

Abstract
Objective
To determine whether 2-(1-{6-[(2-fluorine18–labeled fluoroethyl)methylamino]-2-napthyl}ethylidene) malononitrile ([18F]FDDNP) brain regional values in individuals without dementia predict and correlate with future cognitive change.
Design
Two-year, longitudinal follow-up study.
Setting
A university research institute.
Participants
Volunteer sample of 43 middle-aged and older persons (median age, 64 years), including 21 with mild cognitive impairment (MCI) and 22 with normal aging.
Main Outcome Measures
Longitudinal [18F]FDDNP positron emission tomography (PET) binding values in the medial and lateral temporal, posterior cingulate, parietal, frontal, and global (mean) regions of interest; neuropsychological test battery measuring 5 cognitive domains, including memory, language, attention (and information-processing speed), executive functioning, and visuospatial ability.
Results
For the entire study group (MCI and normal aging), increases in frontal, posterior cingulate, and global binding at follow-up correlated with progression of memory decline (r=−0.32 to −0.37, P=.03 to .01) after 2 years. Moreover, higher baseline [18F]FDDNP binding was associated with future decline in most cognitive domains, including language, attention, executive, and visuospatial abilities (r=−0.31 to −0.56, P=.05 to .002). For the MCI group, frontal and parietal [18F]FDDNP binding yielded the greatest diagnostic accuracy in identifying converters to Alzheimer disease vs nonconverters after 2 years, with an area under the receiver operating characteristic curve of 0.88 (95% CI, 0.72–1.00) compared with 0.68 (95% CI, 0.45–0.91) for medial temporal binding.
Conclusions
[18F]FDDNP PET regional binding patterns are consistent with known neuropathologic patterns of plaque and tangle brain accumulation, spreading from the medial temporal to other neocortical regions as disease progresses. Because binding patterns predict future cognitive decline and increase over time along with clinical decline, [18F]FDDNP PET scanning may have practical utility in identifying people at risk for future cognitive decline and in tracking the effectiveness of novel interventions designed to prevent or delay neurodegeneration and cognitive decline.
Neurodegeneration and cognitive losses afflict millions of people as they age. Nearly 20% of people 65 years or older have mild cognitive impairment (MCI),1 and 10% have dementia.2,3 Such a high prevalence has led to recent research on the development of brain imaging tools to track the neuropathologic changes associated with these conditions. Since the initial positron emission tomography (PET) report4 demonstrating the feasibility of in vivo imaging of amyloid senile plaques and tau neurofibrillary tangles using 2-(1-{6-[(2-fluorine 18–labeled fluoroethyl) methylamino]-2-napthyl}ethylidene) malononitrile ([18F]FDDNP) in living people, several molecular imaging probes (eg, carbon 11–Pittsburgh Compound B and fluorine 18–labeled florbetapir5–8) have been developed to image amyloid in patients with Alzheimer disease (AD) and MCI. Although carbon 11–Pittsburgh Compound B and fluorine 18–labeled florbetapir5–8 measure only amyloid and show relatively higher cortical signals than does [18F]FDDNP in patients with AD compared with controls, [18F]FDDNP is the only molecular PET probe that provides a measure of both amyloid and tau.9,10
Previous cross-sectional studies9–11 have shown that [18F]FDDNP brain binding patterns correspond to the known neuropathologic deposition patterns determined from autopsy studies. Moreover, our previous imaging results with AD, MCI, and normal aging individuals who underwent imaging at one time point demonstrate [18F]FDDNP binding patterns consistent with neuropathologic determinations showing progressive brain accumulation originating in medial temporal regions and spreading to the frontal, parietal, and other cortical areas.12–14 Higher [18F]FDDNP binding values are also associated with older age, APOE4 carrier status, and higher cerebrospinal fluid tau levels.15,16
Using [18F]FDDNP PET, we previously reported the first longitudinal follow-up with only 12 individuals.9 In the present study of middle-aged and older adults without dementia, we extend these initial observations and report results of [18F]FDDNP PET scans and cognitive assessments at baseline and after 2 years of follow-up to assess whether [18F]FDDNP brain regional binding values increase as cognitive decline progresses and whether baseline [18F]FDDNP binding values are predictors of future cognitive decline.
METHODS
STUDY PARTICIPANTS AND CLINICAL ASSESSMENTS
We performed baseline and follow-up cognitive and neuroimaging assessments on 43 individuals selected from a pool of 319 potential volunteers. Volunteers were recruited through advertisements regarding mild memory concerns, media coverage, and referrals from physicians and families. For inclusion, volunteers needed to be willing to participate in a longitudinal study on memory and aging. Although all study participants had noticed mild memory changes, those with any form of dementia at baseline assessments were excluded. From the original pool, volunteers were excluded for the following reasons: medical and psychiatric illnesses (n=131); loss of interest (n=96); use of medications (n=38) that might affect cognition (eg, sedatives) or nonsteroidal anti-inflammatory drugs, which bind to amyloid plaques and may affect [18F]FDDNP in vivo binding values17; and inability to undergo magnetic resonance imaging (MRI) (n=11) because of pacemakers or hip replacement implants.
Participants underwent screening laboratory tests and structural imaging scans (3-dimensional MRI or computed tomography [CT]) to rule out other causes of cognitive impairment (eg, stroke, tumor)18 and for coregistration with PET scans for region-of-interest image analyses. Computed tomography instead of MRI was performed on 4 individuals because they could not tolerate MRI (eg, owing to claustrophobia or metal in the body). Those with vascular lesions apparent on MRI or CT were also excluded from the study. In addition to the Mini-Mental State Examination19 and Hamilton Rating Scale for Depression,20 a neuropsychological test battery21 was administered to assess 5 cognitive domains: (1) memory, including the Wechsler Memory Scale, third edition, logical memory (delayed score), Buschke Selective Reminding Test (total score), and Rey-Osterrieth Complex Figure Recall (3-minute delayed recall score); (2) language, including the Boston Naming Test and the F-A-S and Animal Naming fluency tests; (3) attention and information-processing speed, including Trail-Making A, Stroop color naming (Kaplan version), and Wechsler Adult Intelligence Scale, third edition, digit symbol; (4) executive functioning, including Trail-Making B and Stroop interference (Kaplan version); and (5) visuospatial ability, including Wechsler Adult Intelligence Scale, third edition, block design and Rey-Osterrieth Complex Figure Copy. To ascertain cognitive change in individuals, for each of the cognitive measures, we first calculated change scores (follow-up minus baseline). These raw change scores were converted to z scores by standardizing them to a mean of 0 and an SD of 1. A domain z score was obtained by averaging those z scores belonging to the cognitive tests in that domain. The domain z scores were used to examine associations with [18F]FDDNP brain regional binding levels.
