

Abstract
Chronic vascular inflammation is regarded to have a key role in cardiovascular disease. However, the elicitors of this inflammatory response in the vessel wall are currently not well understood. Excessive amounts of cholesterol, an abundant and fundamental lipid molecule in mammalian cells, can trigger the development and progression of atherosclerosis. Accumulation of cholesterol in early atherosclerotic lesions results in the formation of macrophage foam cells, and crystalline cholesterol is found as a hallmark of advanced atherosclerotic plaques. Cholesterol crystals can activate a multimolecular signaling complex of the innate immune system, the NLRP3 inflammasome, resulting in a caspase-1 mediated activation and secretion of proinflammatory interleukin-1 family cytokines. Furthermore, crystalline cholesterol is thought to induce plaque rupture by physical disruption of the fibrous cap covering atherosclerotic lesions. Here we review the role of cholesterol deposition and crystallization for inflammatory responses in cardiovascular diseases.
Keywords: Cholesterol crystals, Inflammation, NLRP3 inflammasome, Atherosclerosis, Cardiovascular disease, Serum proteins
Introduction
Inflammation results from immune responses towards stimuli that occur during microbial infection and tissue damage. The innate immune system is an evolutionary conserved defense system that relies on germline-encoded pattern recognition receptors (PRRs) expressed on innate immune cells as well as other host cells. These PRR receptors detect unique microbial substances, so called pathogen-associated molecular patterns (PAMPs), and endogenous molecules released from damaged cells, named damage-associated molecular patterns (DAMPs). Activation of these receptors initiates signaling cascades that lead to the transcription of proinflammatory cytokines, chemokines, type I interferons (IFNs), antimicrobial peptides and proteins involved in the modulation of PRR signaling. The resultant inflammatory response is crucial for the clearance of infectious agents and damaged cells, for tissue repair as well as for the initiation of adaptive immunity. Unresolved, chronic inflammation, however, can be harmful to the host and can trigger a number of pathologies [1].
Since the discovery that PRRs can also be involved in the recognition of endogenous self-molecules that appear during cell death or tissue injury, many stimuli causing sterile inflammation have been discovered. Most of these stimuli comprise self-molecules like endogenous nucleic acids, purine metabolites such as ATP, heat shock proteins or extracellular matrix components, which are usually localized in certain cellular compartments but become accessible to immune cells after damage or death of the cell [2]. NLRP3, a member of the NOD-like receptor (NLR) family of PRRs, was linked to the inflammatory response to various aggregated or crystalline materials of endogenous or environmental origin. These substances trigger the formation of the NLRP3 inflammasome, an intracellular multimolecular protein complex containing the adaptor molecule apoptosis-associated speck-like protein containing a CARD (ASC) and pro-caspase-1. Upon its recruitment to ASC autocatalytic activation of pro-caspase-1 occurs and results in the proteolytic activation and release of the pro-inflammatory interleukin (IL)-1β family cytokines [3]. It could be demonstrated, that the NLRP3 inflammasome can contribute to various chronic crystal-induced inflammatory diseases, such as gout, pseudogout, asbestosis, silicosis and more recently atherosclerosis.
While much has been learned about the involvement of the NLRP3 inflammasome in crystal mediated diseases, a unifying molecular mechanism leading to the activation of this multimolecular protein complex has yet to be discovered. Since the NLRP3 inflammasome can be activated by a large number of diverse triggers, an indirect activation mechanism is likely [4-8].
Here, we will discuss the inflammatory potential of crystalline cholesterol and its role in cardiovascular disease.
Physical characteristics of cholesterol
Cholesterol is an essential lipid in vertebrates that is derived from nutrition and from endogenous biosynthesis. As an ubiquitous component of all cellular membranes it plays an essential role in membrane structure and function and it is important for maintaining membrane permeability and cell signaling. In addition, cholesterol metabolites such as bile acids, oxysterols, vitamin D and steroid hormones have essential physiological functions.
