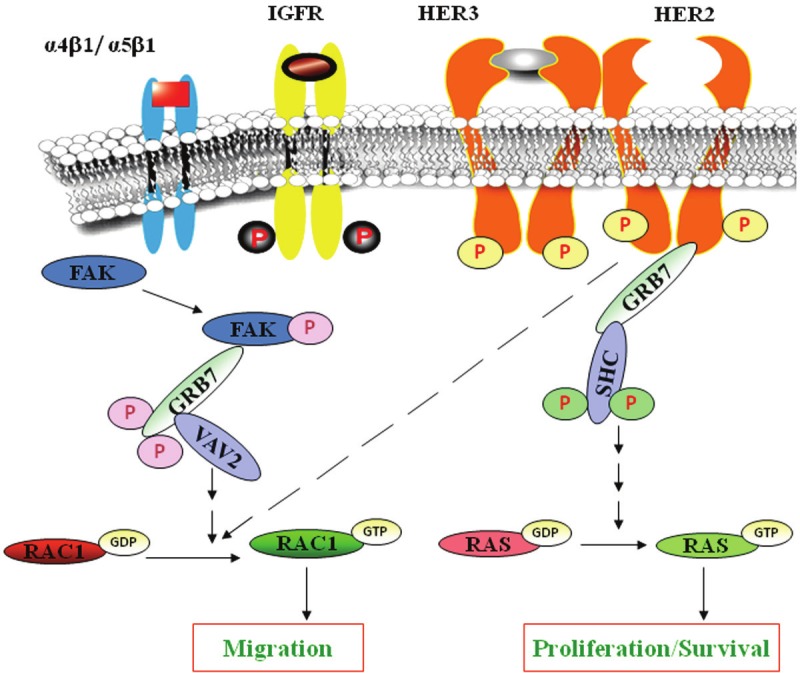
Schematic representation of the central theme of the study to show the mechanisms of proliferation and integrin-mediated migration in HER2-overexpressing breast cancer cells: This schematic diagram shows a summary of our results. Following activation, HER2 receptors can homodimerize/hetrodimerize with members of its family (HER2, HER1, HER3, or HER4) to propagate signals via downstream effector molecules. The current study suggests that a HER2-GRB7-SHC-RAS signaling axis is responsible for proliferation of HER2-overexpressing breast cancer cells. This study also demonstrates that a α4b1/α5b1- FAK- GRB7-VAV2- RAC1 signaling axis is responsible for HER2+ breast cancer cell migration. Both are crucial steps for tumorigenesis.
Materials and methods
Antibodies and reagents
GRB7 inhibitor peptide conjugated with penetratin (G718NATE-penetratin) and control (only penetratin) were gifts from Dr. Stephanie Pero (University of Vermont School of Medicine, Burlington, VT). GRB7 siRNA, control siRNA and Lipofectamine 2000 were bought from Invitrogen Life Tech. (Carlsbad, CA). Rabbit polyclonal antibody against GRB7 and FAK were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA) and used for immunoprecipitation and Western blot analysis. Mouse monoclonal antibodies against RAS, RAC1, phospho FAK (Y397), phosphotyrosine (4G10) and polyclonal antibody for SHC were procured from Upstate Biotechnology/Millipore (Temecula, CA). HER2 antibody was bought from Cell Signaling Tech. Inc. (Danvers, MA). HRP-tagged anti-rabbit IgG, anti-mouse IgG and Chemiluminescence Kit were from Amersham Pharmacia Biotech (Uppsala, Sweden). PAK-1 PBD (for RAC1 assay reagent) and RAF-1RBD (for RAS assay reagent), agarose for pulldown of activated RAC1 and RAS were from Upstate Biotechnology/Millipore (Temecula, CA). Recombinant human heregulin-β1 and fibronectin were purchased from Peprotech Inc. (Rocky Hill, NJ) and Becton Dickinson (Bedford, MA) respectively. All other chemicals were purchased from Sigma (St. Louis, MO) unless otherwise stated.
Cell culture
BT474 and SKBR3 human breast cancer cells were obtained from the American Type Culture Collection (ATCC) and BT474/HR, a trastuzumab resistant derivative obtained by serial passage in the presence of increasing concentrations of trastuzumab (100 μg/ml final maintenance dose), was kindly provided by Dr. Mark Pegram (Division of Hematology/Oncology, Department of Medicine, UCLA School of Medicine, Los Angeles, CA). HER2 overexpressed BT474 and trastuzumab-resistant BT474HR breast cancer cells were cultured in DMEM supplemented with 10% fetal bovine serum, 1% HEPES (Cellgro, Hemdon, VA) with 100 units/ml penicillin and streptomycin (Cellgro, Hemdon, VA) at 37°C in a humidified atmosphere containing 5% CO2. Trastuzumab-resistant cells were maintained with 100 μg/ml of trastuzumab (Genentech Incorporation, San Francisco, CA). HER2 overexpressing SKBR3 cells were cultured in McCoys 5A (Cellgro, Hemdon, VA) supplemented with 10% fetal bovine serum and 100 units/ml penicillin and streptomycin.
RNA isolation and DASL assay
Breast cancer subtypes were differentiated into the HER2, Luminal, and TN (Triple Negative) subtypes by pathology IHC reports of ER- PR-HER2 3+, ER+ and/or PR+, and ER- PR- HER2-, respectively. Formalin-Fixed, Paraffin-Embedded (FFPE) samples were acquired from St. Mary’s Hospital, Montreal Quebec, Canada (Quebec cohort) under Emory IRB protocol # 00006061. Tissue specimens were obtained in three 5 μm sections and analyzed to contain more than 50% tumor by a board certified pathologist. RNA was isolated from FFPE tissues. Tissues were deparaffinized, extracted and purified using commercially available RNA High Pure Kit (Roche, Mannheim, Germany). RNA extraction was conducted according to previously published methods and hybridized to the Illumina standard cancer DASL and the custom Breast Cancer DASL panels [8,20]. RNA concentration and A°260/A°280 ratio were determined using a NanoDrop spectrophotometer. Samples with good quantity (> 0.4 μg) and quality (Ct<29.5) were subjected to the DASL assay which is essentially a multiplexed quantitative RT-PCR hybridized to sentri array matrices, an 8X12 plate microarray according to manufacturer’s protocol (Illumina, San Diego, CA) [7]. In all, 97 patients were expression profiled, including 81 profiled on both the Illumina standard cancer and the custom breast cancer DASL panels. Expression intensities were interpreted in BeadStudio. Differential expression of probes were determined using Significance Analysis of Microarrays (SAM)2 and p-values were calculated using 2 sided Student’s t-tests for unequal variances. Heatmaps were generated using Bioconductor (http://www.bioconductor.org/) in R (http://www.r-project.org/) for which probe level data were z-score normalized.
In vivo knockdown of GRB7 by human specific GRB7 siRNA
GRB7 was knocked down in the HER2 overexpressing breast cancer cell lines (BT474, SKBR3) by transient transfection of GRB7 siRNA using Lipofectamine 2000 following the manufacturer’s instructions. siRNA oligonucleotides were as follows: GRB7 siRNA 5’CGAGUCCAACGUGUACGUGTT3’, 5’CACGUACACGUUGGACUCGTT3’, Control siRNA 5’UUCUCCGAACGUGUCACGUTT3’, 5’ACGUGACACGUUCGGAGAATT3’.
Cell proliferation assay: crystal violet and WST-1
Interference with in vitro growth rate of BT474 cells by GRB7 siRNA was measured using crystal violet or WST assay kits (Roche Diagnostics, Indianapolis, IN). 3X105 BT474 cells were plated in a six-well plate. Twenty-four hours after plating (0 hrs) cells were transfected with control or GRB7 siRNA (40 nM) using Lipofectamine 2000. At different time points (24 hrs, 48 hrs or 72 hrs) cells were fixed with 4% formalin for 10 minutes, then washed twice with distilled water and stained with 0.1% freshly prepared crystal violet for 30 minutes. After washing, the stain was dissolved with 10% acetic acid and subsequently quantified by absorbance at 570 nM [21]. For WST-1-based cell viability assays, 1X104 BT474 cells were seeded in 96 well plates and 24 hrs later, cells were transfected with GRB7 siRNA as described above. The WST-1 colorimetric assays were carried out according to manufacturer’s instructions and were quantified by absorbance at 490 nM at 24, 48 and 72 hrs following the siRNA transfections.
3D ON-TOP Colony assay
This assay was carried out to examine the effect of GRB7 inhibitor peptide on clonogenic growth of HER2+ breast cancer cells. The assay was standardized with little modification from that originally described by Lee et al. [22].
Cell migration assays
Transwell assay
A migration assay was performed toward fibronectin (haptotaxis) using a transwell with polycarbonate membrane from Costar (diameter 6.5 mm, pore size 8μm; Costar Corp., Cambridge, MA). The underside of the membrane to which cells migrate was coated with 20 μg/ml fibronectin in PBS for 1 h. at 37°C. Surfaces were subsequently blocked with heat-denatured BSA. Cells were added at the concentration of 2X105 cells/well (in 100 μl of media containing 0.1% serum) into the upper chamber of the well with or without 10 ng/ml of heregulin and incubated for 4 hours. For negative controls, the wells were coated on both the sides with fibronectin with or without heregulin. The total numbers of cells on the fibronectin-coated side were quantified as described previously [23-25]. Cell adhesion assays were also performed. Briefly, a flat bottom 96-well polystyrene plate (BD Biosciences) was coated with 20 μg/ml of fibronectin in PBS for 1 h. at 37°C. Wells were washed once with PBS, incubated with 20 mg/ml BSA for 1 h. at 37°C for blocking non-specific sites, and again washed twice with PBS. To examine the cell adhesion to the coated surface, 1X105 cells were added to each well and incubated at 37°C for 4 hours. At the end of the incubation, medium and unbound cells were removed by aspiration, and wells were washed carefully with PBS. Adherent cells were fixed with 3.5% formaldehyde and stained with 0.1% crystal violet. The stain was eluted with 10% acetic acid, and absorbance was determined at 600 nm with a microplate reader [23,26].