We used standard diagnostic criteria for amnestic MCI (ie, memory impairment without other cognitive impairments), which include (1) patient awareness of a memory problem, preferably confirmed by another person who knows the patient; (2) memory impairment detected with standard assessment tests; and (3) ability to perform normal daily activities.22 For a broad definition of MCI, we also used guidelines to identify those with other MCI subtypes, including those with memory impairment and additional cognitive deficits.23 The diagnosis was corroborated by clinical judgment22 and included patients with MCI who scored 1 SD or more below the age-corrected norms because this threshold for impairment yields high sensitivity for predicting dementia.24 To balance increased sensitivity with specificity, impairment on at least 2 neuropsychological tests within 1 of the 5 cognitive domains was required.25 Patients in the MCI group did not meet diagnostic criteria for AD,18,26 and the presence of memory concerns was documented using a standardized subjective memory instrument (Memory Functioning Questionnaire)27 and clinical interview.
Volunteers with 1 or more first-degree relatives (ie, sibling or parent) with AD or dementia were classified as having a positive family history of dementia. Prior educational achievement was quantified according to years and months of school completed, beginning with elementary school (ie, first grade).
All clinical assessments were performed within 4 weeks of scanning procedures, and physicians were masked to [18F]FDDNP PET scan results. Written informed consent was obtained in accordance with the University of California, Los Angeles, Human Subjects Protection Committee procedures. Cumulative radiation dosimetry for all scans was below the mandated maximum annual dose and in compliance with state and federal regulations. Two minor adverse events occurred during PET scanning: one individual developed minor bruises at venipuncture sites and another experienced a transient headache.
SCANNING AND IMAGE ANALYSIS PROCEDURES
As previously described, [18F]FDDNP was prepared at high specific activities (>37 GBq/μmol).28 All scans (EXACT HR+ tomograph; Siemens-CTI) were performed with participants in a supine position and with the imaging plane parallel to the orbitomeatal line. A bolus of [18F]FDDNP (320–550 MBq) was injected via an indwelling venous catheter, and consecutive dynamic PET scans were performed for 1 hour. Scans were decay corrected and reconstructed using filtered back-projection (Hann filter, 5.5 mm full-width at half-maximum) with scatter and measured attenuation correction. The resulting images contained 63 contiguous sections with plane separation of 2.42 mm (EXACT HR+).
The [18F]FDDNP binding data were quantified using Logan graphical analysis with the cerebellum as the reference region.9,29 The slope of the linear portion of the Logan plot is the relative distribution volume of the tracer in a region of interest divided by that in the reference region. The relative distribution volume parametric images were generated and analyzed using regions of interest traced on the coregistered MRIs for the left and right parietal, medial temporal (limbic regions, including hippocampus, parahippocampal areas, and entorhinal cortex), lateral temporal, posterior cingulate, and frontal regions, as previously described9 (Figure 1). Each regional relative distribution volume or binding value was expressed as the mean of the left and right regions, and the global region was defined as the mean of these. Rules for region-of-interest drawing were based on the identification of gyral and sulcal landmarks with respect to the atlas of Talairach and Tournoux.30 All PET scans were read and regions of interest drawn by individuals who were masked to clinical assessments.
Figure 1.
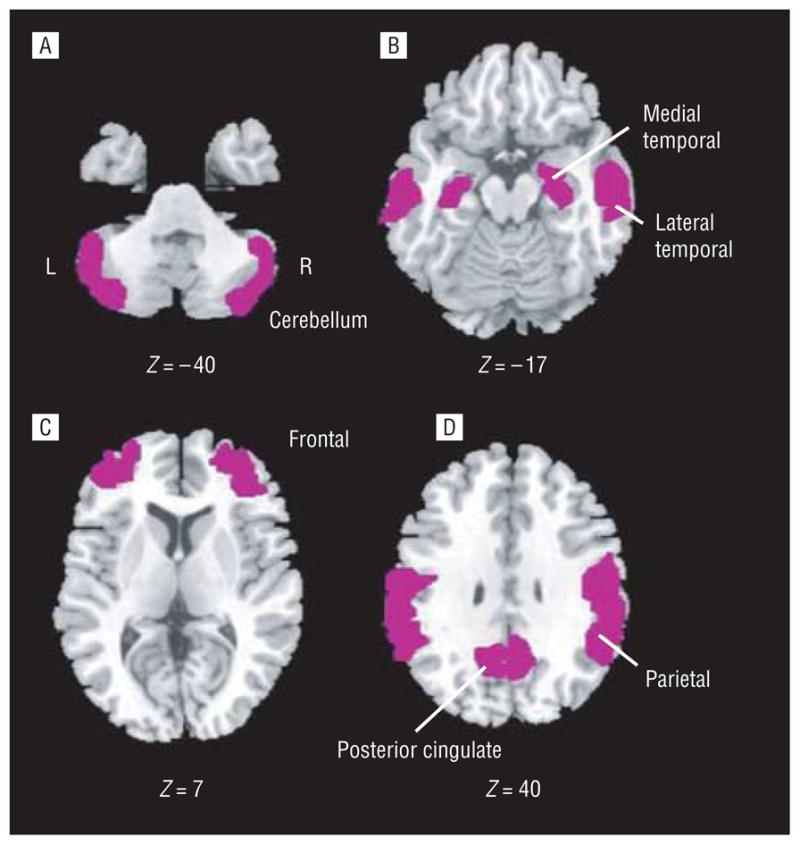
Representative regions of interest (ROIs). The magnetic resonance images show left and right ROIs (shaded in purple) used for the ROI relative distribution volume analyses, superimposed on the Ch2bet template at the indicated Montreal Neurological Institute coordinates. Images were drawn using MRIcro for Windows (version 1.39, build 4; http://www.cabiatl.com/mricro/).
STATISTICAL ANALYSIS
Data were screened for outliers and normality assumptions. Descriptive statistics were computed for the entire sample and for the MCI and the normal aging groups separately. The 2-sample Wilcoxon rank sum test was used to compare the continuous variables of cognitive groups and Fisher exact χ2 test for categorical variables. Within the MCI and normal aging groups, significance of changes in regional [18F]FDDNP binding levels was examined using the nonparametric signed rank test, and between-group comparisons were made using the nonparametric Wilcoxon 2-sample rank sum test, which also was used to compare the MCI with AD converters vs nonconverters.