Free cholesterol has very low solubility in aqueous environments and lipoproteins serve as cholesterol carriers for cholesterol transport in the bloodstream. Cells obtain most exogenous cholesterol from circulating lipoproteins via LDL receptor-mediated endocytosis. LDL-derived cholesterol incorporates in cellular membranes and excess free cholesterol becomes esterified by acyl-coenzyme A:cholesterol acyltransferase (ACAT) which allows cholesterylesters to be stored inside the cells in so-called lipid droplets. Stored cholesterylesters can be converted back into cholesterol by the cellular enzyme cholesterylester hydrolase (CEH). Mammalian cells cannot degrade cholesterol and hence the elimination of excess cholesterol is of particular importance for maintaining cellular cholesterol homeostasis. Efflux of free cholesterol from cells is mainly mediated by high-density lipoprotein (HDL) consisting of apolipoproteins A-I and A-II (apoA-I and apoA-II), phospholipids, cholesterol and cholesterol esters. HDL acquires cholesterol from cells either via passive diffusion following a cholesterol gradient between the cell surface and tangent HDL particles or via an apolipoprotein-mediated transport process involving ABC transporters in the plasma membrane. In particular, ABCA1 transfers cholesterol and phospholipids to lipid-poor apoA-I resulting in the generation of nascent HDL particles, and in macrophages a second transporter, ABCG1, was found to be involved in cholesterol efflux to mature HDL. HDL particles are responsible for retrograde cholesterol transport (RCT) to the liver and steroidogenic tissues, where HDL-derived cholesterol becomes metabolized or prepared for excretion [9].
Although cholesterol biosynthesis, transport and metabolism are tightly controlled, defects in these pathways or disruption of cholesterol homeostasis can result in various pathologies. Atherosclerosis, for instance, is characterized by the accumulation of lipids and immune cells in the arterial intima. In the 1970s Small et al. analyzed the physical properties of cholesterol and he found monohydrate cholesterol crystals at 37°C and he emphasized the presence of extracellular crystalline cholesterol in advanced atherosclerotic lesions [10]. Furthermore, in vitro studies with lipid-laden macrophage foam cells revealed that the formation of cholesterol monohydrate crystals also occurs intracellularly upon accumulation of free cholesterol inside the cells [11, 12]. Taken together, these findings indicated that the accumulation of free, unesterified cholesterol can lead to deposition of cholesterol crystals in vessel walls.
Cholesterol crystal interaction with serum proteins
In the early 1970s it was described that HDL has the potential to remove cholesterol from cells and atherosclerotic tissues. However, it was also noted that the turnover rate of cholesterol in atherosclerosis is relatively slow, which was thought to be a consequence of ineffective phagocytosis and metabolism of the crystalline cholesterol in the arterial walls. Nevertheless, direct interactions between HDL and cholesterol crystals were also analyzed in vitro. The interaction between HDL and crystalline cholesterol resulted in the de novo formation of liposomes on the crystal surface and solubilization of cholesterol crystals. In addition, effective clearance of cholesterol crystals from HDL incubated human atherosclerotic lesions could be demonstrated [13, 14]. Furthermore, an in vivo model in rats showed the capacity of oxysterols to potently solubilize cholesterol crystals from subcutaneous implants in rats. Hence, oxysterols by enhancing the solubility of cholesterol could increase cholesterol clearance from tissues by favoring cholesterol incorporation into HDL particles, thus reducing the atherogenic potential of cholesterol [15]. These studies demonstrated a role of HDL in preventing the accumulation of cholesterol inside cells and in the arterial subendothelial space. In addition, these data indicated that HDL may directly target crystalline cholesterol for dissolution. Complement is another plasma protein that interacts with cholesterol crystals leading to the activation of the complement system. These studies were the first to propose that chrystalline cholesterol was directly involved in activation of the immune system and atherosclerosis progression. High concentrations of cholesterol - when provided as cholesterol-loaded lipid A liposomes - can induce antibodies against cholesterol crystals, demonstrating that cholesterol can indeed be immunogenic [16]. It is also possible that in patients with IgG antibodies against crystalline cholesterol the classical complement pathway can be activated by cholesterol crystals [17]. These experiments provided evidence for the immunostimulatory potential of cholesterol, especially in a crystalline state, and its potential involvement in cardiovascular disease.