Scratch assay
Cells were cultured on 12 well plates coated with 20 μg/ml of fibronectin. Scratches were created in a line across the plates by scraping with a 200 μl standard pipette tip. The scratched monolayers were then washed twice with serum free media to remove all cell debris and incubated with media containing 0.1% serum and 10 ng/ml of heregulin. Photomicrographs were taken at 0 hour and 24 hours. Quantitative analysis of the scratch was determined by measuring the scratch area covered by the migrated cells. Less covered area means less migrated cells [27].
Real-time imaging of live cells
Time-lapse images were acquired with a Perkin Elmer Ultraview ERS confocal system. Bright-field images were acquired with a Hamamatsu Orca-ER camera (10x objective) at 10 minute intervals.
Immunofluorescence studies
To test the colocalization of HER2 and GRB7 in BT474 cells, the cells were seeded onto glass coverslips in 10-cm petri dishes and allowed to attach in culture medium containing 10% FBS as mentioned earlier [28]. Staining was carried out using anti-HER2 (1:50) and anti-GRB7 (1:50) antibodies. Nuclei were counterstained with DAPI. Cells were imaged using a Zeiss (Thornwood, NY) LSM 510 Meta confocal microscope with a 63x (1.4-numerical-aperture) or 100x (1.4-numerical-aperture) Plan-Apochromat oil objective. All images were acquired using Zeiss LSM 510 software and processed using Adobe Photoshop 7.0.
Actin dynamics
BT474 cells were seeded on fibronectin coated cover slips in 6 well plates. Cells were treated with 10 μM G718NATE-penetratin or control for 1 hour and were then processed for Phalloidin 555 staining. Nuclei were stained with DAPI. Stained cells were photomicrographed for actin polymerization using confocal microscopy. Cells were imaged using a Zeiss (Thornwood, NY) LSM 510 Meta confocal microscope with a 63x (1.4-numerical-aperture) or 100x (1.4-numerical-aperture) Plan-Apochromat oil objective. All images were acquired using Zeiss LSM 510 software and processed using Adobe Photoshop 7.0 as described elsewhere [28].
Biochemical analysis
Immunoprecipitation and western blots
Immunoprecipitations were designed to preserve noncovalent protein-protein interactions. Serum starved cells (5X106) in log phase were stimulated with 10 ng/ml heregulin for appropriate times indicated in the individual experiments at 37°C or were plated on a 20 μg/ml fibronectin-coated plate and incubated for 30 minutes at 37°C. At the end of the stimulation time, the medium was removed and solubilized with 500 μl of Triton X containing lysis buffer (1% Triton X 100, 10 mM Tris HCl, pH 7.6, 5 mM EDTA, 50 mM NaCl, 50 mM NaF, 0.1% BSA, 1% aprotinin, 0.2 M sodium orthovanadate, and 0.1 M phenylarsineoxide). For immunoprecipitation of GRB7, clarified lysates were assayed for total protein (Bio-Rad protein assay kit) using BSA as standard. The clear lysates were immunoprecipitated by rabbit polyclonal GRB7 antibody (1 μg/sample) after protein equilibration. Immunoprecipitates were bound to pansorbin, collected by centrifugation, washed in lysis buffer, and resolved in 4-20% gradient or 10% SDS-PAGE. Individual bands were visualized by chemiluminescence reagent ECL.
For Western blots, lysates were prepared and protein was estimated with a Bio-Rad protein assay kit using BSA as a standard. Protein lysates were resolved by SDS-PAGE. Blots were probed with specific antibodies and developed with the ECL method. Beta actin was assayed for the loading control.
GST-fusion protein pull-down assay
The GST-fusion proteins, corresponding to the human RAS binding domain (RBD residues 1-149) of RAF1, or the human PAK1 p21 binding domain (PBD, residues 67-150) were expressed in E. coli. The final protein products were bound to glutathione agarose in liquid suspension, 100 μg of RAF1-RBD in 333 μl of 50% agarose slurry or 300 μg of PAK1-PBD in 20 mM PBS, pH7.4, containing 50% glycerol. The 70-80% confluent cells were kept in serum starvation for 18 hours before heregulin (10 ng/ml) stimulation or integrin engagement in the presence of heregulin (fibronectin-coated plates) for different time periods. Stimulated cells were lysed with extraction buffer (25 mM HEPES pH 7.5, 150 mM NaCl, 1% Igepol CA630, 10 MgCl2, 1 mM EDTA, 10% glycerol, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 1 mM NaF, and 1 mM Na-orthovanadate). Following centrifugation, 10 μl RAF1 RBD/PAK1 PBD (1 μg/ml) was added per lysate sample for 45 minutes at 4°C with gentle rocking. Agarose beads were collected (by pulse centrifugation for 10 seconds at 14,000 rpm), washed in extraction buffer, and resuspended in 30 μl Laemmli sample buffer to resolve protein by 15% SDS-PAGE. The membrane was probed with anti-RAS or anti-RAC1 monoclonal antibody, respectively [29].
Statistical analysis
Results were analyzed using a two-tailed Student’s t test to assess statistical significance. Values of p < 0.05 were considered statistically significant.
Results
GRB7 mRNA and protein overexpression in primary breast tumors and breast cancer cell lines
In an expression study of 97 breast cancer patients, including 9 of the HER2 subtype, we observed concordance between mRNA signal intensities and breast cancer subtype defined by pathology IHC reports (Figure 1A). Tumor expression profiles of patients with HER2+ breast cancer demonstrated upregulation of ERBB2, the mRNA transcript coding for the HER2 protein, GRB7, MED24/THRAP4, TDGF1, MED1/PPARBP, PERLD1, WNT2. Additionally, we noted downregulation of SMAD4, CDKN1B (which codes for p27), IGF1R, MXI1, VHL, and RBBP2 in the HER2 subtype. Analysis of ERBB2 synexpression indicated 17q12-q21 genes most notably GRB7, THRAP4, and MED1/PPARBP co-amplified with HER2/ERBB2 (Figure 1B). mRNA expression levels of a Montreal cohort of breast cancer patients indicated amplification of both ERBB2 and GRB7 in the HER2 breast cancer subtype (Figure 1C). Meta-Analysis of a Stockholm cohort of 159 patients confirmed co-amplification of ERBB2, GRB7, MED24, and PERLD1 in the HER2+ breast cancer subtype [30]. Similar to Giricz and group’s observation [31], our data also show (in Illumina Standard DASL panel) that the expression of GRB7 mRNA in triple negative and luminal subtypes (Figure 1C). Western blot analysis of breast cancer cell lines confirmed that GRB7 overexpression was present in breast cancer cell lines that had HER2/Neu protein overexpression and were known to carry Her2/Neu gene amplification. No GRB7 overexpression was observed in breast cancer cell lines without HER2/Neu protein overexpression or Her2/Neu gene amplification (Figure 1D). Earlier studies showed the co-amplification/co-overexpression of HER2 and GRB7in breast cancer cells [9,32,33]. Our mRNA and protein expression studies demonstrated concordant GRB7 overexpression in breast cancer tissues/breast cancer cell lines with HER2/Neu overexpression, consistent with results from earlier reports.
Figure 1.

GRB7 is co-overexpressed along with HER2 in the HER2 overexpressed breast cancer subset: A. In an expression study of 97 breast cancer patients, including 9 of the HER2 subtype, we observed concordance between mRNA signal intensities and breast cancer subtype defined by pathology IHC reports. Tumor expression profiles of patients with HER2 breast cancer demonstrated upregulation of ERBB2, GRB7, MED24/THRAP4, etc. B. Probes differentially regulated in HER2 breast cancers in 81 of 97 patients profiled on both the Illumina standard cancer and custom breast cancer DASL panels. C. mRNA expression levels of a Montreal cohort of breast cancer patients indicated amplification of both ERBB2 and GRB7 in the HER2 breast cancer subtype. * and ** indicate p-values less than 0.01 and 0.001, respectively, of upregulation of ERBB2 or GRB7 as compared to Triple Negative (TN) or luminal breast cancer subtypes. D. To determine whether GRB7 was co-overexpressed with HER2 in HER2-overexpressing breast cancer cell lines, we performed immunoblot analysis using GRB7 specific antibody. Upper and middle panels show the expression levels of HER2 and GRB7 respectively. A b-actin Western blot was used as a loading control.
GRB7 associates with HER2 and SHC in HER2 overexpressing breast cancer cell lines
GRB7 proteins are known to act as adapter for tyrosine kinase receptor-mediated signaling. Therefore, we examined the interaction of GRB7 with HER2 and other cytosolic adapter following receptor phosphorylation. We also examined whether trastuzumab has any effect on this association. To carry out these experiments, GRB7 was immunoprecipitated from serum starved BT474, BT474HR (trastuzumab-resistant), and SKBR3 cell lines following heregulin stimulation in trastuzumab (humanized mAb for HER2 and first line therapy for HER2 overexpressing breast cancer patients) treated and non-treated conditions. Immunoprecipitation with anti-GRB7, followed by probing with phosphotyrosine-specific antibodies, was able to show heregulin stimulated association of tyrosine phosphorylated-HER2 and tyrosine phosphorylated-SHC with GRB7 (Figure 2A). Data show that basal (NS) tyrosine-phosphorylated HER2 was higher in the trastuzumab-resistant HER2+ cell line (BT474HR) than in parental HER2+ cell lines (BT474 and SKBR3). It was also noted that a small percentage of HER2 or SHC is tyrosine-phosphorylated in these unstimulated cells. This co-immunoprecipitation was not seen with pre-immune serum (data not shown). No other bands were found in the anti-phosphotyrosine blot, suggesting that GRB7 was not phosphorylated at tyrosine following heregulin stimulation and this association (HER2-GRB7-SHC) was not dependent on the phosphorylation of GRB7. Furthermore, unlike tyrosine kinase inhibitors (lapatinib or ZD1839) [34,35] trastuzumab has no effect either on phosphorylation of HER2 or association of HER2 with GRB7 following heregulin stimulation. Earlier it has been reported by other that trastuzumab (Herceptin) has no effect on the phosphorylation pattern of HER2 [35]. Our co-immunoflorescence data further confirmed that HER2 and GRB7 are in binding proximity (Figure 2B). These data suggest that the co-amplification of these two genes up-regulates/activates the HER2-signaling pathway.
Figure 2.

