

Abstract
Focal and segmental glomerulosclerosis (FSGS) is an important cause of steroid resistant nephrotic syndrome in adults and children. It is responsible for 5–20% of all cases of end-stage kidney disease (ESKD) in the United States. The pathogenesis of FSGS has not been fully elucidated; however data from molecular studies of familial cases in the last two decades suggest that FSGS is a defect of the podocyte. The therapeutic agents available for treatment of FSGS are not very effective and only a small percentage of affected individuals will achieve complete remission. Recent data from molecular biology and molecular genetics has provided insight into mechanisms of action of old agents and also identification of other novel therapeutic targets. This review focuses on recent advances in the molecular pathogenesis of FSGS and currently available therapeutic agents as well as potential novel therapies.
Keywords: FSGS, Podocyte, molecular pathogenesis, Therapy
INTRODUCTION
Focal segmental glomerulosclerosis (FSGS) was first described in kidney biopsy of adults with nephrotic syndrome by Fahr in 1925 [1–2]. About thirty two years later, Rich made the observation that the lesion of FSGS in children with nephrotic syndrome classically starts from the corticomedullary junction before involving other parts of the renal cortex [1–2]. FSGS is a clinicopathologic entity that is characterized frequently by steroid-resistant nephrotic syndrome and rapid progression to end-stage kidney disease (ESKD) in the majority of affected individuals. Histologically, the lesion is characterized by focal glomerulosclerosis or tuft collapse, segmental hyalinosis, occasionally IgM staining on immunofluorescence and effacement of foot processes on electron microscopy [3]. Its incidence is estimated at seven per million [4]. FSGS is responsible for 5–20% of all cases of ESKD in the USA and is second only to urogenital and kidney malformations as a cause of ESKD in children [4–6]. The incidence of FSGS appears to be increasing, Kitiyakara et al., reported an 11-fold increase among dialysis patients over 21 years [6]. In every age group, the incidence is higher in blacks than whites and the rate of decline in kidney function is also worse in blacks [4, 6].
CLASSIFICATION
Clinical classification of FSGS is based on presumed etiology (Table 1), however in more than 80% of cases the etiology is unknown and this group is therefore classified as having primary or idiopathic disease. FSGS may be secondary to other disease processes such as sickle cell disease, obesity, heroin use, HIV nephropathy and other glomerulonephritides that are associated with nephron loss. Familial cases of FSGS, both syndromic and non-syndromic have been reported. Although this group is probably responsible for less than 1% of all cases, detailed molecular study of hereditary forms has helped advance understanding of the pathogenesis of FSGS.
Table 1.
Etiology of FSGS
| Primary/Idiopathic FSGS |
| Hereditary diseases (See Table 2) |
| Infections |
| Hepatitis C |
| HIV infection |
| Cytomegalovirus |
| Epstein-Barr virus |
| Parvovirus B19 |
| Drugs/Toxic agents |
| Interferon-α |
| Pamidronate |
| Lithium |
| Gold |
| Heroin |
| Hyperfiltration |
| Bilateral or unilateral renal dysplasia |
| Obesity |
| Reflux nephropathy |
| Other causes of glomerulonephritis associated with nephron loss |
| Aging |
| Ischemia |
| Renal artery stenosis |
| Hypertensive kidney disease |
| Calcineurin inhibitor nephrotoxicity |
| Acute and chronic renal allograft rejection |
| Cholesterol crystal embolism |
| Cyanotic congenital heart disease |
Histopathological findings in FSGS are heterogeneous and until recently, there was no standard sub-classification of FSGS based on morphological features. In order to standardize the pathological diagnosis of FSGS subtypes and possibly relate morphological findings to clinical course, the Columbia classification of FSGS was proposed [7]. In this classification schema, five light microscopic patterns of FSGS have been defined including FSGS not otherwise specified (NOS), perihilar variant, cellular variant, tip variant and a collapsing variant. There are limited data on the correlation between subtypes of FSGS and clinical course of the disease. In a study of adults with FSGS, Stokes et al., [8] reported that collapsing FSGS had the highest rate of renal insufficiency at presentation and progression to CKD compared with the other variants. In the same study, subjects with the tip lesion variant had the highest rate of remission following therapy. In a similar study of 93 European adults with FSGS, the tip lesion was found to be significantly associated with nephrotic syndrome at presentation, and also had a higher remission rate and renal survival after five years of follow up compared with other variants [9]. Studies in children are limited, in a retrospective review of 41 children, Silverstein et al., reported worse outcome in children with collapsing FSGS [10]. The classification scheme is however not a predictor of recurrence of disease in renal allografts following transplantation [11].
PATHOGENESIS
The glomerular filtration barrier
The kidney is responsible for filtration of approximately 180 liters per day of plasma containing over 7,200g grams of albumin; over 99.9% of albumin is retained by combined actions of selective filtration and tubular reuptake [12]. This regulation of filtration of macromolecules is made possible by the glomerular filtration barrier, which is comprised of specialized fenestrated endothelial cells, the glomerular basement membrane (GBM), and glomerular epithelial cells (podocytes) whose distal foot processes are attached to the GBM (Figure 1) [13]. Neighboring podocyte foot processes are connected to each other by networks of specialized cell-cell junctions known as slit diaphragms. In addition, the GBM has an abundant supply of negatively-charged heparan sulfate proteoglycans, resulting in negatively-charged molecules being relatively more restricted from passage than positively-charged molecules of the same size [14]. In health, filtration of macromolecules decreases with increasing molecular size especially with molecules greater than 42 Å in diameter or more than 200 kDa [15]. The maintenance of the GFB depends on structural and functional interaction between the three components [16–20].
Figure 1. The podocyte and the other components of the glomerular filtration barrier.
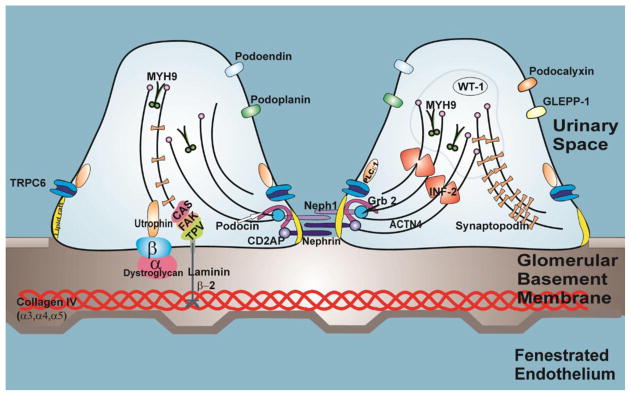
The glomerular filtration barrier is formed by fenestrated endothelial cells, the glomerular basement membrane and podocytes. The podocyte has a unique actin cytoskeleton made up of F-actin and non-muscle myosin such as MYH9. In addition, it has actin-binding proteins such as synaptopodin and α-actinin4 (ACTN4) and actin polymerization regulatory protein inverted formin 2 (INF2). Mutations in ACTN4, INF2 genes are causes of familial FSGS and MYH9 is a complex disease locus in idiopathic FSGS. Wilms’ tumor 1 (WT1) gene is a nuclear transcription factor that is expressed abundantly in the podocyte, mutations in WT1 is a cause of syndromic and non-syndromic nephrotic syndrome. The junctional part of the podocyte (the slit diaphragm) is formed by nephrin, podocin, CD2 aasociated protein (CD2AP) and NEPH1. Podocin associates with lipid rafts, a signaling domain of the slit diaphragm. It recruits nephrin and NEPH1 to form a signaling complex with other molecules such as Transient Receptor Potential Cation Channel, Type 6 (TRPC6), growth factor receptor-bound protein 2 (Grb2) and Phospholipase C epsilon-1 (PLCE1) at the slit diaphragm. Mutations in nephrin, podocin, TRPC6, PLCE1 and CD2AP are known causes of hereditary FSGS and nephrotic syndrome in humans. The apical membrane of the podocyte is formed by negatively charged molecules such as podocalyxin, podoplanin, podoendin and glomerular epithelial protein-1 (GLEPP-1). The basal part of the podocyte contains α3β1 integrin and α and β dystroglycans that anchors the podocyte to the glomerular basement membrane (GBM). Talin, Paxillin and Vincullin (TPV) interact with different laminins in the GBM especially laminin β2. Mutations in laminin β2 is a cause of early onset nephrotic syndrome.
The fenestrated endothelial layer
The glomerular endothelial cells in humans have numerous fenestrae that are approximately 50–100 nm in size. Fenestrae in adults lack diaphragms, which should preclude them from acting as a filtration barrier for macromolecules. However, if the endothelial layer provided no barrier to the macromolecules, movement of albumin and other macromolecules would probably result in the clogging of the glomerular filter [18–19]. In addition there is evidence to suggest that the fenestrae of the endothelial layer are not fully patent, containing dense assemblies of glycoprotein that serve as sieve plugs [17]. Furthermore, during glomerular development podocytes produce angiogenic factors such as vascular endothelial growth factor-A (VEGF-A) and VEGF-C, while the endothelial cells express the receptors for these molecules suggesting that the endothelial layer is important in maintaining the GFB [16, 20].
Glomerular basement membrane
The glomerular basement membrane (GBM) is made up of type IV collagen (α3, 4 and 5), laminin, nidogen/entactin, proteoglycans such as agrin and perlecan and glycoproteins [15]. The GBM is an important part of the GFB because it accounts for most of the restriction of the fluid flux [15]. Furthermore, the early suggestion by Shaloub that nephrotic syndrome results from an abnormality of T-cell function, resulting in secretion of chemical mediators that are toxic to the GBM, supports the hypothesis that the GBM plays a critical role in maintaining the integrity of the GFB [21]. An additional observation which supports this is the report of a FSGS permeability factor by the Savin group in 1996 [22]. The factor is about 30 to 50 kDa in weight and was shown to increase the permeability of the GBM to albumin and induce transient proteinuria in rats. Mutations in the laminin β2 gene, which encodes for a protein highly expressed in the GBM, is characterized by massive proteinuria, suggesting that the GBM is an important barrier to these macromolecules [23]. However, subjects with primary GBM defects such as Alport’s syndrome do not have proteinuria as a prominent early manifestation.
Podocytes
Podocytes are terminally differentiated glomerular visceral epithelial cells. It consists of a cell body, the major or primary processes which ultimately branch into minor foot processes that interdigitate with neighboring podocytes to form a highly specialized interdigitating gap junction, the slit diaphragm. In the early 1970’s Karnovsky and Ainsworth [24] showed by elegant electron microscopy studies that the podocyte slit diaphragm is the most important size selective sieve of the GFB. They described a zipper-like structure with pores that are smaller than the radius of albumin. It is now known that this zipper-like structure is formed by nephrin an important component of the slit diaphragm [25]. The identification of nephrin as the gene mutated in congenital nephrotic syndrome by the Trygvasson group [25] and the discovery of other FSGS and nephrotic syndrome genes provides solid evidence that the podocyte and its slit diaphragm are the most important components of the GFB and that its functional or structural alteration is important in the pathogenesis of FSGS and other glomerular diseases. This has given rise to the concept of podocytopathies as the unifying hypothesis for glomerular diseases [26].