Cholesterol crystals and inflammation
Cholesterol crystals have also been implicated in diseases other than atherosclerosis. For instance, crystalline cholesterol can be found in cholecystolithiasis. In this disorder, a biochemical imbalance of lipids and bile salts in the bile can mediate precipitation of cholesterol leading to gallstones and gallbladder inflammation. Indeed, cholesterol monohydrate crystals form in supersaturated bile and they are regarded as a prerequisite for the development of cholesterol gallstones [18]
Chronic apical periodontitis is another cholesterol-crystal related disease, which bears some analogy to atherosclerosis. Apical periodontitis develops in response to endodontic microbial infection of the root canal system. The conventional treatment involves the elimination of the pathogens followed by orthograde root filling to prevent reinfection, but in rare cases this treatment is not sufficient and a periapical accumulation of cholesterol crystals causes a non-resolving chronic inflammation. Although accumulation of cholesterol crystals in these lesions was regarded as a common histopathological feature in apical periodontitis for a long time, their role in mediating the chronic inflammation could only be deciphered much later. In an experimental study in guinea pigs it could be revealed that cholesterol crystal implants were densely surrounded by macrophages and multinucleate giant cells, suggesting that cholesterol crystals are detected by the host immune system. Even though these cells were not able to eliminate the cholesterol crystals, the induction of an inflammatory response against this crystalline material was found [19]. Furthermore, an in vitro study demonstrated that macrophages exposed to crystalline cholesterol release osteotropic cytokines like IL-1α [20]. Both, the inflammatory response and the bone-resorbing activity were implicated in the impaired healing upon endodontic treatment and the development of this chronic inflammatory disease.
A shared feature between chronic apical periodontitis and cardiovascular disease is the presence of so-called cholesterol crystal clefts that result from the deposition of large cholesterol crystals which are dissolved during histological sample preparation. This analogy suggested that similar inflammatory responses to accumulated crystalline cholesterol can occur in the arterial vessel walls.
Evidence for a role of crystalline cholesterol in cardiovascular inflammation came from studies on ACAT as a potential drug target. It was hypothesized that preventing the formation of macrophage foam cells, a hallmark of early atherosclerotic lesions, could slow down atherosclerosis progression and circumvent atherosclerosis plaque development. Thus, chemical ACAT inhibitors were tested in clinical trials in patients with advanced atherosclerosis. However against expectations, ACAT inhibition did not decrease but rather increased atheroma development [21, 22]. Moreover, experiments in ACAT-1 knockout revealed that ACAT-1 deficiency does not prevent the development of atherosclerotic lesions but results in the accumulation of extracellular crystalline cholesterol accompanied by a marked inflammatory response [23]. These studies indicated that cholesterol phase transition to its crystalline form is linked to inflammation in tissues. Nevertheless, the involvement of cholesterol crystals in cardiovascular inflammation had largely been disregarded since crystalline cholesterol could primarily be found in later stages of atherosclerotic plaque development and thus their appearance was thought to be the consequence of disease progression.
Cholesterol crystal formation in atherosclerosis
Atherosclerosis is a progressive disease starting with the accumulation of lipids, lipoproteins and immune cells in the arterial wall. Disease progression results in the narrowing of the arterial lumen due to continuous plaque growth. Advanced atherosclerotic lesions exhibit a lipid-rich necrotic core that may be covered by a so-called fibrous cap composed of extracellular matrix components and smooth muscle cells. The thinning of this fibrous cap in advanced disease is a critical event as it decreases plaque stability. This favors lesion rupture and luminal thrombosis which can trigger the acute clinical events myocardial infarction and stroke [24].
It is well recognized that cardiovascular inflammation plays a central role in atherosclerosis. Although some viral pathogens and bacterial pathogens have been associated with the development of human atherosclerosis, a study using germ-free mice indicated that the inflammation driving the atherosclerotic process is independent of an infectious organism and suggested that atherogenesis can be triggered by endogenous molecules [25]. A suspect for such an endogenous trigger was cholesterol, which was known to accumulate in atherosclerotic plaques in form of intracellular cholesterol esters in foam cells or as intra- and extracellular crystalline material. Abela and colleagues investigated the role of crystalline cholesterol in advanced atherosclerotic lesions. They observed that crystallization of cholesterol led to volume expansion and could result in sharp-edged cholesterol crystals with the potential to penetrate biological membranes [26]. Thus, cholesterol crystallization was assumed to be an inducer of plaque rupture as they had the potential to perforate the outer layers of atherosclerotic plaques (Figure 1). Indeed, scanning electron microscopy of human coronary arteries provided evidence for this hypothesis [27] and revealed cholesterol crystals perforating the arterial intima in patients that had died from acute coronary syndrome, but not in those that had severe atherosclerosis but no acute cardiac events [28]. These studies were the first to propose that cholesterol crystals can trigger plaque disruption and vascular injury.