GRB7 associates with HER2 and SHC in HER2 overexpressing breast cancer cell lines: A. Tyrosine phosphorylation of HER2, SHC and their association with GRB7 were examined in HER2 overexprssed BT474, SKBR3 and BT474HR (trastuzumab-resistant) cell lines. Anti-GRB7 precipitation was performed on 5x105 cells after heregulin (10 ng/ml) stimulation at 37°C for indicated time points. Lane 1, 5 & 9 represent resting non-stimulated cells (NS), lane 2, 6 & 10 represent cells stimulated with heregulin for 5 minutes, lane 3, 7 & 11 represent cells stimulated with heregulin for 15 minutes and lane 4, 8 & 12 represent cells pretreated with trastuzumab (10 μg/ml) for 30 minutes followed by heregulin stimulation for 15 minutes. The data demonstrate that HER2 is significantly phosphorylated (on tyrosine) in a kinetic manner after HER2 stimulation, constitutively maintaining a stable complex with GRB7 and recruiting tyrosine phosphorylated SHC (KD 46 and 52) into the HER2-GRB7 complex on HER2 stimulation. Pretreatment with trastuzumab neither has any effect on HER2 phosphorylation nor on its association with GRB7 in all three cell lines. B. Subcellular co-immunolocalization of endogenous GRB7 with HER2 in the BT474 cells. Fixed BT474 cells were processed for confocal imaging as mentioned in the Materials and Methods. Photomicrographs show the subcellular distribution of GRB7 in red (A), HER2 in green (B), nuclei in blue (C) and their merged confocal images in yellow (D), respectively. Scale bar, 50 μm. Merged images (inserts) of higher magnification (Scale bar, 10 μm) show a clear change in color, indicating the co-localization (arrows) of these two proteins in the cells.
Suppression of GRB7 protein in BT474 cells by human GRB7 specific siRNA
To address if GRB7 is another possible (along with HER2) therapeutic target for HER2+ breast cancer, we used small interfering RNA (siRNA) to suppress GRB7 expression. The siRNA was transfected with Lipofectamine 2000 (according to the manufacturer’s instructions) into GRB7 overexpressing BT474 or SKBR3 cell lines. Protein lysates were prepared at 24, 48, and 72 hrs after transfection and analyzed by Western blot. Data show that GRB7 expression was suppressed at 48 and 72 hrs and maximum suppression was detected at 72 hrs (Figure 3). Transfection with a non-targeting control siRNA served as a negative control. No effect of GRB7 siRNA was found on GRB2 or other GRB-family protein levels (data not shown).
Figure 3.

Western blot analysis of GRB7 in BT474 cells using RNAi technology: Transfection with siRNA oligonucleotides at a concentration of 40 nM by Lipofectamine 2000 reagent targeting GRB7 mRNA or control. Each cell lysate was prepared at 24, 48, and 72 hrs after transfection. GRB7 expression levels were markedly suppressed at 48 and 72 hrs. Bar diagram shows densitometry scanning analysis (upper panel). A b-actin Western blot was used as a loading control (lower panel).
Knockdown of GRB7 expression in BT474 cells attenuates HER2+ cell proliferation
Both HER2 and GRB7 proteins are overexpressed in BT474 cells (Figure 1D). Knockdown of GRB7 expression was achieved with siRNA transfection. To access the role of GRB7 in proliferation, we examined the cellular proliferation and viability by both crystal violet staining and WST-1 assay following the transfection of GRB7-siRNA in HER2 overexpressing BT474 cells. Data show that proliferation or viability was significantly less in GRB7-siRNA transfected cells compared with control transfected cells at 48 and 72 hours (Figure 4A). Consistent with the proliferation assay, GRB7 inhibitor peptide (G718NATE-penetratin) significantly blocked clonogenic growth of HER2+ BT474 cells on matrigel (3D-assay) and 2D-assay (Figure 4B). Consistent with inhibition of proliferation with GRB7-siRNA, PCNA (proliferating cell nuclear antigen) protein expression was also decreased at 48 and 72 hours time points (Figure 4C) following GRB7-siRNA transfection.
Figure 4.

Proliferation assay after transfection with GRB7 siRNA: A. The growth of BT474 cells transfected with GRB7 siRNA and control siRNA were assessed by crystal violet (i) and WST-1 assays (ii) at different time points (24, 48 and 72 hrs.). 0 Hrs, at the time of transfection, *p<0.005, **p<0.001, compared with control siRNA. B(i). Effect of GRB7 inhibitor peptide (G178NATE-penetratin) on the time course of clonogenic growth of BT474 cells (3D ON-TOP assay). Cells (treated with 10 or 20 μM concentration of GRB7-inhibitor peptide) were plated on growth factor reduced matrigel and colony formation was recorded (Olympus IX71, CellSens, DP72; 10X) after 4 days (96 hours) and 7 days. Data show that GRB7-inhibitor peptide significantly inhibited dose- and time-dependent clonogenic growth of BT474 cells as compared to the control. B(ii). Similar to 3D-ON TOP clonogenic growth of HER2+(BT474) tumor cells, 2D-clonogenic growth was blocked following the treatment with inhibitor peptide. Photomicographs show colony formation of breast tumor cells following treatment using phase contrast microscopy. C. GRB7 siRNA effected a marked reduction of proliferating cells as demonstrated by decreased expression of PCNA at 48 and 72 hrs. From these data, we suggest that GRB7 is required for HER2 overexpressing breast cancer cell proliferation.
Role of GRB7 on the activation of RAS-GTP
To gain insight into the mechanism whereby GRB7 is controlling HER2+ breast cancer cell proliferation, we considered potential downstream effectors of GRB7. Literature suggests that GRB7 has a RAS-binding domain [36] and RAS is one the most important downstream small GTPases of growth factor receptors which controls cell proliferation and survival [37,38]. Considering the wealth of literature indicating the role of RAS-GTP in growth factor induced cell proliferation, we have studied the effect of GRB7 on RAS activation following heregulin stimulation in BT474 cell lines. Heregulin treatment caused a time-dependent increase in the RAS activation in BT474 and BT474HR cells (Figure 5A) and activation was more pronounced in trastuzumab-resistant cell line (BT474-HR) (Figure 5A, comparing lane 3 versus lane 6), this may be due to higher activation of RAS-GTP at basal conditions. This activation was obliterated in BT474 cells either treated with GRB7 inhibitor peptide, G718NATE-penetratin (Figure 5B) or transfected with GRB7-siRNA (Figure 5C [ii]). Interestingly GRB7 inhibitor peptide (G718NATE-penetratin) has no effect in the trastuzumab-resistant cell line (Figure 5B, comparing lane 5 and 6).
Figure 5.

Effect of GRB7 on the downstream effectors of heregulin stimulation in HER2 overexpressing breast cancer cell lines: A. Heregulin-induced activation of RAS. BT474 and trastuzumab-resistant BT474HR cells were treated with 10 ng/ml of heregulin. At different time points (2 and 5 min), lysates were evaluated using a pull-down assay for detection of activated GTP-bound RAS (top panel). Immunoblot of total RAS (bottom panel) was carried out on lysates as loading control. NS, no stimulation. These data demonstrate that RAS activation (GTP-RAS) is significantly elevated following heregulin stimulation at 5 minutes in both the cell lines (lane 3 and 6) and activation of RAS is higher in the resistant cell line (lane 3) compared to parental line (lane 6). B. Effect of GRB7 inhibitor peptide (G178NATE-penetratin) on heregulin-induced RAS activation. HER2-overexpressing, BT474 and trastuzumab-resistant BT474HR cells were pretreated with GRB7 inhibitor peptide (G178NATE-penetratin, lanes 3 & 6) or control peptide (penetratin alone, lanes 2 & 5) at 10 μM for 1 hr followed by heregulin (10 ng/ml) stimulation for 5 minutes at 37°C. Data show that heregulin-induced RAS activation is blocked by GRB7 inhibitor peptide (GG178NATE) only in parental cells (lane 3) but not in trastuzumab-resistant cell line (lane 6). C. Effect of GRB7 on the activation of RAS in BT474 cells. GRB7 was knocked down in GRB7 overexpressing BT474 cells using siRNA [C (i). upper panel]. Cells were transfected with GRB7 specific siRNA or control siRNA and incubated for 72 hrs as described in Materials and Methods. Activation of RAS following heregulin stimulation of GRB7 siRNA transfected (72 hrs) cells was significantly less compared to control siRNA transfected cells [compare lane 2 and lane 4, C (ii)]. Immunoblot of total RAS (bottom panel) was performed on lysates as loading control. NS, no stimulation (lanes 1 & 3), [C (ii)]. Data suggest that heregulin-induced RAS activation (RAS-GTP) is dependent on GRB7 in HER2-overexpressed breast cancer cells.
GRB7 binds with FAK following integrin engagement
Previous studies have shown that upon cell adhesion to β1 integrin FAK is autophosphorylated and associated with a number of SH2 domain containing molecules, such as SRC, GRB2 and PI3K [39-42]. It has been also reported by others that increased expression of FAK and GRB7 has been found in a number of cancers, which correlate with the invasive potential of tumors [43,44]. Since FAK-induced GRB7 tyrosine phosphorylation has also been shown to be important in the regulation of cell migration [45], we investigated the endogenous association of FAK with GRB7 and its phosphorylation by FAK following adhesion to the extra cellular matrix protein, fibronectin (α4β1/α5β1) in BT474, trastuzumab-resistant BT474 and SKBR3 cells. Analysis of the immune complex (following integrin [α4β1/α5β1] engagement and immunoprecipitation with GRB7 antibody) by Western blotting with anti-phospho FAK antibody (Y397) showed that FAK was phosphorylated (Figure 6 upper panel) and bound with GRB7, leading to GRB7 phosphorylation (Figure 6, panel 3 from top). These data may suggest that tyrosine phosphorylation of GRB7 following integrin activation is a FAK dependent phenomenon.
Figure 6.

Association of GRB7 and FAK in HER2 overexpressing breast cancer cell lines following integrin engagement: Co-immunoprecipitation of focal adhesion kinase (FAK) and GRB7 following fibronectin (α4β1/ α5β1) stimulation. BT474HR, BT474 and SKBR3 cells were placed on fibronectin-coated plates. Cell lysates were collected at the times indicated (15 and 30 min.) and immunoprecipitated (IP) by polyclonal anti GRB7 antibody (from Santa Cruz). The immune complexes were analyzed by Western blotting (WB) with phospho-FAK (Y397), total FAK, phosphotyrosine antibody (4G10 from Upstate Biotechnology, for the detection of tyrosine phosphorylated GRB7) and total GRB7.The phosphorylated GRB7 and FAK, total GRB7, and total FAK are marked on the right. NS, no stimulation (lanes 1, 4 & 7). From these data we suggest that tyrosine phosphorylation of GRB7 corresponds to FAK’s tyrosine phosphorylation (Y397, the autophosphorylation site of FAK) in response to integrin activation (lanes 2, 3, 5, 6, 8 & 9). These co-immunoprecipitation data provide evidence that GRB7 tyrosine phosphorylation takes place within the integrin (α4b1/ α5b1)/FAK-mediated signaling pathway.