To accommodate nonlinear relationships among variables, we used rank-based models to study the associations between [18F]FDDNP binding levels and cognitive changes. Thus, to study whether changes in [18F]FDDNP binding were associated with changes in cognition at follow-up, nonparametric analyses of covariance were estimated with cognitive domain change scores as dependent variables and changes in [18F]FDDNP measures as predictors. To limit the number of tests, we used global [18F]FDDNP binding level as the predictor with each of the cognitive domain change scores, and only those domains found to have significant associations were further analyzed to determine region-specific associations. Cognitive status (MCI or normal aging) and the interaction of cognitive status and [18F]FDDNP binding level also were used as predictors to determine differences between the MCI and normal aging groups. For significant associations, findings are presented as Spearman correlation coefficients. We also compared individuals on their cognitive domain z score changes using the Wilcoxon 2-sample rank sum test according to 2 groups: those with increased global binding of more than 0.5 SD (increased [18F]FDDNP group) and those whose binding level changes were within this range (stable [18F]FDDNP group). A cutoff of a 0.5-SD increase was chosen to determine whether subtle changes in [18F]FDDNP binding are associated with cognitive changes. To assess whether MCI and normal aging groups can be pooled together for these correlations, we performed a bootstrap randomization experiment, which suggested that the 2 groups can be pooled together for correlation estimation.
To examine whether baseline [18F]FDDNP cortical binding levels were associated with cognitive changes at follow-up, nonparametric analyses of covariance were estimated with the cognitive domain change scores as dependent variables, but using baseline [18F]FDDNP binding as predictors, in addition to cognitive status and interaction of cognitive status and [18F]FDDNP binding. Global [18F]FDDNP binding was examined first, and follow-up regional analyses were performed for those domains with significant findings.
To explore further the ability of baseline regional [18F]FDDNP binding to predict cognitive change in the MCI group, we used receiver operating characteristic (ROC) curves. We obtained areas under the ROC curves using baseline medial temporal, frontal, and parietal [18F]FDDNP binding values to predict converters to AD vs nonconverters. Sensitivity and specificity of baseline frontal or parietal and medial temporal [18F]FDDNP binding were also calculated to classify MCI patients as converters vs nonconverters. A cutoff value of baseline [18F]FDDNP binding was chosen based on optimal balance between sensitivity and specificity. We selected the best cut point as the point on the ROC curve farthest away from the line connecting the lower left-hand and upper right-hand corners (greatest distance of ROC curve from the line of random prediction).31
Analyses were performed with the SAS statistical software, version 9.2 (SAS Institute, Inc), and StatXact 8 (Cytel). All reported P values were 2-sided.
RESULTS
BASELINE DEMOGRAPHICS
Study participants without dementia (both MCI and normal aging) were middle-aged or older (median age, 64 years; range, 40–87 years) and highly educated (median educational achievement, 16 years; range, 12–24 years). They also showed relatively minimal impairment on cognitive testing (median Mini-Mental State Examination score, 29; range, 24–30). Most of them (72%) had a family history of dementia in at least 1 first-degree relative. Of the 21 participants with MCI, 12 showed memory impairment consistent with amnestic MCI and 9 had amnestic MCI plus deficits in other cognitive areas. The MCI and normal aging groups did not differ significantly according to age, educational achievement, or other demographic characteristics (Table 1). The median interval between baseline and follow-up assessments was 2.0 years (range, 1.4–3.2 years).
Table 1.
Demographic and Clinical Characteristics of Study Participants at Baselinea
| Characteristic | MCI (n=21) | Normal Aging (n=22) |
|---|---|---|
| Mini-Mental State Examination score | 29 (24–30) | 29 (27–30) |
| Age, y | 64 (40–84) | 65.5 (40–86) |
| Educational achievement, y | 16 (12–24) | 17 (12–22) |
| Female sex, No. (%) | 12 (57) | 14 (64) |
| Family history of dementia, No. (%) | 15 (71) | 16 (73) |
| Hamilton Depression Scale score | 1 (0–9) | 2 (0–9) |
| Neuropsychological test scores | ||
| Logical memory, delayed score | 18 (10–40) | 19 (17–45) |
| Buschke Selective Reminding Test, total score | 77 (45–113) | 104.5 (71–134) |
| Rey-Osterrieth Complex Figure Recall, delayed score | 11 (2–16.5) | 16.75 (6.5–24) |
| Boston Naming Test | 56 (36–60) | 58.5 (44–60) |
| F-A-S fluency | 35 (18–65) | 43.5 (23–61) |
| Animal Naming fluency | 17 (9–31) | 20 (11–29) |
| Trail-Making A | 32 (21–66) | 28 (17–46) |
| Stroop color naming | 70 (51–110) | 58.5 (32–87) |
| WAIS-III digit symbol | 55 (28–113) | 65.5 (55–99) |
| Trail-Making B | 95 (39–225) | 68.5 (39–108) |
| Stroop interference | 145 (103–236) | 116.5 (70–160) |
| WAIS-III block design | 35 (10–46) | 40 (18–60) |
| Rey-Osterrieth Complex Figure Copy | 29 (18.5–33) | 30.5 (22–35) |
| Subjective memory concernsb | ||
| Frequency of forgetting | 147 (91–201) | 175 (86–203) |
| Seriousness of forgetting | 74 (46–126) | 97 (55–126) |
| Retrospective functioning | 16 (7–27) | 13 (6–21) |
| Mnemonics use | 22 (11–43) | 19 (8–33) |
Abbreviations: MCI, mild cognitive impairment; WAIS-III, Wechsler Adult Intelligence Scale Third Edition.
Data are presented as median (range) unless otherwise indicated.
From the Memory Functioning Questionnaire; higher scores indicate fewer self-reported memory problems and less use of mnemonics. Mean (SD) norms for people 60 to 69 years old and 70 to 79 years old, respectively: frequency of forgetting, 152 (28) and 149 (29); seriousness of forgetting, 86 (20) and 84 (21); retrospective functioning, 18 (6) and 18 (6); and mnemonics use, 31 (9) and 30 (10).
[18F]FDDNP BINDING CHANGES AT FOLLOW-UP
At the 2-year follow-up assessment, the MCI group (n=21) showed significant increases in [18F]FDDNP binding values in frontal (mean increase, 3.6%; Wilcoxon signed rank statistic [S]=106.5; P<.001), parietal (mean increase, 3.1%; S=86.5; P=.002), posterior cingulate (mean increase, 4.1%; S=101.5; P< .001), and global (mean increase, 2.7%; S=107.5; P<.001) regions, whereas binding values in the medial temporal region, already at high levels, did not increase significantly during that period. The normal aging group did not show significant increases in any region. The 2 groups were significantly different in their [18F]FDDNP binding changes in these same regions: frontal (Wilcoxon statistic [W]=583, P=.005), parietal (W=561, P=.02), posterior cingulate (W=577, P=.01), and global (W=572, P=.01).