FSGS AS A PODOCYTOPATHY
Data from humans and experimental studies have shown that relative or absolute podocyte depletion or changes in its functional integrity is central to the initiation and progression of the lesion seen in FSGS [27–29]. The mechanisms by which podocyte damage evolves into the pathological appearance seen in FSGS have been studied extensively by Kriz and colleagues using different rat models of FSGS [27]. The initial defect is a reduction in podocyte density and the inability of podocytes to completely cover the glomerular tufts. This causes the loss of separation between the glomerular tuft and Bowman’s capsule leading to the formation of synechiae or adhesions between the tuft and the Bowman’s capsule [27]. The perfused capillaries in the tuft adhesion deliver their filtrate into the interstitium instead of Bowman’s space. This misdirected filtration through capillaries lacking podocytes ultimately leads to progression of segmental injury, tubular degeneration and interstitial fibrosis [27]. The role of podocyte depletion as the initiating event in FSGS and other proteinuric kidney diseases is further supported by findings of podocyturia in various glomerular diseases [28]. The quantitative relationship between podocyte number and evolution of FSGS was demonstrated by the Wiggins’ group, using a rat model of diphtheria toxin induced podocyte depletion in which the degree of podocyte loss is regulated [29]. In mild podocyte loss, the remaining podocytes underwent hypertrophy in order to cover the glomerular basement membrane, but with progressive depletion, FSGS and global sclerosis developed [29].
HEREDITARY FSGS
The most compelling evidence to date for the central role of the podocyte and its slit diaphragm in the development of FSGS is the identification of genes mutated in human hereditary FSGS. The products of these genes, without exception localize to the podocyte and its slit diaphragm and most of them participate in signaling events that are essential for maintaining the cytoarchitecture of the podocyte and the slit diaphragm. Additionally, data from animal models and cell based experiments are in agreement with the central role of the podocyte in the pathogenesis of FSGS [30]. The rest of this section will describe recent findings from the positional cloning of genes mutated in hereditary FSGS and also describe how the discovery of these genes contributes to our understanding of the pathogenesis of FSGS.
Nephrin (NPHS1)
The first major breakthrough was the cloning of nephrin (NPHS1) as a cause of congenital nephrotic syndrome (CNS) of the Finnish type [25]. Nephrin is expressed in podocytes and localizes to the podocyte slit diaphragm forming a zipper-like structure by homophilic interaction with adjacent molecules. It functions as a transmembrane receptor complex at the slit diaphragm, forming a complex with the proteins Neph1 and podocin. The lesion caused by the common Fin major (L41fsX90) and Fin minor (R1109X) mutations is characterized by immature glomeruli with cystic changes in the Bowman’s space which later progresses to diffuse interstitial fibrosis. Since the initial report, over 130 different mutations have been reported, including missense mutations causing later onset FSGS and minimal change disease (MCD) [31].
Podocin (NPHS2)
Podocin was positionally cloned by the Antignac group in children with autosomal recessive FSGS that is characterized by early onset, resistance to therapy and rapid progression to ESKD [32]. Podocin is expressed mainly in the podocytes and localizes to the intercellular junction of the podocyte foot processes [33]. Podocin belongs to the stomatin family of proteins. Molecules in this family are known to associate with lipid rafts (a signaling domain) and recruit transmembrane receptors to these rafts. Podocin seems to recruit nephrin, Neph1 and CD2-associated protein (CD2AP) to the lipid raft, thereby forming a complex receptor at the slit diaphragm, with which other components of the slit diaphragm interact [34–36]. This multifunction complex is coupled to the podocyte actin cytoskeleton and also participates in signaling that is responsible for maintaining the functional integrity of the slit diaphragm [37–38]. In one series, podocin mutations were reported to be responsible for up to 20% of all cases of childhood onset FSGS [39], furthermore, the risk of recurrence of disease in renal allograft following transplantation was reduced in individuals with NPHS2 mutations compared to those with no mutations (8% versus 35%) [39]. Recent reports suggest that common variants in podocin may also increase the risk of FSGS in older children and adults [40]. The molecular mechanism by which mutant podocin protein causes FSGS is not completely understood. This is partly because podocin knockout (KO) mice do not survive beyond the first week of life. The KO mice develop severe albuminuria at birth, mesangiolysis and mesangial sclerosis and not classical FSGS seen in humans with podocin mutations. [41]. A conditional podocin inactivation model in mature kidneys using the Cre-loxP technology was recently reported [42]. The mice in this model survived up to eleven weeks and demonstrated the histology of FSGS and clinical features of human nephrotic syndrome [42]. Genome-wide gene expression study of glomeruli from these mice showed a perturbation of the cell-cycle regulation and proliferation pathways, suggesting that a podocyte phenotype switch may be important in the mechanisms of disease in this model [42].
Alpha-actinin-4 (ACTN4)
ACTN4 is an actin-filament cross linking protein and is a member of the spectrin super family [43]. It is expressed abundantly in the podocyte foot process as well as other tissues. Mutations in ACTN4 were reported as a cause of adult onset autosomal dominant FSGS by the Pollak group [43]. These mutations appear to cause a gain of function, as the mutant ACTN4 binds to F-actin more strongly than the wild-type. This strong affinity for F-actin disrupts normal filament assembly and disassembly in the podocyte. Furthermore, it was shown that the mutations also cause protein misfolding and degradation that is partly mediated by the ubiquitin-proteasome pathway [43–44]. In another study, mutant ACTN4 was found to mislocalize within the intracellular compartment. This maldistribution impaired podocyte motility, spreading and peripheral projection [45].
Transient Receptor Potential Cation Channel, Type 6 (TRPC6)
A missense mutation in TRPC6 was reported as a cause of familial FSGS in a large New Zealand family with FSGS by Winn et al., [46]. Subsequently, other families with different TRPC6 mutations have been reported in both adults and children [47–49]. TRPC6 and other TRP ion channels are groups of cation channels with six membrane-spanning domains with both carboxyl- and amino- termini located intracellularly [50]. The TRP channels perform a variety of biologic functions including mechanosensation, ion homeostasis, cell growth and phospholipase C-dependent calcium entry into cells [51]. TRPC6 is widely distributed in the body and is found in the podocyte cell body and the slit diaphragm. Most of the mutations reported to date are gain of function mutations causing increased intracellular calcium influx. The mechanisms, by which the increased calcium influx causes FSGS, are not clear, but possible mechanisms include modification of podocyte contractile structure, increased podocyte apoptosis, or reduced podocyte proliferation during glomerulogenesis [51]. Increased expression of TRPC6 has been reported in kidney biopsy specimens from individuals with acquired kidney disease, suggesting that TRPC6 may have a role in the pathogenesis of the more common idiopathic disease [52]. Recent data from Winn and colleagues showed that the TRPC6 knockout mice developed significantly less proteinuria compared with the wild-type following angiotensin II infusion (Ang II) for 28 days [53]. Furthermore, wild-type mouse primary podocyte cultures showed a change in membrane current in response to Ang II when patch clamping optimized for the TRPC6 channel was performed, whereas TRPC6 null podocytes did not, suggesting that the mechanism of TRPC6-induced injury may be due to changes in ion channel current, likely induced by calcium influx, in response to Ang II (personal communication). TRPC6 KO mice are also protected from puromycin aminonucleoside (PAN) induced renal injury, suggesting that increased apoptosis may be one of the possible mechanisms by which TRPC6 induces glomerular injury [54].
CD2-Associated protein (CD2AP)
CD2AP is an adapter protein with an SH3 domain. It interacts with the T-cell adhesion protein CD2 and is expressed in epithelial and lymphoid cells [55]. In the kidney, CD2AP localizes to the slit diaphragm of podocyte where it interacts with and links podocin and nephrin to the phosphoinositide 3-OH kinase to form a signaling complex in the lipid raft of the slit diaphragm [36, 56]. CD2AP-deficient mice develop severe proteinuria and renal dysfunction and die shortly after birth [57]. On the other hand, heterozygous mice live longer and develop an FSGS-like lesion around the age of nine months [55]. The role of CD2AP in human FSGS is still being elucidated, but individuals with homozygous mutations in CD2AP have been reported to present with early onset FSGS, while those with heterozygous mutations tend to present in adulthood [55, 58–59].
Wilms’ Tumor 1 (WT1)
The WT1 suppressor gene encodes a zinc finger transcription factor that regulates the expression of many genes by DNA binding. It was first identified in the 1990’s as a cause of Wilms’ tumor, aniridia, genitourinary malformations and mental retardation (WAGR) syndrome. Since then, many studies have shown that WT1 is widely expressed in the kidney and it is very important for the development of the genitourinary tract. The spectrum of glomerular diseases associated with WT1 mutations has recently been reviewed by Niaudet and Gubler [60]. WT1 mutations can cause syndromic and non-syndromic glomerular disease. The syndromic forms include Denys-Drash syndrome (early onset nephrotic syndrome with the histology of diffuse mesangial sclerosis, male pseudohermaphroditism and Wilms’ tumor) and Frasier syndrome (male pseudohermaphroditism, FSGS and gonadoblastoma) which is caused by a mutation in the intron 9 splice site of the gene leading to the loss of the +KTS isoform of the protein. Mutations associated with both syndromic and non-syndromic glomerular disease tend to cluster in exons 8 and 9 of WT1 which encode for zinc fingers 2 and 3 [60–61].
Phospholipase C epsilon-1 (PLCE1/NPHS3)
Mutations in PLCE1/NPHS3 were recently reported as a cause of early onset nephrotic syndrome that is characterized predominantly by the histology of diffuse mesangial sclerosis (DMS) [62]. In the original report, families with truncation PLCE1 mutations exhibited the histology of DMS, while a single family with a missense mutation had the onset of FSGS in later childhood. Remarkably, two patients with homozygous truncating mutations responded to steroids or calcineurin inhibitor therapy. Follow-up study showed that mutations in PLCE1 are responsible for about 28% of all cases of isolated DMS [63]. Recent reports suggest that PLCE1 loss of function mutation(s) can also cause FSGS and that some individuals may also remain asymptomatic, implying that there may be modifier genes, yet to be identified, that interact with PLCE1 to cause DMS/FSGS [64–65]. PLCε1 is a member of the phospholipase family of proteins that catalyze the hydrolysis of polyphosphoinositides such as phosphatdylinositol-4,5-bisphosphate (PtdIns(4,5)P2) to generate the second messengers Ins(1,4,5)P3 and diacylglycerol [66]. The products of this reaction initiate a cascade of intracellular responses that result in cell growth and differentiation and gene expression. The mechanisms by which PLCE1 mutations cause nephrotic syndrome have not been completely elucidated. It has been shown however, that PLCε1 is expressed in developing and mature podocytes and that PLCE1 mutations cause glomerular developmental arrest and may reduce nephrin/podocin expression [62].