GRB7 controls heregulin-induced HER2+ breast cancer cell migration on fibronectin
One of the major threats for breast cancer death is metastasis, which account for >90% of breast cancer deaths [46]. Integrin-mediated cell migration is one of the essential steps for metastasis. Evidence suggests a role for GRB7 is in cell migration [27,47]. Here, we examined the role of GRB7 on heregulin-induced, integrin-directed migration of HER2+ breast cancer cells. Treating the cells with heregulin significantly increased the migration of HER2+ cells on fibronectin (Figure 7A). To delineate the involvement of GRB7 in this process, either GRB7 inhibitor peptide (GIP, G718NATE-penetratin) or GRB7-siRNA were tested for heregulin-stimulated, integrin-directed migration on fibronectin. Treatment with GRB7 inhibitor peptide (Figure 7A) or GRB7-siRNA transfection (Figure 7C) significantly blocked haptotaxis in response to heregulin stimulation as compared to heregulin treated control peptide (penetratin alone) or control-siRNA treated BT474 and SKBR3cells (Figure 7A and 7C). Furthermore, inhibition of migration was more pronounced when trastuzumab was used along with GRB7 inhibitor peptide (Figure 7B). However, GRB7 inhibitor peptide (G718NATE-penetratin) has very little effect on heregulin-induced haptotaxis of trastuzumab-resistant cells (BT474HR, Figure 7A). In order to determine whether the change in migration is affected by the capacity of cells to adhere to the substratum (fibronectin), we performed adhesion assays in these cells using similar experimental conditions as those of migration experiments. Adhesion was not affected following the treatment of either GRB7 inhibitor peptide (G718NATE-penetratin) or GRB7-siRNA transfected cells (data not shown). Like the transwell migration assay, the scratch assay (BT474 cells cultured on a 24 well fibronectin coated plate subjected to scratch) also demonstrated that GRB7 reduces cell migration (Figure 8). Finally, we carried out scratch assay on a fibronectin-coated cover slide in a real time manner following treatment with GRB7-inhibitor peptide (Supplementary data video 1 and video 2). Cell motility entails the reorganization of the cytoskeleton and membrane trafficking for effective protrusion. Therefore, we next examined the effect of the GRB7 inhibitor peptide on the organization of actin filaments on fibronectin coated cover slips. Confocal microscopy imaging showed that treatment with GRB7 inhibitor peptide led to alterations in the organization of filamentous actin in BT474 cells (compare between Figure 9A and 9B). From these data, we suggest that GRB7 exerts a positive effect on co-signaling events transmitted through the integrin (α4β1/ α5β1)-HER2/HER3 signaling axis required for migration.
Figure 7.

Effect of GRB7 on heregulin induced, integrin-directed migration in HER2 overexpressing breast cancer cell lines: Transwells with polycarbonate membranes were coated with 20 μg/ml fibronectin for 1 h at 37°C. Cells, 2X105, were added to the upper chamber of the well containing 10 ng/ml of heregulin. Cells which have migrated to the lower membrane were photomicrographed as well as quantified by crystal violet staining. Migration was quantified by counting the migrated cells from 10-12 randomly selected fields. Bars represent mean ± SD representative of 3 -4 independent experiments. A. GRB7 inhibitor peptide (G178NATE-Penetratin) at 25 and 50 μM concentrations significantly inhibited trastuzumab-sensitive (BT474) breast cancer cell migration on fibronectin (p<0.05) but not trastuzumab-resistant cells (BT474 HR). Representative pictures of migration are presented in the side panels. B. Combination of GRB7 inhibitor peptide (25 μm) and trastuzumab (Herceptin) (10 μg/ml) demonstrated marked inhibition of migration over the single agents (either GRB7 inhibitor peptide or trastuzumab alone) in both BT474 (i) and SKBR3 (ii) cells when compared with control group (heregulin treatment alone or compared double treated combination with either Herceptin or GIP treated group). Presence of heregulin (10ng/ml) is required for integrin-directed HER2 overexpressed breast cancer cell migration (see Figure B (i) compare first and second bars in the diagram). C. The dependence of integrin-directed migration of HER2 overexpressing breast cancer cells on GRB7 was further evaluated using GRB7 specific siRNA. HER2 overexpressing breast cancer cells were transiently transfected with control siRNA and GRB7 specific siRNA (from Invitrogen) at a concentration of 20 nM by Lipofectamine 2000 reagent. The cells were harvested and analyzed after 72 hrs. Cell lysates were subjected to immunoblotting with anti GRB7 and b-actin antibodies (see insert). Migration on fibronectin (α4β1/α5β1) of GRB7 siRNA transfected HER2 overexpressing breast cancer cells (BT474 and SKBR3) was significantly less compared to control (p<0.00007 for BT474 and p<0.0003 for SKBR3). From these data we suggest that crosstalk between receptor tyrosine kinase, HER2, integrin (α4β1/ α5β1) and cytosolic adapter protein GRB7 is necessary for integrin-directed HER2 overexpressing cell migration.
Figure 8.
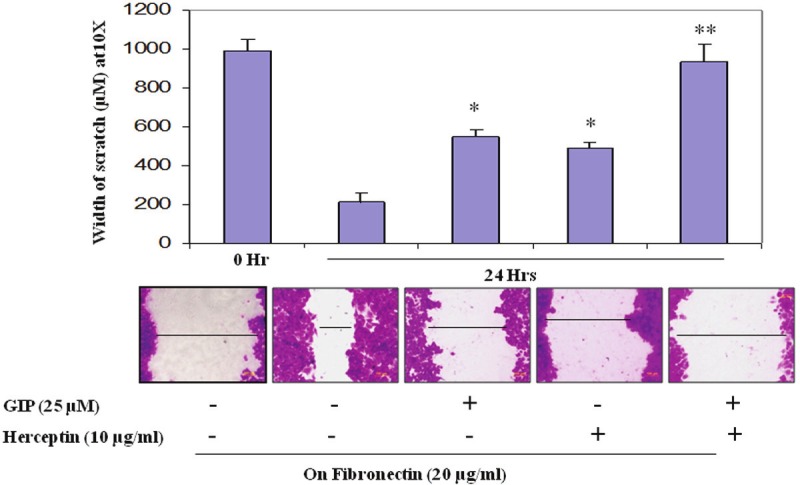
Effects of the combination of GRB7 inhibitor peptide and trastuzumab on fibronectin-mediated BT474 cell migration (in vitro scratch assay): HER2 overexpressing breast cancer cells cultured on a 24 well fibronectin coated plate where subjected to scratch (wound treatment) and then incubated at 37°C, 5% CO2 for 24 hrs in presence of either GRB7 inhibitor peptide (25 μM) or trastuzumab (10 μg/ml) or a combination of both GRB7 inhibitor peptide (25 μM) plus trastuzumab (10 μg/ml). To analyze BT474 cell migration into the scratch area, 0.1% crystal violet stained cells were photomicrographed and the scratch area covered by the migrated cells was measured. Less covered area means less migrated cells. Similar results were obtained in three independent experiments. The scratch assay demonstrated results similar to the transwell assay.
Figure 9.

A and B Gallery of Z sections (1micron) of BT474 cells showing change in the organization of filamentous-actin following the treatment of G178NATE-Penetratin.
GRB7 inhibitor peptide or GRB7-siRNA transfection blocks integrin-induced activation of RAC1
Rac GTPases, small G-proteins widely implicated in metastasis, transduce signals from integrins, receptor tyrosine-kinase, and, G-protein-coupled receptors (GPCRs), and control a number of essential cellular functions including motility. Consistent with the known role for Rho family small GTPases in cell migration, fibronectin engagement caused a time-dependent increase in RAC1 activation in BT474 and BT474HR cells in presence of heregulin (10 ng/ml) (Figure 10A). In absence of heregulin (only on fobronectin-coated plate) RAC1 activation was significantly less in HER2+ breast tumor cells (data not shown). Since RAC1 activation was pronounced at 30 minutes following fibronectin engagement, the effect of GRB7 inhibitor peptide (G718NATE-penetratin) was evaluated at 30 minutes as compared to the non-treated cells after fibronectin-attachment. Figure 10B shows that fibronectin-dependent RAC1 activation is significantly attenuated in BT474 cells but not in trastuzumab-resistant cells (similar to fibronectin-induced migration of BT474HR cells). Similarly, RAC1 activation was abrogated in BT474 and SKBR3 cells transfected with GRB7-siRNA (Figure 10C). From these data we conclude that treatment of cells with GRB7 inhibitor peptide or GRB7-siRNA abrogates fibronectin-mediated RAC1 activation in trastuzumab-sensitive HER2+ breast cancer cells. These results are directly correlated with the capacity of GRB7 to control heregulin-induced HER2+ breast cancer cell migration on fibronectin. Additionally, we tried to understand how GRB7 activates RAC1-GTPase following integrin engagement which is required for HER2+ cell movement. To answer this question, we did a co-immunoprecipitation experiment with GRB7 antibody following fibronectin engagement in BT474 cells. The results demonstrate, for the first time, that GRB7 binds to VAV2 (an exchange factor for RAC1) in a ligand-dependent manner (Figure 10D).
Figure 10.