CORRELATIONS OF [18F]FDDNP BINDING CHANGES AND COGNITIVE CHANGES AT FOLLOW-UP
For the entire study group (N=43), global (t41=−2.55, r=−0.37, P=.01), frontal (t41=−2.56, r =−0.37, P =.01), and posterior cingulate (t41 = −2.18, r = −0.32, P = .03) [18F]FDDNP binding changes correlated with memory domain score changes at follow-up (Figure 2). The interaction term was not significant, indicating that the MCI and normal aging groups did not differ in these associations. We found that 40% of the study participants (12 in the MCI group and 5 in the normal aging group) had increased (>0.5 SD) global [18F]FDDNP binding at follow-up compared with baseline, and this group had comparable baseline cognitive domain scores compared with the 26 individuals with stable [18F]FDDNP binding levels. However, the increased global [18F]FDDNP binding group showed significantly greater memory decline at follow-up compared with the stable group (W=283, P=.03) (Figure 3).
Figure 2.

Plot of global 2-(1-{6-[(2-fluorine 18–labeled fluoroethyl)methyl-amino]-2-napthyl}ethylidene) malononitrile ([18F]FDDNP) binding change vs memory change. For the entire study group (N=43), global [18F]FDDNP binding changes correlated with memory domain score changes at follow-up (t41=−2.55, r =−0.37, P =.01). CTL indicates control; MCI, mild cognitive impairment.
Figure 3.

Mean cognitive change in study participants with stable vs increased global 2-(1-{6-[(2-fluorine 18–labeled fluoroethyl)methyl-amino]-2-napthyl}ethylidene) malononitrile ([18F]FDDNP) binding values. Participants who increased in their global [18F]FDDNP binding had greater memory decline at follow-up compared with those who remained stable. Error bars indicate SDs.
BASELINE [18F]FDDNP BRAIN REGIONAL BINDING AND COGNITIVE CHANGE SCORES
For all study participants, higher baseline global [18F]FDDNP signals were associated with greater decreases in executive function, language, attention and information processing speed, and visuospatial function at follow-up. Additional analyses for the entire study group revealed several regional (frontal, parietal, medial, and lateral temporal) baseline [18F]FDDNP binding level correlations with cognitive domain change scores (Table 2). For executive function domain change, the MCI and normal aging subgroups differed in their associations with baseline [18F]FDDNP binding. For the MCI group, but not the normal aging group, baseline [18F]FDDNP signals (global: t19=−3.13, r =−0.56, P =.01; frontal: t19=−2.06, r=−0.44, P=.05; parietal: t19=−2.88, r =−0.46, P =.03; medial temporal: t19=−2.35, r =−0.44, P =.05) were associated with future decreases in executive function domain scores. For the other domain change scores, the MCI and normal aging groups did not differ in their associations with baseline [18F]FDDNP binding.
Table 2.
Significant Correlations Between Baseline Regional [18F]FDDNP Binding Values and Cognitive Domain Change Scores
| Cognitive Domain and Region of Interesta | t41 | rb | P Valueb |
|---|---|---|---|
| Executive functioning | |||
| Global | −2.51 | −0.36 | .02 |
| Frontal | −1.88 | −0.29 | .06 |
| Parietal | −2.03 | −0.30 | .05 |
| Medial temporal | −2.65 | −0.38 | .01 |
| Language | |||
| Global | −2.70 | −0.39 | .01 |
| Parietal | −3.01 | −0.42 | .004 |
| Medial temporal | −2.07 | −0.31 | .05 |
| Visuospatial | |||
| Global | −1.99 | −0.34 | .03 |
| Frontal | −2.99 | −0.45 | .002 |
| Parietal | −2.41 | −0.39 | .01 |
| Attention and information processing | |||
| Global | −2.52 | −0.37 | .01 |
| Lateral temporal | −2.56 | −0.37 | .01 |
Abbreviation: [18F]FDDNP, 2-(1-{6-[(2-fluorine 18–labeled fluoroethyl)methylamino]-2-napthyl}ethylidene) malononitrile.
Memory domain change scores were not significantly associated with baseline global [18F]FDDNP binding and hence were not reported in this table.
Spearman correlation coefficient and the associated 2-sided P value.
Of the 21 individuals with MCI at baseline, 6 converted to AD at follow-up. These 6 converters had higher frontal (W=97, P=.03), parietal (W=94, P=.04), and global (W=96, P=.03) baseline [18F]FDDNP binding values compared with the 15 nonconverters. In the normal aging group, only 3 study participants converted to MCI at follow-up. It is noteworthy that of these 3 participants, 2 had the highest regional [18F]FDDNP signals at baseline among the normal aging group. One of these study participants had the highest binding values in this normal aging group in the medial temporal region, whereas another had the highest values in the frontal and parietal regions, in addition to being in the 75th percentile in medial temporal region [18F]FDDNP binding.
For a more detailed assessment of the ability of [18F]FDDNP binding values to predict future cognitive decline in the MCI group, we further explored the relationship between baseline regional [18F]FDDNP binding values and MCI conversion to AD using ROC analysis. The results of the ROC analysis comparing medial temporal, frontal, parietal, and the mean of frontal and parietal [18F]FDDNP binding found that frontal and parietal binding yielded the greatest diagnostic accuracy to predict converters. The areas under the ROC curve were 0.68 (95% CI, 0.45–0.91) for medial temporal [18F]FDDNP binding, 0.84 (95% CI, 0.64–1.00) for frontal binding, 0.81 (95% CI, 0.60–1.00) for parietal binding, and 0.88 (95% CI, 0.72–1.00) for frontal and parietal binding.
To calculate sensitivity and specificity of [18F]FDDNP binding to classify converters vs nonconverters in the MCI group, the optimal cutoff value for [18F]FDDNP binding determined from the ROC curve was 1.07 for frontal and parietal and 1.14 for medial temporal. All of the 6 MCI to AD converters had frontal and parietal [18F]FDDNP binding higher than this cutoff value, whereas 10 of the 15 nonconverters had frontal and parietal [18F]FDDNP binding lower than this cutoff value (Fisher exact, P=.01), resulting in a sensitivity of 100% and a specificity of 66.7%. For the medial temporal region, 5 of the 6 MCI to AD converters showed binding higher than the cutoff value, and 9 of the 15 nonconverters showed binding lower than the cutoff value (Fisher exact, P=.15), resulting in a sensitivity of 83.3% and a specificity of 60.0%. A plot of baseline regional [18F]FDDNP binding values against executive function change scores (Figure 4) indicates how baseline frontal and parietal binding is a better predictor of conversion to AD in MCI patients than is medial temporal binding. Figure 5 illustrates an MCI patient with low frontal binding who did not convert to AD after 2 years compared with an MCI patient with higher frontal binding who converted to AD.