Laminin β2 (LAMβ2)
Mutations in this gene cause Pierson syndrome (diffuse mesangial sclerosis, microcoria and neurological anomalies) [23]. LAMβ2 is widely expressed in the eyes, glomerular basement membrane and in the developing brain [23]. LAMβ2 is thought to play a major role in anchoring and the differentiation of podocyte foot processes [23]. The typical renal phenotype associated with LAMβ2 mutations is diffuse mesangial sclerosis, however, FSGS and non-specific ocular anomalies have been reported in individuals with missense mutations in LAMβ2 [67].
Inverted formin 2 (INF2)
The Pollak group recently reported a new locus for FSGS on chromosome 14q32 [68]. The gene encoding Inverted Formin 2 (INF2) was sequenced and 2 heterozygous missense mutations R128Q and S186P were found in two unrelated families. Further analysis of about 90 other families with FSGS yielded nine families with eight new variants most of which clustered in exon 4, a region of the gene encoding for the N-terminal regulatory region of the protein. INF2 is a member of the formin family. Formin is one of the three proteins that promote nucleation of actin, a rate limiting step in actin polymerization. Cell transfection experiments showed mislocalization of the mutant INF2 in the podocyte suggesting that these changes probably have the ability to dysregulate the podocyte cytoskeleton. The disease in subjects with INF2 mutations is characterized by onset of FSGS in early adolescence and adulthood. In addition to the classical findings of FSGS, some of the affected individuals also have prominent actin bundles within the podocyte foot process. The mechanisms by which mutant INF2 induces these changes are still being investigated.
Myosin heavy chain 9 (MYH9) and idiopathic FSGS
The genetic risk factors for the complex and more common, idiopathic FSGS are unknown. Recently, using the strategy of mapping by admixture linkage disequilibrium, two studies reported sequence variation in non-muscle myosin heavy chain IIA (MYH9) as a risk factor for FSGS and ESKD in African Americans [69–70]. These studies reported a strong association between three intron 23 SNPS (E-1 haplotype) and the risk of FSGS in African Americans but not in other racial groups. Follow up studies have replicated these results in the Hispanic population but not in Native American Indians [71–72]. MYH9 is a non-muscle myosin type IIA that is strongly expressed in the podocyte; where it is an important component of the podocyte cytoskeleton and presumably contributes to its contractile function. MYH9 mutations are the cause of a group of autosomal dominant disorders termed the MHY9-related diseases. The disorders include May-Hegglin, Sebastian, Fechtner and Epstein syndromes. The phenotypes associated with these disorders include macrothrombocytopenia, sensorineural deafness, neutrophil Dohle-like bodies and glomerular disease [73–74]. The discovery of MYH9 as a common disease risk factor for FSGS is further evidence that podocyte integrity is central to the pathogenesis of FSGS. However, unlike in single gene defects, the disease is not highly penetrant and resequencing of MYH9 in individuals with the disease associated haplotype did not find any disease causing variants suggesting that the effect may be in the regulatory elements of the gene or alternatively, there may be other genetic and environmental factors predisposing the carriers of high risk alleles to the development of FSGS.
Apolipoprotein L1 (APOL1) and idiopathic FSGS
In a follow-up study, the MYH9 locus was found to be in linkage disequilibrium with the locus for APOL1 the gene encoding apolipoprotein L1 [75]. Whole genome sequence showed that two sequence variants in APOL1 (G1: rs73885319, G2: rs71785313) are more common in individuals of African descent (Yorubas of South Western Nigeria) compared with Europeans. Furthermore, the disease associated alleles were more common in African Americans (AA) with FSGS compared with AA with no disease. The APOL1 locus is located in a region of the genome that is in linkage disequilibrium with variants that have shown signals of recent natural selection [75]. In addition apolipoprotein L1 has the ability to lyse trypanosomes [75]. It is therefore possible that the variant is a survival factor for trypanosomiasis but a risk factor for renal disease [75]. Future studies will hopefully address the mechanisms by which APOL1 variants may predisposes to FSGS.
Others
FSGS may occur as a component of other single gene defect syndromes. The genetic defects associated with FSGS are listed in Table 2 [76–78] and the role of the products of these genes and other genes identified in maintaining the functional integrity of the podocyte and slit diaphragm is shown in a schematic diagram in Figure 1.
Table 2.
Genetic causes of FSGS and nephrotic syndrome
| Genes (Inheritance) | Protein localization | Locus | Phenotype |
|---|---|---|---|
| NPHS1/nephrin (AR) | Podocyte and slit diaphragm | 19q13.1 | Congenital nephrotic syndrome |
| NPHS2/Podocin (AR) | Podocyte and slit diaphragm | 1q25-q31 | Early onset FSGS |
| CD2AP (AD) | Podocyte and slit diaphragm | 6p12.3 | Adult onset FSGS |
| WT1 (AD) | Podocyte | 11p13 | Syndromic DMS, Syndromic and isolated FSGS |
| α-actinin 4 (AD) | Podocyte | 19q13 | Adult onset FSGS |
| TRPC6 (AD) | Podocyte | 11q21-q22 | Adult onset FSGS |
| PLCE1 (AR) | Podocyte | 10q23-q24 | Non syndromic DMS, FSGS |
| LMX1B (AD)76 | Podocyte | 9q34.1 | Syndromic NS and skeletal dysplasia |
| SMARCAL1 (AR)77 | Podocyte | 2q34-q36 | Syndromic immune complex nephritis and skeletal defect |
| LAMB2 (AR) | Glomerular basement membrane | 3p21 | Syndromic DMS, isolated FSGS |
| SCARB2 (AR)78 | Lysosome | 4q21.1 | Syndromic FSGS |
| MYH9(Complex) | Podocyte | 22p | Idiopathic FSGS |
| INF2 (AD) | Podocyte | 14q32 | Adult onset FSGS |
| Unknown | Unknown | Unknown | Galloway Mowat syndrome |
| Multiple | Unknown | Multiple | Charcot-Marie-Tooth disease |
TREATMENT OF FSGS
The major goals of therapy of FSGS are to achieve complete remission of proteinuria and to preserve kidney function. However, there are no therapeutic regimens that induce remission in all cases. Most agents used to treat FSGS are immunomodulators. The rationale for this is that most cases of idiopathic FSGS are thought to be part of the immune-mediated minimal change disease/FSGS disease spectrum. There are presently no evidence-based guidelines for the use of these agents, in part due to rarity of the disease and inadequately powered randomized control trials. The largest trial to date is the FSGS trial sponsored by the NIH (FSGS-CT, NCT00135811). This study is a randomized control trial of cyclosporine and mycophenolate mofetil (MMF) + dexamethasone in the treatment of steroid resistant FSGS. The results of this trial are eagerly awaited.
In addition to immunomodulatory agents, supportive therapy, such as control of edema with diuretics, management of hyperlipidemia and control of proteinuria with angiotensin-converting enzyme inhibitors and angiotensin receptor blockers, may improve the quality of life and may also slow the rate of progression to ESKD. This section will give an update on current therapy of FSGS and recent data explaining the rationale for their use. In addition, new experimental therapy and future therapeutic options will also be discussed. Figure 2 shows possible mechanisms of action of current treatment modalities as well as possible future novel therapeutic targets
Figure 2. New insight into mechanisms of action of FSGS therapy and novel therapeutic targets.

1. In addition to their immunomodulatory role, corticosteroids may also ameliorate kidney injury by increasing actin (A) polymerization through GTPase RhoA [83] 2. Anti-proteinuric effects of calcineurin inhibitors may be as a result of inhibition of dephosphorylation of synaptopodin (S) [85] 3. Inhibition of TRPC6 (T) channel may be useful in the therapy of familial and idiopathic FSGS [46, 52] 4. Galactose may block the binding site or change the configuration of the free soluble factor preventing it from binding to the podocytes [103] 5. Phosphorylation of nephrin (N) and Neph-1 (N1) by Fyn kinase may induce actin polymerization [105, 106]. Modulation of this pathway may be of therapeutic benefit in proteinuric kidney disease. 6. Modulation of sialylation pathway is a potential therapeutic target because glomerular proteinuria induced by mutation in key enzyme of sialic acid synthesis (pathway responsible for sialylation of major podocyte protein such as podocalyxin [P]) is rescued by N-acetylmannosamine [105, 107].
Corticosteroids
Corticosteroids are the mainstay of treatment for idiopathic nephrotic syndrome, however; the International Study of Kidney Disease in Children study (ISKDC) established that only 30% of children with FSGS histology will achieve remission with steroids [79]. Prolonged high dose intravenous methlyprednisolone in conjunction with oral prednisolone and alkylating agents may increase the remission rate to 60% [80–81]. However, these studies were not randomized control trials.
The mechanisms of action of corticosteroids in idiopathic NS and FSGS have not been fully elucidated. The initial rationale for their use was based on the premise that FSGS is an immunological disease and corticosteroids act by suppressing a T-lymphocyte mediated response. However, with the discovery of key podocyte genes, research in this area is now focused on the effects of corticosteroids on the podocyte and its cytoskeleton. Dexamethasone has been shown to upregulate the expression of nephrin and tubulin-α in an immortalized podocyte cell line [82]. Dexamethasone has also been shown to enhance podocyte survival and downregulate cyclin kinase inhibitor p21, a kinase that is upregulated in podocyte injury diseases. In a similar study, Ransom et al., [83] showed that dexamethasone protected and enhanced recovery of podocytes in a primary murine culture treated with puromycin amino nucleoside (PAN) by increasing actin polymerization through the actin-regulating GTPase RhoA. Overall, these data suggest that in addition to the corticosteroids’ effect on T-cell function, they may also have an effect on the cytoskeleton which may explain their benefit in nonimmune-mediated FSGS.