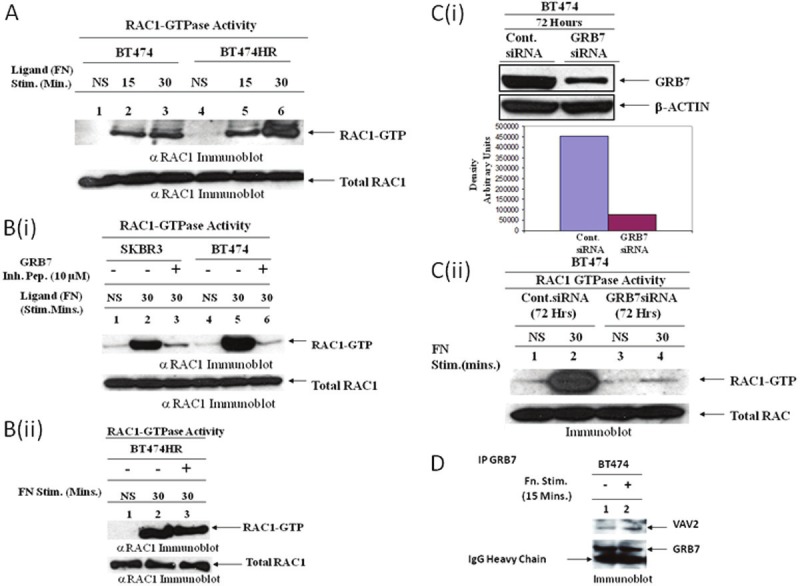
Effect of GRB7 on the downstream effectors of integrin stimulation in HER2 overexpressed breast cancer cell lines: A. Integrin (α4β1/α5β1)-induced activation of RAC1. BT474 overexpressing HER2 and trastuzumab-resistant BT474HR cells were plated on fibronectin (20 μg/ml)-coated plates. At different time points (15 and 30 minutes), lysates were evaluated using a pull-down assay for detection of activated GTP-bound RAC1 (top panel). Immunoblot of total RAC-1 (bottom panel) was done on lysates as a loading control. NS, no stimulation. Data demonstrate that RAC1 activation (GTP-RAC1) is significantly higher following integrin engagement at 30 minutes in both cell lines (lane 3 and 6) and activation of RAC1 is substantially higher in the resistant cell line (lane 6) compared to the sensitive cell line (lane 3). B (i). Effect of GRB7 inhibitor peptide (G178NATE-penetratin) on integrin-induced RAC1 activation. HER2-overexpressing SKBR3 and BT474 cells were pretreated with GRB7 inhibitor peptide (G178NATE-penetratin, lanes 3 & 6) or control peptide (penetratin alone, lanes 2 & 5) at 10 μM for 1 hr followed by plating on fibronectin-coated plates (20 μg/ml) for 30 minutes at 37°C (ii) A similar experiment was carried out in trastuzumab-resistant BT474HR cells. Data show that integrin-induced RAC1 activation was blocked by GRB7 inhibitor peptide (G178NATE) in sensitive cells [lanes 3 and 6 in Figure B (i)] but not in the trastuzumab-resistant cell line [lane 3 in Figure B (ii)]. C. Effect of GRB7 on the activation of RAC1 in BT474 cells. GRB7 was knocked down in GRB7 overexpressing BT474 cells using siRNA [C (i), upper panel]. Cells were transfected with GRB7 specific siRNA or control siRNA and incubated for 72 hrs as described in Materials and Methods. Activation of RAC1 following integrin engagement of GRB7 siRNA transfected (72 hrs) cells was substantially less compared to control siRNA transfected cells (compare lanes 2 and lane 4). Immunoblot of total RAC1 (bottom panel) was done on lysates as a loading control. NS, no stimulation (lane 1 & 3), [C (ii)]. Data suggest that integrin-induced RAC1 activation (RAC1-GTP) is dependent on GRB7 in HER2-overexpressed breast cancer cells. D. Co-immunoprecipitation of VAV2 (exchange factor for RAC1-GTPase activation) and GRB7 following fibronectin (α4β1/α5β1) stimulation.
Discussion
It is likely that complex interactions exist between receptor tyrosine kinases, cytosolic adapter proteins and small GTPases to transmit proliferative and migratory signals following growth factor stimulation and integrin engagement. Crosstalk between HER2 and integrins has been reported in various cell types [48,49]. In this report, our gene expression data sets (from FFPE samples) have uncovered HER2+ breast cancer “pathways” which ultimately could become targets for new pathway-specific drugs. We set out to determine if the co-amplification of GRB7 and HER2 has any impact on HER2+ breast cancer progression. Our results demonstrated that: 1) GRB7 co-immunoprecipitaes with receptor tyrosine kinase, HER2, and non-receptor tyrosine kinase, FAK, following heregulin stimulation and integrin engagement respectively, 2) GRB7 controls HER2+ breast cancer cell proliferation, most likely through downstream activation of RAS-GTP, and 3) GRB7 also controls heregulin-induced HER2+ cell migration on fibronectin, probably through downstream activation of RAC1-GTP. These findings imply that the presence of GRB7 is necessary for HER2 to drive breast tumor phenotypes, i.e. proliferation and integrin-directed migration.
HER2/Neu overexpressing breast cancer is characterized by poor survival due to a high proliferative and metastatic rate, and identifying downstream targets of HER2 should facilitate novel therapies for this disease. Consistent with published literature [50,51], our expression data, using the Illumina DASL platform, showed co-amplification of Grb7 along with Her2 at chromosome 17q12-21 (mRNA correlation, r2=0.83). Concurrent with mRNA data, immunoblot analyses revealed high expression of GRB7 protein in HER2 overexpressing breast cancer cell lines (Figure 1). Although, the literature indicates that GRB7 plays a definitive role in HER2+ breast cancer [12,52], the exact cellular mechanism of action of GRB7 in the HER2+ tumor cells has not been systematically analyzed to date. Interestingly, GRB7 is included on Oncotype DX® (Genomic Health, Redwood City, California), a commercially available multigene molecular assay which can provide individualized risk estimates for patients with breast cancer, based on the expression levels of 16 cancer-related genes in reference to 5 invariant genes [53]. The Oncotype DX® test is a diagnostic test that helps identify which women with early-stage, estrogen-receptor-positive and lymph-node-negative breast cancer are more likely to benefit from adding chemotherapy to their hormonal treatment. This test also helps assess the likelihood that an individual woman’s breast cancer will return. The only mechanism of resistance to endocrine therapy for which clinical data exist is overexpression of ErbB2/HER2 protooncogene [54,55]. Here, we hypothesize that HER2 and GRB7 act together (in the same signaling cascade), and it may be possible to control GRB7’s function(s) thus controlling HER2-mediated breast tumor cell phenotypes. Recently Nadler et al have shown that coexpression of HER2 and GRB7 was associated with worse prognosis than high HER2 alone and high GRB7 protein expression was strongly associated with decreased survival in the entire cohort (P= 0.0034) [10]. In the same line, Ramsey and group have also reported that GRB7 protein overexpression is associated with larger tumor size/grade, more lymph node involvement, and inferior breast cancer free survival (57.4% for GRB7 overexpresser versus 78.2% for GRB7 non-overexpresser tumors) (hazard ration: 1.69) [56]. Laboratory based studies and clinical trials revealed that co-amplification of topoisomerase II alpha with Her2/Neu (in chromosome 17q) predicts benefit from adjuvant anthracyclin-based therapy in HER2+ breast cancer [57-60].
GRB7 is not only co-overexpressed with HER2 in breast cancer, but also exists in a tight complex with HER2. In HER2+ (BT474 and SKBR3) and trastuzumab-resistant (BT474HR) breast cancer cells, we co-immunoprecipitated a large amount of tyrosine phosphorylated HER2 with GRB7 antibody following heregulin stimulation (Figure 2). Fiddse et al. demonstrated that GRB7 was co-immunoprecipitated with both HER3 and HER4 following heregulin-mediated receptor activation in HER-293 cells stably transfected with hGBR7/pRcCMV [61]. Here, we also demonstrate that GRB7 is not co-immunoprecipitaed with HER2 alone but with another phosphoprotein following heregulin stimulation at 46 and 52 KD. We confirmed that a co-migrating protein is phospho-SHC by probing with SHC-specific antibody, which suggests that HER2-GRB7 forms a tertiary complex with another cytoslic adapter protein, SHC (Figure 2). It has been also reported by others that BT474 cells expresses p46 and p52 but not p66 of isoforms of SHC [62]. It has also been reported by Stein et al. that an SH2 domain containing protein forms a tight complex with HER2 in breast cancer cells and SHC is also recruited into this HER2-GRB7 complex [51]. It is known that GRB2 is the SHC binding partner in many cells including breast cancer cells. However, in HER2 overexpressing cells where GRB7 is co-overexpressed, there is a possibility that GRB7 is the primary binding partner for SHC not GRB2. We argue that GRB7 and GRB2 may compete with each other to bind to SHC, since the cytosolic pool of GRB7 is much higher than GRB2 (GRB7>GRB2), GRB7 becomes the preferred binding partner for SHC in HER2+ breast cancer cells. Alternatively, GRB7 may bind to other tyrosine phosphorylated sites of SHC but not Y317, a GRB2 binding site. Our current focus is on the downstream effector(s) of the HER2-GRB7-SHC complex, which may be responsible for major biological function(s) relevant to HER2+ breast tumor progression. An important functional consequence of competition between GRB7 and GRB2 for shared binding sites may be the modulation of RAS signaling. SH2 domains of adapter proteins (such as GRB7) bind to tyrosine phosphorylated growth factor receptors and are found in proteins that serve as substrates for tyrosine kinases, e.g. RAS-GTPase activating protein and PLCγ [63], and GRB7 contains a RAS-associating (RA) domain [36]. A positive correlation has also been observed between the expression of GRB7 and KRAS2, a gene encoding a small G-protein of the RAS family in testicular germ cells [64].
Along the same lines, our data (both siRNA and GRB7 inhibitor peptide data) demonstrate that GRB7 is necessary for RAS activation following heregulin stimulation in HER2+ breast cancer cells (Figure 5) and hence, may control HER2+ breast cancer cell proliferation (Figure 4). In line with our results, a recent study by Wang et al. demonstrated that exogenous overexpression of GRB7 promotes ovarian cancer cell proliferation through extracellular-signal regulated kinase (ERK) [65]. An earlier study showed knockdown of GRB7 expression in breast cancer cells with naturally occurring Her2/Neu amplification led to a decrease in cell proliferation [52]. GRB7 inhibitor peptide (G7-18NATE) has been reported to inhibit breast cancer cell proliferation [11], one of the most important phenotypes for tumor progression.