Fig 1.
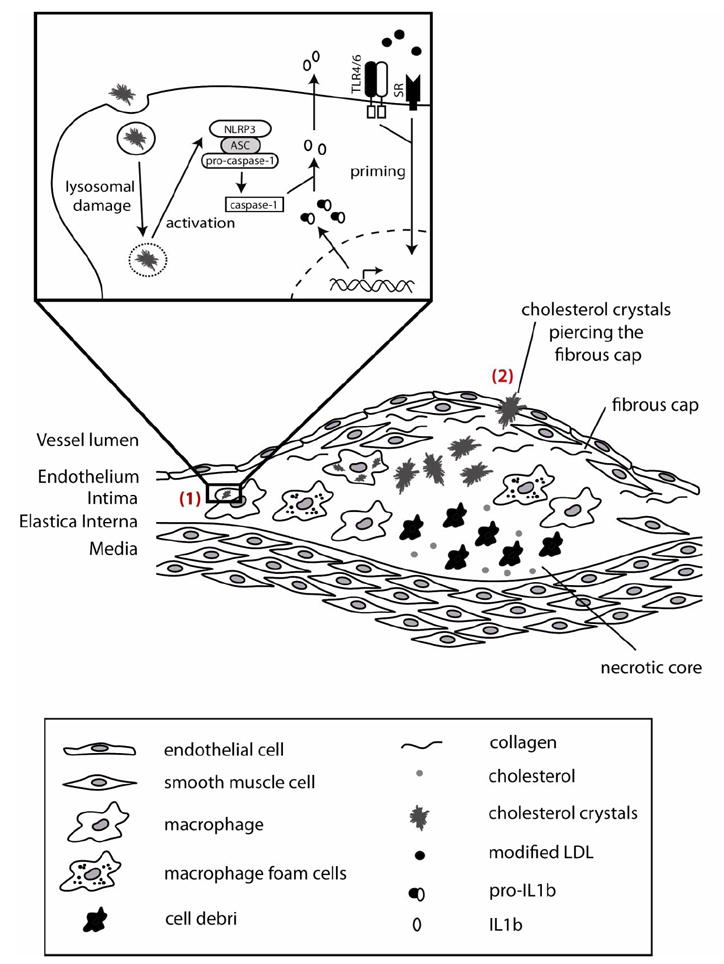
Model of cholesterol crystal induced inflammation in atherosclerosis. Two ways of cholesterol crystal induced inflammation contributing to cardiovascular disease are proposed. (1) Cholesterol crystals phagocytosed by macrophages trigger the secretion of pro-inflammatory cytokines in atherosclerotic lesions (1). Macrophages are primed by oxidized LDL which is recognized via TLRs or scavenger receptors resulting in the expression of pro-IL-1β. Subsequent phagocytosis of cholesterol crystals induces the formation of the NLRP3 inflammasome resulting in the activation of caspase-1 which generates mature IL-1β. In advanced plaques cholesterol crystal formation is thought to trigger the physical rupture of the plaque fibrous cap, thus triggering a systemic inflammatory response to vascular wall injury.