Figure 4.
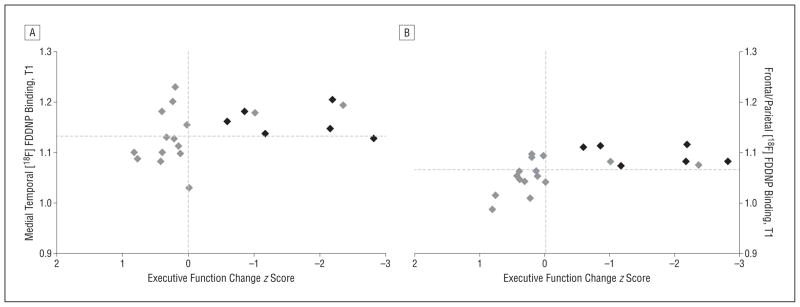
Plot of baseline 2-(1-{6-[(2-fluorine 18–labeled fluoroethyl)methylamino]-2-napthyl}ethylidene) malononitrile ([18F]FDDNP) binding vs executive function change score for mild cognitive impairment (MCI). Gray diamonds indicate MCI patients who remained in the MCI group at follow-up; black diamonds denote MCI patients who converted to Alzheimer disease status at follow-up. The vertical lines indicate zero change (follow-up minus baseline). The horizontal lines indicate the cutoff values for [18F]FDDNP binding chosen based on receiver operating characteristic curves (medial temporal, 1.135; frontal and parietal, 1.07). T1 indicates time 1 (baseline).
Figure 5.
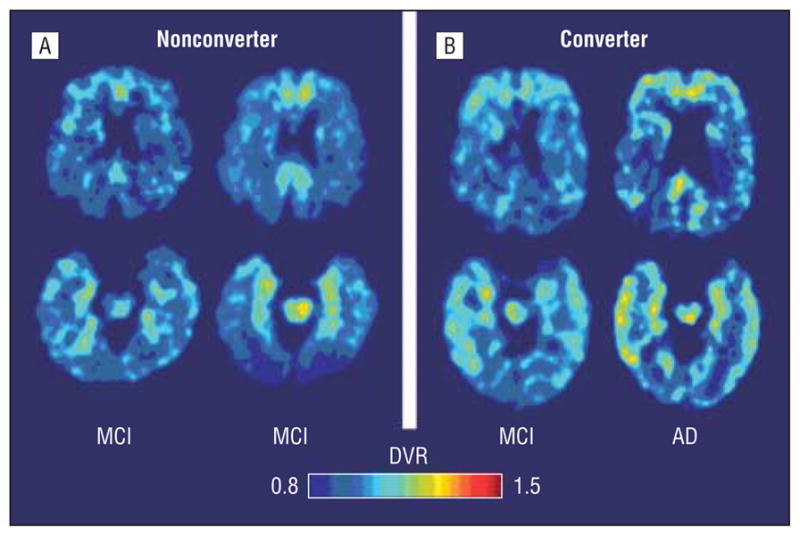
Baseline and follow-up 2-(1-{6-[(2-fluorine 18–labeled fluoroethyl)methylamino]-2-napthyl}ethylidene) malononitrile ([18F]FDDNP) parametric images of a patient with mild cognitive impairment (MCI) who did not convert to Alzheimer disease (AD) after 2 years and a patient who did. A, The parametric [18F]FDDNP scans of the MCI nonconverter showed mild frontal (upper scans) and medial temporal (lower scans) binding at baseline (left) and at follow-up. B, The MCI converter also showed high medial temporal binding at baseline and follow-up but also demonstrated more extensive baseline binding in frontal (upper images) and lateral temporal regions. Warmer colors (yellows, reds) indicate higher binding levels. DVR indicates relative distribution volume.
COMMENT
This 2-year follow-up study of [18F]FDDNP PET in people without dementia and relatively minimal cognitive impairment (overall median Mini-Mental State Examination score of 29) indicates that increases in [18F]FDDNP brain cortical binding values correlate with increases in clinical symptoms of neurodegeneration and regional baseline [18F]FDDNP binding values are significant predictors of future cognitive decline.
In the present study, [18F]FDDNP cortical binding values followed the expected neuropathologic patterns.12,13 The MCI patients who already had high medial temporal binding did not show significant binding increases in this region; thus, medial temporal binding in MCI seems to plateau and remain stable for 2 years, consistent with earlier observations.9 By contrast, a high proportion of the MCI patients with frontal and parietal binding above the ROC established threshold showed cognitive decline after 2 years, whereas none of the MCI patients below this threshold showed cognitive change. Moreover, the results of the ROC analysis indicated that high baseline [18F]FDDNP binding in frontal and parietal regions (corresponding to spread of disease to these regions) is associated with more rapid cognitive decline. Thus, the pattern of [18F]FDDNP binding in MCI, consistent with known disease progression observed at autopsy, may provide useful information for physicians when individual patients are evaluated.32 Our previous work using cluster analysis of [18F]FDDNP binding values in individuals with normal aging and MCI identified a subgroup of individuals with high frontal and parietal binding who may be at high risk for future cognitive decline.33 Moreover, those with this high frontal and parietal pattern in MCI also show fluorodeoxyglucose F18 PET patterns consistent with an increased risk for AD.34
Of the 21 individuals with a diagnosis of MCI at baseline, 12 demonstrated memory impairment consistent with amnestic MCI, whereas the other 9 had amnestic MCI plus deficits in other cognitive areas. These MCI subtypes have a high rate of conversion to dementia,35,36 which provides a useful model for evaluating the predictive ability of molecular imaging probes to determine disease progression. In our study, 6 of the 21 MCI patients (29%) converted to AD at the 2-year follow-up visit, a proportion consistent with the 10% to 15% expected annual risk of conversion of amnestic MCI subtypes.37 The finding that MCI patients with high frontal and parietal binding are more likely to convert to AD after 2 years than those with low binding in these regions further supports the potential utility of [18F]FDDNP PET as a biomarker predictor of cognitive decline in MCI.
We expect that many of the normal aging individuals in this study will eventually progress to MCI, and those with higher baseline [18F]FDDNP binding, particularly in the medial temporal region, might be at the greatest risk for decline within the next few years. In fact, 2 of the 3 normal-aging individuals who converted to MCI at follow-up had the highest baseline regional [18F]FDDNP values in the medial temporal, parietal, and frontal regions. Our results suggest that longer duration of follow-up and larger samples of normal aging individuals would further improve specificity of [18F]FDDNP in predicting future cognitive decline and risk for conversion to a diagnosis of MCI. In less impaired individuals, medial temporal [18F]FDDNP binding might be a more informative predictor of future decline because tau and amyloid deposit accumulation in this area precedes measurable cognitive decline.32,38 Our previous autopsy follow-up study9 of a patient with high medial temporal [18F]FDDNP binding showed both amyloid and tau, with a preponderance of tau tangle deposits in the medial temporal regional.