Several studies have demonstrated that GRB7 was connected to cell motility through its association with focal adhesion kinase, phosphoinositides, ephrin receptor and calmodulin [45,66-68]. Clinicopathological studies have revealed that overexpressed GRB7 significantly associated with metastatic tumor phenotype [69]. Earlier, Shinji et al. have reported that a group of GRB7 and FAK positive hepatocelluar carcinoma patients showed a significantly poorer prognosis than the negative group [70]. These studies led us to examine the mechanism of GRB7-mediated HER2+ cell migration. FAK has been implicated in playing an important role in cellular signaling by integrins, several cytosolic signaling proteins, and with cytoskeletal proteins. It has been reported by others that GRB7 physically interacts with FAK in an integrin-engagement manner [27,47,71]. However, the functional significance of the FAK-GRB7 interaction in HER2+ breast cancer is not fully understood at present. Our co-immunoprecipitation data clearly demonstrated an interaction between FAK and GRB7 in HER2+ cells following fibronectin engagement (Figure 6). It has been reported that phosphorylation of GRB7 could not take place in the absence of FAK following integrin stimulation [72]. This suggests that GRB7 is potentially a direct substrate of FAK. In in vitro experiments, the GRB7 inhibitor peptide blocked binding to FAK and phosphorylation of endogenous GRB7 protein in human pancreatic cancer cells [71]. This FAK dependent phosphorylation of GRB7 (Figure 6) is regulated by integrin (α4β1/ α5β1) signaling that leads to the activation of an unknown functional downstream effector for cell migration. Our in vitro data showed that GRB7 regulates integin-mediated HER2+ cell migration (Figures 7 and 8). FAK-GRB7 complex formation and its correlation with increased cell invasion were also reported in esophageal carcinoma [73]. Several reports suggest that activation of RAC-GTP is necessary for integrin-mediated cell migration [29,74,75]. Accumulating evidence suggests that the RAC’s immediate downstream effector PAK1 is implicated in breast tumor progression. Indeed, more than 50% of breast tumors show overexpression and/or hyperactivation of PAK1 [76]. Our in vitro siRNA and GRB7 peptide inhibitor (G7-18NATE) data indicate that GRB7 may control HER2+ breast cancer cell migration through the activation of small GTPase, RAC1 (Figure 10). Our data also suggest that GRB7 physically associates with Guanine nucleotide Exchange Factor (GEF), VAV2, which is required for the activation state of RAC (GTP bound form of RAC) (Figure 10D). It is known that the FAK-GRB7 complex is independent of the FAK-SRC complex that targets p130CAS-CRK-DOCK-RAC signaling [77], but it has also been reported that PI3 Kinase activation could cooperate with the FAK-GRB7 complex in the stimulation of cell migration [67]. PI3 Kinase is a possible upstream regulator of the RAC signaling pathway [78,79]. PREL1 another homologue of GRB7 was found to bind to the actin cytoskeleton via ENA/VSPP, an important player for actin polymerization [80]. Our initial combination study (trastuzumab plus G7-18NATE) suggests that co-targeting HER2 and GRB7 provides an additive effect on reducing migration beyond targeting HER2 or GRB7 alone, at least in the context of BT474 cells (Figures 7 and 8). Our initial finding, along with Pero’s reported data (GRB7 peptide inhibitor in combination with trastuzumab enhances the inhibitory effect on SKBR3 cell proliferation) [11], suggest that co-targeting of both GRB7 and HER2 would likely provide additional benefit over HER2- targeted therapy alone for patients with a tumor carrying 17q12 amplification.
In conclusion, our study shows that coexpression of GRB7 with HER2 impacts both proliferative and invasive potential of HER2+ breast cancer cells by direct binding with HER2 and FAK, respectively. These data strongly suggest that careful biomarker analysis for each patient is the key for the right treatment for the right patient. Our work supports examining GRB7 protein overexpression for HER2+ breast cancer patients risk stratification. The clinical utility of this or other biomarker(s) in predicting response to therapy or therapies remains of interest, and definitely warrants additional investigations. These studies predict that co-targeting HER2 in addition to GRB7 will be effective in breast tumors that rely on HER2 signaling pathways for survival (Figure 11).
Acknowledgments
This work was supported by grants to BLJ from the National Institute of Health (R21 CA139428-01) and Edith Sanford Breast Cancer, Sanford Research/USD, Sioux Falls, SD. We thank to Dr. Stephanie Pero, University of Vermont School of Medicine, and Burlington, VT for providing GRB7 inhibitor peptide and control peptide. We acknowledge Integrated Cellular Imaging, the microscopy core facility of Winship Cancer Institute at Emory University. The suggestions and thoughtful discussion by Dr. Brian Smith and Dr. Keith Miskimins are also acknowledged.
Conflict of interest statement
None.
Video1
Video2
References
- 1.Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, Levin WJ, Stuart SG, Udove J, Ullrich A, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989;244:707–712. doi: 10.1126/science.2470152. [DOI] [PubMed] [Google Scholar]
- 2.Cardoso F, Piccart MJ, Durbecq V, Di Leo A. Resistance to trastuzumab: a necessary evil or a temporary challenge? Clin Breast Cancer. 2002;3:247–257. doi: 10.3816/CBC.2002.n.028. discussion 258-249. [DOI] [PubMed] [Google Scholar]
- 3.Vogel CL, Cobleigh MA, Tripathy D, Gutheil JC, Harris LN, Fehrenbacher L, Slamon DJ, Murphy M, Novotny WF, Burchmore M, Shak S, Stewart SJ, Press M. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J. Clin. Oncol. 2002;20:719–726. doi: 10.1200/JCO.2002.20.3.719. [DOI] [PubMed] [Google Scholar]
- 4.Lan KH, Lu CH, Yu D. Mechanisms of trastuzumab resistance and their clinical implications. Ann N Y Acad Sci. 2005;1059:70–75. doi: 10.1196/annals.1339.026. [DOI] [PubMed] [Google Scholar]
- 5.Bange J, Zwick E, Ullrich A. Molecular targets for breast cancer therapy and prevention. Nat Med. 2001;7:548–552. doi: 10.1038/87872. [DOI] [PubMed] [Google Scholar]
- 6.Petricoin EF 3rd, Hackett JL, Lesko LJ, Puri RK, Gutman SI, Chumakov K, Woodcock J, Feigal DW Jr, Zoon KC, Sistare FD. Medical applications of microarray technologies: a regulatory science perspective. Nat Genet. 2002;32(Suppl):474–479. doi: 10.1038/ng1029. [DOI] [PubMed] [Google Scholar]
- 7.Abramovitz M, Leyland-Jones B. A systems approach to clinical oncology: focus on breast cancer. Proteome Sci. 2006;4:5. doi: 10.1186/1477-5956-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abramovitz M, Ordanic-Kodani M, Wang Y, Li Z, Catzavelos C, Bouzyk M, Sledge GW Jr, Moreno CS, Leyland-Jones B. Optimization of RNA extraction from FFPE tissues for expression profiling in the DASL assay. Biotechniques. 2008;44:417–423. doi: 10.2144/000112703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bai T, Luoh SW. GRB-7 facilitates HER-2/Neu-mediated signal transduction and tumor formation. Carcinogenesis. 2008;29:473–479. doi: 10.1093/carcin/bgm221. [DOI] [PubMed] [Google Scholar]
- 10.Nadler Y, Gonzalez AM, Camp RL, Rimm DL, Kluger HM, Kluger Y. Growth factor receptor-bound protein-7 (Grb7) as a prognostic marker and therapeutic target in breast cancer. Ann Oncol. 2010;21:466–473. doi: 10.1093/annonc/mdp346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pero SC, Shukla GS, Cookson MM, Flemer S Jr, Krag DN. Combination treatment with Grb7 peptide and Doxorubicin or Trastuzumab (Herceptin) results in cooperative cell growth inhibition in breast cancer cells. Br J Cancer. 2007;96:1520–1525. doi: 10.1038/sj.bjc.6603732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Han DC, Shen TL, Guan JL. The Grb7 family proteins: structure, interactions with other signaling molecules and potential cellular functions. Oncogene. 2001;20:6315–6321. doi: 10.1038/sj.onc.1204775. [DOI] [PubMed] [Google Scholar]
- 13.Lucas-Fernandez E, Garcia-Palmero I, Villalobo A. Genomic organization and control of the grb7 gene family. Curr Genomics. 2008;9:60–68. doi: 10.2174/138920208783884847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Daly RJ, Sanderson GM, Janes PW, Sutherland RL. Cloning and characterization of GRB14, a novel member of the GRB7 gene family. J Biol Chem. 1996;271:12502–12510. doi: 10.1074/jbc.271.21.12502. [DOI] [PubMed] [Google Scholar]
- 15.Margolis B. The GRB family of SH2 domain proteins. Prog Biophys Mol Biol. 1994;62:223–244. doi: 10.1016/0079-6107(94)90013-2. [DOI] [PubMed] [Google Scholar]
- 16.Ooi J, Yajnik V, Immanuel D, Gordon M, Moskow JJ, Buchberg AM, Margolis B. The cloning of Grb10 reveals a new family of SH2 domain proteins. Oncogene. 1995;10:1621–1630. [PubMed] [Google Scholar]
- 17.Pero SC, Daly RJ, Krag DN. Grb7-based molecular therapeutics in cancer. Expert Rev Mol Med. 2003;5:1–11. doi: 10.1017/S1462399403006227. [DOI] [PubMed] [Google Scholar]