Calcineurin inhibitors
Cyclosporine A and more recently tacrolimus are the two major calcineurin inhibitors used in the therapy of FSGS. Calcineurin inhibitors act on T-helper cells to downregulate the transcription of a number of cytokine genes, especially interleukin-2 (IL-2). They also inhibit the proliferation of cytotoxic T-cells and B-cells in response to T-helper cell signaling. This T-cell effect of calcineurin inhibitors is thought to be the mechanism of its action in treating FSGS, although there are also data to suggest that the proteinuria lowering effect of calcineurin inhibitors may also be through alterations in glomerular hemodynamics [84]. Recent data suggest that the anti-proteinuric effect of cyclosporine is from blocking the calcineurin mediated dephosphorylation of synaptopodin [85]. As stated earlier, synaptopodin stabilizes the actin cytoskeleton of the podocytes by its regulation of RhoA GTPases. The implication of these findings suggests that calcineurin inhibitors may have a role in both immune mediated FSGS and familial FSGS that are often due to disruption of the podocyte cytoskeleton.
The clinical data on the use of these agents in the treatment of FSGS are limited. In a recent Cochrane collaboration metanalysis of three randomized trials involving 49 patients with steroid resistant nephrotic syndrome and histology that was mainly MCD and FSGS, cyclosporine was shown to significantly increase the number of children who achieved complete remission compared with placebo or no treatment [86]. In this series, the major adverse effects reported were infection (23%) and hypertension (8%). Combining the data from this study and others, cyclosporine combined with prednisolone and ACEI can induce complete or partial remission in up to 60% of children with FSGS [87–88]. Limited data suggest that tacrolimus may be equally effective as cyclosporine [89]. However, there is a high rate of relapse with withdrawal of calcineurin inhibitors in the therapy of FSGS that has to be balanced with the increased risk of nephrotoxicity with prolonged therapy [88, 90]. Randomized controlled trials are needed to develop evidence-based guidelines addressing indications, duration of therapy, target therapeutic levels, and potential combination therapy in the treatment of FSGS.
Anti proliferative agents
Mycophenolate mofetil (MMF) is an important ant-proliferative agent. It is used extensively for immunosuppresion in solid organ transplantation. MMF acts through its active metabolite mycophenolic acid (MPA) as a non-competitive inhibitor of the enzyme inosine monophosphate dehydrogenase (IMPDH) that preferentially inhibits B and T lymphocyte proliferation. Its mechanism of action in the treatment of glomerular disease is not fully known, but it has been shown in both human and experimental studies that it may act by suppressing lymphocyte proliferation and antibody production [91–94]. MMF may also inhibit mesangial proliferation [93]. It also decreases interleukin 2, interleukin 4 and adhesion molecule expression in the kidney [91–94]. Limited clinical data seem to suggest that it may induce complete or partial remission in steroid and cyclosporine resistant FSGS without inducing the side effects of nephrotoxicity seen with calcineurin inhibitor therapy [95–96]. The rate of relapse after withdrawing MMF therapy and the long term side effects, such as malignancy, are unknown. The results of the FSGS trial will hopefully clarify the usefulness of MMF and cyclosporine in the treatment of FSGS.
Monoclonal antibodies
Monoclonal antibodies are increasingly being used in the treatment of steroid-resistant and steroid-dependent FSGS. The rationale for this therapy is based on the premise that some cases of FSGS and nephrotic syndrome result from T-cell dysregulation and that podocytes express different patterns of cytokines and chemokines during relapse and remission of FSGS. Additional justification for the use of these biologic agents is the identification of soluble human FSGS factors that can induce increased permeability in isolated rodent glomeruli [22]. The main advantage of these agents is that they are directed towards specific cell surface ligands, soluble complement components and cytokines and they therefore produce a more targeted action [97]. Some of the agents that have been used in the treatment of FSGS are:
Rituximab
This is a chimeric monoclonal antibody that inhibits CD20 mediated B lymphocyte proliferation and differentiation. The efficacy of rituximab in the treatment of FSGS has not been well defined; however, there are isolated reports and case series that suggest that it may have a therapeutic role in FSGS. An open prospective study of 22 children comprised of 19 patients with minimal change disease and three children with FSGS was recently reported from France [98]. Children in this cohort were treated with weekly infusions of 375mg/m2 of rituximab for two to four weeks. This regime induced remission in three out of seven patients who were in relapse at the time of the study and also produced sustained remission in 19 of the 22 patients studied. The results of this study should be interpreted with caution because the participants were on other immunosuppressive agents in addition to the rituximab; it is therefore difficult to ascribe the effects seen solely to rituximab. In another study, only two out of eight adults with idiopathic FSGS achieved sustained remission following treatment with rituximab [99]. There is a need for randomized studies to define the efficacy of rituximab in the treatment of FSGS and also its side effects and the clinical predictors of a therapeutic response.
Adalimumab
Adalimumab is a human monoclonal antibody directed against tumor necrosis factor α (TNF-α). The rationale for its use in FSGS treatment is based on the observation that TNF-α is upregulated in both human FSGS and also in experimental models 100–101]. An ongoing phase 1 trial by the novel therapies for resistant FSGS (FONT) study group in children with therapy resistant FSGS showed that adalimumab was well tolerated and after sixteen months of follow up, more than half of the cohort showed stabilization of renal function and reduced proteinuria [100–101]. This observation suggests that adalimumab may have a role in slowing the progression of FSGS, but further studies are needed to confirm this.
Others
Rosiglitazone
This is a peroxisome-proliferator activated receptor-γ agonist that increases insulin sensitivity. It is licensed for the treatment of diabetes mellitus. It has been shown to have anti-fibrotic effects in experimental FSGS. A recent phase 1 trial by the FONT study group showed that rosiglitazone was well tolerated in children with therapy-resistant FSGS and after 16 months of follow up, 71% of participants had stable GFR and reduced proteinuria [101–102]. A comprehensive review of the roles and mechanism of actions of monoclonal antibodies in the therapy of FSGS and other podocytopathies can be found in the review by Marasa and Kopp [97].
Novel therapy
Galactose
A recent observation by Savin group showed that the FSGS soluble factor found in recurrent FSGS has an affinity for galactose in column chromatography experiments [103]. Based on this finding, they postulated that free soluble factor may have galactose binding sites that interact with the transmembrane protein of the glycocalyx leading to activation of signal transduction in podocytes [103]. Galactose may block the binding site or change the configuration of the free soluble factor preventing it from binding to the podocytes [103]. In a single patient with recurrent FSGS, oral galactose reduced the plasma activity of the FSGS soluble factor. An adult patient with therapy resistant FSGS had reduced proteinuria following prolonged high dose oral galactose therapy [103–104]. While these 2 case reports are tantalizing, larger studies are needed to evaluate the role and side effects of this novel approach to the therapy of FSGS. Other possible therapeutic targets for FSGS are shown in Figure 2 and were, recently reviewed by Lavin et al [105] and other groups [106–107].
Future therapy
Podocyte stem cells
As described in the previous section, the initiating event in FSGS may be absolute or relative podocytopenia. Podocytes are however terminally differentiated cells with limited capacity to divide. There are data to suggest that throughout life, we constantly shed podocytes into the urine, yet symptomatic FSGS is not a common aging phenomenon in the general population [108]. It is therefore plausible to hypothesize that there exist cells in the body that are capable of regenerating podocytes throughout life. One possible source of cells for podocyte regeneration is bone marrow derived stem cells. These cells were shown in one study to be capable of migrating to the glomerular tuft in a mouse model of Alport’s syndrome [109]. As attractive as this is, the efficiency of such a mechanism is in doubt as the cells have to cross an anatomical barrier (GBM) to reach the glomerular tufts. Also the bone marrow derived progenitor cells were shown to be only a tiny fraction of the new podocytes in this study, implying that there are other progenitor cells that are probably native to the kidney. Another possible source of progenitor cells are the glomerular parietal epithelial cells (GPEC) lining Bowman’s capsule. These cells are located in the same compartment as the podocyte and are in direct continuity with the glomerular tuft at the vascular pole, they therefore do not have to cross anatomical barriers (Figure 3). Two recent studies using different approaches showed that these cells are capable of switching to the podocyte phenotype and may therefore be a source of “podocyte stem cells” [110–112].
Figure 3. Glomerular parietal epithelial cells (GPEC) as podocyte “stem cells”.
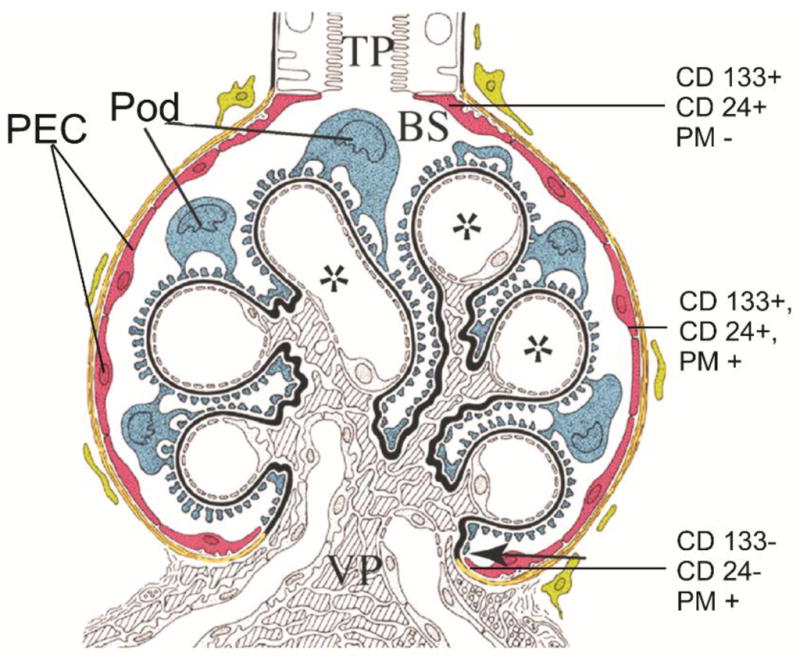
A schematic representation of the glomerulus showing the visceral glomerular epithelial cell or podocyte (Pod; blue) and glomerular parietal epithelial cells (PEC; red). Note that PEC and Pod are in direct continuity at the vascular pole (VP). There are three populations of PEC cells, the least differentiated cells (CD133+, podocyte marker (PM−) and CD24+) are found closer to the urine or tubular pole (TP) and the terminally differentiated cells (CD133−, CD24−, PM+) are found at the VP, and intermediate plueripotent cells (CD133+, CD24+, PM+) cells are found midway between the TP and the VP. Injection of CD133+, CD24+ and PM− cells ameliorate the clinical course of adriamycin-induced proteinuria [110]. Adapted and reproduced with permission from Appel et al. [111].