In the entire study group and the MCI subgroup, baseline medial temporal [18F]FDDNP binding values predicted future executive function decline, an observation that could be explained by disruptions in neural circuitry between medial temporal and frontal regions. Other studies of normal aging indicate neural circuitry disconnections between these 2 cortical areas, including lower entorhinal cortical thickness associated with decreased anterior cingulate and medial frontal activation during a memory retrieval task.39,40 Thus, our observation that high baseline medial temporal [18F]FDDNP binding predicts executive function decline is consistent with tau-mediated disruption of neuronal circuits projecting to pre-frontal regions that control executive functioning.
A useful neuroimaging biomarker for neurodegeneration would not only provide visualizations of relevant disease pathophysiologic characteristics and predict the course of disease but would also demonstrate correlations with disease progression over time.10,41 Our findings indicate that [18F]FDDNP PET demonstrates such utility. For example, for the entire study group (MCI and normal aging), increases in frontal, posterior cingulate, and global binding at follow-up correlated with progression of memory decline. These results are consistent with those of Shin and coworkers42 and Tolboom and colleagues,43 who have reported that higher [18F]FDDNP binding levels are associated with episodic memory impairments.
Finding practical in vivo measures of neurodegeneration for early disease detection and predicting and tracking disease progression are major challenges to the field. Such noninvasive measures of disease progression might assist in testing and monitoring new preventive treatments for protecting neuronal integrity before significant neural damage emerges.10,44–46 The encouraging longitudinal findings with [18F]FDDNP presented in this work will be useful in future clinical trials for successful monitoring of treatments designed to eliminate or prevent the deposition of amyloid or tau or both.
Recent clinical trials of novel treatments have targeted fibrillar amyloid, but efficacy results have been negative,47 pointing to limitations in treatment strategies based solely on the amyloid hypothesis. Amyloid and tau aggregates are clearly important in vivo diagnostic targets, but tau aggregates are associated with both neuronal and cognitive losses and are better indicators of disease progression.48,49 Using an in vivo tau marker for detection and tracking of neurodegenerative diseases is critically important given findings that severity of tau neurofibrillary tangle load, rather than amyloid plaque burden, correlates with rates of tissue loss and neuronal decline.50,51 Even if current antitau treatments prove negative, an in vivo marker for tau aggregates constitutes a useful method to track disease progression at its earlier stages.51 Thus far, [18F]FDDNP is the only available imaging probe that provides in vivo measures of tau in humans.9,10
As in all imaging studies, methodologic limitations should be considered for appropriate interpretation of results. Important considerations include partial volume effects,14 errors introduced from head motion during scanning (particularly with patients with dementia),52 and study population selection (eg, educated samples that may not represent the general population). Our MCI sample was relatively younger than other samples reported in the literature, which might reflect our recruitment focus on normal aging rather than populations with dementia. We have found that head motion error is more likely in more severe forms of cognitive impairment observed in patients with dementia, and this can be corrected effectively.52 Also, the accuracy of cutoffs in the ROC analyses were maximized for this data set, so results from other populations might differ. It is important to cross-validate these results in a larger sample. In addition, we have used combined left and right regions in our analyses. Future studies will examine the contribution of the left and right regions separately.
Our findings indicate that in vivo regional [18F]FDDNP binding patterns are consistent with known patterns of disease deposition and associated with future disease course. Using [18F]FDDNP PET may not only assist in predicting future cognitive decline and identifying individuals more likely to benefit from prevention treatments, but it may also track the effectiveness of such treatments to accelerate drug discovery efforts.
Acknowledgments
Funding/Support: This study was supported by grants P01-AG025831, AG13308, P50 AG 16570, MH/ AG58156, MH52453, AG10123, and M01-RR00865 from the National Institutes of Health; contract DE-FC03-87-ER60615 from the Department of Energy; General Clinical Research Centers Program; the Fran and Ray Stark Foundation Fund for Alzheimer’s Disease Research; the Ahmanson Foundation; the Larry L. Hillblom Foundation; the Lovelace Foundation; the Sence Foundation; the McMahan Foundation; the Judith Olenick Elgart Fund for Research on Brain Aging; and the Elizabeth and Thomas Plott Endowment in Gerontology.
Role of the Sponsors: No company provided support of any kind for this study.
Footnotes
Financial Disclosure: The University of California, Los Angeles, owns a US patent (6,274,119) entitled “Methods for Labeling β-Amyloid Plaques and Neurofibrillary Tangles” that uses the approach outlined in this article. Drs Small, Huang, and Barrio are among the inventors, have received royalties, and may receive royalties on future sales. Dr Small reports having served as a consultant and/or having received lecture fees from Dakim, Eisai, Forest, Lily, Medivation, Novartis, and Pfizer. Dr Small also reports having received stock options from Dakim. Dr Lavretsky reports having received lecture fees from Eisai, Janssen, and Pfizer and having received a grant from Forest. Dr Huang reports having received lecture fees from GlaxoSmithKline. Dr Barrio reports having served as a consultant and having received lecture fees from Nihon Medi-Physics Co, Bristol-Myers Squibb, PETNet Pharmaceuticals, and Siemens. Drs Ercoli, Siddarth, Burggren, Kepe, Kim, Miller, and Bookheimer have no financial conflicts of interest.
Previous Presentations: Presented in part at the American College of Neuropsychopharmacology Annual Meeting; December 9, 2009; Hollywood, Florida.
Additional Contributions: Anel Dzmura, BA, and Colin Shinn, MA, provided help with study participant recruitment, data management, and study coordination, and Gerald Timbol and Anasheh Halabi provided help with image processing.
Author Contributions: Study concept and design: Small, Siddarth, and Kepe. Acquisition of data: Small, Kepe, Ercoli, Burggren, Bookheimer, Miller, Kim, and Lavretsky. Analysis and interpretation of data: Small, Siddarth, Kepe, Ercoli, Burggren, Bookheimer, Huang, and Barrio. Drafting of the manuscript: Small, Siddarth, Miller, Kim, Lavretsky, and Barrio. Critical revision of the manuscript for important intellectual content: Small, Siddarth, Kepe, Ercoli, Burggren, Bookheimer, Lavretsky, Huang, and Barrio. Statistical analysis: Siddarth and Barrio. Obtained funding: Small, Barrio, Ercoli, and Lavretsky. Administrative, technical, and material support: Small, Ercoli, Burggren, Bookheimer, Miller, Kim, Lavretsky, and Huang. Study supervision: Small, Burggren, and Barrio.