- 18.Leavey SF, Arend LJ, Dare H, Dressler GR, Briggs JP, Margolis BL. Expression of Grb7 growth factor receptor signaling protein in kidney development and in adult kidney. Am J Physiol. 1998;275:F770–776. doi: 10.1152/ajprenal.1998.275.5.F770. [DOI] [PubMed] [Google Scholar]
- 19.Kasus-Jacobi A, Bereziat V, Perdereau D, Girard J, Burnol AF. Evidence for an interaction between the insulin receptor and Grb7. A role for two of its binding domains, PIR and SH2. Oncogene. 2000;19:2052–2059. doi: 10.1038/sj.onc.1203469. [DOI] [PubMed] [Google Scholar]
- 20.Abramovitz M, Barwick BG, Willis S, Young B, Catzavelos C, Li Z, Kodani M, Tang W, Bouzyk M, Moreno CS, Leyland-Jones B. Molecular characterisation of formalin-fixed paraffin-embedded (FFPE) breast tumour specimens using a custom 512-gene breast cancer bead array-based platform. Br J Cancer. 2011;105:1574–1581. doi: 10.1038/bjc.2011.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Serra V, Markman B, Scaltriti M, Eichhorn PJ, Valero V, Guzman M, Botero ML, Llonch E, Atzori F, Di Cosimo S, Maira M, Garcia-Echeverria C, Parra JL, Arribas J, Baselga J. NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res. 2008;68:8022–8030. doi: 10.1158/0008-5472.CAN-08-1385. [DOI] [PubMed] [Google Scholar]
- 22.Lee GY, Kenny PA, Lee EH, Bissell MJ. Three-dimensional culture models of normal and malignant breast epithelial cells. Nat Methods. 2007;4:359–365. doi: 10.1038/nmeth1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pradip D, Peng X, Durden DL. Rac2 specificity in macrophage integrin signaling: potential role for Syk kinase. J Biol Chem. 2003;278:41661–41669. doi: 10.1074/jbc.M306491200. [DOI] [PubMed] [Google Scholar]
- 24.Klemke RL, Cai S, Giannini AL, Gallagher PJ, de Lanerolle P, Cheresh DA. Regulation of cell motility by mitogen-activated protein kinase. J Cell Biol. 1997;137:481–492. doi: 10.1083/jcb.137.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sander EE, ten Klooster JP, van Delft S, van der Kammen RA, Collard JG. Rac downregulates Rho activity: reciprocal balance between both GTPases determines cellular morphology and migratory behavior. J Cell Biol. 1999;147:1009–1022. doi: 10.1083/jcb.147.5.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suzuki T, Shoji S, Yamamoto K, Nada S, Okada M, Yamamoto T, Honda Z. Essential roles of Lyn in fibronectin-mediated filamentous actin assembly and cell motility in mast cells. J Immunol. 1998;161:3694–3701. [PubMed] [Google Scholar]
- 27.Chu PY, Huang LY, Hsu CH, Liang CC, Guan JL, Hung TH, Shen TL. Tyrosine phosphorylation of growth factor receptor-bound protein-7 by focal adhesion kinase in the regulation of cell migration, proliferation, and tumorigenesis. J Biol Chem. 2009;284:20215–20226. doi: 10.1074/jbc.M109.018259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dey N, De PK, Wang M, Zhang H, Dobrota EA, Robertson KA, Durden DL. CSK controls retinoic acid receptor (RAR) signaling: a RAR-c-SRC signaling axis is required for neuritogenic differentiation. Mol Cell Biol. 2007;27:4179–4197. doi: 10.1128/MCB.01352-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dey N, Howell BW, De PK, Durden DL. CSK negatively regulates nerve growth factor induced neural differentiation and augments AKT kinase activity. Exp Cell Res. 2005;307:1–14. doi: 10.1016/j.yexcr.2005.02.029. [DOI] [PubMed] [Google Scholar]
- 30.Pawitan Y, Bjohle J, Amler L, Borg AL, Egyhazi S, Hall P, Han X, Holmberg L, Huang F, Klaar S, Liu ET, Miller L, Nordgren H, Ploner A, Sandelin K, Shaw PM, Smeds J, Skoog L, Wedren S, Bergh J. Gene expression profiling spares early breast cancer patients from adjuvant therapy: derived and validated in two population-based cohorts. Breast Cancer Res. 2005;7:R953–964. doi: 10.1186/bcr1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Giricz O, Calvo V, Pero SC, Krag DN, Sparano JA, Kenny PA. GRB7 is required for triple-negative breast cancer cell invasion and survival. Breast Cancer Res Treat. 2012;133:607–615. doi: 10.1007/s10549-011-1822-6. [DOI] [PubMed] [Google Scholar]
- 32.Kauraniemi P, Kallioniemi A. Activation of multiple cancer-associated genes at the ERBB2 amplicon in breast cancer. Endocr Relat Cancer. 2006;13:39–49. doi: 10.1677/erc.1.01147. [DOI] [PubMed] [Google Scholar]
- 33.Vinatzer U, Dampier B, Streubel B, Pacher M, Seewald MJ, Stratowa C, Kaserer K, Schreiber M. Expression of HER2 and the coamplified genes GRB7 and MLN64 in human breast cancer: quantitative real-time reverse transcription-PCR as a diagnostic alternative to immunohistochemistry and fluorescence in situ hybridization. Clin Cancer Res. 2005;11:8348–8357. doi: 10.1158/1078-0432.CCR-05-0841. [DOI] [PubMed] [Google Scholar]
- 34.Konecny GE, Pegram MD, Venkatesan N, Finn R, Yang G, Rahmeh M, Untch M, Rusnak DW, Spehar G, Mullin RJ, Keith BR, Gilmer TM, Berger M, Podratz KC, Slamon DJ. Activity of the dual kinase inhibitor lapatinib (GW572016) against HER-2-overexpressing and trastuzumab-treated breast cancer cells. Cancer Res. 2006;66:1630–1639. doi: 10.1158/0008-5472.CAN-05-1182. [DOI] [PubMed] [Google Scholar]
- 35.Moulder SL, Yakes FM, Muthuswamy SK, Bianco R, Simpson JF, Arteaga CL. Epidermal growth factor receptor (HER1) tyrosine kinase inhibitor ZD1839 (Iressa) inhibits HER2/neu (erbB2)-overexpressing breast cancer cells in vitro and in vivo. Cancer Res. 2001;61:8887–8895. [PubMed] [Google Scholar]
- 36.Depetris RS, Wu J, Hubbard SR. Structural and functional studies of the Ras-associating and pleckstrin-homology domains of Grb10 and Grb14. Nat Struct Mol Biol. 2009;16:833–839. doi: 10.1038/nsmb.1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barbacid M. ras genes. Annu Rev Biochem. 1987;56:779–827. doi: 10.1146/annurev.bi.56.070187.004023. [DOI] [PubMed] [Google Scholar]
- 38.Vojtek AB, Der CJ. Increasing complexity of the Ras signaling pathway. J Biol Chem. 1998;273:19925–19928. doi: 10.1074/jbc.273.32.19925. [DOI] [PubMed] [Google Scholar]
- 39.Xing Z, Chen HC, Nowlen JK, Taylor SJ, Shalloway D, Guan JL. Direct interaction of v-Src with the focal adhesion kinase mediated by the Src SH2 domain. Mol Biol Cell. 1994;5:413–421. doi: 10.1091/mbc.5.4.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chan PY, Kanner SB, Whitney G, Aruffo A. A transmembrane-anchored chimeric focal adhesion kinase is constitutively activated and phosphorylated at tyrosine residues identical to pp125FAK. J Biol Chem. 1994;269:20567–20574. [PubMed] [Google Scholar]
- 41.Chen HC, Appeddu PA, Isoda H, Guan JL. Phosphorylation of tyrosine 397 in focal adhesion kinase is required for binding phosphatidylinositol 3-kinase. J Biol Chem. 1996;271:26329–26334. doi: 10.1074/jbc.271.42.26329. [DOI] [PubMed] [Google Scholar]
- 42.Schlaepfer DD, Hanks SK, Hunter T, van der Geer P. Integrin-mediated signal transduction linked to Ras pathway by GRB2 binding to focal adhesion kinase. Nature. 1994;372:786–791. doi: 10.1038/372786a0. [DOI] [PubMed] [Google Scholar]
- 43.Weiner TM, Liu ET, Craven RJ, Cance WG. Expression of focal adhesion kinase gene and invasive cancer. Lancet. 1993;342:1024–1025. doi: 10.1016/0140-6736(93)92881-s. [DOI] [PubMed] [Google Scholar]
- 44.Tanaka S, Mori M, Akiyoshi T, Tanaka Y, Mafune K, Wands JR, Sugimachi K. Coexpression of Grb7 with epidermal growth factor receptor or Her2/erbB2 in human advanced esophageal carcinoma. Cancer Res. 1997;57:28–31. [PubMed] [Google Scholar]
- 45.Han DC, Shen TL, Guan JL. Role of Grb7 targeting to focal contacts and its phosphorylation by focal adhesion kinase in regulation of cell migration. J Biol Chem. 2000;275:28911–28917. doi: 10.1074/jbc.M001997200. [DOI] [PubMed] [Google Scholar]
- 46.Liu S, Goldstein RH, Scepansky EM, Rosenblatt M. Inhibition of rho-associated kinase signaling prevents breast cancer metastasis to human bone. Cancer Res. 2009;69:8742–8751. doi: 10.1158/0008-5472.CAN-09-1541. [DOI] [PubMed] [Google Scholar]
- 47.Han DC, Guan JL. Association of focal adhesion kinase with Grb7 and its role in cell migration. J Biol Chem. 1999;274:24425–24430. doi: 10.1074/jbc.274.34.24425. [DOI] [PubMed] [Google Scholar]
- 48.Kuwada SK, Kuang J, Li X. Integrin alpha5/beta1 expression mediates HER-2 down-regulation in colon cancer cells. J Biol Chem. 2005;280:19027–19035. doi: 10.1074/jbc.M410540200. [DOI] [PubMed] [Google Scholar]
- 49.Shimizu H, Seiki T, Asada M, Yoshimatsu K, Koyama N. Alpha6beta1 integrin induces proteasome-mediated cleavage of erbB2 in breast cancer cells. Oncogene. 2003;22:831–839. doi: 10.1038/sj.onc.1206203. [DOI] [PubMed] [Google Scholar]
- 50.Tomasetto C, Regnier C, Moog-Lutz C, Mattei MG, Chenard MP, Lidereau R, Basset P, Rio MC. Identification of four novel human genes amplified and overexpressed in breast carcinoma and localized to the q11-q21.3 region of chromosome 17. Genomics. 1995;28:367–376. doi: 10.1006/geno.1995.1163. [DOI] [PubMed] [Google Scholar]