Ronconi et al., [110] showed that there are at least three different populations of cells lining Bowman’s capsule. Cells at the urinary pole are the least differentiated and they are CD133+(marker of hematopoietic and other adult tissue stem cells), CD24+ (marker of human stem cells) but podocalyxin- (PM−: podocyte marker negative); whereas cells that are CD133+, CD24+, PM+ are more differentiated and are found between the urinary and the vascular pole. The third population of cells are terminally differentiated cells that are CD133− and CD24− and are found mainly at the vascular pole. The CD133+, CD24+, PM− cells are capable of transforming into podocytes and tubular cells; whereas the CD133+, CD24+, PM+ are only capable of differentiating into podocytes. The three different GPEC groups were injected into SCID mice with adriamycin-induced renal injury and only the mice injected with CD133+, CD24+, PM− cells showed reduced proteinuria and reduced glomerular damage, suggesting that this cell line has the capacity to ameliorate glomerular injury. In another study, Appel et al., [111], using transgenic animals that differentially identified GPEC and podocytes showed that there are podocyte progenitor cells lining Bowman’s capsule and these cells have the ability to switch to the podocyte phenotype and migrate to the glomerular tuft. If these observations are confirmed by subsequent studies, identification of an on/off switch for transformation of GPEC to podocyte would be a promising therapeutic target for FSGS.
Deoxyspergualin
Buffalo/Mna rats have been reported to develop spontaneous FSGS and nephrotic syndrome and may also develop recurrence of the disease after kidney transplantation as seen in 30% of humans with idiopathic FSGS [113]. A recent report showed that treatment of these rats with deoxyspergualin, a potent immunosuppressive medication known to block T and B lymphocyte differentiation, induced complete remission and regression of FSGS in both native and transplanted kidneys [113]. The effect of this agent in human disease is unknown but this observation may lead to the development of new agents for the treatment of FSGS.
CONCLUSION
FSGS remains an important cause of renal failure worldwide. Advances in molecular genetics and cell biology have contributed significantly to our understanding of the biology of the podocyte and its slit diaphragm. Defects in podocyte function play an important role in the pathogenesis of FSGS. However, this increase in knowledge has not been matched by therapeutic advances. There is a need for further understanding of the mechanisms of actions of agents currently being used in the treatment of FSGS and exploration of other therapeutic options such as podocyte regeneration by native renal cells. Further studies of inherited forms of FSGS will continue to help in unraveling the mechanisms of this disease and possibly lead to the identification of new and better targeted therapies.
Acknowledgments
Funding: NIH K08DK082495-01 and grants from Nephcure foundation to RG. RG is a recipient of a Doris Duke Clinical Scientist Development Award.
References
- 1.Cameron JS. Focal segmental glomerulosclerosis in adults. Nephrol Dial Transplant. 2003;18:vi45–51. doi: 10.1093/ndt/gfg1058. [DOI] [PubMed] [Google Scholar]
- 2.Rich AR. A hitherto undescribed vulnerability of the juxtamedullary glomeruli in lipoid nephrosis. Bull Johns Hopkins Hosp. 1957;100:173–86. [PubMed] [Google Scholar]
- 3.Churg J, Habib R, White RH. Pathology of the nephrotic syndrome in children: a report for the International Study of Kidney Disease in Children. Lancet. 1970;760:1299–302. doi: 10.1016/s0140-6736(70)91905-7. [DOI] [PubMed] [Google Scholar]
- 4.Kitiyakara C, Kopp JB, Eggers P. Trends in the epidemiology of focal segmental glomerulosclerosis. Semin Nephrol. 2003;23:172–82. doi: 10.1053/snep.2003.50025. [DOI] [PubMed] [Google Scholar]
- 5.USRDS report 2007
- 6.Kitiyakara C, Eggers P, Kopp JB. Twenty-one-year trend in ESRD due to focal segmental glomerulosclerosis in the United States. Am J Kidney Dis. 2004;44:815–25. [PubMed] [Google Scholar]
- 7.D’Agati VD, Fogo AB, Bruijn JA, Jennette JC. Pathologic classification of focal segmental glomerulosclerosis: a working proposal. Am J Kidney Dis. 2004;43:368–82. doi: 10.1053/j.ajkd.2003.10.024. [DOI] [PubMed] [Google Scholar]
- 8.Stokes MB, Valeri AM, Markowitz GS, D’Agati VD. Cellular focal segmental glomerulosclerosis: Clinical and pathologic features. Kidney Int. 2006;70:1783–92. doi: 10.1038/sj.ki.5001903. [DOI] [PubMed] [Google Scholar]
- 9.Deegens JK, Steenbergen EJ, Borm GF, Wetzels JF. Pathological variants of focal segmental glomerulosclerosis in an adult Dutch population--epidemiology and outcome. Nephrol Dial Transplant. 2008;23:186–92. doi: 10.1093/ndt/gfm523. [DOI] [PubMed] [Google Scholar]
- 10.Silverstein DM, Craver R. Presenting features and short-term outcome according to pathologic variant in childhood primary focal segmental glomerulosclerosis. Clin J Am Soc. 2007;2:700–7. doi: 10.2215/CJN.00230107. [DOI] [PubMed] [Google Scholar]
- 11.Canaud G, Dion D, Zuber J, Gubler MC, Sberro R, Thervet E, Snanoudj R, Charbit M, Salomon R, Martinez F, Legendre C, Noel LH, Niaudet P. Recurrence of nephrotic syndrome after transplantation in a mixed population of children and adults: course of glomerular lesions and value of the Columbia classification of histological variants of focal and segmental glomerulosclerosis (FSGS) Nephrol Dial Transplant. 2010;55:558–65. doi: 10.1093/ndt/gfp500. [DOI] [PubMed] [Google Scholar]
- 12.Johnstone DB, Holzman LB. Clinical impact of research on the podocyte slit diaphragm. Nat Clin Pract Nephrol. 2006;2:271–82. doi: 10.1038/ncpneph0180. [DOI] [PubMed] [Google Scholar]
- 13.Smoyer WE, Mundel P. Regulation of podocyte structure during the development of nephrotic syndrome. J Mol Med. 1998;76:172–83. doi: 10.1007/s001090050206. [DOI] [PubMed] [Google Scholar]
- 14.White RH, Glasgow EF, Mills RJ. Clinicopathological study of nephrotic syndrome in childhood. Lancet. 1970;1:1353–9. doi: 10.1016/s0140-6736(70)91268-7. [DOI] [PubMed] [Google Scholar]
- 15.Haraldsson B, Nyström J, Deen WM. Properties of the glomerular barrier and mechanisms of proteinuria. Physiol Rev. 2008;88:451–87. doi: 10.1152/physrev.00055.2006. [DOI] [PubMed] [Google Scholar]
- 16.Partanen TA, Arola J, Saaristo A, Jussila L, Ora A, Miettinen M, Stacker SA, Achen MG, Alitalo K. VEGF-C and VEGF-D expression in neuroendocrine cells and their receptor, VEGFR-3, in fenestrated blood vessels in human tissues. FASEB J. 2000;14:2087–96. doi: 10.1096/fj.99-1049com. [DOI] [PubMed] [Google Scholar]
- 17.Rostgaard J, Qvortrup K. Sieve plugs in fenestrae of glomerular capillaries--site of the filtration barrier? Cells Tissues Organs. 2002;170:132–8. doi: 10.1159/000046186. [DOI] [PubMed] [Google Scholar]
- 18.Weinbaum S, Tarbell JM, Damiano ER. The structure and function of the endothelial glycocalyx layer. Annu Rev Biomed. 2007;9:121–67. doi: 10.1146/annurev.bioeng.9.060906.151959. [DOI] [PubMed] [Google Scholar]
- 19.Ballermann BJ, Stan RV. Resolved: capillary endothelium is a major contributor to the glomerular filtration barrier. J Am Soc Nephrol. 2007;18:2432–8. doi: 10.1681/01.asn.0000926880.23301.cc. [DOI] [PubMed] [Google Scholar]
- 20.Vaughan MR, Quaggin SE. How do mesangial and endothelial cells form the glomerular tuft? J Am Soc Nephrol. 2008;19:24–33. doi: 10.1681/ASN.2007040471. [DOI] [PubMed] [Google Scholar]
- 21.Shalhoub RJ. Pathogenesis of lipoid nephrosis: a disorder of T-cell function. Lancet. 1974;2:556–60. doi: 10.1016/s0140-6736(74)91880-7. [DOI] [PubMed] [Google Scholar]
- 22.Savin VJ, Sharma R, Sharma M, McCarthy ET, Swan SK, Ellis E, Lovell H, Warady B, Gunwar S, Chonko AM, Artero M, Vincenti F. Circulating factor associated with increased glomerular permeability to albumin in recurrent focal segmental glomerulosclerosis. N Engl J Med. 1996;334:878–83. doi: 10.1056/NEJM199604043341402. [DOI] [PubMed] [Google Scholar]
- 23.Zenker M, Aigner T, Wendler O, Tralau T, Müntefering H, Fenski R, Pitz S, Schumacher V, Royer-Pokora B, Wühl E, Cochat P, Bouvier R, Kraus C, Mark K, Madlon H, Dötsch J, Rascher W, Maruniak-Chudek I, Lennert T, Neumann LM, Reis A. Human laminin beta2 deficiency causes congenital nephrosis with mesangial sclerosis and distinct eye abnormalities. Hum Mol Genet. 2004;13:2625–32. doi: 10.1093/hmg/ddh284. [DOI] [PubMed] [Google Scholar]
- 24.Karnovsky MJ, Ainsworth SK. The structural basis of glomerular filtration. Adv Nephrol Necker Hosp. 1972;2:35–60. [PubMed] [Google Scholar]