Crystalline cholesterol can be found in all stages of atherogenesis and is present in early atherosclerotic lesions [6, 10]. Two independent studies showed that cholesterol crystals can be phagocytosed by murine or human macrophages. In phagocytic cells that engulf abundant amounts of cholesterol crystals lysosomal damage is induced, which results in the activation of the NLRP3 inflammasome with subsequent activation of caspase-1 and activation and secretion of IL-1β family cytokines [6, 7]. Of note, oxidized LDL particles, which accumulate in early atherosclerotic lesions, can activate macrophage TLRs and scavenger receptors and mediate cellular priming resulting in NLRP3 and pro-IL-1β expression, which are prerequisites for NLRP3 inflammasome mediated IL-1β secretion [6] (Figure 1). IL-1b family cytokines have long been described as potent pro-atherogenic cytokines and their appearance and activation correlated to disease severity [29]. Bone-marrow transplantation from mice deficient in NLRP3 into irradiated atherosclerosis-prone LDL-receptor (LDLR) knockout mice revealed decreased lesion development upon receiving a high-cholesterol diet [6]. Together with the findings that IL-1β-deficiency in an apolipoprotein E (ApoE) knockout atherosclerosis model attenuates disease progression [30], these results suggested that cholesterol crystal-induced NLRP3 inflammasome activation and subsequent IL-1β production can drive atherogenesis. Another study, however, in which ApoE-deficient mice lacking NLRP3 inflammasome components were tested did not show differences in atherogenesis [31]. The reasons for the difference in outcome are not entirely clear. ApoE-deficient mice are known to have a more severe atherogenic phenotype than LDLR deficient mice [32]. In addition, the ApoE-deficient mice employed in the latter study [31] were fed a much stronger atherogenic diet (about eightfold more cholesterol) for a longer time than the LDLR deficient mice in the other study [6]. It is known that several redundant mechanisms contribute to atherosclerosis development and thus experimental conditions could have a large effect on the gene-dose effect. A recent study investigating the role of autophagy in atherosclerosis provided futher evidence for the importance of inflammasome activation on cardiovascular disease progression [33]. Macrophage-specific ATG5-null (ATG5-mϕKO) mice, a common mouse model for autophagy deficiency, showed enlarged atherosclerotic plaques with increased cholesterol crystal content. It could furthermore be demonstrated that macrophages deficient in autophagy had increased cholesterol crystal-mediated inflammasome activation resulting in an enhanced proatherogenic IL-1β response.
Collectively, the studies presented above [6, 7, 33] indicate that the inflammatory response caused by cholesterol crystal-induced NLRP3 inflammasome activation represents one of the driving forces for atherosclerosis development and progression.
Potential therapeutic approaches based on CC induced inflammation
Despite the progress in disease prevention and advances in the medical and surgical treatment of atherosclerosis during the past decades, cardiovascular disease remains to be the leading cause of mortality worldwide. Therefore, novel therapeutic strategies to treat atherosclerosis and associated diseases are urgently needed. The findings that cholesterol crystals and the subsequently induced inflammation are critical in the early and late atherosclerotic process [6, 34], indicate that reducing plaque cholesterol crystal content or blocking the inflammasome pathway could decrease the initiation or progression of atherosclerosis.
Prevention of cholesterol crystal formation might be achieved by lowering the plaque cholesterol load and enhancing the retrograde cholesterol transport, for instance by raising plasma levels of HDL or by increasing the expression of ABC transporters to promote HDL function. As low concentrations of HDL in the blood are among the most prominent risk factors for cardiovascular disease [35], intense efforts are under way to develop methods to increase HDL levels. In particular, cholesteryl ester transfer protein (CETP) is regarded as a potential drug target as it can significantly increase HDL concentrations while decreasing LDL levels [36]. However, initial clinical trials evaluating the effects of the CETP inhibitor torcetrapib revealed disappointing results. Despite beneficial changes in lipoprotein levels major cardiac events and the overall mortality rate were significantly increased, presumably due to off-target effects such as elevated blood pressure, resulting in the premature termination of the ILLUMINATE study. Nevertheless, CETP remains a promising drug target since another CETP inhibitor (anacetrapib) and a CETP modulator (dalcetrapib) do not possess these unfavorable off-target effects like torcetrapib [37]. However, a phase III trial assessing the effectiveness of dalcetrapib (dal-OUTCOMES) in preventing atherosclerosis related morbidity has recently been halted because of futility [38]. Another clinical trial, REVEAL, which tests the effectiveness of anacetrapib, is still ongoing.