- 51.Stein D, Wu J, Fuqua SA, Roonprapunt C, Yajnik V, D’Eustachio P, Moskow JJ, Buchberg AM, Osborne CK, Margolis B. The SH2 domain protein GRB-7 is co-amplified, overexpressed and in a tight complex with HER2 in breast cancer. EMBO J. 1994;13:1331–1340. doi: 10.1002/j.1460-2075.1994.tb06386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kao J, Pollack JR. RNA interference-based functional dissection of the 17q12 amplicon in breast cancer reveals contribution of coamplified genes. Genes Chromosomes Cancer. 2006;45:761–769. doi: 10.1002/gcc.20339. [DOI] [PubMed] [Google Scholar]
- 53.Paik S, Shak S, Tang G, Kim C, Baker J, Cronin M, Baehner FL, Walker MG, Watson D, Park T, Hiller W, Fisher ER, Wickerham DL, Bryant J, Wolmark N. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med. 2004;351:2817–2826. doi: 10.1056/NEJMoa041588. [DOI] [PubMed] [Google Scholar]
- 54.De Laurentiis M, Cancello G, Zinno L, Montagna E, Malorni L, Esposito A, Pennacchio R, Silvestro L, Giuliano M, Giordano A, Caputo F, Accurso A, De Placido S. Targeting HER2 as a therapeutic strategy for breast cancer: a paradigmatic shift of drug development in oncology. Ann Oncol. 2005;16(Suppl 4):iv7–13. doi: 10.1093/annonc/mdi901. [DOI] [PubMed] [Google Scholar]
- 55.Ellis MJ, Tao Y, Young O, White S, Proia AD, Murray J, Renshaw L, Faratian D, Thomas J, Dowsett M, Krause A, Evans DB, Miller WR, Dixon JM. Estrogen-independent proliferation is present in estrogen-receptor HER2-positive primary breast cancer after neoadjuvant letrozole. J. Clin. Oncol. 2006;24:3019–3025. doi: 10.1200/JCO.2005.04.3034. [DOI] [PubMed] [Google Scholar]
- 56.Katz E, Dubois-Marshall S, Sims AH, Faratian D, Li J, Smith ES, Quinn JA, Edward M, Meehan RR, Evans EE, Langdon SP, Harrison DJ. A gene on the HER2 amplicon, C35, is an oncogene in breast cancer whose actions are prevented by inhibition of Syk. Br J Cancer. 2010;103:401–410. doi: 10.1038/sj.bjc.6605763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Coon JS, Marcus E, Gupta-Burt S, Seelig S, Jacobson K, Chen S, Renta V, Fronda G, Preisler HD. Amplification and overexpression of topoisomerase IIalpha predict response to anthracycline-based therapy in locally advanced breast cancer. Clin Cancer Res. 2002;8:1061–1067. [PubMed] [Google Scholar]
- 58.Arriola E, Rodriguez-Pinilla SM, Lambros MB, Jones RL, James M, Savage K, Smith IE, Dowsett M, Reis-Filho JS. Topoisomerase II alpha amplification may predict benefit from adjuvant anthracyclines in HER2 positive early breast cancer. Breast Cancer Res Treat. 2007;106:181–189. doi: 10.1007/s10549-006-9492-5. [DOI] [PubMed] [Google Scholar]
- 59.Knoop AS, Knudsen H, Balslev E, Rasmussen BB, Overgaard J, Nielsen KV, Schonau A, Gunnarsdottir K, Olsen KE, Mouridsen H, Ejlertsen B. retrospective analysis of topoisomerase IIa amplifications and deletions as predictive markers in primary breast cancer patients randomly assigned to cyclophosphamide, methotrexate, and fluorouracil or cyclophosphamide, epirubicin, and fluorouracil: Danish Breast Cancer Cooperative Group. J. Clin. Oncol. 2005;23:7483–7490. doi: 10.1200/JCO.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 60.Tanner M, Isola J, Wiklund T, Erikstein B, Kellokumpu-Lehtinen P, Malmstrom P, Wilking N, Nilsson J, Bergh J. Topoisomerase IIalpha gene amplification predicts favorable treatment response to tailored and dose-escalated anthracycline-based adjuvant chemotherapy in HER-2/neu-amplified breast cancer: Scandinavian Breast Group Trial 9401. J. Clin. Oncol. 2006;24:2428–2436. doi: 10.1200/JCO.2005.02.9264. [DOI] [PubMed] [Google Scholar]
- 61.Fiddes RJ, Campbell DH, Janes PW, Sivertsen SP, Sasaki H, Wallasch C, Daly RJ. Analysis of Grb7 recruitment by heregulin-activated erbB receptors reveals a novel target selectivity for erbB3. J Biol Chem. 1998;273:7717–7724. doi: 10.1074/jbc.273.13.7717. [DOI] [PubMed] [Google Scholar]
- 62.Yokote K, Margolis B, Heldin CH, Claesson-Welsh L. Grb7 is a downstream signaling component of platelet-derived growth factor alpha- and beta-receptors. J Biol Chem. 1996;271:30942–30949. doi: 10.1074/jbc.271.48.30942. [DOI] [PubMed] [Google Scholar]
- 63.Margolis B, Silvennoinen O, Comoglio F, Roonprapunt C, Skolnik E, Ullrich A, Schlessinger J. High-efficiency expression/cloning of epidermal growth factor-receptor-binding proteins with Src homology 2 domains. Proc Natl Acad Sci U S A. 1992;89:8894–8898. doi: 10.1073/pnas.89.19.8894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McIntyre A, Summersgill B, Spendlove HE, Huddart R, Houlston R, Shipley J. Activating mutations and/or expression levels of tyrosine kinase receptors GRB7, RAS, and BRAF in testicular germ cell tumors. Neoplasia. 2005;7:1047–1052. doi: 10.1593/neo.05514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang Y, Chan DW, Liu VW, Chiu P, Ngan HY. Differential functions of growth factor receptor-bound protein 7 (GRB7) and its variant GRB7v in ovarian carcinogenesis. Clin Cancer Res. 2010;16:2529–2539. doi: 10.1158/1078-0432.CCR-10-0018. [DOI] [PubMed] [Google Scholar]
- 66.Han DC, Shen TL, Miao H, Wang B, Guan JL. EphB1 associates with Grb7 and regulates cell migration. J Biol Chem. 2002;277:45655–45661. doi: 10.1074/jbc.M203165200. [DOI] [PubMed] [Google Scholar]
- 67.Shen TL, Han DC, Guan JL. Association of Grb7 with phosphoinositides and its role in the regulation of cell migration. J Biol Chem. 2002;277:29069–29077. doi: 10.1074/jbc.M203085200. [DOI] [PubMed] [Google Scholar]
- 68.Li H, Sanchez-Torres J, del Carpio AF, Nogales-Gonzalez A, Molina-Ortiz P, Moreno MJ, Torok K, Villalobo A. The adaptor Grb7 is a novel calmodulin-binding protein: functional implications of the interaction of calmodulin with Grb7. Oncogene. 2005;24:4206–4219. doi: 10.1038/sj.onc.1208591. [DOI] [PubMed] [Google Scholar]
- 69.Shen TL, Guan JL. Grb7 in intracellular signaling and its role in cell regulation. Front Biosci. 2004;9:192–200. doi: 10.2741/1229. [DOI] [PubMed] [Google Scholar]
- 70.Itoh S, Taketomi A, Tanaka S, Harimoto N, Yamashita Y, Aishima S, Maeda T, Shirabe K, Shimada M, Maehara Y. Role of growth factor receptor bound protein 7 in hepatocellular carcinoma. Mol Cancer Res. 2007;5:667–673. doi: 10.1158/1541-7786.MCR-06-0282. [DOI] [PubMed] [Google Scholar]
- 71.Tanaka S, Pero SC, Taguchi K, Shimada M, Mori M, Krag DN, Arii S. Specific peptide ligand for Grb7 signal transduction protein and pancreatic cancer metastasis. J Natl Cancer Inst. 2006;98:491–498. doi: 10.1093/jnci/djj105. [DOI] [PubMed] [Google Scholar]
- 72.Sieg DJ, Ilic D, Jones KC, Damsky CH, Hunter T, Schlaepfer DD. Pyk2 and Src-family protein-tyrosine kinases compensate for the loss of FAK in fibronectin-stimulated signaling events but Pyk2 does not fully function to enhance FAK- cell migration. EMBO J. 1998;17:5933–5947. doi: 10.1093/emboj/17.20.5933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tanaka S, Mori M, Akiyoshi T, Tanaka Y, Mafune K, Wands JR, Sugimachi K. A novel variant of human Grb7 is associated with invasive esophageal carcinoma. J Clin Invest. 1998;102:821–827. doi: 10.1172/JCI2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dey N, Crosswell HE, De P, Parsons R, Peng Q, Su JD, Durden DL. The protein phosphatase activity of PTEN regulates SRC family kinases and controls glioma migration. Cancer Res. 2008;68:1862–1871. doi: 10.1158/0008-5472.CAN-07-1182. [DOI] [PubMed] [Google Scholar]
- 75.Hage B, Meinel K, Baum I, Giehl K, Menke A. Rac1 activation inhibits E-cadherin-mediated adherens junctions via binding to IQGAP1 in pancreatic carcinoma cells. Cell Commun Signal. 2009;7:23. doi: 10.1186/1478-811X-7-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Balasenthil S, Sahin AA, Barnes CJ, Wang RA, Pestell RG, Vadlamudi RK, Kumar R. p21-activated kinase-1 signaling mediates cyclin D1 expression in mammary epithelial and cancer cells. J Biol Chem. 2004;279:1422–1428. doi: 10.1074/jbc.M309937200. [DOI] [PubMed] [Google Scholar]
- 77.Cary LA, Han DC, Polte TR, Hanks SK, Guan JL. Identification of p130Cas as a mediator of focal adhesion kinase-promoted cell migration. J Cell Biol. 1998;140:211–221. doi: 10.1083/jcb.140.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Eriksson A, Cao R, Roy J, Tritsaris K, Wahlestedt C, Dissing S, Thyberg J, Cao Y. Small GTP-binding protein Rac is an essential mediator of vascular endothelial growth factor-induced endothelial fenestrations and vascular permeability. Circulation. 2003;107:1532–1538. doi: 10.1161/01.cir.0000055324.34758.32. [DOI] [PubMed] [Google Scholar]
- 79.Paruchuri S, Broom O, Dib K, Sjolander A. The pro-inflammatory mediator leukotriene D4 induces phosphatidylinositol 3-kinase and Rac-dependent migration of intestinal epithelial cells. J Biol Chem. 2005;280:13538–13544. doi: 10.1074/jbc.M409811200. [DOI] [PubMed] [Google Scholar]
- 80.Jenzora A, Behrendt B, Small JV, Wehland J, Stradal TE. PREL1 provides a link from Ras signalling to the actin cytoskeleton via Ena/VASP proteins. FEBS Lett. 2005;579:455–463. doi: 10.1016/j.febslet.2004.10.110. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.