References
- 1.Lopez OL, Jagust WJ, DeKosky ST, et al. Prevalence and classification of mild cognitive impairment in the Cardiovascular Health Study Cognition Study: part 1. Arch Neurol. 2003;60(10):1385–1389. doi: 10.1001/archneur.60.10.1385. [DOI] [PubMed] [Google Scholar]
- 2.Bachman DL, Wolf PA, Linn RT, et al. Incidence of dementia and probable Alzheimer’s disease in a general population: the Framingham Study. Neurology. 1993;43(3 pt 1):515–519. doi: 10.1212/wnl.43.3_part_1.515. [DOI] [PubMed] [Google Scholar]
- 3.Demirovic J, Prineas R, Loewenstein D, et al. Prevalence of dementia in three ethnic groups. Ann Epidemiol. 2003;13(6):472–478. doi: 10.1016/s1047-2797(02)00437-4. [DOI] [PubMed] [Google Scholar]
- 4.Shoghi-Jadid K, Small GW, Agdeppa ED, et al. Localization of neurofibrillary tangles and β-amyloid plaques in the brains of living patients with Alzheimer disease. Am J Geriatr Psychiatry. 2002;10(1):24–35. [PubMed] [Google Scholar]
- 5.Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in Alzheimer disease with Pittsburgh Compound-B. Ann Neurol. 2004;55(3):306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 6.Verhoeff NP, Wilson AA, Takeshita S, et al. In-vivo imaging of Alzheimer disease β-amyloid with [11C]SB-13 PET. Am J Geriatr Psychiatry. 2004;12(6):584–595. doi: 10.1176/appi.ajgp.12.6.584. [DOI] [PubMed] [Google Scholar]
- 7.Vandenberghe R, Van Laere K, Ivanoiu A, et al. 18F-flutemetamol amyloid imaging in Alzheimer disease and mild cognitive impairment: a phase 2 trial. Ann Neurol. 2010;68(3):319–329. doi: 10.1002/ana.22068. [DOI] [PubMed] [Google Scholar]
- 8.Clark CM, Schneider JA, Bedell BJ, et al. AV45-A07 Study Group. Use of florbetapir-PET for imaging β-amyloid pathology [published correction appears in JAMA. 2011;305(11):1096] JAMA. 2011;305(3):275–283. doi: 10.1001/jama.2010.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Small GW, Kepe V, Ercoli LM, et al. PET of brain amyloid and tau in mild cognitive impairment. N Engl J Med. 2006;355(25):2652–2663. doi: 10.1056/NEJMoa054625. [DOI] [PubMed] [Google Scholar]
- 10.Small GW, Bookheimer SY, Thompson PM, et al. Current and future uses of neuroimaging for cognitively impaired patients. Lancet Neurol. 2008;7(2):161–172. doi: 10.1016/S1474-4422(08)70019-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Braskie MN, Klunder AD, Hayashi KM, et al. Plaque and tangle imaging and cognition in normal aging and Alzheimer’s disease. Neurobiol Aging. 2008;31(10):1669–1678. doi: 10.1016/j.neurobiolaging.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82(4):239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 13.Braak H, Braak E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging. 1997;18(4):351–357. doi: 10.1016/s0197-4580(97)00056-0. [DOI] [PubMed] [Google Scholar]
- 14.Protas HD, Huang S-C, Kepe V, et al. FDDNP binding using MR derived cortical surface maps. Neuroimage. 2010;49(1):240–248. doi: 10.1016/j.neuroimage.2009.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Small GW, Siddarth P, Burggren AC, et al. Influence of cognitive status, age, and APOE-4 genetic risk on brain FDDNP positron-emission tomography imaging in persons without dementia. Arch Gen Psychiatry. 2009;66(1):81–87. doi: 10.1001/archgenpsychiatry.2008.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tolboom N, van der Flier WM, Yaqub M, et al. Relationship of cerebrospinal fluid markers to 11C-PiB and 18F-FDDNP binding. J Nucl Med. 2009;50(9):1464–1470. doi: 10.2967/jnumed.109.064360. [DOI] [PubMed] [Google Scholar]
- 17.Agdeppa ED, Kepe V, Petri A, et al. In vitro detection of (S)-naproxen and ibu-profen binding to plaques in the Alzheimer brain using the positron emission tomography molecular imaging probe 2-(1-[6-[(2-[(18)F]fluoroethyl)(methyl) amino]-2-naphthyl]ethylidene)malononitrile. Neuroscience. 2003;117(3):723–730. doi: 10.1016/s0306-4522(02)00907-7. [DOI] [PubMed] [Google Scholar]
- 18.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34(7):939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 19.Folstein MF, Folstein SE, McHugh PR. Mini-mental state: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 20.Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23:56–62. doi: 10.1136/jnnp.23.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lezak M, Howieson D, Loring D. Neuropsychological Assessment. 4. New York: New York University Press; 2004. [Google Scholar]
- 22.Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med. 2004;256(3):183–194. doi: 10.1111/j.1365-2796.2004.01388.x. [DOI] [PubMed] [Google Scholar]
- 23.Winblad B, Palmer K, Kivipelto M, et al. Mild cognitive impairment—beyond controversies, towards a consensus: report of the International Working Group on Mild Cognitive Impairment. J Intern Med. 2004;256(3):240–246. doi: 10.1111/j.1365-2796.2004.01380.x. [DOI] [PubMed] [Google Scholar]
- 24.de Jager CA, Budge MM. Stability and predictability of the classification of mild cognitive impairment as assessed by episodic memory test performance over time. Neurocase. 2005;11(1):72–79. doi: 10.1080/13554790490896820. [DOI] [PubMed] [Google Scholar]
- 25.Busse A, Hensel A, Gühne U, Angermeyer MC, Riedel-Heller SG. Mild cognitive impairment. Neurology. 2006;67(12):2176–2185. doi: 10.1212/01.wnl.0000249117.23318.e1. [DOI] [PubMed] [Google Scholar]
- 26.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4. Washington, DC: American Psychiatric Association; 2000. (Text Revision) [Google Scholar]
- 27.Gilewski MJ, Zelinski EM. Questionnaire assessment of memory complaints. In: Poon LW, editor. Handbook for Clinical Memory Assessment of Oder Adults. Washington, DC: American Psychological Association; 1986. pp. 93–107. [Google Scholar]