Further therapeutic strategies for cardiovascular disease involve the reduction of vessel wall inflammation. As mentioned above, proinflammatory IL-1 cytokines play a pivotal role in atherogenesis. Under physiological conditions the proinflammatory activity of IL-1 cytokines is controlled by a naturally occurring IL-1 receptor agonist (IL-1Ra). This structural homologue of IL-1 competes with IL-1 cytokines for binding to the IL-1 receptor (IL-1R) without inducing its activation [39]. The proatherogenic character of IL-1 cytokines was demonstrated in various animal studies. The deficiency of IL-1β in an atherosclerosis mouse model decreased atherosclerosis progression [30], whereas IL-1Ra-deficiency promoted atherosclerotic lesion development [40]. Furthermore, overexpression IL-1Ra [41] or administration of recombinant IL-1Ra [42] reduced the size of atherosclerotic lesions. Finally, a clinical study using canakinumab, a human monoclonal neutralizing IL-1β antibody is under way. The Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS) will evaluate the impact of IL-1β inhibition on major cardiac events in patients with preexisting coronary artery disease [43]. IL-1β could, however, also exert protective functions in advanced atherosclerosis as it can mediate pro-fibrotic tissue responses. Indeed, it was recently demonstrated that atherosclerotic plaques of IL-1R deficient ApoE knockout mice placed on a high-cholesterol diet show features of plaque instability in advanced lesions, such as reduced plaque smooth muscle cell, plaque collagen content and increased intraplaque hemorrhage [44]. These effects could be beneficial in maintaining stable plaques. Nevertheless, it is also very established that atherogenesis and lesion size is dramatically reduced in mice with altered IL-1 signaling and the benefits of anti-IL-1β likely outweighs potential risks.
While there is hope for the success of the mentioned clinical trials, further therapeutic approaches might be directed towards directly targeting the elicitors of vascular inflammation. In particular, potential agents mediating the dissolution of cholesterol crystals and thus promoting the efflux of excess cholesterol from atherosclerotic lesions might have beneficial effects. Furthermore, a better understanding of the molecular mechanisms resulting in NLRP3 inflammasome activation by cholesterol crystals might reveal novel potential drug targets for the treatment of atherosclerosis.
Conclusions
The immune system recognizes when endogenous molecules such as uric acid or cholesterol change their physical state from soluble to crystalline. These mechanisms likely have evolved to recognize sterile tissue damage. After tissue damage uric acid and potentially other molecules are misplaced from intracellular pools to the extracellular environment and the capacity of cells to control the amount of free cholesterol diminishes. Immune cells in the tissues, such as tissue macrophages of dendritic cells can recognize these danger signals and alert other immune cells via the production of cytokines of the IL-1b family. The mechanisms that are at play to recognize sterile tissue damage can, however, also lead to chronic inflammatory responses, if the danger signals appear chronically or are not efficiently cleared such as seen in arteries in hypercholesteremic individuals or joints of hyperuricemic patients. It is likely that pharmacological prevention of cholesterol crystallization in early stages of disease or the reduction of cholesterol crystal plaque content in advanced stages might reduce atherogenesis. Therefore, cholesterol crystals themselves might represent a new target for approaches to treat atherosclerosis.
Footnotes
Disclosure No potential conflicts of interest relevant to this article were reported.
References
- 1.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140(6):805–20. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 2.Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 2010;10(12):826–37. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stutz A, Golenbock DT, Latz E. Inflammasomes: too big to miss. J Clin Invest. 2009;119(12):3502–11. doi: 10.1172/JCI40599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dostert C, et al. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320(5876):674–7. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hornung V, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 2008;9(8):847–56. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6••.Duewell P, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464(7293):1357–61. doi: 10.1038/nature08938. Demonstration that cholesterol crystals can activate the NLRP3 inflammasome and contribute to atherosclerosis development in a murine model of atherosclerosis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7•.Rajamaki K, et al. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PLoS One. 2010;5(7):e11765. doi: 10.1371/journal.pone.0011765. This report demonstrates that human immune cells can be activated by cholesterol crystals to secrete IL-1β cytokines. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boya P, Kroemer G. Lysosomal membrane permeabilization in cell death. Oncogene. 2008;27(50):6434–51. doi: 10.1038/onc.2008.310. [DOI] [PubMed] [Google Scholar]
- 9.Chang TY, et al. Cholesterol sensing, trafficking, and esterification. Annu Rev Cell Dev Biol. 2006;22:129–57. doi: 10.1146/annurev.cellbio.22.010305.104656. [DOI] [PubMed] [Google Scholar]
- 10.Small DM. George Lyman Duff memorial lecture. Progression and regression of atherosclerotic lesions Insights from lipid physical biochemistry. Arteriosclerosis. 1988;8(2):103–29. doi: 10.1161/01.atv.8.2.103. [DOI] [PubMed] [Google Scholar]
- 11.Tangirala RK, et al. Formation of cholesterol monohydrate crystals in macrophage-derived foam cells. J Lipid Res. 1994;35(1):93–104. [PubMed] [Google Scholar]
- 12.Kellner-Weibel G, et al. Crystallization of free cholesterol in model macrophage foam cells. Arterioscler Thromb Vasc Biol. 1999;19(8):1891–8. doi: 10.1161/01.atv.19.8.1891. [DOI] [PubMed] [Google Scholar]
- 13.Adams CW, Abdulla YH. The action of human high density lipoprotein on cholesterol crystals. Part 1. Light-microscopic observations. Atherosclerosis. 1978;31(4):465–71. doi: 10.1016/0021-9150(78)90142-9. [DOI] [PubMed] [Google Scholar]
- 14.Abdulla YH, Adams CW. The action of human high density lipoprotein on cholesterol crystals. Part 2. Biochemical observations. Atherosclerosis. 1978;31(4):473–80. doi: 10.1016/0021-9150(78)90143-0. [DOI] [PubMed] [Google Scholar]
- 15.Krut LH. Clearance of subcutaneous implants of cholesterol in the rat promoted by oxidation products of cholesterol. A postulated role for oxysterols in preventing atherosclerosis. Atherosclerosis. 1982;43(1):105–18. doi: 10.1016/0021-9150(82)90103-4. [DOI] [PubMed] [Google Scholar]
- 16.Swartz GM, Jr, et al. Antibodies to cholesterol. Proc Natl Acad Sci U S A. 1988;85(6):1902–6. doi: 10.1073/pnas.85.6.1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hammerschmidt DE, et al. Cholesterol and atheroma lipids activate complement and stimulate granulocytes. A possible mechanism for amplification of ischemic injury in atherosclerotic states. J Lab Clin Med. 1981;98(1):68–77. [PubMed] [Google Scholar]
- 18.Sedaghat A, Grundy SM. Cholesterol crystals and the formation of cholesterol gallstones. N Engl J Med. 1980;302(23):1274–7. doi: 10.1056/NEJM198006053022302. [DOI] [PubMed] [Google Scholar]
- 19.Nair PN, Sjogren U, Sundqvist G. Cholesterol crystals as an etiological factor in non-resolving chronic inflammation: an experimental study in guinea pigs. Eur J Oral Sci. 1998;106(2 Pt 1):644–50. doi: 10.1046/j.0909-8836.1998.eos106206.x. [DOI] [PubMed] [Google Scholar]
- 20.Sjogren U, et al. Bone-resorbing activity from cholesterol-exposed macrophages due to enhanced expression of interleukin-1alpha. J Dent Res. 2002;81(1):11–6. doi: 10.1177/002203450208100104. [DOI] [PubMed] [Google Scholar]
- 21.Nissen SE, et al. Effect of ACAT inhibition on the progression of coronary atherosclerosis. N Engl J Med. 2006;354(12):1253–63. doi: 10.1056/NEJMoa054699. [DOI] [PubMed] [Google Scholar]
- 22.Meuwese MC, et al. ACAT inhibition and progression of carotid atherosclerosis in patients with familial hypercholesterolemia: the CAPTIVATE randomized trial. JAMA. 2009;301(11):1131–9. doi: 10.1001/jama.301.11.1131. [DOI] [PubMed] [Google Scholar]
- 23.Accad M, et al. Massive xanthomatosis and altered composition of atherosclerotic lesions in hyperlipidemic mice lacking acyl CoA:cholesterol acyltransferase 1. J Clin Invest. 2000;105(6):711–9. doi: 10.1172/JCI9021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lusis AJ. Atherosclerosis. Nature. 2000;407(6801):233–41. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wright SD, et al. Infectious agents are not necessary for murine atherogenesis. J Exp Med. 2000;191(8):1437–42. doi: 10.1084/jem.191.8.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abela GS, Aziz K. Cholesterol crystals cause mechanical damage to biological membranes: a proposed mechanism of plaque rupture and erosion leading to arterial thrombosis. Clin Cardiol. 2005;28(9):413–20. doi: 10.1002/clc.4960280906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abela GS, Aziz K. Cholesterol crystals rupture biological membranes and human plaques during acute cardiovascular events--a novel insight into plaque rupture by scanning electron microscopy. Scanning. 2006;28(1):1–10. doi: 10.1002/sca.4950280101. [DOI] [PubMed] [Google Scholar]