- 25.Kestilä M, Lenkkeri U, Männikkö M, Lamerdin J, McCready P, Putaala H, Ruotsalainen V, Morita T, Nissinen M, Herva R, Kashtan CE, Peltonen L, Holmberg C, Olsen A, Tryggvason K. Positionally cloned gene for a novel glomerular protein--nephrin--is mutated in congenital nephrotic syndrome. Mol Cell. 1998;1:575–82. doi: 10.1016/s1097-2765(00)80057-x. [DOI] [PubMed] [Google Scholar]
- 26.Wiggins RC. The spectrum of podocytopathies: a unifying view of glomerular diseases. Kidney Int. 2007;71:1205–14. doi: 10.1038/sj.ki.5002222. [DOI] [PubMed] [Google Scholar]
- 27.Kriz W. The pathogenesis of ‘classic’ focal segmental glomerulosclerosis-lessons from rat models. Nephrol Dial Transplant. 2003;(Suppl 6):vi39–44. doi: 10.1093/ndt/gfg1064. [DOI] [PubMed] [Google Scholar]
- 28.Sato Y, Wharram BL, Lee SK, Wickman L, Goyal M, Venkatareddy M, Chang JW, Wiggins JE, Lienczewski C, Kretzler M, Wiggins RC. Urine podocyte mRNAs mark progression of renal disease. J Am Soc Nephrol. 2009;20:1041–52. doi: 10.1681/ASN.2007121328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wharram BL, Goyal M, Wiggins JE, Sanden SK, Hussain S, Filipiak WE, Saunders TL, Dysko RC, Kohno K, Holzman LB, Wiggins RC. Podocyte depletion causes glomerulosclerosis: diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J Am Soc Nephrol. 2005;16:2941–52. doi: 10.1681/ASN.2005010055. [DOI] [PubMed] [Google Scholar]
- 30.Patrakka J, Tryggvason K. New insights into the role of podocytes in proteinuria. Nat Rev Nephrol. 2009;5:463–8. doi: 10.1038/nrneph.2009.108. [DOI] [PubMed] [Google Scholar]
- 31.Santín S, García-Maset R, Ruíz P, Giménez I, Zamora I, Peña A, Madrid A, Camacho JA, Fraga G, Sánchez-Moreno A, Cobo MA, Bernis C, Ortiz A, de Pablos AL, Pintos G, Justa ML, Hidalgo-Barquero E, Fernández-Llama P, Ballarín J, Ars E, Torra R FSGS Spanish Study Group . Nephrin mutations cause childhood- and adult-onset focal segmental glomerulosclerosis. Kidney Int. 2009;76:1268–76. doi: 10.1038/ki.2009.381. [DOI] [PubMed] [Google Scholar]
- 32.Boute N, Gribouval O, Roselli S, Benessy F, Lee H, Fuchshuber A, Dahan K, Gubler MC, Niaudet P, Antignac C. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet. 2000;24:349–54. doi: 10.1038/74166. [DOI] [PubMed] [Google Scholar]
- 33.Roselli S, Gribouval O, Boute N, Sich M, Benessy F, Attié T, Gubler MC, Antignac C. Podocin localizes in the kidney to the slit diaphragm area. Am J Pathol. 2002;160:131–9. doi: 10.1016/S0002-9440(10)64357-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barletta GM, Kovari IA, Verma RK, Kerjaschki D, Holzman LB. Nephrin and Neph1 co-localize at the podocyte foot process intercellular junction and form cis hetero-oligomers. J Biol Chem. 2003;278:19266–71. doi: 10.1074/jbc.M301279200. [DOI] [PubMed] [Google Scholar]
- 35.Simons M, Schwarz K, Kriz W, Miettinen A, Reiser J, Mundel P, Holthöfer H. Involvement of lipid rafts in nephrin phosphorylation and organization of the glomerular slit diaphragm. Am J Pathol. 2001;159:1069–77. doi: 10.1016/S0002-9440(10)61782-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schwarz K, Simons M, Reiser J, Saleem MA, Faul C, Kriz W, Shaw AS, Holzman LB, Mundel P. Podocin, a raft-associated component of the glomerular slit diaphragm, interacts with CD2AP and nephrin. J Clin Invest. 2001;108:1621–9. doi: 10.1172/JCI12849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harder T. Lipid raft domains and protein networks in T-cell receptor signal transduction. Curr Opin Immunol. 2004;16:353–9. doi: 10.1016/j.coi.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 38.Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol. 2000;1:31–9. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- 39.Ruf RG, Lichtenberger A, Karle SM, Haas JP, Anacleto FE, Schultheiss M, Zalewski I, Imm A, Ruf EM, Mucha B, Bagga A, Neuhaus T, Fuchshuber A, Bakkaloglu A, Hildebrandt F Arbeitsgemeinschaft Für Pädiatrische Nephrologie Study Group. Patients with mutations in NPHS2 (podocin) do not respond to standard steroid treatment of nephrotic syndrome. J Am Soc Nephrol. 2004;15:722–32. doi: 10.1097/01.asn.0000113552.59155.72. [DOI] [PubMed] [Google Scholar]
- 40.Machuca E, Hummel A, Nevo F, Dantal J, Martinez F, Al-Sabban E, Baudouin V, Abel L, Grünfeld JP, Antignac C. Clinical and epidemiological assessment of steroid-resistant nephrotic syndrome associated with the NPHS2 R229Q variant. Kidney Int. 2009;75:727–35. doi: 10.1038/ki.2008.650. [DOI] [PubMed] [Google Scholar]
- 41.Roselli S, Heidet L, Sich M, Henger A, Kretzler M, Gubler MC, Antignac C. Early glomerular filtration defect and severe renal disease in podocin-deficient mice. Mol Cell Biol. 2004;24:550–60. doi: 10.1128/MCB.24.2.550-560.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mollet G, Ratelade J, Boyer O, Muda AO, Morisset L, Lavin TA, Kitzis D, Dallman MJ, Bugeon L, Hubner N, Gubler MC, Antignac C, Esquivel EL. Podocin inactivation in mature kidneys causes focal segmental glomerulosclerosis and nephrotic syndrome. J Am Soc Nephrol. 2009;20:2181–9. doi: 10.1681/ASN.2009040379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaplan JM, Kim SH, North KN, Rennke H, Correia LA, Tong HQ, Mathis BJ, Rodríguez-Pérez JC, Allen PG, Beggs AH, Pollak MR. Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat Genet. 2000;24:251–6. doi: 10.1038/73456. [DOI] [PubMed] [Google Scholar]
- 44.Yao J, Le TC, Kos CH, Henderson JM, Allen PG, Denker BM, Pollak MR. Alpha-actinin-4-mediated FSGS: an inherited kidney disease caused by an aggregated and rapidly degraded cytoskeletal protein. PLoS Biol. 2004;2:e167. doi: 10.1371/journal.pbio.0020167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Michaud JL, Chaisson KM, Parks RJ, Kennedy CR. FSGS-associated alpha-actinin-4 (K256E) impairs cytoskeletal dynamics in podocytes. 2006;70:1054–61. doi: 10.1038/sj.ki.5001665. [DOI] [PubMed] [Google Scholar]
- 46.Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, Hawkins AF, Daskalakis N, Kwan SY, Ebersviller S, Burchette JL, Pericak-Vance MA, Howell DN, Vance JM, Rosenberg PB. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science. 2005;308:1801–4. doi: 10.1126/science.1106215. [DOI] [PubMed] [Google Scholar]
- 47.Reiser J, Polu KR, Möller CC, Kenlan P, Altintas MM, Wei C, Faul C, Herbert S, Villegas I, Avila-Casado C, McGee M, Sugimoto H, Brown D, Kalluri R, Mundel P, Smith PL, Clapham DE, Pollak MR. TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat Genet. 2005;37:739–44. doi: 10.1038/ng1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Heeringa SF, Möller CC, Du J, Yue L, Hinkes B, Chernin G, Vlangos CN, Hoyer PF, Reiser J, Hildebrandt F. A novel TRPC6 mutation that causes childhood FSGS. PLoS One. 2009;4:e7771. doi: 10.1371/journal.pone.0007771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhu B, Chen N, Wang ZH, Pan XX, Ren H, Zhang W, Wang WM. Identification and functional analysis of a novel TRPC6 mutation associated with late onset familial focal segmental glomerulosclerosis in Chinese patients. Mutat Res. 2009:66484–90. doi: 10.1016/j.mrfmmm.2008.11.021. [DOI] [PubMed] [Google Scholar]
- 50.Clapham DE, Runnels LW, Strübing C. The TRP ion channel family. Nat Rev Neurosci. 2001;2:387–96. doi: 10.1038/35077544. [DOI] [PubMed] [Google Scholar]
- 51.Winn MP. 2007 Young Investigator Award: TRP’ing into a new era for glomerular disease. J Am Soc Nephrol. 2008;19:1071–5. doi: 10.1681/ASN.2007121292. [DOI] [PubMed] [Google Scholar]
- 52.Möller CC, Wei C, Altintas MM, Li J, Greka A, Ohse T, Pippin JW, Rastaldi MP, Wawersik S, Schiavi S, Henger A, Kretzler M, Shankland SJ, Reiser J. Induction of TRPC6 channel in acquired forms of proteinuric kidney disease. J Am Soc Nephrol. 2007;18:29–36. doi: 10.1681/ASN.2006091010. [DOI] [PubMed] [Google Scholar]
- 53.Eckel J, Mukerji N, Lavin P, Ferimazova N, Gbadegesin R, Damodaran T, Bowling B, Wu G, Homstad A, Barisoni L, Bartkowiak B, Winn M. TRPC6 deficiency does not cause glomerulosclerosis. J Am Soc Nephrol. 2009:314A. [Google Scholar]
- 54.Gbadegesin RA, Damodaran T, Homstad A, Bartkowiak B, Bowling B, Wu G, Lavin P, Eckel J, Mukerji N, Winn M. TRPC6 gene dose ameliorates the course of puromycin induced kidney injury. J Am Soc Nephrol. 2009:318A. [Google Scholar]
- 55.Kim JM, Wu H, Green G, Winkler CA, Kopp JB, Miner JH, Unanue ER, Shaw AS. CD2-associated protein haploinsufficiency is linked to glomerular disease susceptibility. Science. 2003;300:1298–300. doi: 10.1126/science.1081068. [DOI] [PubMed] [Google Scholar]
- 56.Wolf G, Stahl RA. CD2-associated protein and glomerular disease. Lancet. 2003;362:1746–8. doi: 10.1016/S0140-6736(03)14856-8. [DOI] [PubMed] [Google Scholar]
- 57.Shih NY, Li J, Karpitskii V, Nguyen A, Dustin ML, Kanagawa O, Miner JH, Shaw AS. Congenital nephrotic syndrome in mice lacking CD2-associated protein. 1999;286:312–5. doi: 10.1126/science.286.5438.312. [DOI] [PubMed] [Google Scholar]