- 28.Liu J, Kepe V, Zabjek A, et al. High-yield, automated radiosynthesis of 2-(1-6-[(2-[18F]fluoroethyl)(methyl)amino]-2-naphthylethylidene)malononitrile ([18F]FDDNP) ready for animal or human administration. Mol Imaging Biol. 2007;9(1):6–16. doi: 10.1007/s11307-006-0061-4. [DOI] [PubMed] [Google Scholar]
- 29.Logan J, Fowler JS, Volkow ND, Wang GJ, Ding YS, Alexoff DL. Distribution volume ratios without blood sampling from graphical analysis of PET data. J Cereb Blood Flow Metab. 1996;16(5):834–840. doi: 10.1097/00004647-199609000-00008. [DOI] [PubMed] [Google Scholar]
- 30.Talairach J, Tournoux P. Coplanar Stereotaxic Atlas of the Human Brain: Three-Dimensional Proportional System: An Approach to Cerebral Imaging. New York, NY: Thieme; 1988. [Google Scholar]
- 31.Riffenburgh RH. Statistics in Medicine. 2. San Diego, CA: Elsevier Academic Press; 2006. [Google Scholar]
- 32.Price JL, Morris JC. Tangles and plaques in nondemented aging and “preclinical” Alzheimer disease. Ann Neurol. 1999;45(3):358–368. doi: 10.1002/1531-8249(199903)45:3<358::aid-ana12>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 33.Ercoli LM, Siddarth P, Kepe V, et al. Differential FDDNP PET patterns in nondemented middle-aged and older adults. Am J Geriatr Psychiatry. 2009;17(5):397–406. doi: 10.1097/JGP.0b013e318198750b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Small GW, Suthana NA, Kepe V, et al. Combining Neuroimaging and Genetic Measures for Presymptomatic Detection of Neurodegeneration. Brentwood, TN: American College of Neuropsychopharmacology; 2010. [Google Scholar]
- 35.Griffith HR, Netson KL, Harrell LE, Zamrini EY, Brockington JC, Marson DC. Amnestic mild cognitive impairment. J Int Neuropsychol Soc. 2006;12(2):166–175. doi: 10.1017/S1355617706060267. [DOI] [PubMed] [Google Scholar]
- 36.Maioli F, Coveri M, Pagni P, et al. Conversion of mild cognitive impairment to dementia in elderly subjects: a preliminary study in a memory and cognitive disorder unit. Arch Gerontol Geriatr. 2007;44(suppl 1):233–241. doi: 10.1016/j.archger.2007.01.032. [DOI] [PubMed] [Google Scholar]
- 37.Landau SM, Harvey D, Madison CM, et al. Alzheimer’s Disease Neuroimaging Initiative. Comparing predictors of conversion and decline in mild cognitive impairment. Neurology. 2010;75(3):230–238. doi: 10.1212/WNL.0b013e3181e8e8b8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Petersen RC, Parisi JE, Dickson DW, et al. Neuropathologic features of amnestic mild cognitive impairment. Arch Neurol. 2006;63(5):665–672. doi: 10.1001/archneur.63.5.665. [DOI] [PubMed] [Google Scholar]
- 39.Braskie MN, Small GW, Bookheimer SY. Entorhinal cortex structure and functional MRI response during an associative verbal memory task. Hum Brain Mapp. 2009;30(12):3981–3992. doi: 10.1002/hbm.20823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shin J, Kepe V, Small GW, Phelps ME, Barrio JR. Multimodal imaging of Alzheimer pathophysiology in the brain’s default mode network. Intl J Alzheim Dis. 2011;687:945. doi: 10.4061/2011/687945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Katz R. Biomarkers and surrogate markers: an FDA perspective. NeuroRx. 2004;1(2):189–195. doi: 10.1602/neurorx.1.2.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shin J, Lee SY, Kim SH, Kim YB, Cho SJ. Multitracer PET imaging of amyloid plaques and neurofibrillary tangles in Alzheimer’s disease. Neuroimage. 2008;43(2):236–244. doi: 10.1016/j.neuroimage.2008.07.022. [DOI] [PubMed] [Google Scholar]
- 43.Tolboom N, van der Flier WM, Yaqub M, et al. Differential association of [11C]PIB and [18F]FDDNP binding with cognitive impairment. Neurology. 2009;73(24):2079–2085. doi: 10.1212/WNL.0b013e3181c679cc. [DOI] [PubMed] [Google Scholar]
- 44.The Ronald and Nancy Reagan Research Institute of the Alzheimer’s Association and the National Institute on Aging Working Group. Consensus report of the working group on: Molecular and Biochemical Markers of Alzheimer’s Disease. Neurobiol Aging. 1998;19(2):109–116. [PubMed] [Google Scholar]
- 45.Coley N, Andrieu S, Delrieu J, Voisin T, Vellas B. Biomarkers in Alzheimer’s disease: not yet surrogate endpoints. Ann N Y Acad Sci. 2009;1180:119–124. doi: 10.1111/j.1749-6632.2009.04947.x. [DOI] [PubMed] [Google Scholar]
- 46.Vellas B, Andrieu S, Sampaio C, Wilcock G European Task Force Group. Disease-modifying trials in Alzheimer disease. Lancet Neurol. 2007;6(1):56–62. doi: 10.1016/S1474-4422(06)70677-9. [DOI] [PubMed] [Google Scholar]
- 47.Strobel G. 12th International Conference on Alzheimer Disease (ICAD), Vienna, Austria. J Alzheimers Dis. 2009;18(4):973–990. doi: 10.3233/JAD-2009-1250. [DOI] [PubMed] [Google Scholar]
- 48.Spires-Jones TL, Stoothoff WH, de Calignon A, Jones PB, Hyman BT. Tau pathophysiology in neurodegeneration: a tangled issue. Trends Neurosci. 2009;32 (3):150–159. doi: 10.1016/j.tins.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 49.Josephs KA, Whitwell JL, Ahmed Z, et al. β-Amyloid burden is not associated with rates of brain atrophy. Ann Neurol. 2008;63(2):204–212. doi: 10.1002/ana.21223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Giannakopoulos P, Herrmann FR, Bussière T, et al. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer disease. Neurology. 2003;13(60):1495–1500. doi: 10.1212/01.wnl.0000063311.58879.01. [DOI] [PubMed] [Google Scholar]
- 51.Thal LJ, Kantarci K, Reiman EM, et al. The role of biomarkers in clinical trials for Alzheimer disease. Alzheimer Dis Assoc Disord. 2006;20(1):6–15. doi: 10.1097/01.wad.0000191420.61260.a8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wardak M, Wong KP, Shao W, et al. Movement correction method for human brain PET images. J Nucl Med. 2010;51(2):210–218. doi: 10.2967/jnumed.109.063701. [DOI] [PMC free article] [PubMed] [Google Scholar]