- 28.Abela GS, et al. Effect of cholesterol crystals on plaques and intima in arteries of patients with acute coronary and cerebrovascular syndromes. Am J Cardiol. 2009;103(7):959–68. doi: 10.1016/j.amjcard.2008.12.019. [DOI] [PubMed] [Google Scholar]
- 29.Galea J, et al. Interleukin-1 beta in coronary arteries of patients with ischemic heart disease. Arterioscler Thromb Vasc Biol. 1996;16(8):1000–6. doi: 10.1161/01.atv.16.8.1000. [DOI] [PubMed] [Google Scholar]
- 30.Kirii H, et al. Lack of interleukin-1beta decreases the severity of atherosclerosis in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 2003;23(4):656–60. doi: 10.1161/01.ATV.0000064374.15232.C3. [DOI] [PubMed] [Google Scholar]
- 31.Menu P, et al. Atherosclerosis in ApoE-deficient mice progresses independently of the NLRP3 inflammasome. Cell Death Dis. 2011;2:e137. doi: 10.1038/cddis.2011.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wouters K, et al. Understanding hyperlipidemia and atherosclerosis: lessons from genetically modified apoe and ldlr mice. Clin Chem Lab Med. 2005;43(5):470–9. doi: 10.1515/CCLM.2005.085. [DOI] [PubMed] [Google Scholar]
- 33•.Razani B, et al. Autophagy links inflammasomes to atherosclerotic progression. Cell Metab. 2012;15(4):534–44. doi: 10.1016/j.cmet.2012.02.011. This paper demonstrates that autophagy regulates NLRP3 inflammasome activation by cholesterol crystals in atherosclerotic plaques. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abela GS. Cholesterol crystals piercing the arterial plaque and intima trigger local and systemic inflammation. J Clin Lipidol. 2010;4(3):156–64. doi: 10.1016/j.jacl.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 35.Gordon T, et al. High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. Am J Med. 1977;62(5):707–14. doi: 10.1016/0002-9343(77)90874-9. [DOI] [PubMed] [Google Scholar]
- 36.Redondo S, et al. Emerging therapeutic strategies to enhance HDL function. Lipids Health Dis. 2011;10:175. doi: 10.1186/1476-511X-10-175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hewing B, Fisher EA. Rationale for cholesteryl ester transfer protein inhibition. Curr Opin Lipidol. 2012;23(4):372–6. doi: 10.1097/MOL.0b013e328353ef1d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schwartz GG, et al. Effects of Dalcetrapib in Patients with a Recent Acute Coronary Syndrome. N Engl J Med. 2012 doi: 10.1056/NEJMoa1206797. [DOI] [PubMed] [Google Scholar]
- 39.Nicklin MJ, et al. Arterial inflammation in mice lacking the interleukin 1 receptor antagonist gene. J Exp Med. 2000;191(2):303–12. doi: 10.1084/jem.191.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Isoda K, et al. Lack of interleukin-1 receptor antagonist modulates plaque composition in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2004;24(6):1068–73. doi: 10.1161/01.ATV.0000127025.48140.a3. [DOI] [PubMed] [Google Scholar]
- 41.Devlin CM, et al. Genetic alterations of IL-1 receptor antagonist in mice affect plasma cholesterol level and foam cell lesion size. Proc Natl Acad Sci U S A. 2002;99(9):6280–5. doi: 10.1073/pnas.092324399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Elhage R, et al. Differential effects of interleukin-1 receptor antagonist and tumor necrosis factor binding protein on fatty-streak formation in apolipoprotein E-deficient mice. Circulation. 1998;97(3):242–4. doi: 10.1161/01.cir.97.3.242. [DOI] [PubMed] [Google Scholar]
- 43•.Ridker PM, et al. Interleukin-1beta inhibition and the prevention of recurrent cardiovascular events: rationale and design of the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS) Am Heart J. 2011;162(4):597–605. doi: 10.1016/j.ahj.2011.06.012. Design of the CANTOS study testing the effect of anti-IL-1β therapy in atherosclerosis. The first human test of the inflammation hypothesis for atherogenesis. [DOI] [PubMed] [Google Scholar]
- 44.Alexander MR, et al. Genetic inactivation of IL-1 signaling enhances atherosclerotic plaque instability and reduces outward vessel remodeling in advanced atherosclerosis in mice. J Clin Invest. 2012;122(1):70–9. doi: 10.1172/JCI43713. [DOI] [PMC free article] [PubMed] [Google Scholar]