- 58.Löwik MM, Groenen PJ, Pronk I, Lilien MR, Goldschmeding R, Dijkman HB, Levtchenko EN, Monnens LA, van den Heuvel LP. Focal segmental glomerulosclerosis in a patient homozygous for a CD2AP mutation. Kidney Int. 2007;72:1198–203. doi: 10.1038/sj.ki.5002469. [DOI] [PubMed] [Google Scholar]
- 59.Gigante M, Pontrelli P, Montemurno E, Roca L, Aucella F, Penza R, Caridi G, Ranieri E, Ghiggeri GM, Gesualdo L. CD2AP mutations are associated with sporadic nephrotic syndrome and focal segmental glomerulosclerosis (FSGS) Nephrol Dial Transplant. 2009;24:1858–64. doi: 10.1093/ndt/gfn712. [DOI] [PubMed] [Google Scholar]
- 60.Niaudet P, Gubler MC. WT1 and glomerular diseases. Pediatr Nephrol. 2006;21:1653–60. doi: 10.1007/s00467-006-0208-1. [DOI] [PubMed] [Google Scholar]
- 61.Mucha B, Ozaltin F, Hinkes BG, Hasselbacher K, Ruf RG, Schultheiss M, Hangan D, Hoskins BE, Everding AS, Bogdanovic R, Seeman T, Hoppe B, Hildebrandt F Members of the APN Study Group . Mutations in the Wilms’ tumor 1 gene cause isolated steroid resistant nephrotic syndrome and occur in exons 8 and 9. Pediatr Res. 2006;59:325–31. doi: 10.1203/01.pdr.0000196717.94518.f0. [DOI] [PubMed] [Google Scholar]
- 62.Hinkes B, Wiggins RC, Gbadegesin R, Vlangos CN, Seelow D, Nürnberg G, Garg P, Verma R, Chaib H, Hoskins BE, Ashraf S, Becker C, Hennies HC, Goyal M, Wharram BL, Schachter AD, Mudumana S, Drummond I, Kerjaschki D, Waldherr R, Dietrich A, Ozaltin F, Bakkaloglu A, Cleper R, Basel-Vanagaite L, Pohl M, Griebel M, Tsygin AN, Soylu A, Müller D, Sorli CS, Bunney TD, Katan M, Liu J, Attanasio M, O’toole JF, Hasselbacher K, Mucha B, Otto EA, Airik R, Kispert A, Kelley GG, Smrcka AV, Gudermann T, Holzman LB, Nürnberg P, Hildebrandt F. Positional cloning uncovers mutations in PLCE1 responsible for a nephrotic syndrome variant that may be reversible. Nat Genet. 2006;38:1397–405. doi: 10.1038/ng1918. [DOI] [PubMed] [Google Scholar]
- 63.Gbadegesin R, Hinkes BG, Hoskins BE, Vlangos CN, Heeringa SF, Liu J, Loirat C, Ozaltin F, Hashmi S, Ulmer F, Cleper R, Ettenger R, Antignac C, Wiggins RC, Zenker M, Hildebrandt F. Mutations in PLCE1 are a major cause of isolated diffuse mesangial sclerosis (IDMS) Nephrol Dial Transplant. 2008;23:1291–7. doi: 10.1093/ndt/gfm759. [DOI] [PubMed] [Google Scholar]
- 64.Gilbert RD, Turner CL, Gibson J, Bass PS, Haq MR, Cross E, Bunyan DJ, Collins AR, Tapper WJ, Needell JC, Dell B, Morton NE, Temple IK, Robinson DO. Mutations in phospholipase C epsilon 1 are not sufficient to cause diffuse mesangial sclerosis. Kidney Int. 2009;75:415–9. doi: 10.1038/ki.2008.573. [DOI] [PubMed] [Google Scholar]
- 65.Boyer O, Benoit G, Gribouval O, Nevo F, Pawtowski A, Bilge I, Bircan Z, Deschênes G, Guay-Woodford LM, Hall M, Macher MA, Soulami K, Stefanidis CJ, Weiss R, Loirat C, Gubler MC, Antignac C. Mutational analysis of the PLCE1 gene in steroid resistant nephrotic syndrome. J Med Genet. 2010;47:45–52. doi: 10.1136/jmg.2009.076166. [DOI] [PubMed] [Google Scholar]
- 66.Wing MR, Bourdon DM, Harden TK. PLC-epsilon: a shared effector protein in Ras-, Rho-, and G alpha beta gamma-mediated signaling. Mol Interv. 2003;3:273–80. doi: 10.1124/mi.3.5.273. [DOI] [PubMed] [Google Scholar]
- 67.Hasselbacher K, Wiggins RC, Matejas V, Hinkes BG, Mucha B, Hoskins BE, Ozaltin F, Nürnberg G, Becker C, Hangan D, Pohl M, Kuwertz-Bröking E, Griebel M, Schumacher V, Royer-Pokora B, Bakkaloglu A, Nürnberg P, Zenker M, Hildebrandt F. Recessive missense mutations in LAMB2 expand the clinical spectrum of LAMB2-associated disorders. Kidney Int. 2006;70:1008–12. doi: 10.1038/sj.ki.5001679. [DOI] [PubMed] [Google Scholar]
- 68.Brown EJ, Schlöndorff JS, Becker DJ, Tsukaguchi H, Uscinski AL, Higgs HN, Henderson JM, Pollak MR. Mutations in the formin gene INF2 cause focal segmental glomerulosclerosis. Nat Genet. 2010;42:72–6. doi: 10.1038/ng.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kopp JB, Smith MW, Nelson GW, Johnson RC, Freedman BI, Bowden DW, Oleksyk T, McKenzie LM, Kajiyama H, Ahuja TS, Berns JS, Briggs W, Cho ME, Dart RA, Kimmel PL, Korbet SM, Michel DM, Mokrzycki MH, Schelling JR, Simon E, Trachtman H, Vlahov D, Winkler CA. MYH9 is a major-effect risk gene for focal segmental glomerulosclerosis. Nat Genet. 2008;40:1175–84. doi: 10.1038/ng.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kao WH, Klag MJ, Meoni LA, Reich D, Berthier-Schaad Y, Li M, Coresh J, Patterson N, Tandon A, Powe NR, Fink NE, Sadler JH, Weir MR, Abboud HE, Adler SG, Divers J, Iyengar SK, Freedman BI, Kimmel PL, Knowler WC, Kohn OF, Kramp K, Leehey DJ, Nicholas SB, Pahl MV, Schelling JR, Sedor JR, Thornley-Brown D, Winkler CA, Smith MW, Parekh RS. Family Investigation of Nephropathy and Diabetes Research Group. MYH9 is associated with nondiabetic end-stage renal disease in African Americans. Nat Genet. 2008;40:1185–92. doi: 10.1038/ng.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Behar DM, Rosset S, Tzur S, Selig S, Yudkovsky G, Bercovici S, Kopp JB, Winkler CA, Nelson GW, Wasser WG, Skorecki K. African ancestry allelic variation at the MYH9 gene contributes to increased susceptibility to non-diabetic end-stage kidney disease in Hispanic Americans. Hum Mol Genet. 2010;19:1816–27. doi: 10.1093/hmg/ddq040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Franceschini N, Voruganti VS, Haack K, Almasy L, Laston S, Goring HH, Umans JG, Lee ET, Best LG, Fabsitz RR, MacCluer JW, Howard BV, North KE, Cole SA. The association of the MYH9 gene and kidney outcomes in American Indians: the Strong Heart Family Study. Hum Genet. 2010;127:295–301. doi: 10.1007/s00439-009-0769-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ghiggeri GM, Caridi G, Magrini U, Sessa A, Savoia A, Seri M, Pecci A, Romagnoli R, Gangarossa S, Noris P, Sartore S, Necchi V, Ravazzolo R, Balduini CL. Genetics, clinical and pathological features of glomerulonephritis associated with mutations of nonmuscle myosin IIA (Fechtner syndrome) Am J Kidney Dis. 2003;41:95–104. doi: 10.1053/ajkd.2003.50028. [DOI] [PubMed] [Google Scholar]
- 74.Dong F, Li S, Pujol-Moix N, Luban NL, Shin SW, Seo JH, Ruiz-Saez A, Demeter J, Langdon S, Kelley MJ. Genotype-phenotype correlation in MYH9-related thrombocytopenia. Br J Haematol. 2005;130:620–7. doi: 10.1111/j.1365-2141.2005.05658.x. [DOI] [PubMed] [Google Scholar]
- 75.Genovese G, Friedman DJ, Ross MD, Lecordier L, Uzureau P, Freedman BI, Bowden DW, Langefeld CD, Oleksyk TK, Uscinski Knob AL, Bernhardy AJ, Hicks PJ, Nelson GW, Vanhollebeke B, Winkler CA, Kopp JB, Pays E, Pollak MR. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science. 2010;329:841–5. doi: 10.1126/science.1193032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dreyer SD, Zhou G, Baldini A, Winterpacht A, Zabel B, Cole W, Johnson RL, Lee B. Mutations in LMX1B cause abnormal skeletal patterning and renal dysplasia in nail patella syndrome. Nat Genet. 1998;19:47–50. doi: 10.1038/ng0598-47. [DOI] [PubMed] [Google Scholar]
- 77.Boerkoel CF, Takashima H, John J, Yan J, Stankiewicz P, Rosenbarker L, André JL, Bogdanovic R, Burguet A, Cockfield S, Cordeiro I, Fründ S, Illies F, Joseph M, Kaitila I, Lama G, Loirat C, McLeod DR, Milford DV, Petty EM, Rodrigo F, Saraiva JM, Schmidt B, Smith GC, Spranger J, Stein A, Thiele H, Tizard J, Weksberg R, Lupski JR, Stockton DW. Mutant chromatin remodeling protein SMARCAL1 causes Schimke immuno-osseous dysplasia. Nat Genet. 2002;30:215–20. doi: 10.1038/ng821. [DOI] [PubMed] [Google Scholar]
- 78.Berkovic SF, Dibbens LM, Oshlack A, Silver JD, Katerelos M, Vears DF, Lüllmann-Rauch R, Blanz J, Zhang KW, Stankovich J, Kalnins RM, Dowling JP, Andermann E, Andermann F, Faldini E, D’Hooge R, Vadlamudi L, Macdonell RA, Hodgson BL, Bayly MA, Savige J, Mulley JC, Smyth GK, Power DA, Saftig P, Bahlo M. Array-based gene discovery with three unrelated subjects shows SCARB2/LIMP-2 deficiency causes myoclonus epilepsy and glomerulosclerosis. Am J Hum Genet. 2008;82:673–84. doi: 10.1016/j.ajhg.2007.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.ISKDC . Nephrotic syndrome in children: prediction of histopathology from clinical and laboratory characteristics at time of diagnosis. A report of the International Study of Kidney Disease in Children. Kidney Int. 1978;13:159–65. doi: 10.1038/ki.1978.23. [DOI] [PubMed] [Google Scholar]
- 80.Mendoza SA, Tune BM. Treatment of childhood nephrotic syndrome. J Am Soc Nephrol. 1992;3:889–94. doi: 10.1681/ASN.V34889. [DOI] [PubMed] [Google Scholar]
- 81.Tune BM, Mendoza SA. Treatment of the idiopathic nephrotic syndrome: regimens and outcomes in children and adults. J Am Soc Nephrol. 1997;8:824–32. doi: 10.1681/ASN.V85824. [DOI] [PubMed] [Google Scholar]
- 82.Xing CY, Saleem MA, Coward RJ, Ni L, Witherden IR, Mathieson PW. Direct effects of dexamethasone on human podocytes. Kidney Int. 2006;70:1038–45. doi: 10.1038/sj.ki.5001655. [DOI] [PubMed] [Google Scholar]
- 83.Ransom RF, Lam NG, Hallett MA, Atkinson SJ, Smoyer WE. Glucocorticoids protect and enhance recovery of cultured murine podocytes via actin filament stabilization. Kidney Int. 2005;68:2473–83. doi: 10.1111/j.1523-1755.2005.00723.x. [DOI] [PubMed] [Google Scholar]
- 84.Zietse R, Wenting GJ, Kramer P, Schalekamp MA, Weimar W. Effects of cyclosporin A on glomerular barrier function in the nephrotic syndrome. Clin Sci (Lond) 1992;82:641–50. doi: 10.1042/cs0820641. [DOI] [PubMed] [Google Scholar]
- 85.Faul C, Donnelly M, Merscher-Gomez S, Chang YH, Franz S, Delfgaauw J, Chang JM, Choi HY, Campbell KN, Kim K, Reiser J, Mundel P. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat Med. 2008;14:931–8. doi: 10.1038/nm.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hodson EM, Willis NS, Craig JC. Non-corticosteroid treatment for nephrotic syndrome in children. Cochrane Database Syst Rev. 2008;23:CD002290. doi: 10.1002/14651858.CD002290.pub3. [DOI] [PubMed] [Google Scholar]
- 87.Mahmoud I, Basuni F, Sabry A, El-Husseini A, Hassan N, Ahmad NS, Elbaz M, Moustafa F, Sobh M. Single-centre experience with cyclosporin in 106 children with idiopathic focal segmental glomerulosclerosis. Nephrol Dial Transplant. 2005;20:735–42. doi: 10.1093/ndt/gfh766. [DOI] [PubMed] [Google Scholar]
- 88.Cattran DC, Alexopoulos E, Heering P, Hoyer PF, Johnston A, Meyrier A, Ponticelli C, Saito T, Choukroun G, Nachman P, Praga M, Yoshikawa N. Cyclosporin in idiopathic glomerular disease associated with the nephrotic syndrome : workshop recommendations. Kidney Int. 2007;72:1429–47. doi: 10.1038/sj.ki.5002553. [DOI] [PubMed] [Google Scholar]
- 89.Xia Z, Liu G, Gao Y, Fan Z, Fu Y, Zhang LF, Ren X, Gao C. FK506 in the treatment of children with nephrotic syndrome of different pathological types. Clin Nephrol. 2006;66:85–8. doi: 10.5414/cnp66085. [DOI] [PubMed] [Google Scholar]
- 90.Cattran DC, Appel GB, Hebert LA, Hunsicker LG, Pohl MA, Hoy WE, Maxwell DR, Kunis CL. A randomized trial of cyclosporine in patients with steroid-resistant focal segmental glomerulosclerosis. North America Nephrotic Syndrome Study Group. Kidney Int. 1999;56:2220–6. doi: 10.1046/j.1523-1755.1999.00778.x. [DOI] [PubMed] [Google Scholar]
- 91.Ziswiler R, Steinmann-Niggli K, Kappeler A, Daniel C, Marti HP. Mycophenolic acid: a new approach to the therapy of experimental mesangial proliferative glomerulonephritis. J Am Soc Nephrol. 1998;9:2055–66. doi: 10.1681/ASN.V9112055. [DOI] [PubMed] [Google Scholar]
- 92.Hauser IA, Renders L, Radeke HH, Sterzel RB, Goppelt-Struebe M. Mycophenolate mofetil inhibits rat and human mesangial cell proliferation by guanosine depletion. Nephrol Dial Transplant. 1999;14:58–63. doi: 10.1093/ndt/14.1.58. [DOI] [PubMed] [Google Scholar]
- 93.Penny MJ, Boyd RA, Hall BM. Mycophenolate mofetil prevents the induction of active Heymann nephritis: association with Th2 cytokine inhibition. J Am Soc Nephrol. 1998;9:2272–82. doi: 10.1681/ASN.V9122272. [DOI] [PubMed] [Google Scholar]
- 94.Allison AC, Kowalski WJ, Muller CJ, Waters RV, Eugui EM. Mycophenolic acid and brequinar, inhibitors of purine and pyrimidine synthesis, block the glycosylation of adhesion molecules. Transplant Proc. 1993;25:67–70. [PubMed] [Google Scholar]
- 95.Cattran DC, Wang MM, Appel G, Matalon A, Briggs W. Mycophenolate mofetil in the treatment of focal segmental glomerulosclerosis. Clin Nephrol. 2004;62:405–11. doi: 10.5414/cnp62405. [DOI] [PubMed] [Google Scholar]
- 96.Montané B, Abitbol C, Chandar J, Strauss J, Zilleruelo G. Novel therapy of focal glomerulosclerosis with mycophenolate and angiotensin blockade. Pediatr Nephrol. 2003;18:772–7. doi: 10.1007/s00467-003-1174-5. [DOI] [PubMed] [Google Scholar]
- 97.Marasà M, Kopp JB. Monoclonal antibodies for podocytopathies: rationale and clinical responses. Nat Rev Nephrol. 2009;5:337–48. doi: 10.1038/nrneph.2009.70. [DOI] [PubMed] [Google Scholar]
- 98.Guigonis V, Dallocchio A, Baudouin V, Dehennault M, Hachon-Le Camus C, Afanetti M, Groothoff J, Llanas B, Niaudet P, Nivet H, Raynaud N, Taque S, Ronco P, Bouissou F. Rituximab treatment for severe steroid- or cyclosporine-dependent nephrotic syndrome: a multicentric series of 22 cases. Pediatr Nephrol. 2008;23:1269–79. doi: 10.1007/s00467-008-0814-1. [DOI] [PubMed] [Google Scholar]
- 99.Fernandez-Fresnedo G, Segarra A, González E, Alexandru S, Delgado R, Ramos N, Egido J, Praga M Trabajo de Enfermedades Glomerulares de la Sociedad Española de Nefrología (GLOSEN) . Rituximab treatment of adult patients with steroid-resistant focal segmental glomerulosclerosis. Clin J Am Soc Nephrol. 2009;4:1317–23. doi: 10.2215/CJN.00570109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Joy MS, Gipson DS, Powell L, MacHardy J, Jennette JC, Vento S, Pan C, Savin V, Eddy A, Fogo AB, Kopp JB, Cattran D, Trachtman H. Phase 1 trial of adalimumab in Focal Segmental Glomerulosclerosis (FSGS): II. Report of the FONT (Novel Therapies for Resistant FSGS) study group. Am J Kidney Dis. 2010;55:50–60. doi: 10.1053/j.ajkd.2009.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Peyser A, Machardy N, Tarapore F, Machardy J, Powell L, Gipson DS, Savin V, Pan C, Kump T, Vento S, Trachtman H. Follow-up of phase I trial of adalimumab and rosiglitazone in FSGS: III. Report of the FONT study group. BMC Nephrol. 2010;11:2. doi: 10.1186/1471-2369-11-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Joy MS, Gipson DS, Dike M, Powell L, Thompson A, Vento S, Eddy A, Fogo AB, Kopp JB, Cattran D, Trachtman H. Phase I trial of rosiglitazone in FSGS: I. Report of the FONT Study Group. Clin J Am Soc Nephrol. 2009;4:39–47. doi: 10.2215/CJN.02310508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Savin VJ, McCarthy ET, Sharma R, Charba D, Sharma M. Galactose binds to focal segmental glomerulosclerosis permeability factor and inhibits its activity. Transl Res. 2008;151:288–92. doi: 10.1016/j.trsl.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 104.De Smet E, Rioux JP, Ammann H, Déziel C, Quérin S. FSGS permeability factor-associated nephrotic syndrome: remission after oral galactose therapy. Nephrol Dial Transplant. 2009;24:2938–40. doi: 10.1093/ndt/gfp278. [DOI] [PubMed] [Google Scholar]
- 105.Lavin P, Gbadegesin R, Damodaran TV, Winn MP. Therapeutic targets in focal and segmental glomerulosclerosis. Curr Opin Nephrol Hypertens. 2008;17:386–392. doi: 10.1097/MNH.0b013e32830464f4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Garg P, Verma R, Nihalani D, Johnstone DB, Holzman LB. Neph1 cooperates with nephrin to transduce a signal that induces actin polymerization. Mol Cell Biol. 2007;27:8698–8712. doi: 10.1128/MCB.00948-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Galeano B, Klootwijk R, Manoli I, Sun M, Ciccone C, Darvish D, Starost MF, Zerfas PM, Hoffmann VJ, Hoogstraten-Miller S, Krasnewich DM, Gahl WA, Huizing M. Mutation in the key enzyme of sialic acid biosynthesis causes severe glomerular proteinuria and is rescued by N-acetylmannosamine. J Clin Invest. 2007;117:1585–1594. doi: 10.1172/JCI30954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Vogelmann SU, Nelson WJ, Myers BD, Lemley KV. Urinary excretion of viable podocytes in health and renal disease. Am J Physiol Renal Physiol 2003. 2003;285:F40–8. doi: 10.1152/ajprenal.00404.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Prodromidi EI, Poulsom R, Jeffery R, Roufosse CA, Pollard PJ, Pusey CD, Cook HT. Bone marrow-derived cells contribute to podocyte regeneration and amelioration of renal disease in a mouse model of Alport syndrome. Stem Cells. 2006;24:2448–55. doi: 10.1634/stemcells.2006-0201. [DOI] [PubMed] [Google Scholar]
- 110.Ronconi E, Sagrinati C, Angelotti ML, Lazzeri E, Mazzinghi B, Ballerini L, Parente E, Becherucci F, Gacci M, Carini M, Maggi E, Serio M, Vannelli GB, Lasagni L, Romagnani S, Romagnani P. Regeneration of glomerular podocytes by human renal progenitors. J Am Soc Nephrol. 2009;20:322–32. doi: 10.1681/ASN.2008070709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Appel D, Kershaw DB, Smeets B, Yuan G, Fuss A, Frye B, Elger M, Kriz W, Floege J, Moeller MJ. Recruitment of podocytes from glomerular parietal epithelial cells. J Am Soc Nephrol. 2009;20:333–43. doi: 10.1681/ASN.2008070795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Little MH, Bertram JF. Is there such a thing as a renal stem cell? J Am Soc Nephrol. 2009;20:2112–7. doi: 10.1681/ASN.2009010066. [DOI] [PubMed] [Google Scholar]
- 113.Le Berre L, Bruneau S, Naulet J, Renaudin K, Buzelin F, Usal C, Smit H, Condamine T, Soulillou JP, Dantal J. Induction of T regulatory cells attenuates idiopathic nephrotic syndrome. J Am Soc Nephro. 2009;20:57–67. doi: 10.1681/ASN.2007111244. [DOI] [PMC free article] [PubMed] [Google Scholar]